

1 Introduction
A review of the literature showed that some triterpenes and their derivatives (Fig. 1) have been associated with various pharmacological activities. They have shown good anti-inflammatory [1–2], antitumoral [3], anticancer [4], hypoglycaemic [5], antiviral [6–8], antibacterial [9–12] and antifungal [13–14] activities. These properties as well as the important amount of oleanolic acid 1, isolated in our laboratory from the fruit barks of Periploca laevigata (Asclepiadaceae) [15], motivated us to prepare some derivatives (oxidation derivatives, phosphorized derivatives, esters and amides) in order to study their antibacterial activities and structure–activity relationships.

Structures of the natural triterpenic compounds 1, 2 and 3 and of the acetylated derivatives 1a and 3a.
2 Results and discussion
2.1 Chemistry
The treatment of oleanolic acid 1 with acetic anhydride in pyridine gives the acetyloleanolic acid 1a in 98% yield. Its IR spectrum showed absorptions attributable to acetyloxy (1734 cm–1) and carboxylic (1711 cm–1) carbonyl functions. Its structure was confirmed by the appearance of signals at 2.06 and 4.42 ppm attributable to CH3 of the acetyloxy group and H3, respectively, in the 1H-NMR spectrum. The 13C-NMR spectrum showed signals at 80.9 ppm and 171.0 ppm corresponding to C3 and the carbonyl of the acetyloxy group, respectively. 1a has been used as a starting material for the preparation of most derivatives. The specific epoxidation of Δ12–13 using a mixture KMnO4–CuSO4 in refluxing CH2Cl2 for 4 h, in the presence of a small amount of water and tertiobutanol, did not give the desired product (3-acetyl-12,13-epoxioleanolic acid), but provided the 12α-hydroxy-δ-lactone 4 in 82% yield along with an other secondary product 12-oxo-δ-lactone 5 in 11% yield (Fig. 2). We think that the approach and the stereochemistry of the carboxy group at C17 in 1a allowed the lactonisation via the epoxidation of the C12–C13 double bound. The same reaction, carried out again under the same conditions but for 12 h, gave besides 4 and 5 another secondary product, the 3-acetyl-11-oxo-oleanoic acid 6 in 5%, 60% and 27% yield, respectively (Fig. 3).
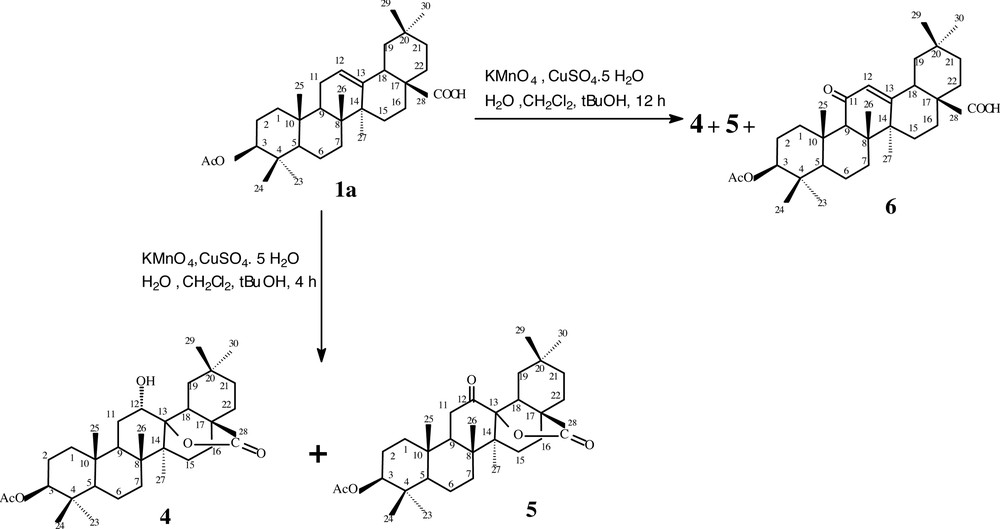
Structures of the prepared oxidizing derivatives 4, 5 and 6.

Structures of the prepared phosphorus derivatives 7 and 8.
The compound 4 was obtained as a white powder. The ESI+ mass spectrum showed ion peak at m/z 515 [M + H]+. The IR spectrum revealed absorptions attributable to a free hydroxyl group (3529 cm–1), δ-lactonic and acetyloxy (1733 cm–1) carbonyl functions. The structure was evidenced by the disappearance in the 1H-NMR spectrum of the signal at 5.19 ppm relative to the ethylenic proton H12 in 1a and the appearance of a signal at 3.80 ppm attributable to the same proton H12, but attached to a carbon-bearing hydroxyl group. The 13C-NMR spectrum reinforced this structure by the disappearance of the two ethylenic carbon signals C12 and C13 in 1a and the appearance of a signal at 90.5 ppm attributed to an oxycarbonyl-lacton-group-bearing quaternary carbon atom, as well as signals at 76.1 and 179.8 ppm attributable to a tertiary carbon bearing a hydroxyl group and a lactone carbonyl function, respectively. The position of the hydroxyl group at C12 was deduced from the COSY spectrum showing H12–H11a and H12–H11b correlations. The HMBC spectrum confirmed the location of this hydroxyl group in C12 by observation of the 2J heteronuclear correlation between H12 and the quaternary carbon C13. The relative stereochemistry of C12 was determined on the basis of the NOESY spectrum, which proved the α-orientation of the hydroxyl group at C12 by the absence of the dipolar coupling between H12 and the β-oriented methyl group CH3-27. This result was also confirmed by the multiplicity (triplet-like) and the weak coupling constant J12–13 deduced from the 1D TOCSY NMR spectrum.
