

1 Introduction
New high-silica zeolite framework types with 10–12-membered-ring (MR) pores are still of key interest in petroleum and chemical industry for separation and catalysis since the pore size and shape influences significantly the selectivity in educt uptake and product release in catalytic processes. A careful study of the reasons crucial for the specific catalytic properties of microporous materials showed that the pore size of the pore opening and the curvature of the internal oxide surface play a fundamental role in the catalytic activity and the shape selectivity of microporous zeolite catalysts [1]. It was concluded that 8–12 MR zeolite frameworks are most effective. Smaller pores can not be entered by organic molecules, larger ring size allows bigger molecules to selectively enter pore space, but product formation becomes more and more equivalent to reactions with flat, amorphous oxide surfaces. ITQ-7 is a very interesting new member of the family of high silica 12 MR zeolites. It is structurally closely related to zeolite beta which has a disordered framework structure. As zeolite beta, ITQ-7 also has a 3-dimensional (3D) channel system made of intersecting 12 MR pores whose pore diameters are almost similar (5.9–6.5 Å diameter). In contrast to beta, however, this material can be obtained with a highly ordered silicate framework structure. It is typically synthesized with a specific structure directing agent (SDA), 1,3,3-trimethylspiro[6-azoniabicyclo [3.2.1]]octane-6,1′-pyrrolidine (TAD), in a narrow synthesis field using the fluoride route [2]. The crystal structure of the calcined material (zeolite framework type ISV [3]) has been reported recently by Villaescusa et al. [2]. In successive papers very interesting catalytic properties have been reported [4–6]. In addition, the substitution of Si by Ge has been studied [7–9]. Of particular interest for the basic understanding of the formation is the selective occupation of T-sites by Ge, in particular in the D4R and the Ge-induced reduction of the synthesis time by two orders of magnitude to 12 h. Although the general features of the crystal structure are known, there is no detailed information on the host guest interaction in the as synthesized material which might be important for the efficiency of the SDA. Here we report on the synthesis of pure silica ITQ-7 with a new, modified SDA and on the structural characterization with solid state NMR and powder XRD. Particular attention has been paid to in situ studies of the synthesis with 19F solid state NMR and on the crystal structure analysis of the as synthesized form using the Rietveld technique.
2 Experimental
ITQ-7 was synthesized from quasi-dry gel preparations in the temperature range 150–170 °C with a starting gel composition of SiO2/SDA/H2O = 1:0.5:2.97–15.90 (Table 1). 1,3,3trimethylspiro[6-azoniabicyclo[3.2.1]]octane-6,1′-piperidinium fluoride (TAT) was used as new SDA for ITQ-7 type materials (Fig. 1). The starting material was transferred into a Teflon lined steel autoclave and kept for 5–10 d in an oven at synthesis temperature.
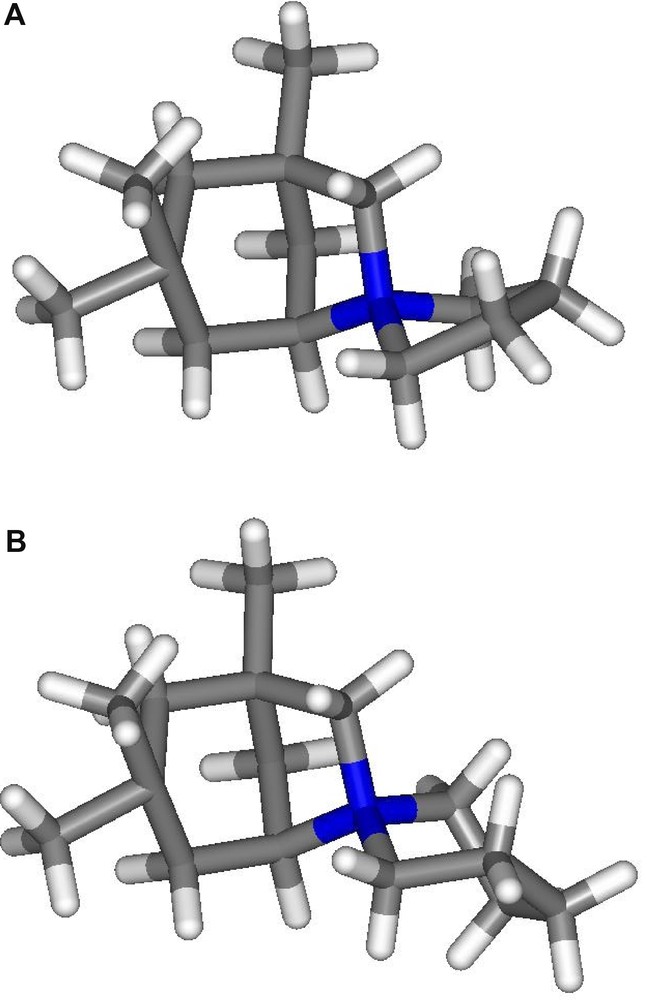
The known (A) and the new (B) SDA used for the synthesis of ISV-type material. (A) 1,3,3-Trimethylspiro[6-azoniabicyclo[3.2.1]]octane-6,1′-pyrrolidinium cation. (B) 1,3,3-Trimethylspiro[6-azoniabicyclo[3.2.1]]octane-6,1′-piperidinium cation.
Scanning electron microscopy (SEM) investigations to study the morphology of the crystals and the homogeneity of the samples were performed using a LEO-1530 Gemini.
X-ray powder diffraction data were collected from a sample of the as synthesized material on a Siemens D5000 diffractometer using primary monochromatized Cu Kα1 radiation and a 6° Braun PSD in the range of 4–94° 2θ and with step width of 0.00777° 2θ. The experiment was performed in transmission mode with the sample kept in a capillary to avoid preferred orientation. Rietveld analysis was performed with the program package FullProf suite, version May 2004, including FULLPROF 2k, WINPOLTR, and GFOUR [10].
Thermal properties were analyzed on a Bähr STA 503 Thermal Analyzer under air in the temperature range from RT to 1030 °C. The heating rate was 5 °C/min. DTA- and TG-signals were recorded simultaneously.
The 13C, 19F and 29Si solid state MAS NMR experiments were performed using a Bruker ASX 400 spectrometer in standard probe heads. For the in situ solid state NMR investigation of the synthesis reaction glass capillaries were used which fit inside the rotor of the Bruker 7 mm probe head. The summary of the experimental condition of the NMR experiments is given in Table 2.
