

1 Introduction
In the last few years an increased interest was devoted to materials combining electrical conductivity and magnetic interactions, with the aim to obtain magnetic coupling between the localized spins of the inorganic part (d electrons) via the mobile electrons of the organic part (π electrons) [1–11]. Some compounds of this kind such as (ET)4[(H3O)Fe(C2O4)3]C6H5CN [2b,2c] (ET = bis(ethylenedithio)tetrathiafulvalene) show a coexistence of superconductivity and paramagnetism. In other cases, as in (ET)3[MnCr(C2O4)3] [7a] and (BO)3[FeCr(C2O4)3] [7b] (BO = bis(ethylenedioxy)tetrathiafulvalene), a metallic state and ferromagnetism coexist. For most of these salts, however, the magnetic order is not mediated by the conducting electrons. Only a few examples, as in λ-(BETS)2FeCl4 (BETS = bis(ethylenedithio)tetraselenafulvalene) [3] and (DMET)2FeBr4 (DMET = dimethyl(ethylenedithio)diselenadithiafulvalene) [8] interactions between π and d electrons have been reported. However, the interactions between the conducting and paramagnetic systems which take place through space between anionic and cationic species are rather weak. Several strategies based on modifications of the organic and inorganic units are underway in order to establish such magnetic and/or structural interactions between the organic and inorganic sublattices. One of them focuses on the use of anions containing π and NCS– ligands, which can enable both S···S and π···π interactions between the conducting and magnetic systems. Thus, materials presenting bulk ferrimagnetism in TTF-based salts with transition temperatures (Tc) ranging from 4.2 to 8.9 K [9] were obtained using isothiocyanato-complexes [MIII(NCS)4(L)n]– where M = Cr and Fe and L = 1,10-phenanthroline (phen) or isoquinoline (isoq). With the same anions combined with tetrathiapentalene (TTP) derived donors, we obtained recently bulk weak ferromagnets formulated as D[M(NCS)4(isoq)2], D = BDH-TTP, DTDH-TTP, BDA-TTP, with Tc ranging from 5.9 to 8.9 K [10]. Another strategy concerns the synthesis of paramagnetic transition metal coordination complexes containing TTF (tetrathiafulvalene) derivatives as ligands such as [MII(hfac)2(TTF-py)2] [M = Cu and Mn; hfac = hexafluoroacetylacetonate] [11] where the conducting and magnetic systems are covalently linked through a conjugated bridge. Finally, a third strategy uses functionalized organic donors such as iodine substituted donors [12] (see Scheme 1) combined with paramagnetic anions containing nitrogen heterocycles (π systems) and CN– ligands, which can enable both –I···N– and π···π interactions between the conducting and magnetic systems [13]. The paramagnetic [Fe(bpca)(CN)3]– anion [14] (see Scheme 1) used in this work possesses three important characteristics of this last strategy: it can give rise to –N···I– and –O···I– interactions thanks to both CN– and bpca ligands and it contains also a π ligand which can lead to π···π overlap between adjacent anions or between the anions and the π system of the donors.
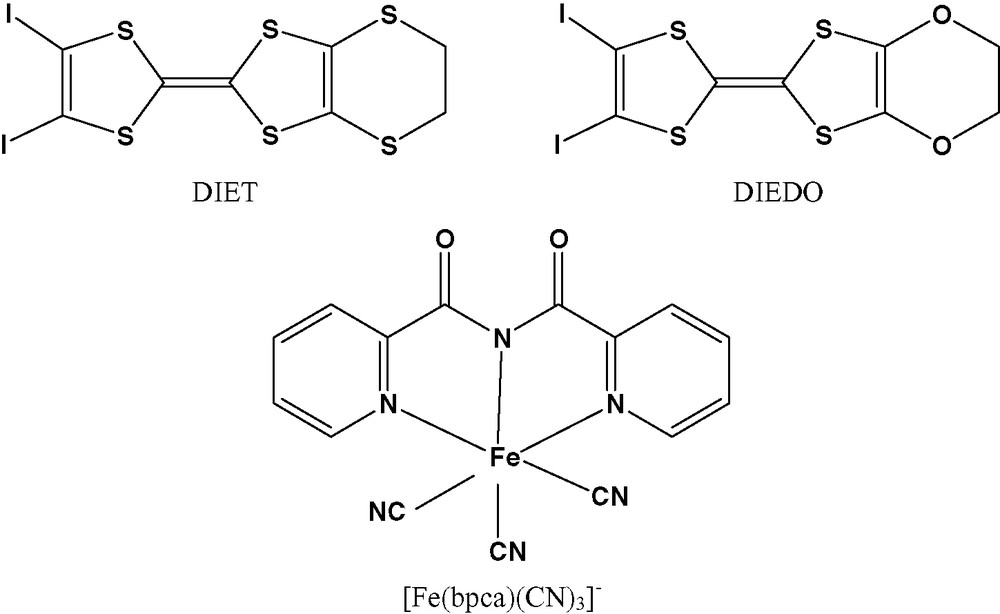
Structure of DIET, DIEDO and [Fe(bpca)(CN)3]– anion.
2 Results and discussion
Here we report the synthesis, X-ray crystal structures, electrical conductivity, magnetic properties, spectroscopic studies and band structure calculations of two new charge transfer salts, namely D2[Fe(bpca)(CN)3], D = diiododithiotetrathiafulvalene (DIET) (1) and diiodoethylenedioxotetrathiafulvalene (DIEDO) (2).
2.1 Crystal structures
The asymmetric units of 1–2 contain two crystallographically independent organic molecules referred A and B and one [Fe(bpca)(CN)3]– entity all in general positions. Fig. 1 shows ORTEP drawings of the molecular structures and atom numbering scheme. Selected bond lengths and bond angles are given in Table 1.