Compound 5 was obtained as a white powder. The ESI+ mass spectrum exhibited a protoned [M + H]+ and a sodiated [M + Na]+ molecular ion at m/z 513 and 535, respectively. The [2 M + Na]+ at m/z 1048 was also observed in the same spectrum. All these data are compatible with the molecular formula C32H48O5 (M = 512). The IR spectrum of 5 showed absorptions attributable to ketonic (1724 cm–1), δ-lactonic and acetyloxy (1734 cm–1) carbonyl functions. On the other hand, the structure of 5 was confirmed by the disappearance in the 1H-NMR spectrum of the signal at 5.19 ppm relative to the ethylenic proton H12 in 1a and the appearance of two signals at 2.31 ppm and 2.48 ppm, attributable to H11 protons. The 13C-NMR spectrum reinforced this structure by the disappearance of the two ethylenic carbon signals C12 and C13 in 1a, and the appearance of signals at 178.9 ppm corresponding to a lactonic carbonyl function, and at 206.4 ppm relative to the ketonic carbonyl function.
The compound 6 was obtained as a white powder. Its ESI+ mass spectrum was recorded and showed a protoned molecular ion [M + H]+ at m/z 513. The IR spectrum exhibited absorptions attributable to an ethylenic system (1667 cm–1), ketonic (1684 cm–1), carboxylic (1708 cm–1) and acetyloxy (1736 cm–1) carbonyl functions. The α,β-unsaturated ketonic moiety was immediately recognized from the following 1H and 13C-NMR data H12(5.56; s), H18(2.86; m), H9(2.30; s), C-11 (200.4), C-28 (182.5), C-13 (163.2) and C-12 (131.2).
Mechanistically, the oxidation of C11 could be initiated by potassium permanganate, which abstracts a hydrogen atom from the activated carbon. The formed radical continues to react with O2 to give a peroxide, which is converted into a ketonic function at C11 with the formation of a water molecule [16].
Acetyl oleanoic acid chloride previously prepared, readily reacts with triethylphosphate to give the Michaelis–Arbusov product: diethyl 3-acetylolean-12-en-28-oxyphosphonate 7 in 93% yield (Fig. 3). It was obtained as a white powder. Its ESI+ mass spectrum showed a protoned [M + H]+ and a sodiated [M + Na]+ molecular ion at m/z 619 and 641, respectively. The IR spectrum of 7 revealed absorptions attributable to ethylenic (1674 cm–1), ketonic (1718 cm–1), acetyloxy (1728 cm–1), P=O (1240 cm–1) and P–O–C (1020 cm–1) systems. The presence of the OCPO(OEt)2 system in compound 7 was ascertained by the observation of a new multiplet (q-like) at 4.20 ppm due to methylenic protons of the two ethyloxy moiety fixed at the phosphorus atom as well as new signals in the high-field region of the 1H-NMR spectrum relative to the methyl groups of the same system. Furthermore, the presence of the ketonic carbonyl function in compound 7 was deduced from its 13C–NMR spectrum (C28: 214.4 ppm). The 31P–NMR spectrum revealed two signals at 0.27 and –0.01 ppm, showing the presence of two conformers anti and syn at the COPO(OEt)2 moiety. The hydrolysis of the compound 7 was carried out with aqueous HCl and gave the diethyl 3-hydroxyolean-28-oxyphosphonate 8 in 92% yield (Fig. 3). The structure elucidation of 8 was made by comparison of its 1H and 13C-NMR spectra with those of compound 7. This comparison let us note the disappearance of the H-3 signal at 4.51 ppm in 7 and the appearance of a new signal at 3.15 ppm attributable to the same proton in 8. Moreover, we have noted the absence of the singlet at 2.05 ppm relative to the methyl group of the acetyloxy system in compound 7. The absence in the compound 8 13C-NMR spectrum of the ester carbonyl signal at 170.9 ppm (present in the case of 7) proved the hydrolysis. This result was reinforced by the absorption revealed from the IR spectrum of 8 at 3420 cm–1 relative to the free hydroxyl group. The 31P-NMR spectrum of 8 showed signals at 0.26 and –0.01 ppm showing the presence, as in the case of 7, of two conformers anti and syn at the COPO(OEt)2 moiety. The absorptions relative to P=O (1242 cm–1) and P–O–C (1012 cm–1) groups deduced from the IR spectrum of 8 confirmed the presence of the phosphorus moiety in the molecule.
The three esters 9, 10 and 11 were prepared by reaction of oleanolic acid 1 with benzoylchloride, 2-hydroxybenzoylchloride and 2-acetyloxybenzoylchloride in pyridine, in 32%, 19% and 15% yield, respectively (Fig. 4).
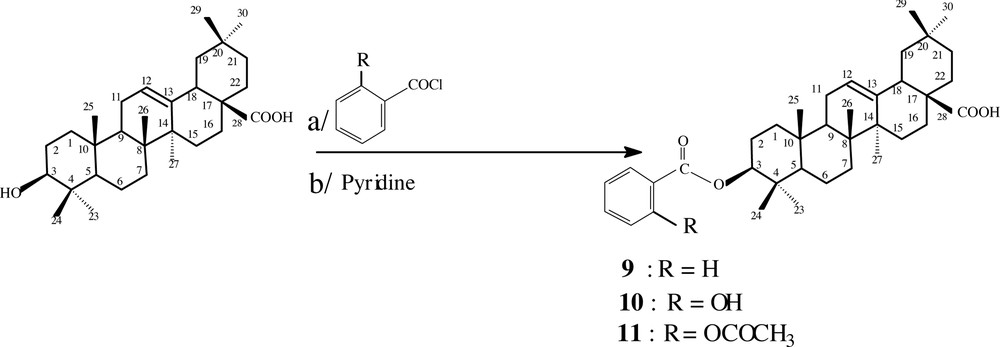
Structures of the prepared esters 9, 10 and 11.
