

1 Introduction
The oxidation of alkanes with hydroperoxides in the presence of transition metals is known since the paper by Fenton [1] and occurs, as a rule, via a chain radical mechanism. However, the reactivity of hydroperoxides and metal complexes depends on the nature of the solvent, metal, and ligand environment [2-4]. The use of metals with the d0 nonbonding configuration opens up pathways for polar peroxide activation [2–7].
Vanadium(V) compounds in AcOH have been shown [8–12] to catalyze H2O2 decomposition leading to the formation of singlet dioxygen 3O2 (reactions (1, 2)) and small amounts of ozone (reaction (3)):
| (1) |
| (2) |
| (3) |
Singlet dioxygen is responsible for the oxidation of anthracene and its derivatives to anthraquinones and for the oxidative degradation of olefins to carbonyl compounds [6,7,9,10,13–16]. The scope of reaction mentioned above has been rationalized within speculative Scheme 1, which is in a good agreement with data published in [6,7,9,10,13–16].
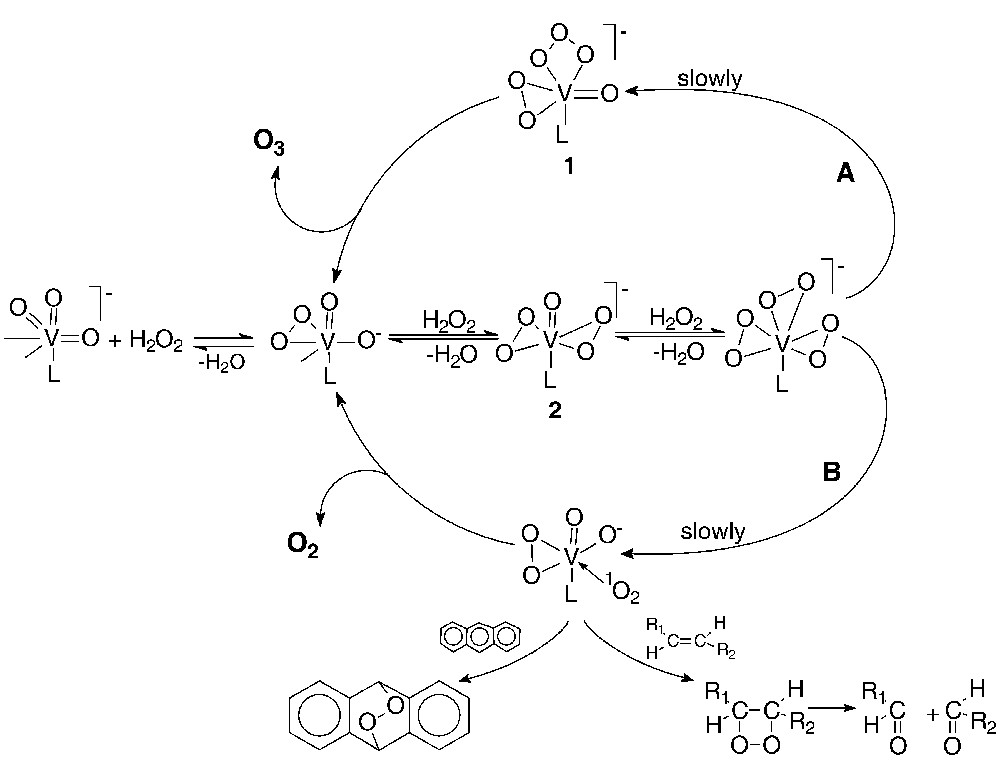
Mechanism of H2O2 decomposition in AcOH solution. Formation of ozone and singlet dioxygen (there is no data on the composition of the coordination sphere. except for kinetic or spectroscopic evidence of O2- and O22- groups; ligand L is assumed to be H2O or AcOH molecules.
Saturated substrates such as cyclohexane and cyclopentane, which are rather inert toward ozone and singlet dioxygen, have been shown to be oxidized, giving rise to the corresponding cycloalkanols and cycloalkanones in the V(V)/H2O2/AcOH system [17], suggesting the involvement of vanadium(V) peroxo complexes in the oxidation reactions.
In this work, we studied the reactions of linear alkanes (C5–C20), isoalkanes – 2,2,4-trimethylpentane (isooctane), 2-methylbutane, 2-methylpentane, 3-methylpentane, 2,3-dimethylbutane, and 2,2-dimethylbutane (neohexane), – in order to elucidate the nature of processes that form the basis for alkane oxidation in the V(V)/H2O2/AcOH system. The active complexes responsible for alkane oxidation have been proved to be capable of molecular nitrogen conversion into N2O.
2 Experimental
2.1 Materials
The following chemicals were used without preliminary purification: glacial acetic acid (chemically pure), hydrogen peroxide (analytical grade, a 16.2 mol l–1 aqueous solution), commercial trifluoroacetic acid, linear alkanes (pentane, hexane, heptane, octane, nonane, dodecane, hexadecane, and eicosane, all of 98% or better purity according to GLC analysis), isooctane (reference, GOST 4374-48), 2-methylpentane, 3-methylpentane, 2,3-dimethylbutane, 2,2-dimethylbutane (98-99%, Aldrich), NH4VO3 (chemically pure), bis(trimethylsilyl)trifluoroacetamide (BSTFA, Merck), methanol (chemically pure), LiAlH4 (pure), BHT (2,6-di-tert-butyl-4-methylphenol, chemically pure), and molecular nitrogen (high-purity grade). Commercial VO(acac)2 was purified by recrystallization from CHCl3.
2.2 Preparation of catalyst solution
Finely ground NH4VO3 (100 mg) was added to AcOH (100 ml) and refluxed for 4–5 h. The undissolved NH4VO3 was filtered off. According to ICP–MS data, the concentration of vanadium(V) in the solution formed was 3.7 × 10–3 mol l–1. The ICP–MS spectra were measured on an HP 4500 ICP–MS instrument (aqueous solution of NH4VO3 as external standard). In some kinetic experiments, VO(acac)2, which is higher soluble in AcOH, was used as catalyst precursor. According to EPR data, the V(IV) signal from VO(acac)2 disappeared immediately after the addition of the first portion of H2O2. The replacement of VO(acac)2 by NH4VO3 did not change the composition and distribution of the reaction products and the reaction rate.
2.3 Experimental procedure for oxidation and analysis of products
The oxidation of linear and branched alkanes was performed in a thermostatic jacketed glass vessel with a magnetic stirrer at 30 °C. In a typical experiment, 16.2 mol l–1 aqueous H2O2 (0.3 ml, 4.86 mmol) was added to a solution of VO(acac)2 (5 mg, 0.019 mmol) and n-hexane (43 mg, 0.5 mmol) in 5 ml of glacial acetic acid. The decomposition of H2O2 was monitored by iodometry. The conversion of substrates and the yields and compositions of the reaction products were determined by GLC and GC–MS. The full list of the oxidation products of branched alkanes detected with GC–MS technique counts more than 60 compounds, including carboxylic acids, diols, and ketoalcohols. To characterize all products and estimate their yields, we shall require all standards. As a consequence, this is an extremely labor-consuming task and a costly job. This has prevented us from getting more information concerning the reaction under the discussion. The discussion of oxidation chemistry of those substrates was restricted by comparison with qualitative data.