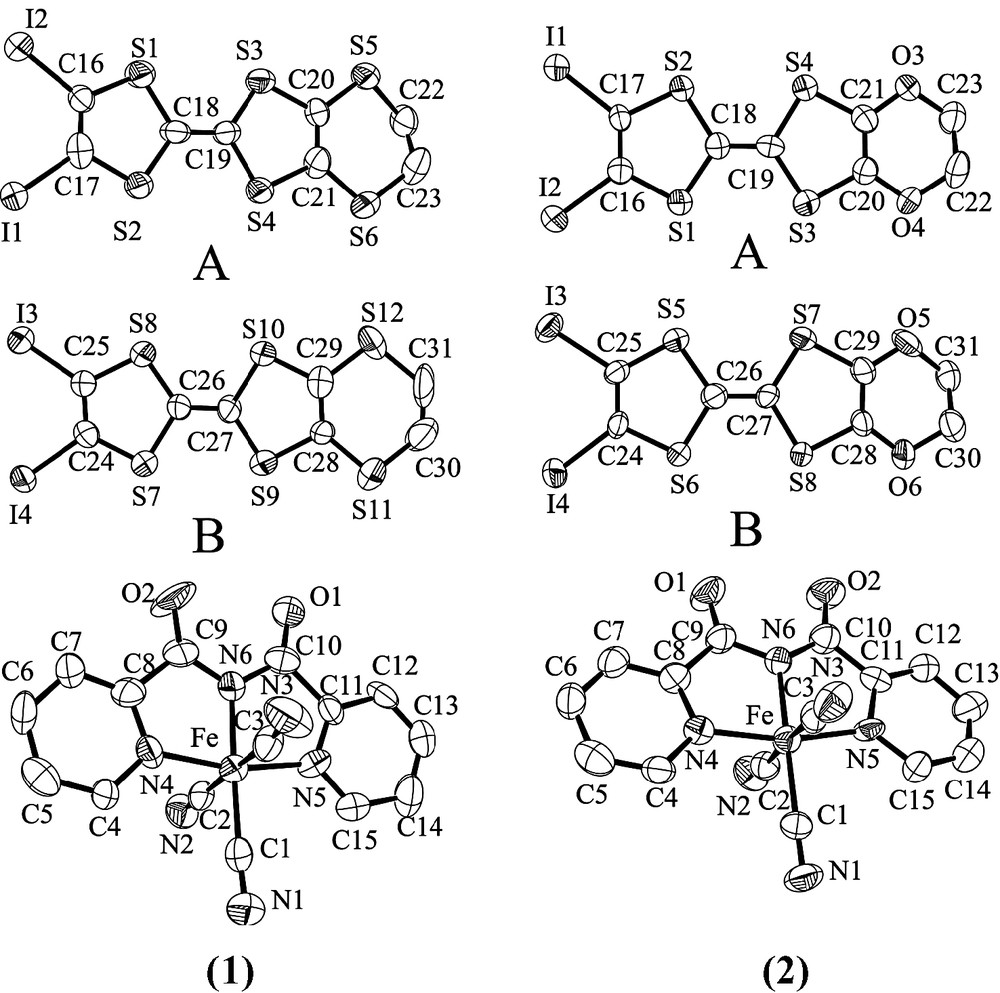
ORTEP diagrams with 50% probability for compounds 1–2.
Selected bond lengths (Å) and bond angles (°) for compounds 1–2
| 1 | 2 | ||
| Fe–C(1) | 1.927(15) | Fe–C(1) | 1.922(11) |
| Fe–C(2) | 1.951(17) | Fe–C(2) | 1.942(13) |
| Fe–C(3) | 1.933(14) | Fe–C(3) | 1.933(13) |
| Fe–N(4) | 1.943(10) | Fe–N(4) | 1.939(8) |
| Fe–N(5) | 1.989(12) | Fe–N(5) | 1.936(8) |
| Fe–N(6) | 1.895(11) | Fe–N(6) | 1.874(8) |
| S(1)–C(18) | 1.769(16) | S(1)–C(18) | 1.740(11) |
| S(1)–C(16) | 1.733(15) | S(1)–C(16) | 1.722(11) |
| S(2)–C(18) | 1.724(15) | S(2)–C(18) | 1.733(11) |
| S(2)–C(17) | 1.751(18) | S(2)–C(17) | 1.746(10) |
| C(18)–C(19) | 1.374(18) | C(18)–C(19) | 1.345(16) |
| S(3)–C(19) | 1.721(15) | S(3)–C(19) | 1.722(10) |
| S(3)–C(20) | 1.735(15) | S(3)–C(20) | 1.717(11) |
| S(4)–C(19) | 1.753(16) | S(4)–C(19) | 1.736(10) |
| S(4)–C(21) | 1.773(16) | S(4)–C(21) | 1.713(12) |
| S(5)–C(20) | 1.742(14) | O(3)–C(21) | 1.351(12) |
| S(6)–C(21) | 1.745(17) | O(4)–C(20) | 1.350(12) |
| C(20)–C(21) | 1.34(2) | C(20)–C(21) | 1.312(15) |
| C(16)–C(17) | 1.35(2) | C(16)–C(17) | 1.341(15) |
| I(1)–C(17) | 2.081(17) | I(1)–C(17) | 2.046(11) |
| I(2)–C(16) | 2.063(15) | I(2)–C(16) | 2.061(11) |
| S(7)–C(26) | 1.775(16) | S(6)–C(26) | 1.736(11) |
| S(7)–C(24) | 1.732(16) | S(6)–C(24) | 1.739(10) |
| S(8)–C(26) | 1.716(16) | S(5)–C(26) | 1.733(11) |
| S(8)–C(25) | 1.771(16) | S(5)–C(25) | 1.723(11) |
| C(26)–C(27) | 1.346(17) | C(26)–C(27) | 1.356(15) |
| S(9)–C(27) | 1.727(16) | S(8)–C(27) | 1.717(10) |
| S(9)–C(28) | 1.776(15) | S(8)–C(28) | 1.739(10) |
| S(10)–C(27) | 1.752(16) | S(7)–C(27) | 1.720(11) |
| S(10)–C(29) | 1.712(17) | S(7)–C(29) | 1.738(12) |
| S(11)–C(28) | 1.714(15) | O(6)–C(28) | 1.333(12) |
| S(12)–C(29) | 1.738(18) | O(5)–C(29) | 1.327(13) |
| C(28)–C(29) | 1.35(2) | C(28)–C(29) | 1.327(15) |
| C(24)–C(25) | 1.35(2) | C(25)–C(24) | 1.346(14) |
| I(3)–C(25) | 2.044(16) | I(3)–C(25) | 2.063(10) |
| I(4)–C(24) | 2.097(14) | I(4)–C(24) | 2.038(11) |
| N(1)–C(1)–Fe | 176.7(13) | N(1)–C(1)–Fe | 178.0(11) |
| N(2)–C(2)–Fe | 178.3(12) | N(2)–C(2)–Fe | 172.1(11) |
| N(3)–C(3)–Fe | 170.3(14) | N(3)–C(3)–Fe | 178.5(10) |
The crystal structures of both compounds (Fig. 2) consist of alternating layers of organic donors and inorganic anions as commonly observed in such type of materials. Despite the two kinds of compounds 1 and 2 crystallize in different space groups, their crystal structures are very similar, the main difference lies in the relative disposition of the inorganic layers in the c direction. The shape of these inorganic layers looks like sinusoidal curves running along the b direction. In compound 1, an in-phase disposition of successive sinusoidal curves, in the c direction is observed while in compound 2, these curves are out-of-phase, i.e. they are displaced in opposite directions with respect to each other along the b axis. This difference results in the doubling of the unit cell parameter c of compound 2.