The compound 9 was obtained as a white crystalline powder. Its EI mass spectrum showed a molecular ion peak at m/z 560 M+•. The 1H-NMR spectrum showed a new triplet at 4.69 ppm (J = 6.7 Hz) attributable to H-3 and new other signals at 7.40– 8.00 ppm corresponding to the aromatic protons. The 13C-NMR spectrum of 9 showed a signal at 81.4 ppm relative to C3, signals at 128.2, 129.4, 130.8 and 132.6 ppm attributable to the aromatic carbons. The same spectrum showed signals at 122.4 and 143.4 ppm corresponding to C12 and C13, respectively. The signals at 166.1 and 183.3 ppm attributable to the ester and carboxylic acid carbonyl functions, respectively, were also observed.
The compound 10 was obtained as a white crystalline powder. Its EI mass spectrum showed an ion peak at m/z 439 [M–C7H5O3]+ and 438 [M–C7H6O3]+• relative to the loss of the benzoyloxy moiety fixed at C-3. The 1H-NMR spectrum exhibited, in particular, signals at 6.80–8.20 ppm attributable to the aromatic protons. It showed also a multiplet at 4.17 ppm relative to H-3.
Compound 11 was obtained as a white crystalline powder. The recorded APCI+ mass spectrum revealed ion peak at m/z 439 [M–C9H7O4]+ corresponding to the loss of the aryloxy system attached to C-3. The 1H-NMR spectrum showed a singlet at 2.07 ppm attributable to the methyl of the acetyloxy group fixed to C-2’ of the aromatic ring. The multiplet observed in the same spectrum at 4.54 ppm was attributed to H-3. The aromatic protons were also identified by the observation of the corresponding signals at 7.00-8.10 ppm.
The reaction of oleanolic acid chloride in refluxing chloroform with 3-aminopyridine, heptylamine, hexylamine, 1-methylpropylamine and decylamine afforded the corresponding N-3-pyridinacetyloleanolic amide 12, N-heptylacetyloleanolic amide 13, N-hexyacetyl-oleanolic amide 14, N-l-methylpropylacetyloleanolic amide 15 and N-decylacetyloleanolic amide 16, in 65%, 74%, 80,%85% and 84% yield, respectively (Fig. 5).
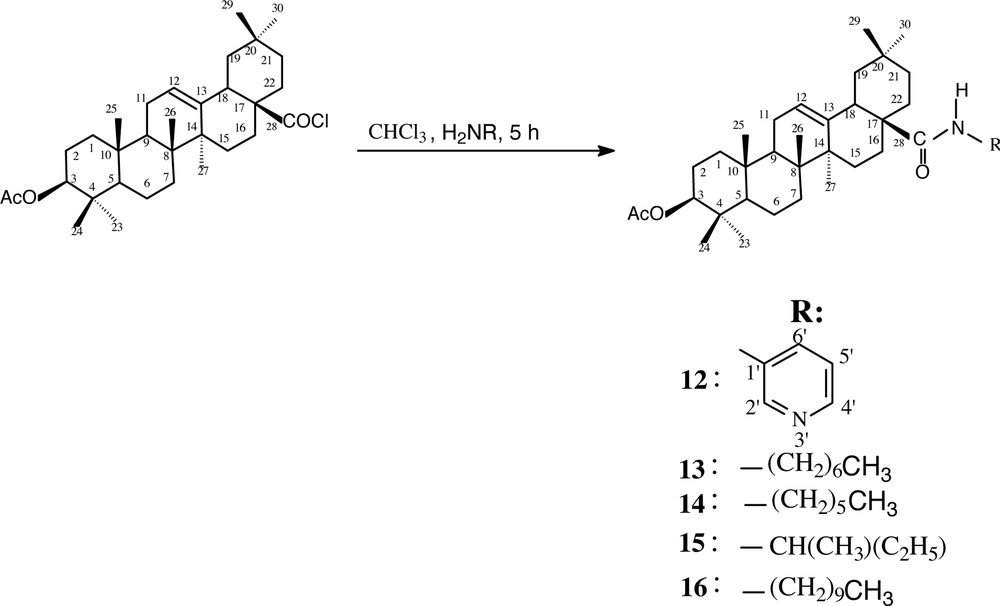
Structures of the prepared amides 12, 13, 14, 15 and 16.
Compound 12 was obtained as a white powder. Its FABMS exhibited [M + H]+ at m/z 575. Its IR spectrum showed absorptions attributable to ketonic (1690 cm–1), acetyloxy (1734 cm–1) carbonyl functions, and C=N aromatic double bounds (1518 cm–1). The 1H–NMR spectrum showed a singlet at 7.75 ppm attributable to H-2′ of the pyridinic system. Three multiplets were also observed at 7.16, 8.15 and 8.22 ppm relative to H-5′, H-6′ and H-4′ of the pyridinic moiety, respectively. The 13C–NMR spectrum confirmed the above spectral data by the observation of five signals at 123.7, 127.1, 134.9, 140.1 and 140.9 ppm relative to C-5′, C-2′, C-4′, C-1′ and C-6′ of the pyridinic system, respectively. The same spectrum showed signals at 171.0 and 177.2 ppm attributable to the carbonyls of the acetyloxy group at C-3 and the amide function at C-17, respectively.