Summary of the experimental conditions of the solid state MAS NMR experiments
| 13C | 23Si | 29F | 19F in situ | |
| Standard | TMS | TMS | NaF | NaF |
| Frequency (MHz) | 100.6 | 79.5 | 376.5 | 376.5 |
| Pulse width (10–6) | 4.1 | 6.5 | 6 | 6 |
| Contact time (10–3 s) | 1 | 5 | ||
| Recycle time (s) | 1 | 1 | 5 | 60 |
| Spinning rate (kHz) | 4 | 3.5 | 8 | 2 |
| Number of scans | 570 | 1200 | 160 | 80 |
3 Results and discussion
In most synthesis runs ITQ-7(TAT) was obtained as a pure phase consisting of transparent, colorless crystals with a plate-like, tetragonal prismatic morphology. A typical synthesis required 6 d at 165 °C synthesis temperature as compared to 15 d at 150 °C in the original preparation [2]. At low temperature (150 °C) and short reaction times (5.3–5.5 d), however, only amorphous products were obtained. The conditions and the results of the synthesis in the simple system 1 SiO2/0.5 TAT/x H2O are summarized in Table 1.
Summary of the synthesis experiments using 1,3,3-trimethylspiro[6-azoniabicyclo[3.2.1]]octane6,1′-piperidinium fluoride as a new SDA
| Sample | Water content | Temperature (°C) | Duration (days) | Products amorphous |
| e5d4 | 3.49 | 150 | 5.3 | Amorphous |
| e5d1 | 8.36 | 150 | 5.3 | Amorphous |
| e5d2 | 15.90 | 150 | 5.5 | Amorphous |
| e5c2 | 4.13 | 150 | 9.7 | ITQ-7 |
| e5c1 | 4.57 | 150 | 9.7 | Beta |
| e5c4 | 8.33 | 150 | 9.7 | ITQ-7 |
| e5a4 | 2.97 | 165 | 6.0 | ITQ-7 |
| e5a3 | 3.92 | 165 | 6.0 | ITQ-7 |
| e5a2 | 4.64 | 165 | 6.0 | Amorphous, ITQ-7 |
| e5a1 | 8.33 | 165 | 6.0 | ITQ-7, amorphous |
| e5e1 | 3.45 | 170 | 6.4 | ITQ-7 |
Unfortunately, no further synthesis experiments could be carried out since the SDA was no longer available on the market.
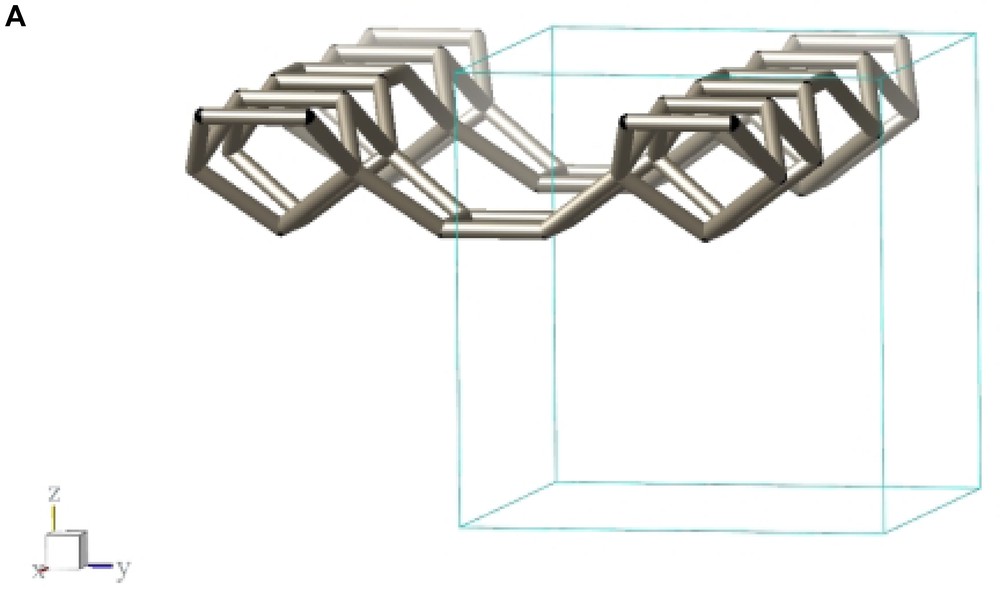
Fig. 2 shows a typical conglomerate of ITQ-7(TAT) crystals. As can be derived from the plate-like morphology of the tetragonal crystals the growth along the tetragonal c-axis is slower than along the prism faces leading to aspect ratios c/a << 1. Considering the silicate framework structure of ITQ-7 which can be built from layer-like building units (LLBU) stacked along the c-axis (Fig. 3A) it seems likely that the creation of new layers along the c-axis is the rate determining step during growth whereas the development of the crystal along a and b continues much faster.

SEM image of ITQ-7(TAT) crystals in the as synthesized form. The tetragonal-prismatic morphology can clearly be seen indicating slow growth along the c-direction and faster growth along the a- and b-direction.
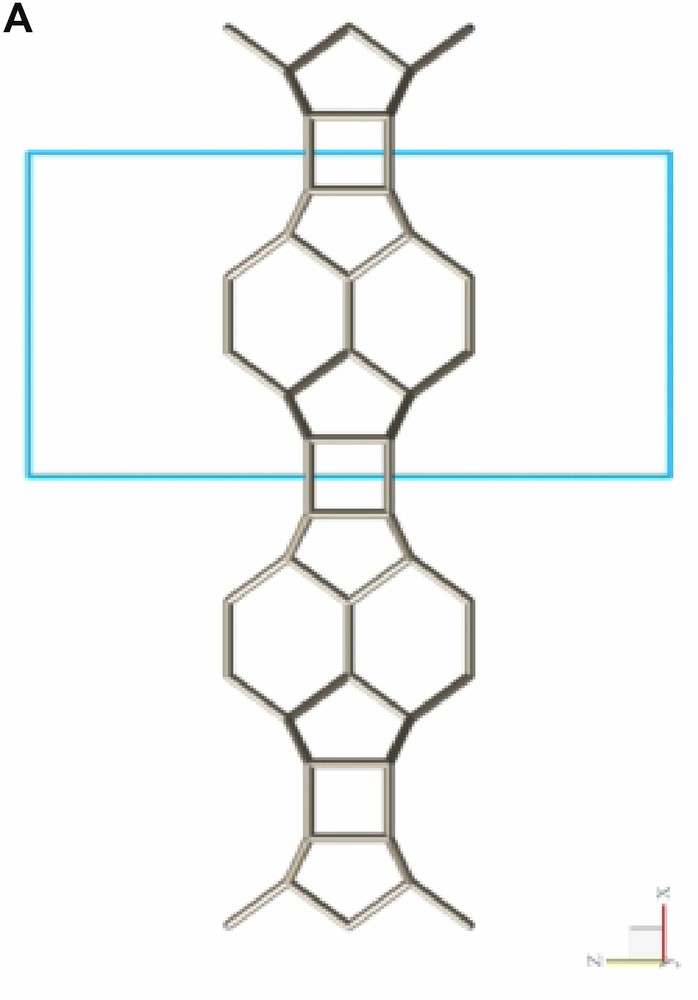
LLBU-1 of the ITQ-7 silica framework. The skeleton represents the connection of Si-atoms. Oxygens are located at ca. the midpoint between two Si-sites and are omitted in the drawing for clarity. As reference the unit cell outline is shown. Neighboring layers are connected via oxygen bridges to give the 3D framework.