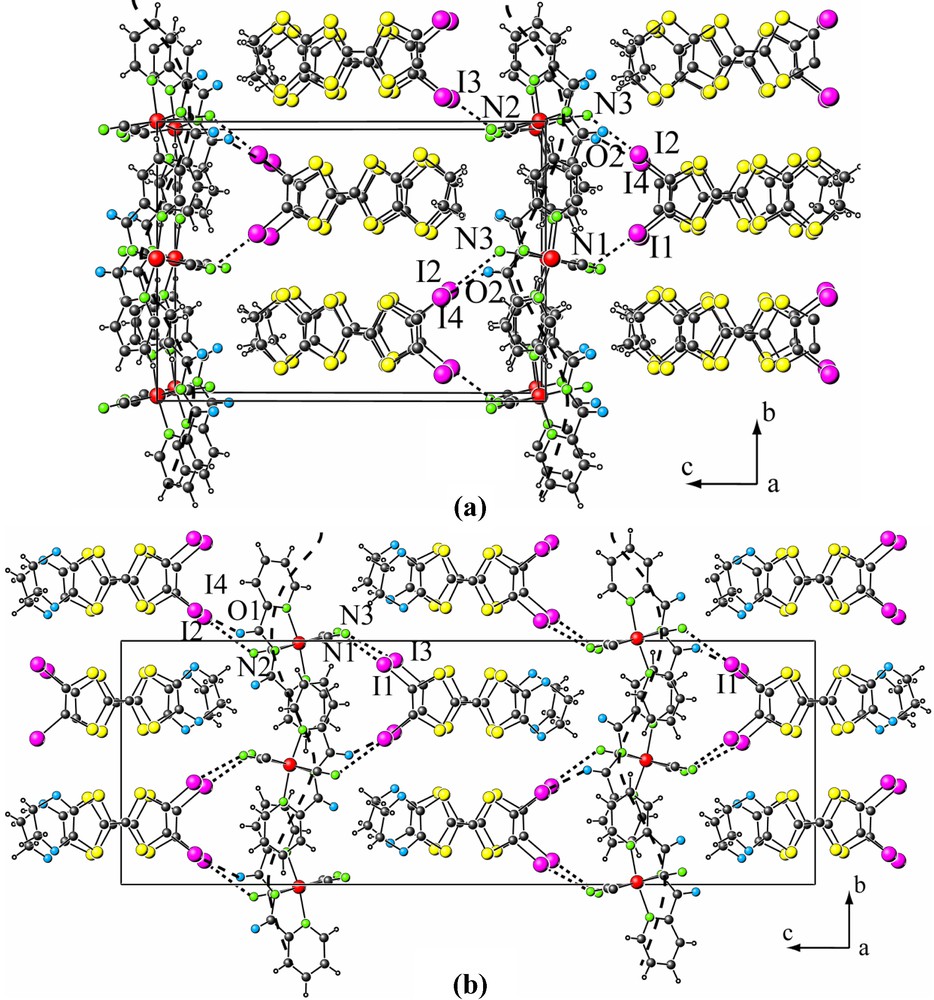
Crystal structures of compounds 1 (a) and 2 (b).
In both compounds, the bond lengths and bond angles within the [Fe(bpca)(CN)3]– units are very similar and compare well with those reported previously for such a unit [14]. The iron atom is in a distorted coordination octahedron. The average values of the Fe–C and Fe–N bond distances are equal to ca. 1.934(17) and 1.929(12) Å [versus 1.945(3), 1.936(2) Å in the precursor] [14]. In the inorganic layer (Fig. 3), the anions form planes parallel to the ab plane. This plane is built from infinite chains of anion in the b direction where all bpca ligands are parallel to each other. This chain alternate with another infinite chain along the [110] direction. The bpca ligands from two neighboring chains form an angle of 7.0(9)° for 1 and 13.2(3)° for 2 with significant overlap between the bpca ligands of two chains, the shortest distances between the pyridine rings of two anions in the a direction are given in Table 2. The difference between d1 and d2 values is due to a larger angle of 10.1(6)° between two adjacent pyridine rings of two anions for 2 compare to that of compounds 1 (6.5(8)°).