The compounds 13, 14, 15 and 16 were obtained as a white powder. Their structures were evidenced by the appearance in the corresponding 1H-NMR spectra signals at 2.80–3.80 ppm relative to –NCH2– and –NCH– protons, at 5.0–6.0 ppm corresponding to HN–CO protons. Moreover, new signals were observed in the high-field region of each spectrum of the above compounds relative to the hydrocarbon chain attached to the corresponding amine. The 13C-NMR spectra of these compounds showed in particular HN–CO signal at 178.1 ppm (13), 178.2 ppm (14), 177.3 ppm (15) and 178.0 ppm (16).
2.2 Antibacterial activity
General analysis of the data from the antibacterial effects of the tested compounds (Table 1) shows that compounds 1, 3, 4 and 5 exhibited interesting activities against Salmonella typhimurium, the most susceptible bacteria, with a minimum bactericidal concentration (MBC) value of 300 μg ml–1 and with minimum inhibitory concentrations (MIC) values of 65, 95, 65 and 97 μg ml–1, respectively. Compounds 1, 1a, 2a, 3, 3a, 5, 6, 8 and 12 showed moderate activities against Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa bacteria, whereas compounds 4, 7, 15 and 16 can be considered inactive against these bacteria.
Minimum inhibitory concentrations (MIC) and minimum bactericidal concentrations (MBC) of triterpenic compounds (expressed as μg ml–1).
| Compounds | Staphylococcus aureus | Escherichia coli | Pseudomonas aeruginosa | Salmonella typhi | ||||
| MBC | MIC | MBC | MIC | MBC | MIC | MBC | MIC | |
| 1 | 700 | 95 | 900 | 95 | — | > 97 | 300 | 65 |
| 1a | 900 | 90 | 900 | 90 | 900 | > 97 | 900 | > 97 |
| 2a | 700 | 90 | 900 | 90 | 900 | 90 | 900 | 90 |
| 3 | — | > 97 | 900 | 97 | 900 | 97 | 300 | 95 |
| 3a | 700 | 90 | 700 | 90 | 500 | 78 | 500 | 95 |
| 4 | — | > 97 | — | > 97 | — | > 97 | 300 | 65 |
| 5 | 900 | 90 | 900 | 90 | — | > 97 | 300 | 97 |
| 6 | 500 | 90 | 900 | 90 | 900 | 97 | 900 | 90 |
| 7 | — | > 97 | — | > 97 | — | > 97 | — | > 97 |
| 8 | 700 | 85 | 900 | 85 | 900 | 90 | 900 | 90 |
| 12 | 900 | 90 | 900 | 90 | 300 | 90 | 700 | 90 |
| 15 | — | > 97 | — | > 97 | — | > 97 | — | > 97 |
| 16 | — | > 97 | — | > 97 | — | > 97 | — | > 97 |
The comparison of the MBC of 1 (300 μg ml–1) with 1a (900 μg ml–1) and that of 3 (300 μg ml–1) with 3a (500 μg ml–1) showed that the free β-hydroxyl group at C3 contributes to the antibacterial activity of 1 and 3. On the other hand, the MBC of 1a (900 μg ml–1) and 3a (500 μg ml–1) let us conclude that the β-oriented carboxyl function at C17 in compound 1 decreases the antibacterial activity, but when it was converted into a C13–C17 lactone function or when the formation of the α-hydroxyl and the ketonic function at C12 in compounds 4 and 5, respectively, occurred, the activity against Salmonella typhimurium was recovered. It is interesting to note the activity observed for N-3-pyridinacetyloleanolic amide 12 against Pseudomonas aeruginosa, one of the most resistant bacteria to antibiotics and antiseptics, with MBC value of 300 μg ml–1 and MIC value of 90 μg ml–1. Comparison of these data with those of the other inactive amides 15 and 16 indicated that the activity is related to the structure of the system attached to the amine.
3 Experimental section
3.1 General experimental procedures
FTIR spectra were measured on a Perkin-Elmer 157G infrared spectrophotometer. Shimadzu QP-1000EX and MS-80RF spectrometers were used in the EI, FAB, ESI+ and APCI+ experiments, respectively. 1H (200, 300 and 400 MHz) and 13C (50, 75 and 100 MHz) one- and two-dimensional NMR spectra were recorded on a Bruker AM-200, AM-300, ARX-400 and WM-400 spectrometers, using CDCl3 and DMSOd6 as solvents and TMS as internal standard. Melting points were determined on a Büchi 510 apparatus using capillary tubes.
3.2 Preparation of compounds 4 and 5
A mixture of KMnO4 (0.8 g) and CuSO4·5 H2O (0.4 g) was ground to fine powder. Water (0.04 ml) was added, and the slightly wet mixture transferred to the reaction flask. To a stirred suspension of this mixture in CH2Cl2 (2 ml), 200 mg of the acetylated oleanolic acid (0.4 mmol) are added, followed by the addition of tert-butanol (0.2 ml) [17]. After stirring for 4 h, the resulting solution was filtered, dried over anhydrous Na2SO4 and concentrated in vacuo. The resulting residue was separated by silica gel column chromatography (petroleum ether: EtOAc 9:1 then 8:2) to give 4 (0.166 g, 82%) and 5 (0.022 g, 11%).
3.3 Preparation of compound 6
3-acetyl-11-oxo-oleanoic acid 6 was prepared as indicated above (see compounds 4 and 5). In this case, the stirring time of the reaction mixture was extended to 12 h. The separation of the resulting residue by column chromatography on silica gel (petroleum ether: EtOAc 9:1 then 8:2) gave 4 (0.01 g, 5%), 5 (0.123 g, 60%) and 6 (0.055 g, 27%).