The thermal properties upon calcination of the as synthesized ITQ-7 are shown in Fig. 4. At temperatures close to 400 °C the breakdown of the SDA sets in. The oxidative decomposition indicated by two exothermic peaks at ~ 430 and ~450 °C together with a weight loss of almost 20% shows the break down of the organic molecule and the expulsion of the degradation products. Since no further DTA-signal is observed, the calcined form of silica ITQ-7 should be stable up to at least 1000 °C.
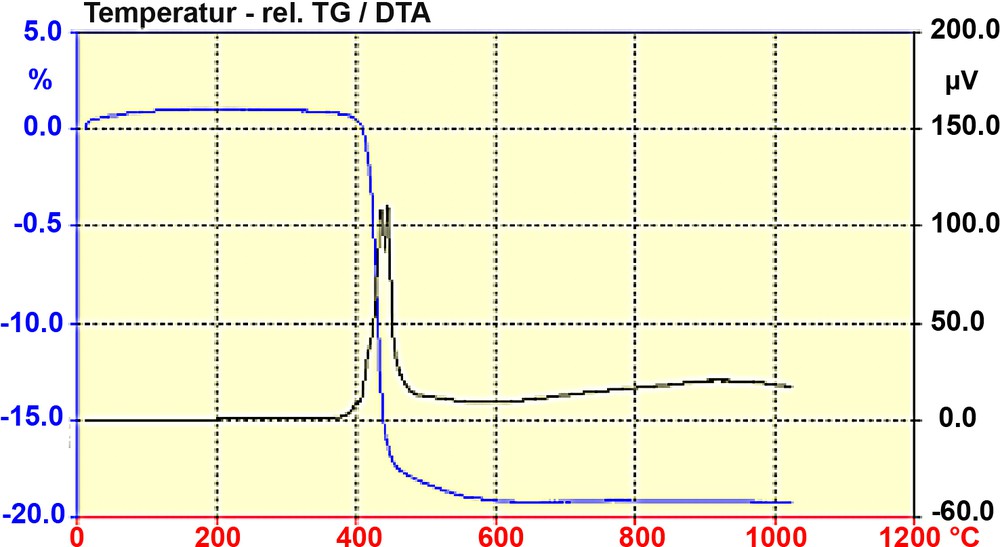
TG and DTA diagram of ITQ-7(TAT). The decomposition of the SDA is indicated by the almost 20% weight loss (TG) and the exothermic peaks (DTA) at ca. 430 and 450 °C. The lack of further exothermic events up to 1000 °C indicates the thermal stability of the silica framework of ITQ-7.
Using solid-state NMR techniques the role of the fluoride anion, the state of the SDA and the crystallinity of the silica framework have been investigated. The 1H–29Si CP MAS NMR spectrum shows a typical spectrum for Q4(4Si) framework silicas (Fig. 5). The cross polarization experiment does not reflect the intensity ratios of the nuclei quantitatively. However, in the case of lattice defects which would introduce silanol carrying Q3-silicate species, the intensity of the defect site would greatly be enhanced. Since there is no signal in the range between –95 and –105 ppm the framework structure is defect free. Three signals at –107, –112 and –117 ppm are observed representing the eight symmetrically inequivalent T-sites of the crystal structure. As often observed for silica zeolites prepared with the fluoride route there is no indication of five-coordinated Si, at least at room temperature [11]. The rather broad signals indicate distorted local geometries of the [SiO4]-tetrahedra in addition to signal overlap from different T-sites. The ITQ-7(TAT) sample investigated, therefore, might deviate from the highest possible space group symmetry for the ITQ-7 framework, P42/mmc, possessing five inequivalent T-sites.
Room temperature 1H–29Si CP MAS NMR spectrum of as synthesized ITQ-7(TAT). The spectrum shows that the silica framework has no silanol defects and also no five-coordinate silicon typical for fluoride-assisted synthesis.
The 13C CP MAS spectrum of the ITQ-7(TAT) (Fig. 6) proves that the intact TAT cation has been incorporated inside the zeolite void system.
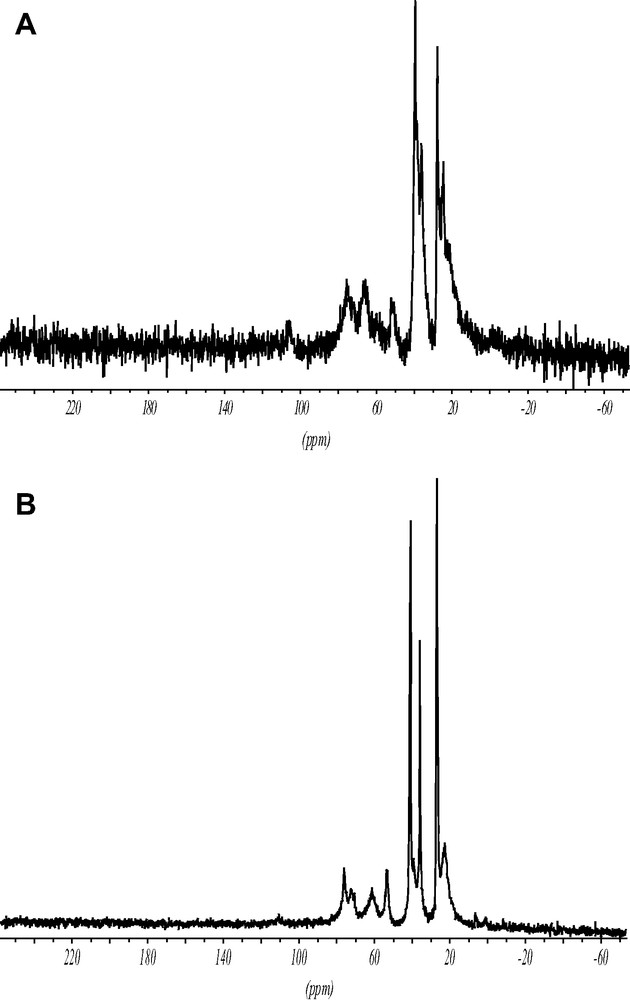
1H–13C CP MAS spectra showing the intact SDA cation inside as made ITQ-7(TAT) (A) and the solid free template (B).