To measure the linear substrate conversion, a 0.1-mol l–1 solution of acetonitrile in acetic acid (0.1 ml, internal standard) was added to 0.1 ml of the reaction mixture and the resulting mixture (0.5 μl) was analyzed by GLC.
A sample for the identification of alkane oxidation products was prepared as follows: after the complete consumption of H2O2, CHCl3 or diethyl ether (2 ml) and water (5 ml) were added to 1 ml of the reaction mixture. The organic phase was separated, washed with 5% aqueous Na2CO3 solution (1 ml) and water (1 ml), and underwent to the GLC and/or GC–MS analysis.
In experiments with isooctane, the above sample preparation was changed because the water-soluble components (such as ethanol and 2-propanol) cannot be extracted completely from aqueous solutions by diethyl ether. After the treatment of the oxidation products mixture with diethyl ether, the extragent was removed by slow evaporation at ~20 °C in a nitrogen flow to prevent the evaporation of other volatile components as far as possible.
2-Propanol, 2-methyl-2-propanol, 2-butanol, 2-butanone, 2-methyl-1-propanol, and ethyl acetate were separated from the reaction mixture by using chromatographic columns of different polarity and identified by a comparison of the chromatographic retention times and mass spectra of the sample components with those of standard substances.
A diethyl ether solution of the oxidation products of 3-methylpentane or isooctane was treated with LiAlH4 to reduce ketones and esters to the corresponding alcohols prior to analyses. A comparison of the chromatograms of the initial and reduced samples obtained by using chromatographic columns of different polarity, as well as the mass spectra, was used to elucidate the structures of the sample components. An alpha-Dex 120 enantioselective chromatographic column was used when the retention times and mass spectra of components were similar (e.g., for 2,4,4-trimethyl-1-pentanol and 2,2,4-trimethyl-1-pentanol). The oxidation was found not to be enantioselective. Only 2,4,4-trimethyl-1-pentanol of the above two isomers has an asymmetric center, and the chromatogram of this alcohol exhibits two chromatographic peaks with equal intensities, while the mass spectra of these peaks are identical.
An Automass 150 GC–MS instrument (Delsy Nermag, France) was used for recording mass spectra under the following conditions: EI = 70 eV; the ion source temperature was 100 °C; the mass spectrometer was adjusted using FC43 (reference peaks, m/z: 18, 28, 69, 100, 131, 219, 264, 414, and 502); the spectrum range was 30–300 amu; the scan time was 300 ms; a fused-silica capillary column (0.25 mm × 25 m; df = 0.3 μm) (Rescom) with OV-1, CPWax-58, or alpha-Dex 120 was used; the split ratio was 1/40; Pinj = 0.6 atm (at Tcol = 50 °C); He; Tinj = 250 °C. The Lucy (ver. 2.10) and AMDIS (ver. 2.1) softwares and the NIST mass-spectral database (1987, 1995) were used for the GC–MS data acquisition and processing (see examples in the supplementary material section).
A 3700 chromatograph (Russia) equipped with an HP-1 capillary column (0.25 mm × 25 m; df = 0.3 μm) was used for the quantitative GLC analysis of the initial substances and oxidation products; the split ratio was 1/40; P = 1 atm (at Tinitial = 50 °C); He; Tinitial was varied depending on the substrate; the heating rate was 10 K min–1; Tfinal = 250 °C; FID. An L-241 analog-to-digital converter (20 bit) and the Multichrom 1.39 software (Ampersand Ltd) were used for the chromatographic data acquisition and processing (see Table 1 of the supplementary material section).
Oxidation of n-alkanes in the V(V)/H2O2/AcOH system. 30 °C, [VV] = 4 × 10–3 M, [H2O2]0 = 1 M.
| Nº | Substrate (S) | [S]0 (mol l–1) | Conversion (%) | [Alcohol]summ × 104 (mol l–1) | [Keton]summ × 103 (mol l–1) |
| 1 | n-pentane | 0.1 | 49 | 45a | 40b |
| 2 | n-pentane | 0.1 | 47 | 43 | 38 |
| BHT | 0.01 | ||||
| 3 | n-hexane | 0.1 | 32 | 37c | 25d |
| 4 | n-heptane | 0.1 | 32 | 24e | 18f |
| 5 | n-octane | 0.1 | 30 | 32g | 20h |
| 6 | n-nonane | 0.05 | 31 | 18 | 9k |
| 7 | n-decane | 0.05 | 29 | 22 | 8 |
| 8 | n-tetradecane | 0.05 | 21 | 33 | 6 |
| 9 | n-pentadecane | 0.05 | 16 | 23 | 5 |
| 10 | n-hexadecane | 0.05 | 15 | 18 | 5 |
| 11 | n-nonadecane | 0.05 | 11 | 21 | 3 |
a [2-pentanol]/[3-pentanol]=1.6/1;
b [2-pentanone]/[3-pentanone]=1.9/1;
c [2-hexanol]/[3-hexanol]=0.7/1;
d [2-hexanone]/[3-hexanone]=0.8/1;
e [2-heptanol]/[3-heptanol]/[4-heptanol]=0.8/2/1;
f [2-heptanone]/[3-heptanone]/[4-heptanone]=1.4/1.6/1;
g [2-octanol]/[3-octanol]/[4-octanol]=0.3/1/2;
h [2-octanone]/[3-octanone]/[4-octanone]=0.7/1.2/1;
k [2-nonanone]/[3-nonanone]/([4-nonanone]+[5-nonanone])=1.3/1/1.
The starting isooctane (analytical grade) was analyzed with a 3700 chromatograph (Russia) equipped with an SPB-1 capillary column (0.25 mm × 100 m; df = 0.25 μm) (Supelco); the split ratio was 1/40; P = 0.32 atm; Tinj = 250 °C; and Tcol = 60 °C. According to the GLC data, the purity of isooctane was 99.86 ± 0.04%. The following impurities were identified based on the retention times [18]: 2,3-dimethylpentane (0.06%), 3-ethylpentane (0.02%), and 2,2-dimethylhexane (0.05%). The overall concentration of the other impurities was at most 0.01%.
In N2 oxidation, an N2 flow (30–60 ml min–1) was passed through the reactor containing a solution of H2O2 (1 mol l–1) and VO(acac)2 (10–2 mol l–1) in trifluoroacetic acid at 0–20 °C. The outcoming gas mixture was collected above an aqueous solution of NaCl or NaOH and analyzed by GC–MS (Automass 150 GC–MS; Supel-QPlot column (30 m ( 0.32 mm)) (Supelco); –10 °C; Pinj = 0.5 atm; split ratio 1/40; sample volume 100 µl; EI = 70 eV; mass range 25–100 amu; and scan time 50 ms).