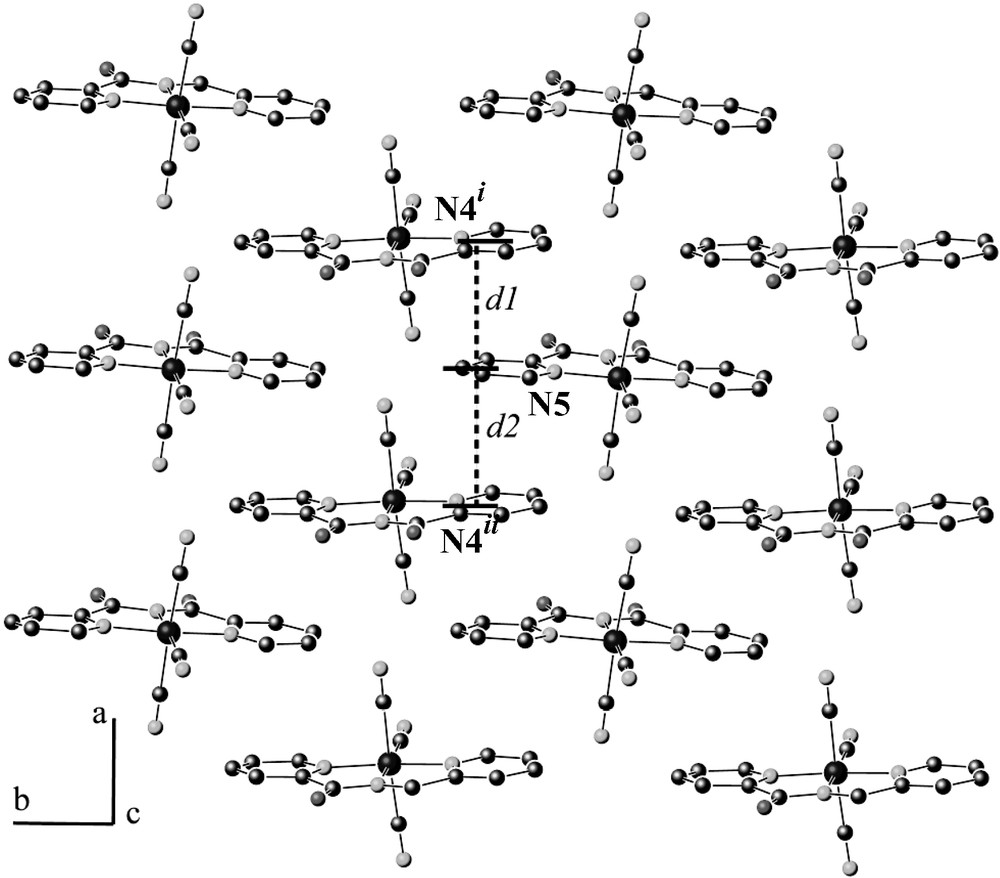
Projection of the inorganic layer in the ab plane for compound 1, see Table 2 for d1 and d2 values. i: –x, 1/2 + y, –z; ii: –1 – x, 1/2 + y, –z. for 1; i: 2 – x, y – 1/2, 1/2 – z; ii: 1 – x, y – 1/2, 1/2 – z for 2.
Interplanar separations (Å) for compounds 1–2. d1–d2: between pyridine rings along a direction: from mean plane (N5–C11–C12–C13–C14–C15) and N4 belonging to (N4–C4–C5–C6–C7–C8) pyridine rings (see Fig. 3) ; d3–d4 between TTF skeleton (S1–S2–C18–C19–S3–S4) and C26 (see Fig. 4)
| Compound | 1 | 2 |
| d1 | 3.457(23) | 3.917(50) |
| d2 | 3.563(23) | 2.986(51) |
| d3 | 3.853(17) | 3.746(12) |
| d4 | 3.592(16) | 3.513(12) |
Fig. 4 gives the details of the organic sublattice. The terminal dithiolane rings of the organic radical cations have a conformational disorder due to their non-planarity. Donor molecules present dimerized slipped stacks along the a axis with the –AB AB AB– sequence, the intra- and inter-dimer separations are given in Table 2. The structure of the organic layer is reminiscent to the β-type of packing usually observed in the BEDT-TTF based charge transfer salts [15]. The shortest intrachain contacts are given in Fig. 4. The side view of the chains with the shortest intermolecular contacts are shown in Fig. 4b,c for 1 and 2, respectively. A head to head arrangement of the molecules is observed for all compounds. The positive charges on each independent organic donor were assumed equal to +0.5 from the stoichiometry, since their estimation was not obvious from the examination of the intramolecular bond lengths.
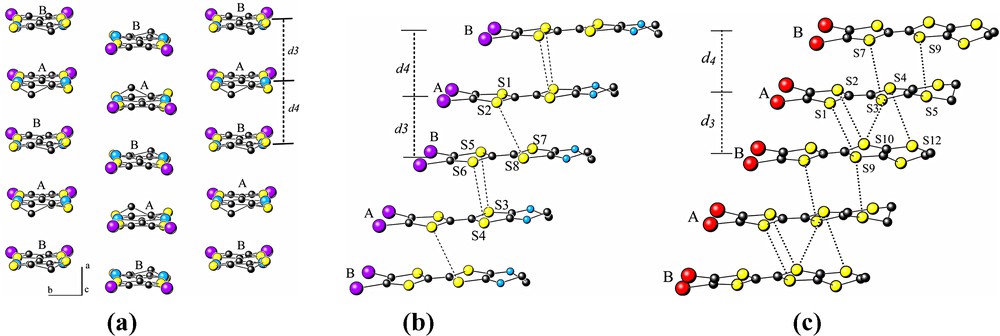
A view of the organic layer in the ab plane (a). Side views of the organic layer in compound 1 (b) and 2 (c), respectively (see Table 2 for d3 and d4 values).
In both compounds, the donor molecules and the anion are fully connected through –N···I– (shortest 2.933(14) Å) and –O···I– (shortest 2.760(16) Å) contacts (see Table 3 and Fig. 5). These contacts are about 10–15% shorter than the sum of the van der Waals radii for nitrogen and iodine (3.35 Å) and oxygen and iodine (3.32 Å) atoms.
Short –N···I– and –O···I– (Å) contacts in 1–2
| Compounds | 1 | 3 | |
| N(1)–I(1) | 2.989(16) | N(1)–I(1) | 3.022(10) |
| N(2)–I(3) | 2.933(14) | N(2)–I(2) | 3.279(11) |
| N(3)–I(2) | 3.332(20) | N(3)–I(3) | 3.009(10) |
| O(2)–I(4) | 2.760(16) | O(1)–I(4) | 2.794(8) |
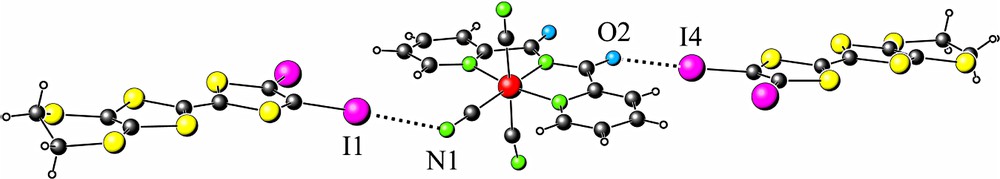
–N···I– and –O···I contacts in 1–2 (see Table 3).