3.4 Preparation of compound 7
To a solution of acetyloleanolic acid chloride (0.130 g, 0.252 mmol), previously prepared by the general procedure [18], in toluene (2.5 ml) was added 2 equiv of triethyphosphate (0.09 ml, 0.504 mmol) and the mixture was stirred at 80–90 °C for 1 h [19]. After removal of the solvent under reduced pressure, the residue was purified by silica gel column chromatography (petroleum ether: EtOAc 7.5: 2.5) to afford 7 (0.145 g, 93%).
3.5 Preparation of compound 8
To a solution of 7 (0.07 g, 1.2 mmol) in (CH3)2CO–H2O 1:1, was added 1 ml of aqueous HCl (3 M). The resulting mixture was stirred at 40 °C for 8 h. After removal of (CH3)2CO in vacuo, the reaction mixture was extracted twice with CHCl3. The organic layer was washed with an excess of 10% NaHCO3 then with water and dried over anhydrous Na2SO4. The resulting product precipitated from MeOH to give 8 (0.064 g, 92%).
3.6 Preparation of esters 9, 10 and 11
The acid chlorides were prepared according to the general procedure [18], using excess of thionylchloride in refluxing dry pyridine.
To a mixture of oleanolic acid (0.05 g, 0.11 mmol) in refluxing pyridine, the appropriate acid chloride (1 equiv) was added and the mixture was refluxed for 4 h. The solvent was then removed under reduced pressure. The resulting mixture was washed with water to remove salts, then extracted with CHCl3. The organic layer was dried over Na2SO4. CHCl3 was removed in vacuo and the resulting product precipitated from EtOH to give the corresponding esters: 9 (0.02 g, 32%), 10 (0.012 g, 19%) and 11 (0.01 g, 15%).
3.7 Preparation of amides 12, 13, 14, 15 and 16
A mixture of acetyloleanolic acid chloride and the appropriate amine (2.5 equiv mol) in dry CHCl3 (4 ml) was refluxed for 5 h. The reaction mixture was diluted with ice and water, then extracted with CHCl3. The organic layer was washed with water, dried over anhydrous Na2SO4 and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether: EtOAc 9:1) to afford the corresponding amides 12 (0.09 g, 65%), 13 (0.106 g, 74%), 14 (0.121 g, 80%), 15 (0.124 g, 85%) and 16 (0.061 g, 84%).
3.7.1 Oleanolic acid 1
White powder; mp 310 °C; [α]20D +60.0 (c = 0.01, CHCl3); IR (cm–1); νO–H = 3500, νCH3 = 2950, νCH2 = 2850, νC=O = 1715; EIMS m/z (relative intensity) 456 (M+•) (5), 412 (3), 248 (100), 203 (50), 167 (25), 44 (51). 1H–NMR (DMSOd6, 250 MHz) δ 0.75 (3H, s, CH3-26), 0.87 (3H, s, CH3-24), 0.89 (3H, s, CH3-23), 0.91 (3H, s, CH3-25), 0.96 (3H, s, CH3-30), 1.12 (3H, s, CH3-27), 2.87 (1H, m, H-18), 3.23 (1H, m, H-3), 5.28 (1H, m, H-12); 13C-NMR (DMSOd6, 62.5 MHz) δ 15.1 (C-25), 15.9 (C-24), 16.8 (C-26), 18.0 (C-6), 22.6 (C-16), 22.9 (C-11), 23.4 (C-30), 26.9 (C-15), 27.2 (C-2), 28.2 (C-23), 30.4 (C-20), 32.4 (C-21), 32.9 (C-29), 33.4 (C-7), 36.6 (C-10), 38.1 (C-1), 38.4 (C-4), 41.3 (C-14), 45.5 (C-8 and C-17), 47.1 (C-9), 54.9 (C-5), 76.9 (C-3), 121.5 (C-12), 143.7 (C-13), 178.6 (C-28).
3.7.2 Oleanolic acid acetate 1a
White powder; mp 220–222 °C; IR (cm–1); νCH3 = 2924, νCH2 = 2860, νC=O = 1734, νC=O (COOH) = 1711, νC–O–C = 1242; 1H-NMR (CDCl3, 400 MHz) δ 0.67 (3H, s, Me-26), 0.79 (3H, s, Me-29), 0.87 (3H, s, Me-24), 0.88 (3H, s, Me-23), 1.00 (3H, s, Me-30), 1.05 (3H, s, Me-25), 1.18 (3H, s, Me-27), 2.06 (3H, s,OAc-31), 2.78 (1H, m, H-18), 4.42 (1H, m, H-3), 5.19 (1H, m, H-12); 13C-NMR (CDCl3, 100 MHz) δ 15.5 (C-25), 16.7 (C-24), 17.2 (C-26), 18.1 (C-6), 22.8 (COO), 23.4 (C-16), 23.6 (C-30), 24.0 (C-11), 27.7 (C-2), 28.0 (C-15), 29.7 (C-23), 30.7 (C-20), 32.8 (C-7), 33.1 (C-29), 33.8 (C-21), 37.7 (C-10), 38.0 (C-1), 39.0 (C-4), 41.5 (C-14), 46.6 (C-17), 48.0 (C-9), 55.3 (C-5), 80.9 (C-3), 122.5 (C-12), 143.6 (C-13), 171.0 (CH3), 178.6 (C-28).