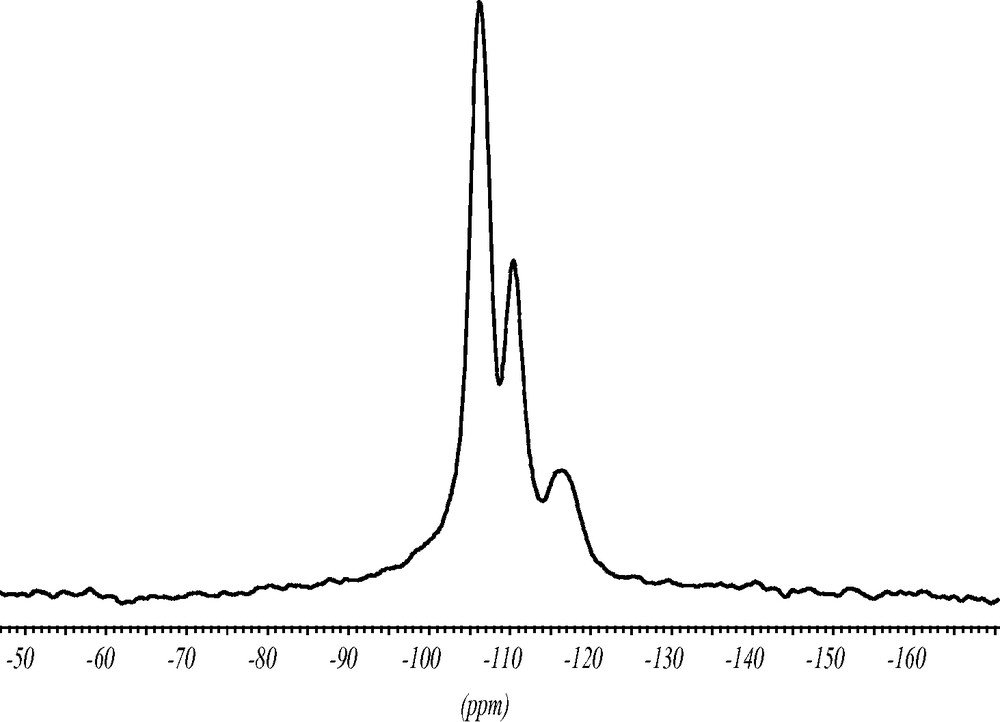
The 19F MAS NMR spectrum contains two strong signals indicating that there are two symmetrically inequivalent charge balancing fluoride anions (Fig. 7). The chemical shifts of the signals at –38.5 and –39 ppm are typical for the fluoride anion inside double-four-ring (D4R) units made of pure silica. The splitting of the signal supports the structure analysis with two inequivalent fluoride anions in two sets of D4R of the silica framework. In addition, there are minor intensities in the 19F NMR spectrum at ca. –70 and –130 ppm which might be due to fluoride species occluded in amorphous impurities of the synthesis batch. Since the chemical shift of the fluoride signal correlates with the void volume of the cavity it occupies [12] this species should be in a more open environment.
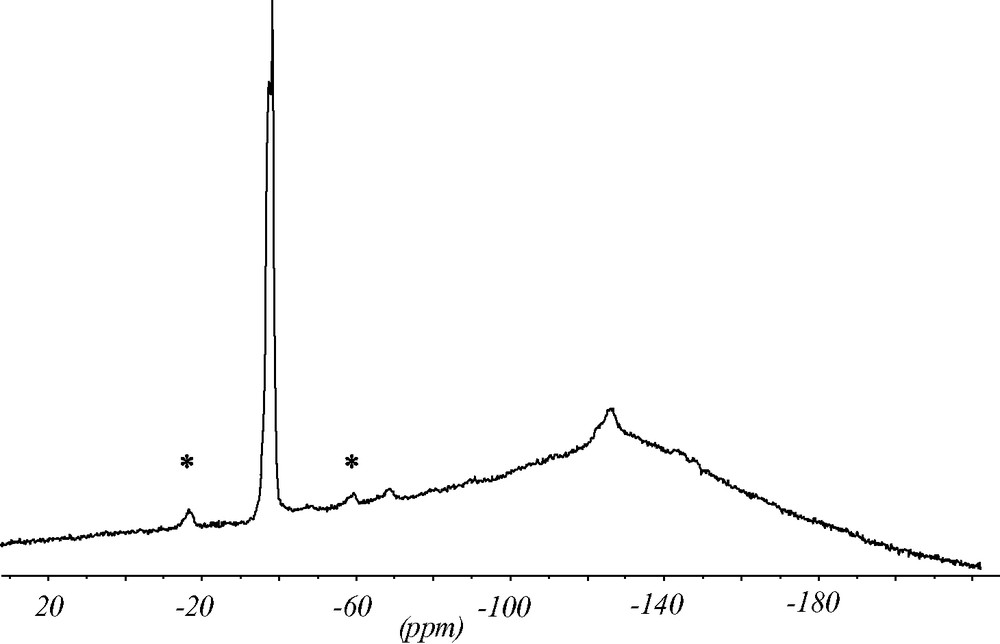
19F MAS NMR spectrum of as synthesized ITQ-7(TAT). The two strong signals at –38.5 and –39 ppm are characteristic for the fluoride anion inside the D4R unit. The signals at ca. –70 and –130 ppm indicate fluoride anions in more open sites. They are attributed to impurities. * Design spinning side bands.
In order to find out more about details of the synthesis process, in situ NMR has been performed. The synthesis dry gel was sealed in a silica glass capillary and kept at synthesis temperature of 150 °C inside the 7 mm rotor of the NMR probe head. In regular time intervals 19F spectra were recorded. After 2 d, for the first time the signal at ~–39 ppm appeared, long before X-ray analysis detected crystalline ITQ-7(TAT) (see Table 1). This indicates the formation of nuclei or species for the nucleation of the zeolite framework involving a complex which includes the fluoride anion together with its D4R cavity. The observation also supports the co-templating hypothesis for the fluoride anion [13]. The interaction with the charge compensating organic cation finally leads to the crystallization of the zeolite framework which is specific for the particular organic SDA. Additional NMR experiment using the 13C, 29Si, and 1H nuclei for observation did not provide any further information. For our sample with the 13C- and 29Si-nuclei in natural abundance no signal was observed in the solid and no significant changes were visible for the protons.
The crystal structure of the as synthesized material was refined with the aim to analyze the specific interaction of the new SDA with the silicate framework. Careful analysis of the powder pattern did not reveal any indication that in the as synthesized form the SDA has changed the space group symmetry of ITQ-7(TAT) to lower symmetry than that of the silica framework in the calcined sample [2]. All attempts to refine the structure in lower symmetry space group and to localize the SDA inside the channel system in an ordered arrangement with the silica framework failed. Therefore, the Rietveld refinement was carried out in the tetragonal space group P42/mmc with lattice parameters a = 12.827(1) Å and c = 25.179(1) Å and the unit cell content of |(C15H29N)4 F4| [Si64O128]. The details of the Rietveld analysis are summarized in Table 3. The observed and calculated powder XRD together with the difference plot are shown in Fig. 8.