3 Results and discussion
3.1 Oxidation of linear alkanes
Similarly to our earlier experiments with cyclohexane [17], no detectable consumption of the substrates within several hours at room temperature was found when the H2O2/AcOH solutions contained no catalyst. After the addition of the catalyst (~10–2 mol/l), H2O2 consumption began and practically completed (H2O2 exhausted) in ~1 h at 30°C. The amount of the oxidized alkane depended on the initial H2O2 concentration ([H2O2]0) and monotonically increased as this concentration was increased. More than a tenfold excess of H2O2 was required for attaining ~90% conversion of alkane as far as hydrogen peroxide was decomposed in parallel to the oxidation reaction. The major portion of H2O2 was converted into H2O and O2.
The reactivity of n-alkanes slightly decreases with carbon chain length in the order pentane > hexane > decane > dodecane.
The secondary alcohols and ketones with the carbon chain lengths equal to the chain length of the starting hydrocarbon were identified as linear hydrocarbon oxidation products (see Table 1). The oxidation of linear alkanes involves the Csec–H bonds only. Neither the oxidative degradation of the substrate nor the oxidation of the terminal methyl groups was observed. For instance, 2-heptanol, 3-heptanol, 4-heptanol, 2-heptanone, 3-heptanone, and 4-heptanone were formed in the oxidation of n-heptane (Scheme 2). Neither 1-heptanol nor heptanal were detected in the oxidation products of n-heptane. The products of skeletal isomerization were also not found.
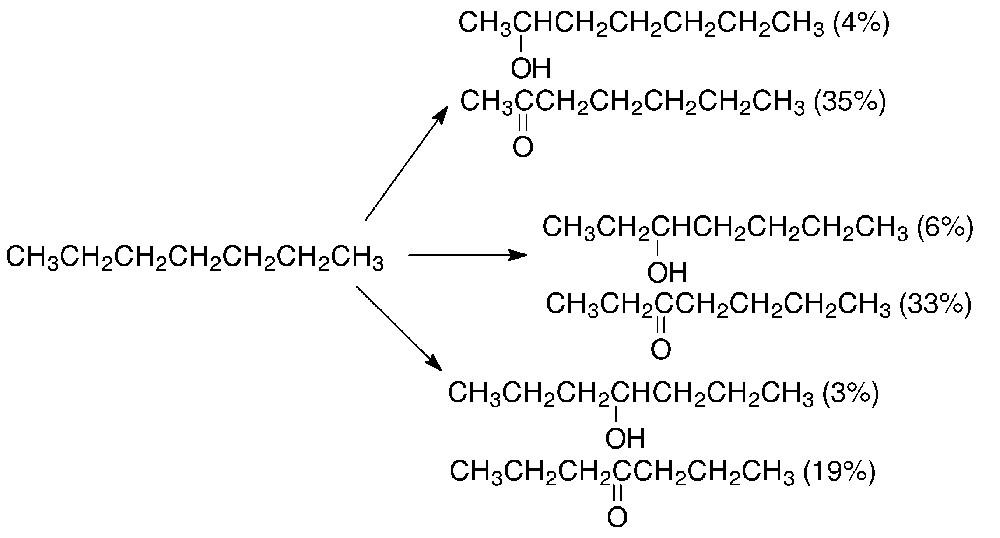
Identified products of n-heptane oxidation in the V(V)/H2O2/AcOH system (relative product yields are given in parentheses).
The total yield of alcohols and ketones was at most 60% at substrates conversion of ~35%. The ketone content in the oxidation products was much higher than that of alcohol (see Table 1). Ketoalcohols were not found among the oxidation products of n-heptane and lower alkanes.
The kinetic curves for the ketones and alcohols accumulation during the oxidation of alkanes reveal no induction period or singular points. Traditional free-radical inhibitors, such as BHT and para-benzoquinone, exerted no effect on the composition of the alkane oxidation products (Table 1, Nos. 1, 2) or the shape of the kinetic curves. The yield of the oxidation products, the ratio [ketone]/[alcohol], and the yield of positional isomers were also not influenced by the inhibitors. These data allows one to rule out the free-radical chain mechanism of alkane oxidation in the V(V)/H2O2/AcOH system, similarly to the earlier studied cyclohexane oxidation [17].
The prevailing of ketones in the reaction products could have been rationalized within a reaction scheme involving the following consequence: alkane → alcohol → ketone. To test and verify hypothesis, we studied the competitive oxidation of n-octane and 3-pentanol. In the competitive one-pot oxidation of 3-pentanol (0.01 mol l –1) and n-octane (0.1 mol l–1) at [H2O2]0 = 1 mol l –1, the conversion of 3-pentanol was found to be negligibly low, while the conversion of n-octane reached 1–5%. In analogous experiments, 2-butanol (0.01 mol l –1) was oxidized competitively with n-octane (0.1 mol l–1). The final concentration of 2-butanone was at most 0.001 mol l–1 even after the complete decomposition of H2O2 ([H2O2]0 = 1 mol l –1) and consumption of 3% n-octane. All these facts indicate that alcohols are not intermediates in the formation of ketones. Alcohols and ketones are formed in parallel reactions, and they are the final products of the oxidation of linear alkanes.
Hence, the following similarities between the oxidation of linear alkanes and cyclohexane [17] were found:
- • (1) hydrocarbon skeleton remains unchanged upon oxidation;
- • (2) all the substrates are oxidized to the corresponding alcohols and ketones;
- • (3) alcohols and ketones are formed in parallel reactions;
- • (4) linear alkanes and cyclohexane are oxidized via a non-chain mechanism.
Detailed analysis of the scope of data available for cyclohexane oxidation allowed a conclusion that a common oxidant (presumably, complex 1, Scheme 1) is responsible for the formation of both cyclohexanone and cyclohexanol [17].
The competitive experiments in which individual linear alkanes and cyclohexane were one-pot oxidized showed that the relative rate constants of oxidation of the substrates are in the range 0.6–1.7. Thus, the reactivity of cyclohexane is commensurable with the reactivity of the tested n-alkanes, except for n-pentane 1 (Table 2).
Relative rate constants of alkane and cyclohexane oxidation (kA/kcyclohexane) in the V(V)/H2O2/AcOH system at 30 °C
| Alkane | kA/kcyclohexane |
| n-pentane | 1.71 ± 0.05 |
| n-heptane | 0.88 ± 0.05 |
| n-octane | 0.64 ± 0.04 |
| n-nonane | 0.83 ± 0.05 |
| n-decane | 0.72 ± 0.04 |
3.2 Oxidation of branched alkanes
3.2.1 Oxidation of 2,2-dimethylbutane
The oxidation of 2,2-dimethylbutane in the V(V)/H2O2/AcOH system gives rise to the expected products of the methylene group oxidation, 3,3-dimethyl-2-butanone (4) and 3,3-dimethyl-2-butanol (5). However, the primary alcohols, 3,3-dimethyl-1-butanol (6) and 2,2-dimethyl-1-butanol (7), which are the products of the methyl group hydroxylation, were found among the reaction products. Moreover, the appearance of 2-methyl-2-propanol (8), 2,2-dimethyl-1-propanol (9), 2-methyl-2-butanol (10) in the reaction products seems to be quite unusual as far as the carbon chains of these alcohols are shorter than that of the starting alkane (Scheme 3) 2.