2.2 Physical properties
2.2.1 Electrical conductivity
Fig. 6 shows the temperature dependence of electrical resistivities for compounds 1–2. Compound 1 (the DIET salt) shows semiconductive behavior from room temperature. Compound 2 is clearly semiconductive at low temperature but, at seen inset Fig. 6, it is weakly metallic around room temperature. The degree of the metallic behavior depends on the samples and the transition temperature varies in the range 270–290 K.
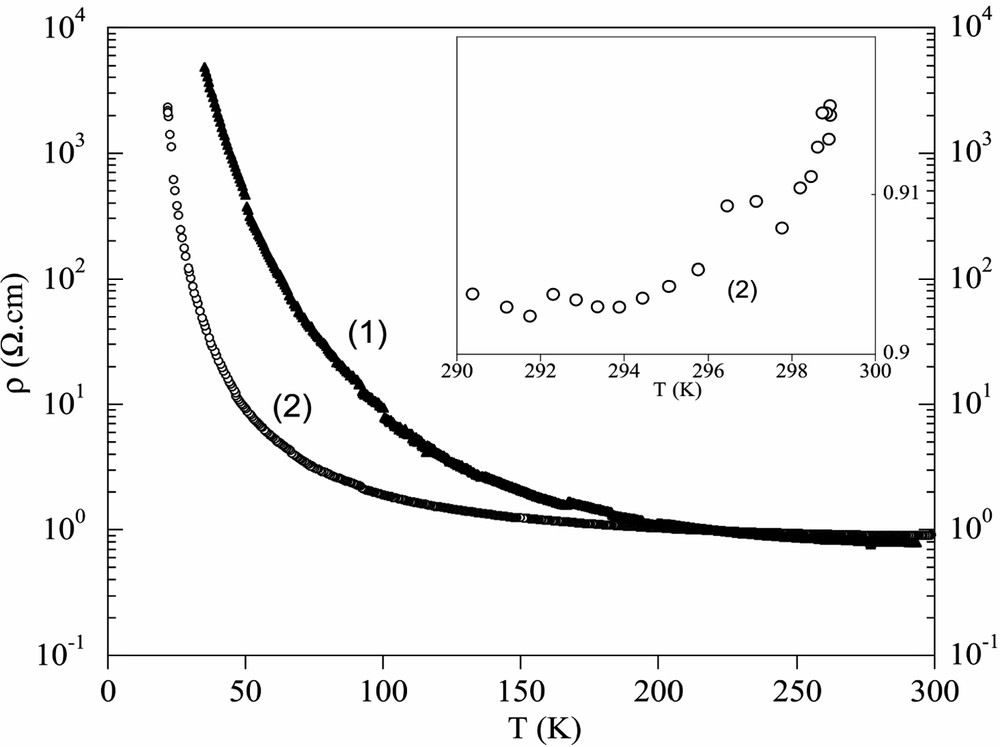
Temperature dependence of electrical resistivities for 1–2. Resistivity of 2 at room temperature is given inset.
2.2.2 Spectroscopic properties
Spectroscopic studies can provide information about interactions between donors and acceptors as well as electronic properties of conducting layers. The crystallographic data show that donor and acceptor molecules interact through N···I and O···I contacts, therefore it is to be expected that suitable bands attributed to vibrations of C≡N, C=O or C–I should be modified. On the other hand, it is well known that vibrational spectroscopy is a valuable tool to investigate charge distribution in conducting layers (Raman) and effects of electron—molecular vibration coupling (IR).
We began our investigations with the measurement of IR and Raman spectra of neutral donor molecules and (PPh4)[Fe(bpca)(CN)3]·H2O. Preliminary assignments of the most important vibrational bands and their intensity classification (w – weak, m – medium, s – strong) were performed. They are given as supporting information.
The Raman spectra of 1–2 within the frequency region below 1700 cm–1 are shown in Fig. 7. The IR spectra of the salts 1 and 2 are displayed in Fig. 8.
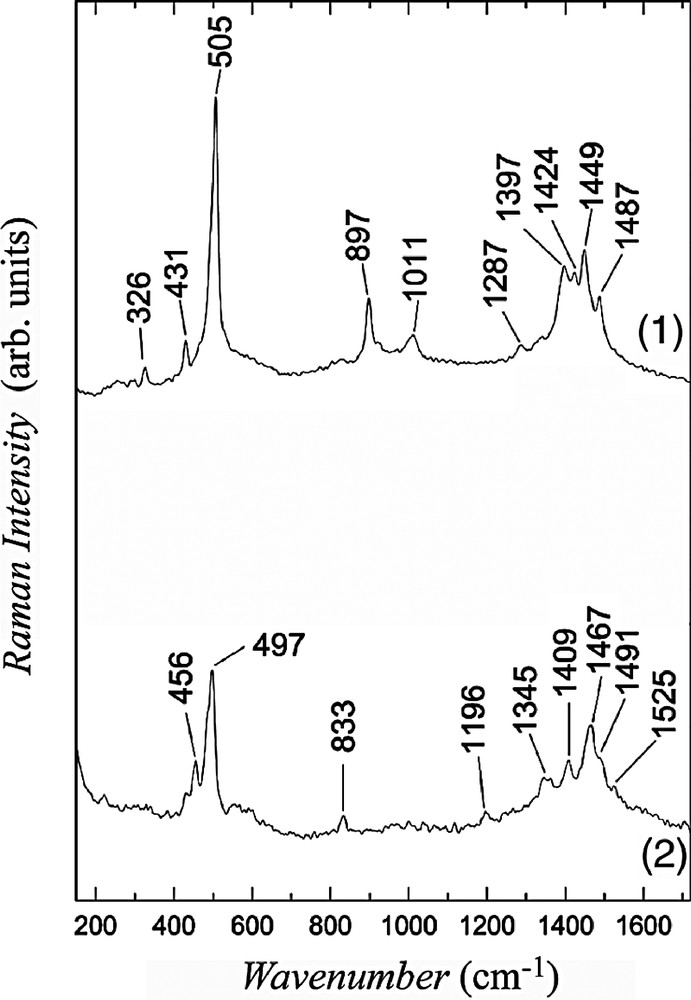
FT-NIR Raman spectra of 1–2 (excitation λ = 1064 nm).