3.7.3 12-hydroxy-δ-lactone 4
White powder from (EtOAc-petroleum ether); mp = 285–287 °C ; IR (cm–1): νO–H = 3529, νC=O(δ-lactone) and (OAc) = 1733, νC–O–C = 1246; ESI + m/z (relative intensity) 1052 [2 M + Na]+ (13), 537 [M + Na]+ (54), 515 [M + H]+ (100), 469 (58), 455 (22), 437 (19), 409 (13), 301 (8), 247 (14), 220 (11), 205 (22), 189 (24), 135 (16); 1H-NMR (CDCl3, 200 MHz) δ 0.76 (3H, s, CH3-25), 0.79 (3H, s, CH3-23), 0.85 (3H, s, CH3-30), 0.86 (3H, s, CH3-24), 0.93 (3H, s, CH3-29), 1.08 (3H, s, CH3-26), 1.07 (1H, m, H2b), 1.25 (3H, s, CH3-27), 1.45 (1H, m, H11b), 1.63 (1H, m, H2a), 2.02 (3H, s, CH3COO), 2.00 (1H, m, H11a), 2.04 (1H, m, H18), 3.80 (1H, m, H12), 4.41 (1H, m, H3); 13C-NMR (CDC13, 50 MHz) δ 16.2 (C-24), 16.3 (C-25), 17.5 (C-6), 18.4 (C-27 and C-26), 21.0 (C-16), 21.2 (COO), 23.4 (C-2), 23.7 (C-30), 27.3 (C-22), 27.8 (C-23), 27.9 (C-15), 28.7 (C-11), 29.5 (C-20), 33.1 (C-29), 33.7 (C-7), 34.0 (C-21), 36.2 (C-10), 37.6 (C-1), 39.2 (C-4), 41.9 (C-14), 42.1 (C-8), 44.3 (C-9), 44.5 (C-17), 50.9 (C-18), 55.1 (C-5), 76.1 (C-12), 80.6 (C-3), 90.5 (C-13), 170.9 (OCH3), 179.8 (C-28).
3.7.4 12-oxo-δ-lactone 5
White powder from (EtOAc-petroleum ether); mp = 288 °C; IR (cm–1): νCH3 = 2932, νCH2 = 2868, νC=O(δ-lactone) and (OAc) = 1734, νC=O(ketone) = 1724, νC–O–C = 1242; ESI + m/z (relative intensity) 1048 [2M + Na]+ (23), 535 [M + Na]+ (100), 513 [M + H]+ (36), 398 (18), 301 (18), 279 (12), 220 (12), 150 (24); 1H-NMR (CDCl3, 300 MHz) δ 0.60 (9H, s, CH3-25, CH3-24 and CH3-23), 0.61 (3H, s, CH3-29), 0.70 (3H, s, CH3-26), 0.75 (3H, s, CH3-30), 1.12 (3H, s, CH3-27), 1.83 (3H, s, CH3COO), 2.14 (1H, dd, J1 = 14.4 Hz and J2 = 3 Hz, H18), 2.31 (1H, t-like, H11a), 2.48 (1H, t-like, H11b), 4.25 (1H, m, H3); 13C-NMR (CDC13, 75 MHz) δ 51.4 (C-9), 55.4 (C-5), 80.6 (C-3), 91.4 (C-13), 171.4 (OCH3), 178.9 (C-28), 206.4 (C-12).
3.7.5 Acetyl-11-oxooleanolic acid 6
White powder from (EtOAc-petroleum ether); mp = 335 °C ; IR (cm–1): νCH3 = 2938, νCH2 = 2862, νC=O(OAc) = 1736, νC=O(COOH) = 1708, νC=O(ketone) = 1684, νC=C = 1667, νC–O–C = 1234; ESI + m/z (relative intensity) 513 [M + H]+ (10), 512 (M)+• (35), 511 [M–H]+ (100), 467 (6), 424 (4), 351 (34), 301 (6), 270 (6), 250 (4), 239 (6), 185 (67), 169 (6), 135 (12), 127 (20), 119 (9), 111 (12); 1H-NMR (CDCl3, 300 MHz) δ 0.70 (6H, s, CH3-24 and CH-25), 0.80 (3H, s, CH3-23), 0.86 (3H, s, CH3-29), 0.95 (3H, s, CH3-30), 1.15 (3H, s, CH3-27), 2.05 (3H, s, CH3COO), 2.15 (1H, m, H19b), 2.30 (1H, s, H9), 2.38 (1H, m, H19a), 2.86 (1H, m, H18), 4.51 (1H, dd, J1 = 11.4 Hz and J2 = 5.6 Hz, H3), 5.56 (1H, s, H12); 13C-NMR (CDC13, 75 MHz) δ 200.0 (C-11), 182.5 (C-28), 171.4 (OCH3), 163.2 (C-13), 131.2 (C-12), 81.0 (C-3).
3.7.6 Compound 7
White powder from (EtOAc-petroleum ether ); mp = 124–128 °C; IR (cm–1): νCH3 = 2926, νCH2 = 2856, νC=O(OAc) = 1728, νC=O = 1718, νC=C = 1674, νP=O = 1240, νP–O–C = 1020; ESI + m/z (relative intensity) 641 [M + Na]+ (52), 619 [M + H]+ (100), 559 [MH+–(CH3 and OCH2CH3)] (44); 1H–NMR (CDCl3, 400 MHz) δ 0.75 (3H, s, CH3-25), 0.87 (3H, s, CH3-24), 0.88 (3H, s, CH3-23), 0.92 (3H, s, CH3-29), 0.94 (3H, s, CH3-26), 0.95 (3H, s, CH3-30), 1.15 (3H, s, CH3-27), 1.36 (6H, m, H2′), 2.05 (3H, s, CH3COO), 3.05 (1H, m, H18), 4.20 (4H, q-like, H1’), 4.51 (1H, m, H3), 5.35 (1H, m, H12); 13C-NMR (CDC13, 75 MHz) δ. 15.4 (C-26), 16.3 (C-2’), 16.4 (C-24), 16.9 (C-25), 18.1 (C-6), 21.2 (C-16 and COO), 23.4 (C-2), 23.5 (C-30), 23.5 (C-11), 25.6 (C-27), 28.0 (C-23), 32.6 (C-21 and C-29), 37.6 (C-10), 41.7 (C-8), 47.4 (C-9), 55.2 (C-5), 63.3 (C-1’), 80.8 (C-3), 122.6 (C-12), 143.2 (C-13), 170.9 (OCH3), 215 and 214.4 (C-28); 31P-NMR (CDCl3, 121.48 MHz) δ (0.27, 75.9%) and (–0.01, 24.1%).