3.2.2 Oxidation of 2-methylpentane
The oxidation of 2-methylpentane was expected to attack the C–H bonds of the methyl, methylene, and methine groups. Indeed, the corresponding oxidation products were identified in the reaction mixture: 4-methyl-1-pentanol (11), 2-methyl-1-pentanol (12), 2-methyl-3-pentanol (13), 2-methyl-2-pentanol (14), 4-methyl-2-pentanol (15), 2-methyl-3-pentanone (16), 4-methyl-2-pentanone (17), and 2-methylpentanal (18). Meanwhile, 2-pentanol (19) and 2-pentanone (20) were detected along with the above oxidation products. Note that the carbon skeletons of these compounds are shorter than that of the initial hydrocarbon (Scheme 4).
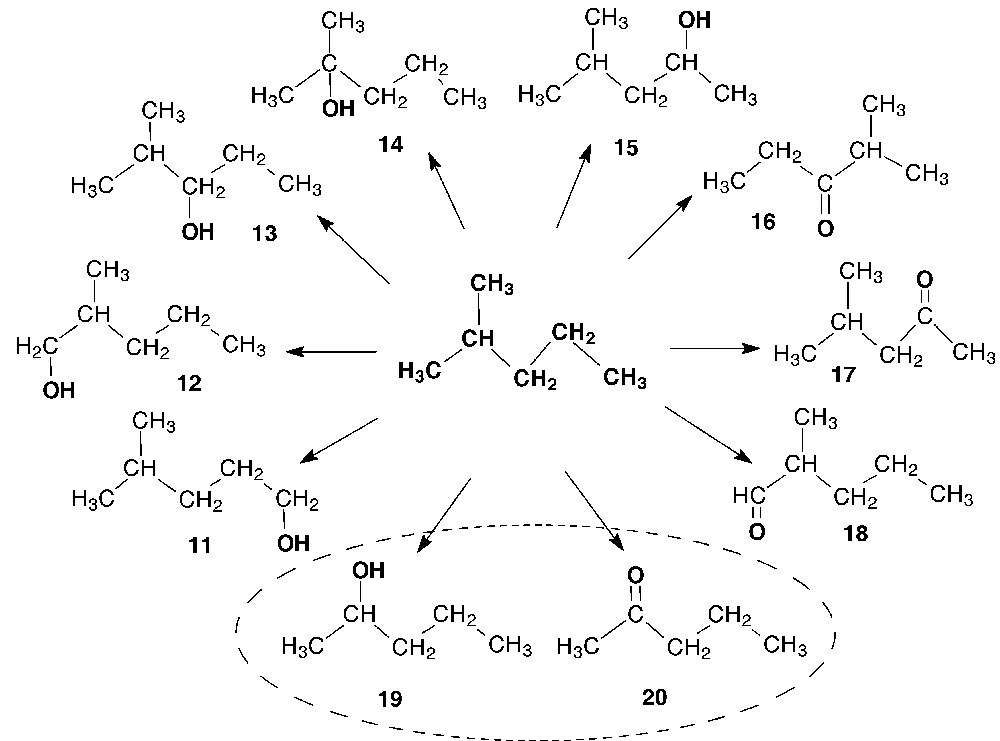
Identified products of 2-methylpentane oxidation in the V(V)/H2O2/AcOH system.
3.2.3 Oxidation of 3-methylpentane
The predominant products of 3-methylpentane oxidation in the system under study are 3-methyl-2-pentanone (21) and 3-methyl-3-pentanol (22). Other hydrocarbon oxidation products, in which the carbon skeleton of the substrate was retained (3-methyl-2-pentanol (23), 3-methyl-1-pentanol (24), 2-ethyl-1-butanol (25), and 3-methylpentanal (26)), were also detected. In addition to the expected oxidation products, the following ketones, alcohols, and their esters with shorter carbon skeletons were found and identified by GC-MS: 3-pentanol (27), 3-pentanone (28), 2-butanone (29), 2-butanol (30), 2-butyl acetate, ethanol (31), and ethyl acetate (Scheme 5).
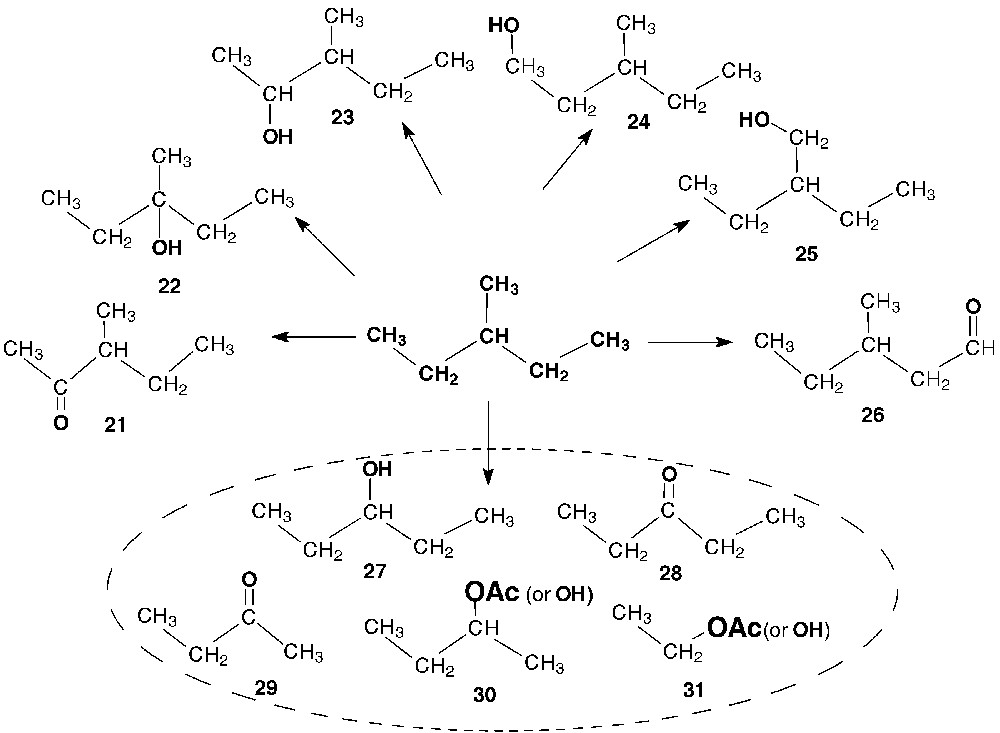
Identified products of 3-methylpentane oxidation in the V(V)/H2O2/AcOH system.