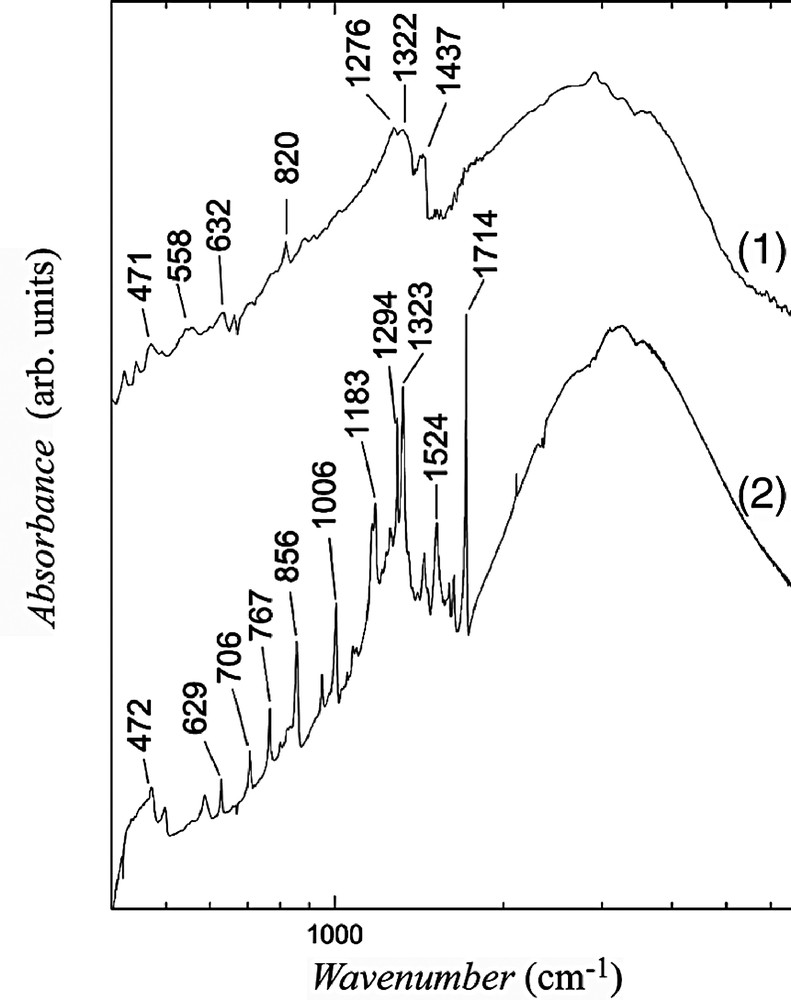
Infrared absorption spectra of 1 and 2. Note the logarithmic frequency scale.
Within the region of C≡N stretching, in the spectra of (PPh4)[Fe(bpca)(CN)3]·H2O we observed three IR bands at 2136, 2127, 2116 cm–1 (the band at 2116 cm–1 is stronger than the other two) and analogous three Raman bands at 2135, 2128, 2117 cm–1 (the bands 2128 and 2117 cm–1 are of equal intensity and they are stronger than the third one). Weak C≡N bands were only detected for the salt 2: in IR spectrum at 2111 cm–1 and in Raman spectrum at 2121 cm–1. The C=O bands, for (PPh4)[Fe(bpca)(CN)3] at 1718 cm–1 (IR) and 1715 cm–1 (Raman), were also found only for the salt 2 at 1714 and 1703 cm–1, respectively. The interaction between donor and acceptor has no considerable influence on these vibrational bands, nevertheless, one can suggest that the C=O groups interact slightly stronger with donors than the C≡N groups. At this stage of investigation, however, we cannot assign unambiguously donor bands related to C–I stretching, which should be located within the frequency region 500–600 cm–1.
It is well known that for molecules derived of TTF the frequencies of C=C vibrations are sensitive on the ionization degree: their positions shift strongly towards lower frequencies when the average charge grows. For example, such dependence upon the ionization degree was determined for BEDT–TTF [16,17] and BEDO–TTF [18] salts. A comparison of the Raman data for neutral molecules and salts shows that considerable frequency shift exists also for studied donors. The Raman bands related to the C=C stretching are collected in Table 4. We assume that in charge-transfer salts the C=C bands are enhanced by resonance Raman effect (resonance with charge-transfer transition), therefore, the C=C bands of acceptor molecule are not observed.
Frequencies of Raman bands within the region of C=C stretching vibrations (cm–1)
| Neutral molecules | Compounds | (PPh4) [Fe(pbca)(CN)3] | ||
| DIET | DIEDO | 1. (DIET) | 2. (DIEDO) | |
| 1552 | 1653 | 1524 | 1633 | |
| 1536 | 1540 | 1487 | 1491 | 1603 |
| 1515 | 1530 | 1449 | 1467 | 1586 |
| 1495 | 1497 | 1424 | 1409 | 1567 |
| 1397 | 1345 | 1472 |
A strong electronic absorption due to charge transfer between charged and neutral molecules in the conducting stacks and some vibrational features is observed in the IR spectra of the investigated compounds. Strong and broad vibrational features at 1500–1000 cm–1 for 1 and 1400–1200 cm–1 for 2 are consequence of the coupling of C=C vibrations with charge transfer. For 1 these bands are smeared and broad, what suggest stronger charge delocalization in 1 in comparison with 2.