Experimental conditions and crystallographic parameters for the structure refinement of ITQ-7(TAT)
| Diffractometer | Siemens D5000 with 6° PSD |
| Wavelength (Å) | 1.5406 |
| 2θ Range of data collection (°) | 4.05–93.99 |
| Step size (°2θ) | 0.007769 |
| Number of steps | 11,579 |
| Number contributing reflections | 1113 |
| Number of distance restraints | |
| d(Si–Si) = 3.10(4) Å, 14 × | |
| d(O–O) = 2.64(2) Å, 27 × | |
| d(Si–O) = 1.610(4) Å, 18 × | 59 |
| Number of structural parameters | 57 |
| Number of profile parameters | 7 |
| FWHM at 21.8° 2θ (°2θ) | 0.11 |
| RBragg | 5.2 |
| Rwp | 9.2 |
| Rexp | 5.1 |
| χ2 | 3.3 |
| Space group | P42/mmc |
| a0 (Å) | 12.827(1) |
| c0 (Å) | 25.179(1) |
| VUC (Å3) | 4142.7(1) |
| Unit cell content | [Si64O128](C15H29N)4F4 |
| FD | 15.5 |
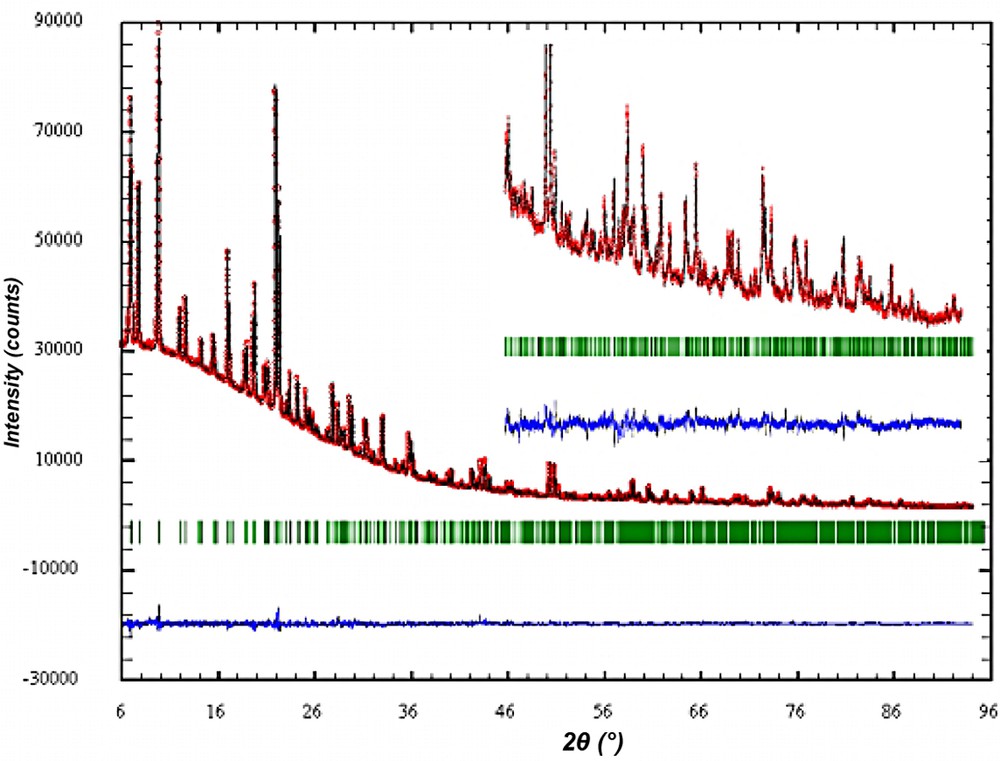
Rietveld plot of the observed calculated and diffracted XRD powder diagram of as synthesized ITQ-7.
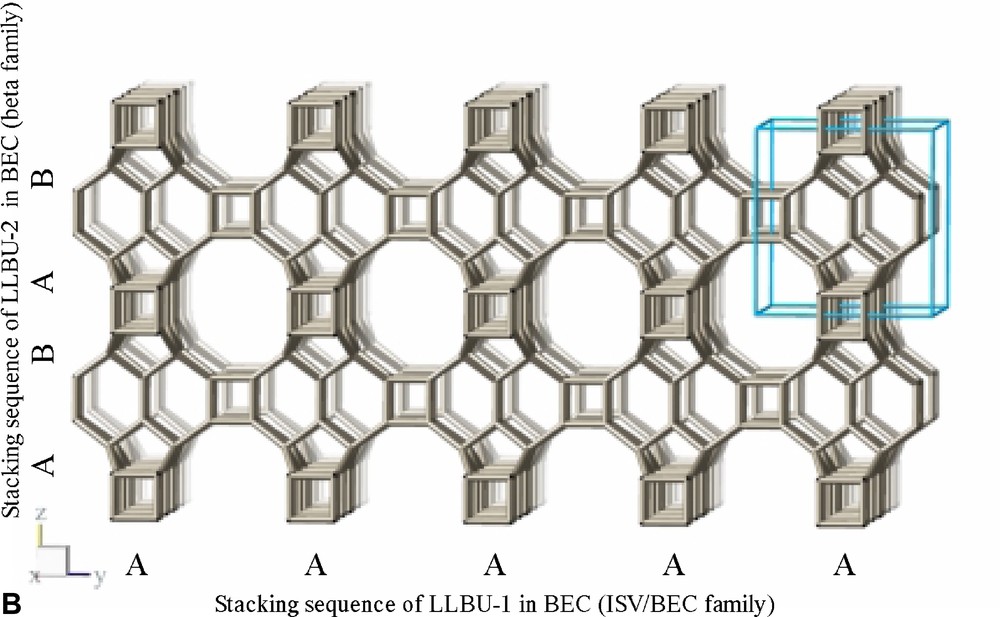
The refinement confirms the framework topology of ITQ-7 as described by Villaescusa et al. [2] (Fig. 3B). ITQ-7 has a 3D channel system with 12 MR openings. ITQ-7 (TAT) possesses five symmetrically independent silicon sites, 14 independent oxygen sites and two independent fluoride sites. The fractional coordinates are given in Table 4. The values of d(Si–Si), d(Si–F), d(O–O), and d(Si–O) as well as the ∠(Si–O–Si) are as expected for a microporous silica framework (see Table 5). The ∠(O–Si–O) scatters in the range between 102° and 115° also typical for Rietveld analyses of porous silica framework. Since NMR is more sensitive for the local order of the nucleus studied than results from diffraction analyses and the 29Si chemical shift values measured for ITQ-7(TAT) are typical for silica frameworks, no exceptional distortion of the [SiO4]-tetrahedra is observed. The microporous framework structure contains four intersection points of the channels per unit cell and a total of four D4R per unit cell, two per set of inequivalent fluoride anions.