3.7.7 Compound 8
White powder from (EtOAc-petroleum ether); mp = 153 °C; IR (cm–1): νO–H = 3420, νCH3 = 2918, νCH2 = 2860, νC=O = 1716, νP=O = 1242, νP–O–C = 1012; FABMS m/z (relative intensity) [MH]+ 577 (4), 559 (3), 411 (100), 393 (21), 269 (5), 255 (11), 241 (12), 215 (26), 203 (49), 189 (59), 187 (44); 1H-NMR (CDCl3, 300 MHz) δ 0.62 (3H, s, CH-25), 0.68 (3H, s, CH3-24), 0.71 (3H, s, CH3-23), 0.82 (3H, s, CH3-29), 0.85 (3H, s, CH3-26), 0.90 (3H, s, CH3-30), 0.95 (3H, s, CH3-27), 1.31 (6H, m, H2’), 2.97 (1H, m, H18), 3.15 (1H, m, H3), 4.15 (4H, q-like, H1’), 5.27 (1H, m, H12); 13C-NMR (CDC13, 75 MHz) δ 63.8 (C-1’), 79.3 (C-3), 123.0 (C-12), 143.6 (C-13), 215.0 (C-28); 31P-NMR (CDCl3, 121.48 MHz) δ (0.26, 75%) and (–0.01, 25%).
3.7.8 Product 9
White crystalline powder from (EtOAc-petroleum ether); EI m/z (relative intensity) 560 (M+•) (6), 514 [M–HCOOH]+• (21), 438 [M-C7H6O2]+• (6), 248 (100), 203 (43), 190 (28), 105 [C7H5O]+• (33); 1H-NMR (CDCl3, 400 MHz) δ 0.70 (3H, s, CH3-25), 0.85 (3H, s, CH3-30), 0.87 (3H, s, CH3-23), 0.89 (3H, s, CH3-29), 1.02 (3H, s, CH3-26), 1.04 (3H, s, CH3-24), 1.17 (3H, s, CH3-27), 2.76 (1H, m, H18), 4.69 (1H, m, H3), 5.23 (1H, m, H12), 7.40 (2H, m, H4’ and H6’), 7.49 (1H, m, H5’), 8.00 (2H, m, H3’ and H7’); 13C–NMR (CDC13, 100 MHz) δ 55.2 (C-5), 81.4 (C-3), 122.4 (C-12), 128.2 (C-4’ and C-6’), 129.4 (C-3’ and C-7’), 130.8 (C-2’), 132.6 (C-5’), 143.4 (C-13), 166.1 (C-1’), 183.3 (C-28).
3.7.9 Product 10
White crystalline powder from (EtOAc-petroleum ether); EI m/z (relative intensity) 439 [M–C7H5O3]+ (8), 438 [M-C7H6O3]+• (7), 248 (100), 203 (53), 121 [C7H5O2]+• (67); 1H-NMR (CDCl3, 200 MHz) δ 2.78 (1H, m, H18), 4.17 (1H, m, H3), 5.21(1H, m, H12), 6.80–8.20 (4H, m, 4Harom).
3.7.10 Product 11
White crystalline powder from (EtOAc–petroleum ether); APCI + m/z (relative intensity) 439 [M-C9H7O4]+ (100), 440 [M–C9H7O4+H]+ (28), 281 (25), 149 (92); 1H-NMR (CDCl3, 400 MHz) δ 0.77 (3H, s, CH3-25), 0.85 (6H, s, CH3-30 and CH3-23), 0.94 (3H, s, CH3-29), 0.96 (3H, s, CH3-26), 1.15 (3H, s, CH3-24), 1.26 (3H, s, CH3-27), 2.07 (3H, s, CH3COO), 2.86 (1H, m, H18), 4.54 (1H, m, H3), 5.33 (1H, m, H12), 7.00–8.10 (4H, m, 4Harom.).
3.7.11 Compound 12
White powder from (EtOAc-petroleum ether); mp = 122 °C ; IR (cm–1): νCH3 = 2922, νCH2 = 2860, νC=O(OAc) = 1734, νC=O(ketone) = 1690, νC=C arom = 1634, νC=N arom = 1518, νC–O–C = 1242; FABMS m/z (relative intensity) 575 [MH]+ (79), 562 (68), 515 (12), 484 (14), 470 (12), 396 (15), 394 (14), 350 (71), 313 (24), 279 (23), 236 (100); 1H-NMR (CDCl3, 200 MHz) δ 0.60 (3H, s, CH3-25), 0.74 (3H, s, CH3-24), 0.76 (3H, s, CH3-23), 0.81 (3H, s, CH3-29), 0.85 (3H, s, CH3-26), 0.89 (3H, s, CH3-30), 1.11 (3H, s, CH3-27), 1.97 (3H, s, CH3COO), 2.62 (1H, m, H18), 4.41 (1H, m, H3), 5.49 (1H, m, H12), 7.16 (1H, m, H5’), 7.75 (1H, s, H2’), 8.15 (1H, m, H6’), 8.22 (1H, m, H4’), 8.32 (1H, m, NCO); 13C–NMR (CDC13, 50 MHz) δ 55.2 (C-5), 80.8 (C-3), 123.6 (C-12), 123.7 (C-5’), 127.1 (C-2’), 134.9 (C-4’), 140.1 (C-1’), 140.9 (C-6’), 144.9 (C-13), 171.0 (OCH3), 177.2 (CONH).