2.2.3 Magnetic properties of 2 and 3
The thermal dependence of the χMT product of compound 2 together with that of the low-spin iron(III) mononuclear complex (PPh4)[Fe(bpca)(CN)3] (3) [χM being in both cases the magnetic susceptibility per mol of Fe(III)] in the temperature range 205–1.9 K are shown in Fig. 9a. The magnetic properties of complex 3, whose preparation and structural characterization were reported elsewhere [14] were included here in order to visualize the magnetic behavior of the magnetically isolated low-spin iron(III) unit [Fe(bpca)(CN)3]–, this entity being also present in complex 2. The value of χMT at 205 K for complex 3 is 0.64 cm3 mol–1 K and it decreases smoothly upon cooling to reach a value close to 0.49 cm3 mol–1 K at 1.9 K. This behavior is the expected one for a low-spin distorted octahedral iron(III) (S = 1/2) with spin-orbit coupling of the 2T2g ground term [19,20]. χMT for compound 2 at 205 K is 0.89 cm3 mol–1 K, a value which lies somewhat above that of complex 3 due to the presence of a part of DIEDO as a radical. Upon cooling, χMT for 3 strongly decreases and matches the χMT plot of 3 at T < 30 K (see inset of Fig. 9a). The magnetization versus H plots at 2.0 K for compounds 2 and 3 (Fig. 9b) are identical and correspond to what is expected for a low-spin iron(III) complex (S = 1/2). These features indicate that a strong antiferromagnetic interaction occurs in 2 between the DIEDO units, the [Fe(bpca)(CN)3]– entity remaining magnetically isolated. The very small χMT values resulting from the difference between the χMT plots of 2 and 3 prevent an accurate evaluation of the exchange coupling in the DIEDO chain along the a axis. The crystal structure shows a regular chain of organic donors dimers with one spin S = 1/2 per dimer which are strongly antiferromagnetically coupled. Such relatively large antiferromagnetic interactions have been reported in previous magneto-structural studies concerning related types of charge transfer salts [21].
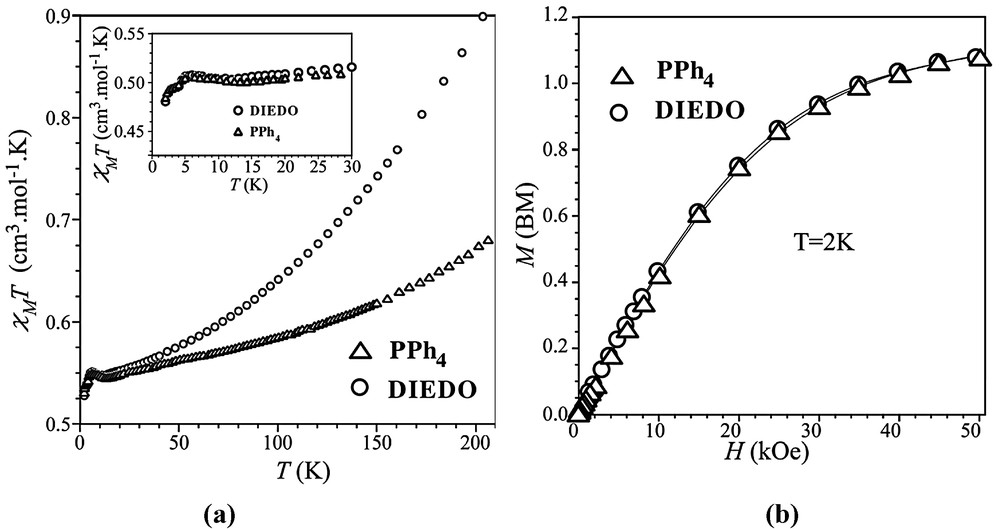
(a) Temperature dependence of the χMT product for compounds 2 and 3; (b) magnetization at 2 K for compound 2 and 3.
2.2.4 Electronic band calculations
The donor arrangement of 1 viewed along the molecular long axis is depicted in Fig. 10. There exist donor columns along the crystallographic a-axis and packing motif within the donor layer is so-called β-type. Calculated intermolecular overlap integrals of HOMOs of the DIET molecules indicate that the donor molecules are dimerized within the column and inter-column interactions are weak. Simple tight-binding band calculations indicate that there exist two pairs of quasi one-dimensional opened Fermi surfaces and this low dimensionality is in accordance with the semiconductive behavior at low temperature. Two symmetry related donor layers are included in the unit cell of the salt 2 and Fig. 11 shows one of the layers viewed along the molecular long axis. The temperature dependence of the electrical resistivity of 2 shows a weak metallic behavior from room temperature. This inconsistency between the results of electronic band calculations and electrical resistivity measurements may be due to the large on-site coulombic repulsion energy of the DIEDO molecule.
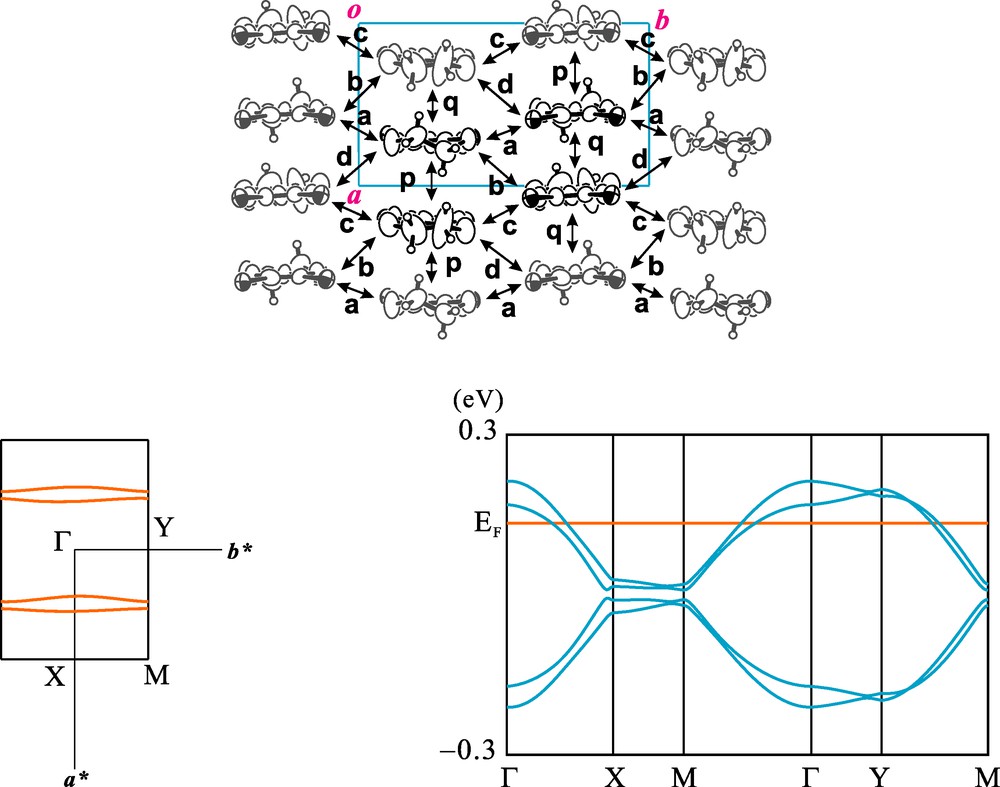
Calculated electronic band structure of 1. Overlap integrals (×103): p = −10.02, q = −9.08, a = −0.21, b = −0.61, c = 0.08, d = −1.35.