3.2.4 Oxidation of 2,3-dimethylbutane
2,3-Dimethyl-2-butanol (32) dominates among the oxidation products of 2,3-dimethylbutane. In addition, the following compounds with a shorter molecular skeleton were also found among the oxidation products of 2,3-dimethylbutane: 2-propanol (36), 3-methyl-2-butanol (37), and 3-methyl-2-butanone (38). The oxidation products, in which the parent carbon skeleton was retained, were also detected in the reaction solutions: 2,3-dimethyl-1-butanol (33), 2,3-dimethylbutanal (34), and 2,3-dimethyl-2-butene (35) (Scheme 6).
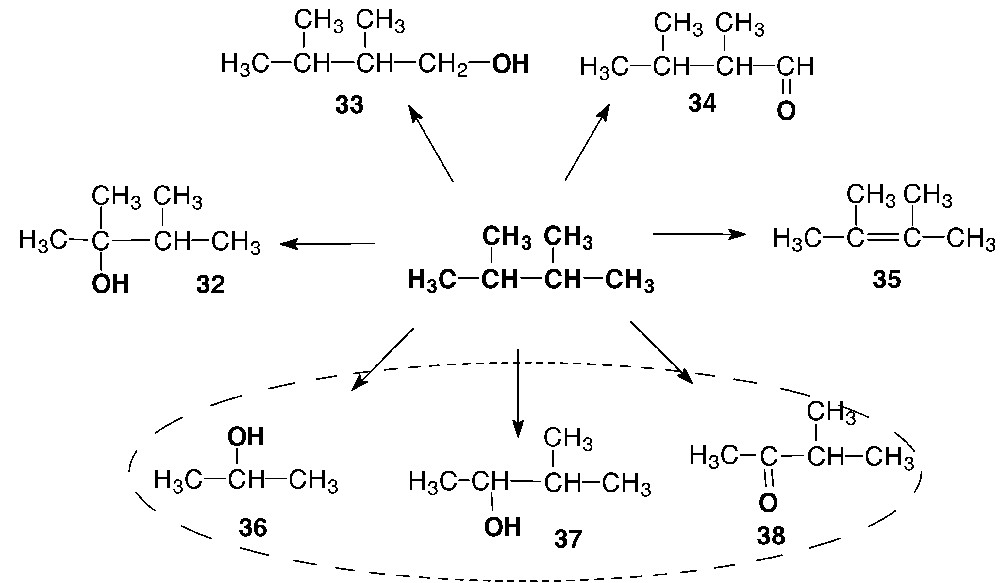
Identified products of 2,3-dimethylbutane oxidation in the V(V)/H2O2/AcOH system.
3.2.5 Oxidation of isooctane
The fullest information on the reactivity of various C–C bonds can be best demonstrated by the oxidation of isooctane, whose molecule contains the primary, secondary, tertiary, and quaternary carbon atoms. Therefore, the oxidation of this hydrocarbon was studied more detailed. More than 12 products have been detected by using GC–MS technique. Up to 30% of the starting isooctane was converted into oxidation products.
2,2,4-Trimethyl-3-pentanone (39, the main product), 2,2,4-trimethyl-3-pentanol (40), 2,2,4-trimethyl-1-pentanol (41), 2,4,4-trimethyl-1-pentanol (42), and 2,4,4-trimethyl-1-pentene (43), as well as compounds with shortened hydrocarbon chains (4,4-dimethyl-2-pentanol (44), 2,4-dimethyl-2-pentanol (45), 4,4-dimethyl-2-pentanone (46), 1,1-dimethylethanol (47), 2-methyl-1-propanol (48), 2-methyl-1-propyl acetate, 2,2-dimethyl-1-propanol (49), 2,2-dimethyl-1-propyl acetate, 4-methyl-2-pentanone (50), and 4-methylpentan-4-ol-2-one (51)) were found and identified by GC–MS in the reaction products of isooctane oxidation. Additionally, traces of 2-propanol were also detected (see Scheme 7).
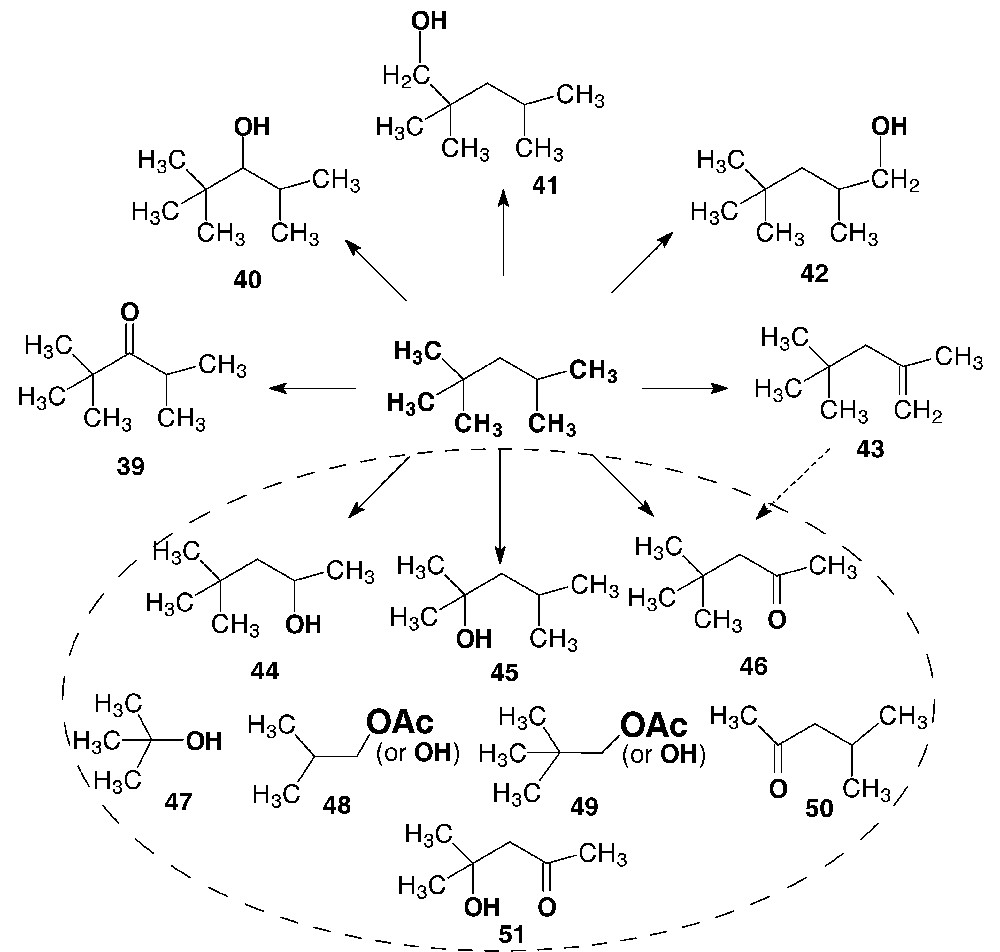
Identified products of isooctane oxidation in the V(V)/H2O2/AcOH system.
Free-radical chain reaction inhibitors, such as BHT and para-benzoquinone, exerted no effect the product composition of isooctane oxidation in the V(V)/H2O2/AcOH system, suggesting the free-radical chain oxidation of this and above-mentioned hydrocarbons to be excluded from consideration.