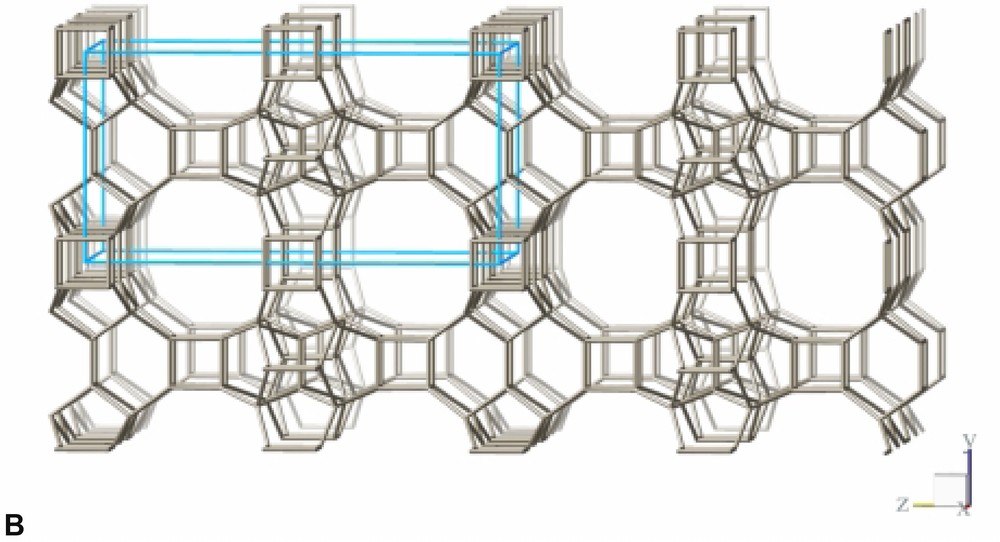
Framework structure of ITQ-7 built from LLBU, LLBU-1. Neighboring layers are stacked in ABAB sequence and rotated by 90° about c with respect to each other. The unit cell of ITQ-7 is given as reference.
Fractional coordinates for ITQ-7(TAT) as obtained from the Rietveld analysis of X-ray powder data. ESD’s are corrected for linear correlation of step scan intensity data
| Atom | x | y | z | B(equ) | SOF |
| Si1 | 0.11749 (017) | 0.11732 (018) | 0.06144 (008) | 1.299 (17) | 1 |
| Si2 | 0.30417 (015) | 0.25141 (016) | 0.90328 (008) | 1.299 (17) | 1 |
| Si3 | 0.5 | 0.11920 (022) | 0.06318 (010) | 1.299 (17) | 0.5 |
| Si4 | 0.38166 (018) | 0.38186 (018) | 0.18992 (007) | 1.299 (17) | 1 |
| Si5 | 0.37574 (020) | 0.37928 (022) | 0 | 1.299 (17) | 0.5 |
| O1 | 0.14297 (063) | 0.14270 (065) | 0 | 1.891 (37) | 0.5 |
| O2 | 0.14968 (052) | 0 | 0.07541 (029) | 1.891 (37) | 0.5 |
| O3 | 0 | 0.14519 (055) | 0.07684 (029) | 1.891 (37) | 0.5 |
| O4 | 0.19334 (031) | 0.19267 (039) | 0.09352 (017) | 1.891 (37) | 1 |
| O5 | 0.39684 (025) | 0.16994 (030) | 0.91210 (018) | 1.891 (37) | 1 |
| O6 | 0.31095 (036) | 0.33714 (031) | 0.94849 (012) | 1.891 (37) | 1 |
| O7 | 0.30882 (043) | 0.30573 (044) | 0.84440 (014) | 1.891 (37) | 1 |
| O8 | 0.5 | 0 | 0.08033 (031) | 1.891 (37) | 0.25 |
| O9 | 0.5 | 0.14635 (063) | 0 | 1.891 (37) | 0.25 |
| O10 | 0.5 | 0.35699 (067) | 0 | 1.891 (37) | 0.25 |
| O11 | 0.34173 (066) | 0.5 | 0 | 1.891 (37) | 0.25 |
| O12 | 0.35125 (030) | 0.35125 (030) | 0.25 | 1.891 (37) | 0.5 |
| O13 | 0.5 | 0.35043 (061) | 0.17492 (030) | 1.891 (37) | 0.5 |
| O14 | 0.35686 (064) | 0.5 | 0.17375 (031) | 1.891 (37) | 0.5 |
| F1 | 0 | 0 | 0 | 1.72 (17) | 0.125 |
| F2 | 0.5 | 0.5 | 0.25 | 1.72 (17) | 0.125 |
| C1 | 0.06700 (161) | 0.45413 (232) | 0.07038 (052) | 30.33 (68) | 0.656 |
| C2 | 0 | 0.40321 (240) | 0.18985 (149) | 30.33 (68) | 0.656 |
| C3 | 0 | 0.36272 (290) | 0.27388 (137) | 30.33 (68) | 0.656 |
| C4 | 0.05826 (328) | 0.28837 (235) | 0.26892 (191) | 30.33 (68) | 0.656 |
| C5 | –0.06375 (224) | 0.17509 (124) | 0.27965 (098) | 30.33 (68) | 0.656 |
| C6 | 0.08649 (198) | 0.5 | 0.15270 (085) | 30.33 (68) | 0.656 |
| C7 | 0 | 0.33730 (280) | 0.20588 (147) | 30.33 (68) | 0.656 |
| C8 | 0.10279 (150) | 0.5 | 0.23133 (108) | 30.33 (68) | 0.656 |
Ranges of angles and distances as obtained from the Rietveld structure refinement
| d(Si–O) | 1.59–1.64 Å |
| d(O–O) | 2.49–2076 Å |
| d(Si–Si) | 3.01–3.19 Å |
| d(Si–F) | 2.62–2.63 Å |
| ∠(Si–O–Si) | 140°–160° |
| ∠(O–Si–O) | 103°–118° |
After refining the framework structure difference Fourier maps were calculated which show the remaining electron density inside the channel voids representing the occluded organic guest ion. The observed electron density indicated that there is no specific host guest interaction. There is no distinct molecular orientation of the guest cation with respect to the silica framework but a statically or dynamically disordered arrangement. The electron density distribution, therefore, is regarded as a superposition of all possible orientations of the organic guest cation inside the channels void. The disorder of the SDA precludes the unambiguous determination of the host guest interaction by X-ray diffraction structure refinements. However, from 13C MAS NMR experiments and crystal chemical reasoning it is clear that the guest molecule is intact and balances the negative charge of the fluoride anion in the D4R. Therefore, the refinement of the structure of the as made ITQ-7(TAT) yielded only a superposition of all possible molecular positions of the SDA represented in the electron density. For the structure refinement the scattering contribution has been simulated by using eight symmetrically inequivalent carbon atoms as scatterers. A representation of the SDA inside the channel is shown in Fig. 9.