3.7.12 Compound 13
White powder from (EtOAc-petroleum ether); 1H-NMR (CDCl3, 200 MHz) δ 0.70 (3H, s, CH3-25), 0.79 (3H, s, CH3-24), 0.83 (3H, s, CH3-23), 0.87 (3H, s, CH3-29), 1.01 (3H, s, CH3-26), 1.08 (3H, s, CH3-30), 1.20 (3H, s, CH3-27), 2.01 (3H, s, CH3COO), 2.42 (1H, m, H18), 2.96 (1H, m, Hl’b), 3.26 (1H, m, Hl’a), 4.42 (1H, m, H3), 5.25 (1H, m, H12), 5.83 (1H, m, NCO); 13C-NMR (CDC13, 50 MHz) δ 55.2 (C-5), 80.8 (C-3), 122.6 (C-12), 145.2 (C-13), 171.0 (OCH3), 178.1 (CONH).
3.7.13 Compound 14
White powder from (EtOAc-petroleum ether); 1H-NMR (CDCl3, 200 MHz) δ 0.69 (3H, s, CH3-25), 0.78 (3H, s, CH3-24), 0.82 (3H, s, CH3-23), 0.86 (3H, s, CH3-29), 1.01 (3H, s, CH3-26), 1.04 (3H, s, CH3-30), 1.20 (3H, s, CH3-27), 1.98 (3H, s, CH3COO), 2.70 (1H, m, H18), 2.92 (1H, m, Hl’b), 3.28 (1H, m, H1’a), 4.40 (1H, m, H3), 5.19 (1H, m, H12), 5.86 (1H, m, HNCO); 13C-NMR (CDC13, 50 MHz) δ 55.3 (C-5), 80.9 (C-3), 122.5 (C-12), 145.2 (C-13), 171.0 (OCH3), 178.2 (CONH).
3.7.14 Compound 15
White powder from (EtOAc-petroleum ether); 1H-NMR (CDCl3, 200 MHz) δ 0.72 (3H, s, CH3-25), 0.76 (3H, s, CH3-24), 0.80 (3H, s, CH3-23), 0.84 (3H, s, CH3-29), 0.95 (3H, s, CH3-26), 1.05 (3H, s, CH3-30), 1.18 (3H, s, CH3-27), 2.02 (3H, s, CH3COO), 2.50 (1H, m, H18), 3.76 (1H, m, Hl’), 4.39 (1H, m, H3), 5.20 (1H, m, H12), 5.52 (1H, m, HNCO); 13C-NMR (CDC13, 50 MHz) δ 55.2 (C-5), 80.8 (C-3), 122.4 (C-12), 144.8 (C-13), 171.0 (OCH3), 177.3 (CONH).
3.7.15 Compound 16
White powder from (EtOAc-petroleum ether); 1H-NMR (CDCl3, 300 MHz) δ 0.77 (3H, s, CH3-25), 0.85 (3H, s, CH3-24), 0.87 (3H, s, CH3-23), 0.89 (3H, s, CH3-29), 0.94 (3H, s, CH3-26), 1.15 (3H, s, CH3-30), 1.25 (3H, s, CH3-27), 2.01 (3H, s, CH3COO), 2.27 (1H, m, H18), 3.00 (1H, m, Hl’b), 3.34 (1H, m, Hl’a), 4.42 (1H, m, H3), 5.38 (1H, m, H12), 5.90 (1H, m, HNCO); 13C-NMR (CDC13, 75 MHz) δ 55.2 (C-5), 80.8 (C-3), 122.5 (C-12), 145.2 (C-13), 171.0 (OCH3), 178.0 (CONH).
3.8 Antibacterial activity
3.8.1 Microbial cultures growth conditions
Test microorganisms included the following Gram negative bacteria such as Escherichia coli (ATCC 25922), Pseudomonas aeruginosa (ATCC 27853), Salmonella typhimurium and Gram-positive strain Staphylococcus aureus (ATCC 25923). Cultures of these bacteria were done on Nutrient Mueller–Hinton medium produced by ‘Institut Pasteur (Tunis)’ and were incubated at 37 °C.
3.8.2 Experimental
All tested compounds are not water-soluble. To overcome this problem, we have included an emulsifier, Tween 80 at the concentration of 5% (v/v) [20].
The compounds were dissolved in sterile physiological water with 5% Tween 80 and were tested for antimicrobial activity using the microdilution method on liquid media to determine the minimal inhibitory concentration by using the following concentrations mentioned in Table 1. The MIC was considered as the lowest concentration of the sample that prevented visible growth.
The test of antibacterial activity is done by using the decimal dilution method in both Mueller–Hinton media. The minimal bactericidal concentrations were determined by streaking the Mueller–Hinton nutrients that were incubated for 24 h at 37 °C, in the nutritive agar plates. The MBC was defined as the lowest concentration of the tested compound at which 99.99% of bacteria have been killed.
Acknowledgements
The authors wish to thank Dr Amina Bakhrouf, ‘Département de microbiologie, Faculté de Pharmacie de Monastir’, Tunisia for assistance in antibacterial essays, and Dr Féthia Harzallah-Skhiri (‘École supérieure d’horticulture de Chott Mériem’, Sousse, Tunisia) for the botanical classification of the plant material.