The oxidation of the sole methylene group of isooctane should give rise to 2,2,4-trimethyl-3-pentanone (39) and 2,2,4-trimethyl-3-pentanol (40) analogously to the oxidation of cyclohexane [17] and n-alkanes. Besides these compounds, the products of methyl group hydroxylation were also observed: 2,2,4-trimethyl-1-pentanol (41) and 2,4,4-trimethyl-1-pentanol (42). Surprisingly, 2,2,4-trimethyl-2-pentanol (product of hydroxylation at the tertiary carbon atom of isooctane) was not found among the products, in spite of expectation based on the free-radical mechanism and mechanistic schemes involving the abstraction of the hydride ion from the alkane. It is likely that this alcohol or the corresponding acetate is converted via dehydration or AcOH elimination into 2,4,4-trimethyl-1-pentene (43), which was detected in the reaction mixture, e.g.: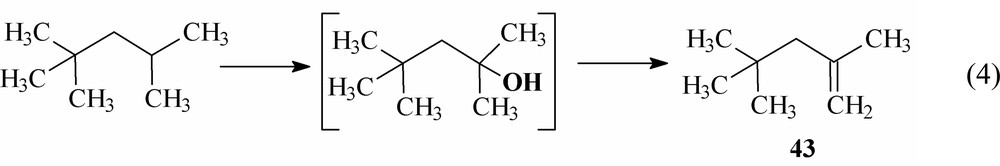
Both the oxidative cleavage of 2,4,4-trimethyl-1-pentene (43) and direct degradation of the C(2)H-CH3 bond can be considered as possible pathways for the formation of 4,4-dimethyl-2-pentanon (46). The alkene oxidative cleavage is well known for the V(V)/H2O2/AcOH system [13,14,17] (see Scheme 1).
The formation of 4,4-dimethyl-2-pentanol (44) and 2,4-dimethyl-2-pentanol (45) seems to indicate that isooctane oxidation involves the C–C bond cleavage. This is the least expected route of the reaction under discussion. The presence of the C4–C7 hydrocarbon derivatives (44–51, Scheme 7) among the reaction products cannot be assigned to the availability of the corresponding impurities in the initial substrate. The GLC analysis of the initial hydrocarbon demonstrated that the isooctane purity was at least 99.8%, the concentration of 2,2-dimethylpentane and 2,4-dimethylpentane was at most 0.02%, and the concentration of C4–C6 hydrocarbons was lower than 0.01%. Hence, we can conclude with confidence that the alcohols and ketones with the shortened carbon chains (1,1-dimethylethanol (47), 2-methyl-1-propanol (48), 2,2-dimethyl-1-propanol (49), 4-methyl-2-pentanone (50), 4-methylpentan-4-ol-2-one (51), and 2-propanol) are formed from isooctane via the C–C bond cleavage.3
Both linear and branched alkanes undergo oxidation primarily at the C–H bonds of the substrate with the retention of the molecular carbon skeleton. The primary C–H bonds have been proved to be much less reactive than the secondary and tertiary bonds. The difference between activities of the primary and secondary C–H bonds is less pronounced in the oxidation of branched alkanes. The ketone concentration in the reaction products was always higher than those of the corresponding alcohols (see Table 1).
A similar relation between ketonization and hydroxylation reactions was observed in alkane oxidation with dioxygen catalyzed by CoII/CoIII in a solution of CF3COOH; this reaction is supposed to include a one-electron transfer from the hydrocarbon molecule to metal cation [19,20].
The oxidation of saturated branched-chain hydrocarbons seemingly differs remarkably from the reactions of linear alkanes and cyclohexane.
- • (1) Primary alcohols and aldehydes were found in the oxidation products of branched alkanes, whereas the terminal methyl groups of linear alkanes remained unaffected. Note, however, that the number of the CH3 groups per branched hydrocarbon molecule is much higher than that in linear hydrocarbons.
- • (2) Oxidation of linear alkanes does not produce alcohols and ketones with carbon chains shortened compared to the parent hydrocarbon, whereas the yield of the shorter-chain alcohols and ketones in the oxidation products of branched hydrocarbons is comparable with the amount of the ‘normal’ hydroxylation and ketonization products. Moreover, oxidative degradation primarily occurs at the Ctert–C bond.
- • (3) Esters of alcohols with shortened carbon chains have been found, whereas esters were not detected in the products of hydroxylation of cyclohexane and linear alkanes (molecular carbon skeleton was retained).
Nevertheless, two latter discrepancies can be rationalized within the framework of a mechanistic scheme involving a common oxidant species for all of the alkane conversion routes (hydroxylation, ketonization, and oxidative degradation). The scope of kinetic data on the cyclohexane and anthracene oxidation with the V(V)/H2O2/AcOH system suggests that a possible candidate for such an oxidant can be complex 1 (see Scheme 1) [17].
Diperoxo vanadium(V) complex 2 predominates among the vanadium(V) complexes under the oxidation conditions (see Scheme 1) [6,7]. The kinetic data (zero order in alkane concentration, first order in [V(V)] and [H2O2]) suggest that complex 2 is converted into an active oxidant (supposedly complex 1), as shown in Scheme 1.