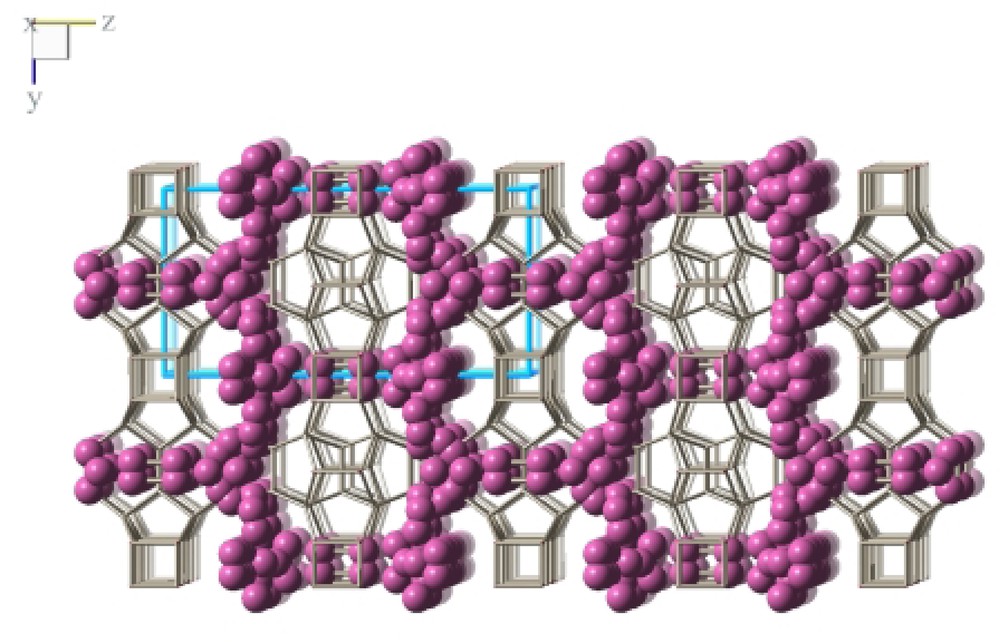
Representation of the distribution of the SDA inside the pore system of ITQ-7(TAT). The positions of the electron density maxima as obtained from the structure refinement are presented as spheres. The superposition of maxima indicates orientational disorder of the SDA.
As expected from the 19F MAS NMR experiments, the D4R units are both occupied with fluoride anions which compensate the charge of the organic cations (Fig. 10). Based on the structure refinement there is no indication for a bonding interaction of the fluoride anion with silicon atoms (Table 5). This is in contrast to a series of other zeolites synthesized with the fluoride technique. Here, fivefold coordination of Si has been observed with the fluoride anion as additional bonding partner increasing the coordination number [9]. In ITQ-7(TAT) both fluoride anions occupy the center of the respective D4R cage with equal mean distances to the framework silicon of ~2.64 Å. The crystallographic analysis, therefore, supports the assignment of the 19F NMR signal of the crystalline material and also supports the conclusions drawn from the in situ NMR experiments which indicate that the D4R unit as entities are formed long before long-range crystalline order is achieved.
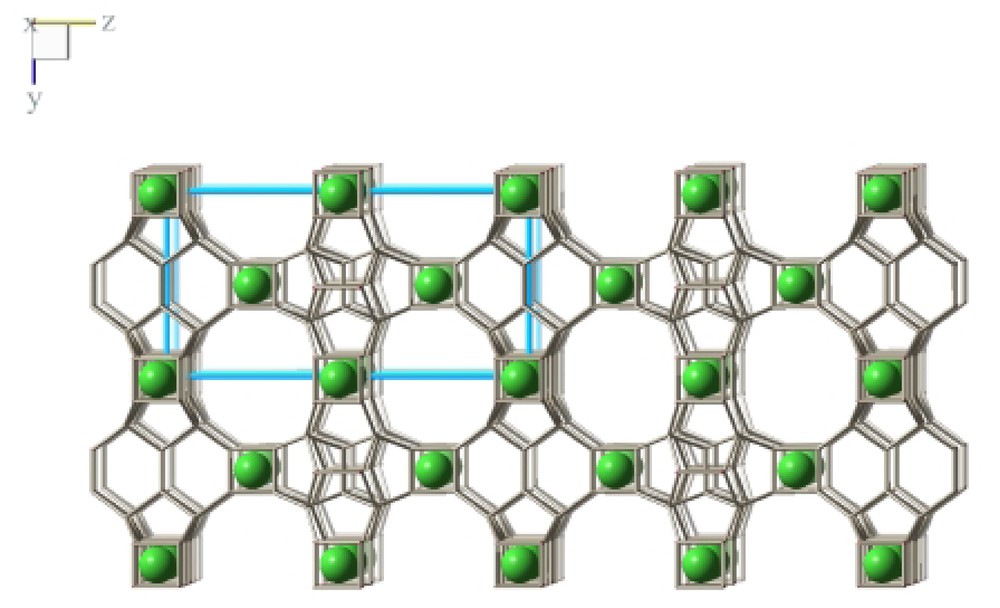
The fluoride co-template shown occupying the D4R units in ITQ-7(TAT).
The framework structure of ITQ-7 (zeolite framework type ISV) is closely related to the framework structure of polymorph C of the beta family of disordered zeolites [14] (zeolite framework type BEC). ISV and BEC contain the same LLBU-1 (Fig. 3A and Fig. 11B), however, which is different from the LLBU-2 of the beta family (Fig. 11A). In ISV the LLBU-1 is stacked in AB sequence along the tetragonal c-axis with layer B rotated 90° about the c-axis with respect to layer A. This leads to a tetragonal framework structure with the highest possible symmetry P42/mmc with the 42-axis perpendicular to the LLBU-1. In BEC the stacking of this layer is along the tetragonal a- or b-axis and is oriented perpendicular to the LLBU-2 used for the description for the beta family. The stacking sequence in BEC is AA and also leads to a tetragonal framework structure. The tetragonal 42-axis in BEC, however, is running parallel to the LLBU-1.
(A) LLBU-2 of the beta disorder family. (B) Skeletal model of the BEC-type framework. The LLBUs used to describe the structure as members of the beta disorder family as well as of the ISV/BEC disorder family are indicated.