A detailed analysis of the data available showed that the hydroxylation of the alkane (O atom insertion into the C–H bond) and its ketonization fall into two categories of the electron transfer reactions [17]. Within the mechanistic scheme under discussion, complex 1 will be active in both alkane reactions. The hydroxylation is assigned to a two-electron process (see Eq. (5)):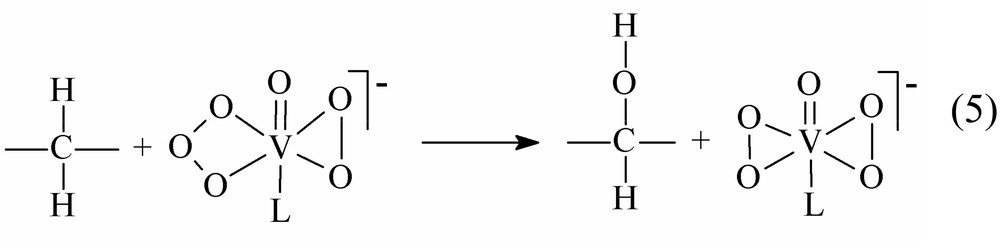
In this reaction, the coordinated oxidant group donates an O atom to the substrate molecule and the ligand transforms into the coordinated peroxo group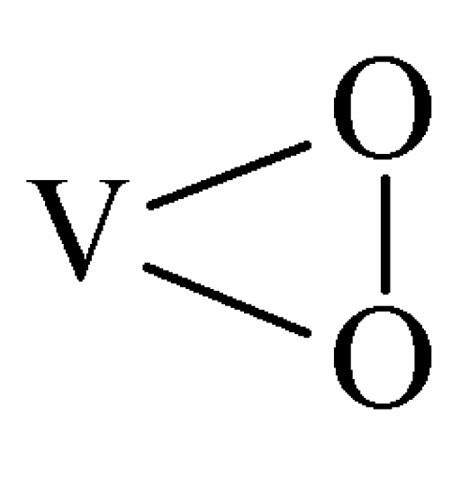
For simplicity sake reaction (5) can be represented as the sum of two redox reactions (Eqs. (6)–(8)):
| (6) |
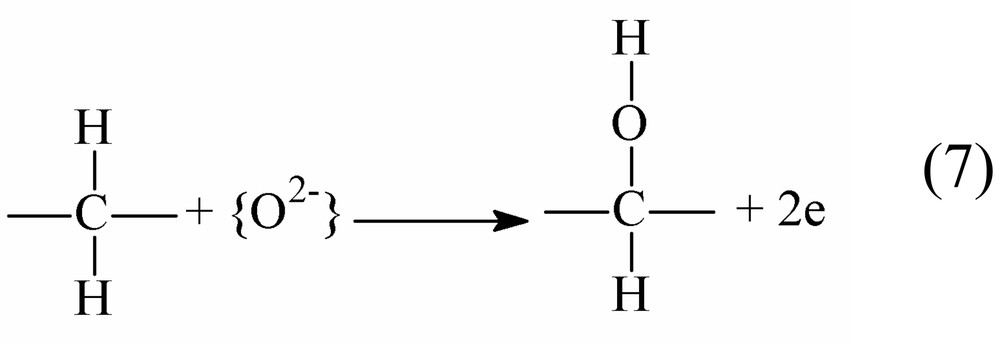
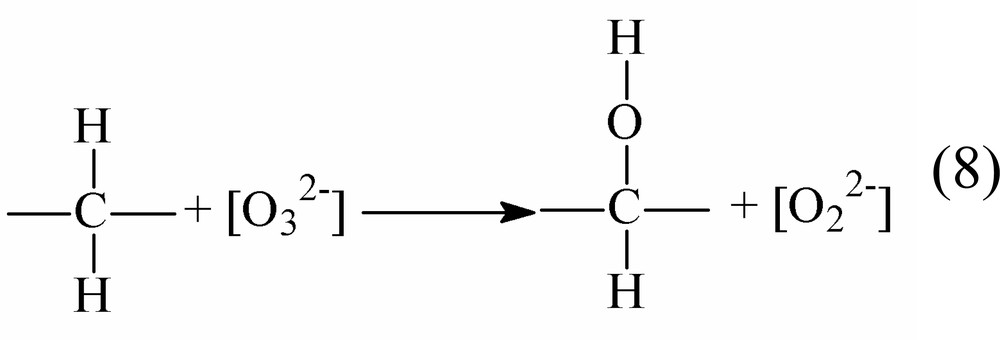
Both [] and [] are coordinated ligands, {O2–} is a specie formally transferred to the substrate after the two electrons removal.
Contrary to that, all the data available suggest ketonisation to be a three-electron oxidation (see Eq. (9)) [17]: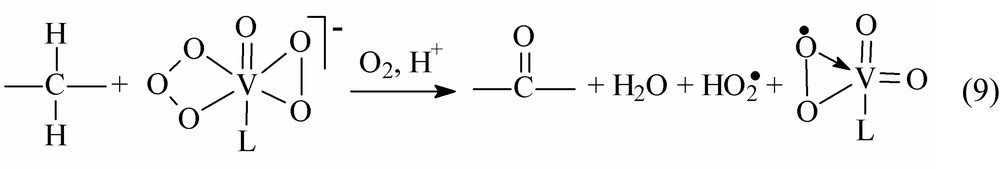
Two H-atoms of the methylene group undergoing ketonization should form the water molecule.
The reaction can be rationalized as the transfer of the oxygen cation radical O+• from the coordinated oxidant ligand () to the substrate molecule in the transition oxidation state. Both the [] and [] ligands are to be destroyed to produce the coordinated oxo-ligand and superoxide anion. As evidenced by EPR spectra, the vanadium(V) complexes with the coordinated superoxide anion always occur in the reaction solutions [6–8]. Factors affecting the competition between the O-atom insertion and O+• substrate/oxidant transfer are unknown. The mode of the H–C–H vibration symmetrical vs antisymmetrical could be of significance. The hypothesis on the participation of the oxygen radical cation [O+•] in oxidation reactions catalyzed by copper complexes has been forwarded by Shilov et al. [22].
Within the mechanistic scheme under question, the difference between hydroxylation and ketonization reactions consist in the nature of the species transferred to the hydrocarbon from complex 1 containing the ozonide dianion . The transfer of one oxygen atom from the ligand to the substrate molecule is expected to produce an alcohol. The transfer of the O+• species from to the substrate molecule is responsible for ketone formation from the linear alkane. Remarkably, no products of the O-atom insertion into the C–C bond, such as >C–O–C<, were observed. This fact is in contradiction with mechanistic schemes involving the formal O atom insertion into the C–C bond as the starting point for the C–C bond degradation.
The insertion of O+• species into the C–H bond can be shown by Eq. (10):
Structure 3B is to dominate in the gas phase. However, the interaction of the specie 3 with solvent molecules, like H2O and AcOH, will contribute to the relative weight of structure 3A in the initial product of the formal O+• insertion into the C–H bond. The removal of H+ as H3O+ from intermediate cation radical 3 will give rise to a radical species, which will react with dioxygen to produce ketone and the radical rapidly converted into H2O2.
Carbon tetrachloride, a scavenger of the C-centered free radicals [23], does not affect the rate of alcohol formation [17]. The oxidation of alkanes in the system under study does not involve radical chain reactions, and alkane hydroxylation can be considered as a polar insertion of one of the oxygen atoms of the ligand into the hydrocarbon C–H bond.
Nevertheless, according to Eq. (10), a C-centered free radical is expected to form in the course of hydrocarbon ketonization. This radical cannot serve as chain carrier. However, chloroform and C2Cl6 were detected among the products of cyclohexane oxidation in the presence of CCl4 [17]. These products indicate a possible involvement of the intermediate 1-hydroxy-1-cyclohexyl radical, the product of insertion of the O+• species into the C–H bond of cyclohexane (see Eq. (11)).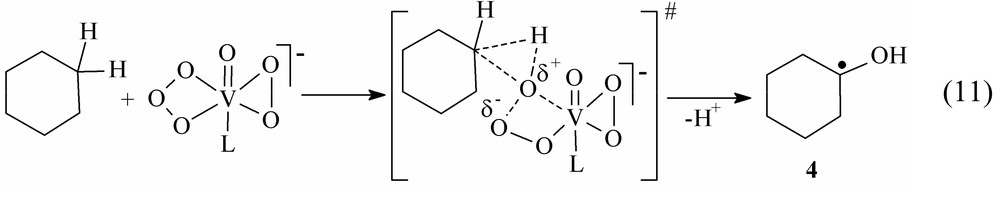
Radical 4 will react with oxygen giving rise to ketone and the radical, which is known to be inactive in hydrocarbon oxidation. The formation of chloroform and hexachloroethane in the oxidation of cyclohexane and CCl4 can be rationalized within the framework of this hypothesis (see Eq. (12)):
The KIE (kH/kD) for the formation of cyclohexanone in the system under study is much higher than that for cyclohexanol and equal to 2.45 ± 0.09 [17]. This figure is in good agreement with the proposed mechanism, which presumes the insertion of the oxygen radical cation into the C–H bond and the splitting off a proton from the same carbon atom (reaction 12). In this case these processes are likely to occur synchronously.