In order to investigate the influence of stacking disorder on diffraction experiments in the BEC/ISV family DIFFaX [15] simulations of X-ray powder diagrams have been performed. Fig. 12A shows the result of the simulation of the X-ray powder diagrams of the calcined silica framework of the intergrowth of BEC and ISV-framework-type materials. The bottom diagram represents the silica framework of BEC. With increasing proportions of ISV in steps of 10%, the influence of the intergrowth becomes evident in the series of diagrams until pure silica ISV is reached. However, there are only few significant changes in the powder diagrams of this disorder series. For high ISV contents (from about 80% of ISV) the two peaks at 2θ angles of 7–8° 2θ for Cu Kα radiation coalesce (Fig. 12A). Below 70% ISV content the disorder shows up only in an asymmetric peak broadening of the first peak at 7° 2θ and is difficult to evaluate. For the analysis of the intergrowth below 70% ISV content, the set of six reflections between 20° and 24° 2θ shows sensitive changes indicative for the relative proportions of BEC- and ISV-type frameworks (Fig. 12B). Because of the subtle changes in the powder diagrams it is expected that disordered materials have already been synthesized, however, not been recognized yet.
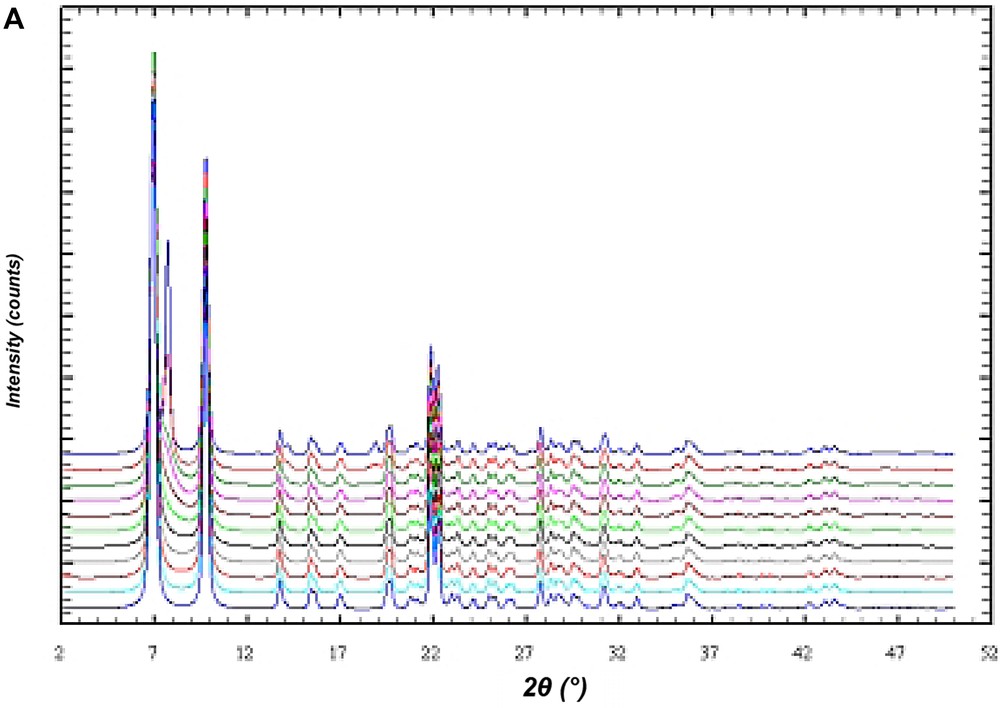
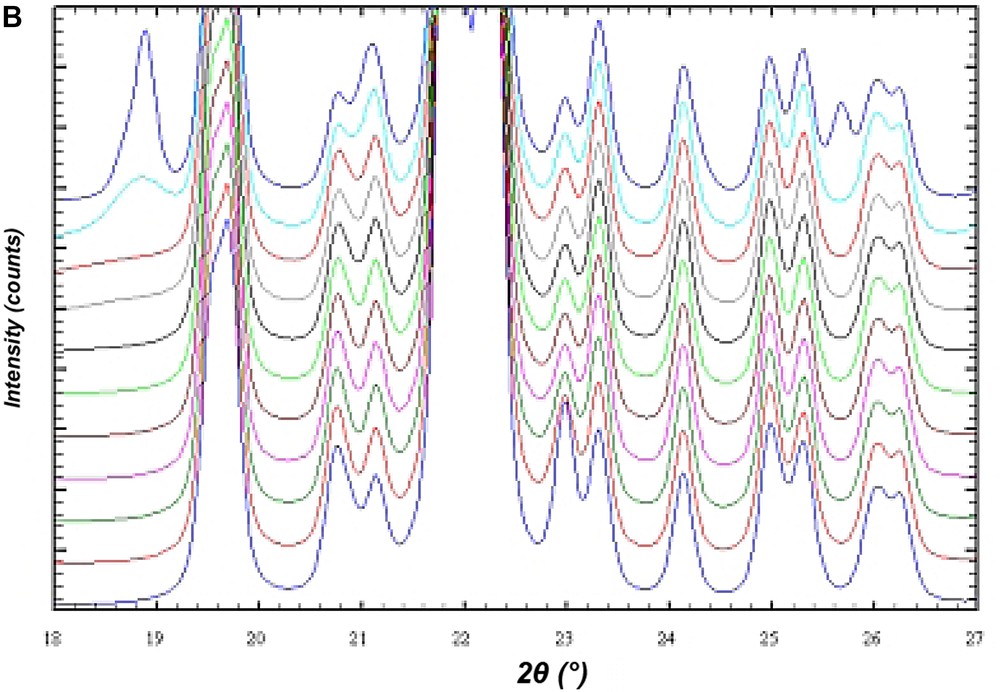
DIFFaX simulations of the X-ray powder pattern of the ISV/BEC disorder family. The bottom diagram represents the pure silica form of the BEC-type framework. The top diagram is for the respective ISV-type framework. The diagrams in between are for disordered materials calculated in 10% steps from 100% BEC/0% ISV to 0% BEC/100% ISV. (A) Shows an overview in the two-theta range from 2° to 50° 2θ. (B) Shows an enlarged section between 18° and 27° 2θ.
An interesting consequence of the study of the disorder of the two families, beta and the BEC/ISV disorder family, is that there are different layer-like periodic building units and, therefore, stacking sequences which relate the corresponding two end-members. From crystal growth considerations the crystallization, obviously, follows different mechanisms in the two systems which lead to the respective stacking of PerBUs. This might explain why no intergrowth between BEC and BEA has been observed so far, despite the close relationships between the respective framework types.
4 Conclusion
- • 1,3,3-Trimethylspiro[6-azoniabicyclo[3.2.1]]octane-6,1′-piperidinium fluoride proved to be a highly efficient SDA for the formation of pure silica ITQ-7.
- • The structure analysis showed that unfaulted ITQ-7 was obtained with the new template. This is in contrast to the SDA used by Villaescusa et al. [2] which led in only a very narrow range of synthesis conditions (concentration, temperature, ratio of components) to ordered ITQ-7.
- • The Rietveld analysis confirms the structure model published by Villaescusa et al. [2].
- • In situ 19F NMR evidenced the formation of D4R-units housing the fluoride anion long before crystalline order can be detected in diffraction experiments. This confirms the co-templating effect of the fluoride anion [13].