Within the concept under discussion, the transfer of the O+• species to the branched alkane molecule can be accompanied with the C–C bond cleavage and substrate degradation. The insertion of the O+• species into the C–H bond of the branched hydrocarbon molecule will give rise to a positively charged radical species whose spin density is delocalized over the center of the attack and groups bound to it: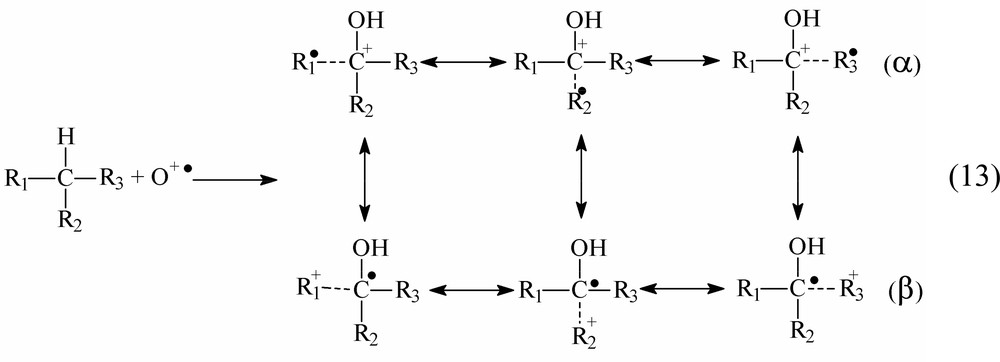
The contribution of resonance structures in the α and β rows seems to be of significance for the product distribution. The interaction of the intermediates with a solvent and the capability of dioxygen to react with radical species converting them into peroxo radicals or oxidizing them into carbocations should be taken into consideration.
- (1) If the relative weight of structures α predominates, an alcohol and/or its ester and a carbonyl compound with the shortened chain (aldehyde (Ri = H) or ketone (Ri = alkyl)) should be expected.
- (2) If the weight of structures (β) predominates, a nucleophilic attack on the +Ri group results in alcohol formation. The dioxygen attack on ‘free-radical’ fragments gives rise to peroxy radicals, which could propagate chain oxidation. However, the radicals are not active as initiators [23]. In fact, our experiments showed that the addition of BHT, an inhibitor of free-radical chain reactions, exerted no effect on the composition of oxidation products.
Both alcohols and ketones with the shortened carbon skeletons were found among the products of branched alkanes oxidation (see Schemes 3–7). Moreover, a number of acetates (see, for example, Schemes 5 and 7) of shorter-chain alcohols were found, suggesting that the parent cation radicals react with the solvent as intermediate species.
3.3 Oxidation of molecular nitrogen
Detailed analysis of the above results [17,22] led us to the conclusion that the vanadium(V) complex containing the ligand, which is responsible for alkane oxidation, can donate a substrate molecule with either the oxygen atom (in two-electron oxidation) or the oxygen radical cation (O+•) (in three-electron oxidation). Both the O+• and O(1P) atoms transferred to a substrate in the activated complex are strong acids (in terms of the Usanovich theory [24,25]). One can expect that these species could react even with such a weak base as molecular nitrogen.
Indeed, we found experimentally that molecular nitrogen was converted in the system under question to N2O under mild conditions (1 atm; 0–20 °C):
| (14) |
The oxidation of N2 to N2O or NO with molecular oxygen in the ground 3O2(3Σg) and excited 1O2(1Δg) states is thermodynamically impossible under conditions close to standard ones.
The reaction of molecular nitrogen with ozone is unknown but thermodynamically allowed [26]:
| (15) |
The oxidation of molecular nitrogen with hydrogen peroxide is also not forbidden thermodynamically:
| (16) |
| (17) |
| (18) |
Our experiments showed that when gaseous N2 contacted with a V(V)/H2O2/CF3COOH solution it underwent oxidation with hydrogen peroxide (see for preliminary communication [27]). The GC–MS analysis detected the formation of N2O4. The N2O content in exhaust gases did not exceed 0.1%.
The samples of gaseous nitrogen used in experiments were proved not to contain N2O. Nitrogen(I) oxide was not detected when the catalyst was absent. It also was absent from the gaseous products of hydrogen peroxide decomposition in helium atmosphere in the V(V)/CF3COOH system. Molecular nitrogen does not react with ozone in CF3COOH solution containing V(V) compounds.
Hence, the vanadium(V) complex is believed to be responsible for the oxidation of molecular nitrogen with H2O2. The reaction found is the first example of the involvement of molecular nitrogen in the catalyzed hydroperoxide oxidations under mild conditions.
4 Conclusions
Our studies of catalytic properties of vanadium(V) compounds in carboxylic acid solutions revealed a number of novel phenomena in the chemistry of peroxide oxidation. The reactions of branched alkanes with the C–C bond cleavage were observed for the first time. The involvement of the terminal methyl groups is also unusual for this area of chemistry. The appearance of esters as hydroperoxide oxidation products seems to be an equally novel and unexpected phenomenon as well.
A number of complexes are known to exist in the systems H2O2/transition metal/solvent [6–10,17,21,28–38]. The results of the study suggest that active intermediates form in the reaction of hydrogen peroxide with vanadium complexes. The oxidation reactions of the difficult-to-oxidize substrates like linear and branched alkanes and molecular nitrogen point to the formation of high-energy species in the V(V)/H2O2/RCOOH systems. The data available are consistent with hypothetical schemes involving the O+• cation radical and/or O(1P) atom transfer from the oxidant to a substrate.
The forwarded mechanisms can be used as the basis for designing new catalyst systems and further studies.
Acknowledgments
This work was supported by the Program of President, Russian Federation, for the Support of Leading Scientific Schools (grant 1764.2003.3) and the Program ‘Fundamentals of Energochemical Technologies’ of the Russian Academy of Sciences.
1 The consumption of low-boiling n-pentane can be slightly overestimated because of its removal from the reaction vessel by the oxygen flow that formed in the decomposition of hydrogen peroxide (reaction temperature is 30 °C, and the boiling temperature of n-pentane is 35–36 °C).
2 The oxidation products containing lower numbers of carbon atoms than that in the parent substrate were detected as alcohols and esters in the reaction mixture. However, for simplicity, the corresponding alcohols are shown in reaction schemes.
3 2-Propanol is fairly soluble in water. For this reason, in the course of sample preparation by extraction with diethyl ether, a considerable portion of 2-propanol remained in water to underestimate the apparent yield of this compound.
4 The procedure used in this work for the detection of products did not allow us to find other conceivable N2 oxidation products: NO, NO2, and HNO3.