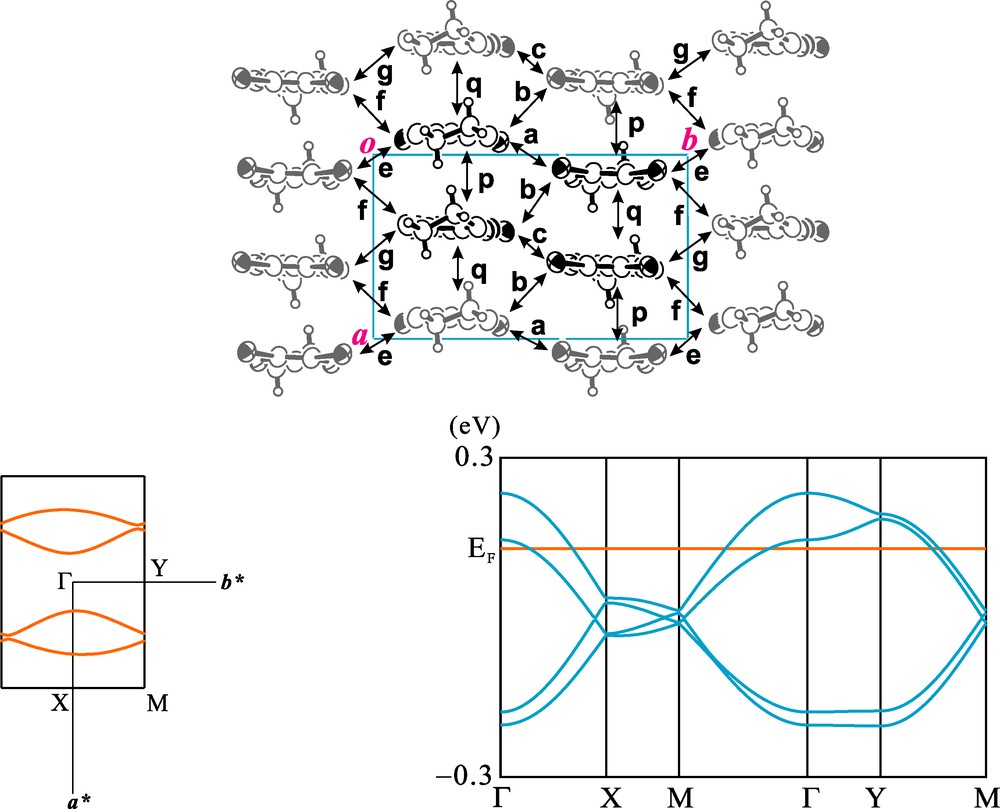
Calculated electronic band structure of 2. Overlap integrals (×103): p = −9.58, q = −9.41, a = −0.56, b = −1.63, c = −0.07, e = −1.33, f = −1.17, g = −1.21.
3 Conclusion
Two charge transfer salts composed of iodine substituted organic π-electron donors, DIET, DIEDO, and the paramagnetic [FeIII(bpca)(CN)3]– anion were investigated. Compound 1 is a semiconductor and compound 2 is a weak metal from room temperature to 250 K. The differences of the conducting behavior between these two salts are not completely consistent with the dimensionality of the calculated electronic band structures but mainly dominated by the on-site coulombic repulsion energy of donor molecules or band width of the conduction band, i.e. electronic correlation effects. The crystal structures consist or alternate layers of organic and inorganic units, the organic layer contains dimerized donor chains. Structural interactions between conducting and paramagnetic layers were established through very short –I(donor)···N(anion)– and –I(donor)···O(anion)– contacts. The magnetic properties of 2 are dominated by a strong antiferromagnetic coupling between the donor spins in the dimerized organic chain preventing any interactions with the paramagnetic inorganic units.
4 Experimental section
4.1 Synthesis
All experiments were conducted under nitrogen or argon. The solvents were distilled before use and the starting reagents were used as received. Donors DIET, DIEDO [12] and (PPh4)[Fe(bpca)(CN)3]·H2O [14] (3) were prepared following published methods. Black elongated plate crystals of 1–2 were obtained on platinum wire electrodes (∅ = 1 mm) by galvanostatic oxidation (I = ca. 1.0 μA) of DIET (10 mg) and DIEDO (10 mg), respectively, for 1 and 2, using (PPh4)[Fe(bpca)(CN)3]·H2O (100 mg) in CH2Cl2 (20 ml) as a supporting electrolyte. The stoichiometries of target materials were checked by X-ray crystal structure analysis.
4.2 Crystallographic data collection and structure determination
Single crystals of the title compounds were mounted on a Nonius four circle diffractometer (CDFIX, Université Rennes-1) equipped with a CCD camera and a graphite monochromated MoKα radiation source (λ = 0.71073 Å). Data collection was performed at room temperature for 1 and 120 K for 2. Effective absorption correction was performed (SCALEPACK [22]). Structures were solved with SHELXS-97 [23] and refined with SHELXL-97 [23] programs by full-matrix least-squares method on F2. Crystallographic data are summarized in Table 5.
Crystal data for compounds D2[Fe(bpca)(CN)3] (1–2)
| Compound | 1 | 2 |
| Formula | C31H16FeI4N6O2S12 | C31H16FeI4N6O6S8 |
| Formula weight (g mol–1) | 1452.67 | 1388.43 |
| Temperature (K) | 293(2) | 120(2) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21 | P21/c |
| a (Å) | 8.8238(2) | 8.6870(1) |
| b (Å) | 13.2891(3) | 12.6122(2) |
| c (Å) | 18.5042(5) | 36.0277(11) |
| β (°) | 91.1150(1) | 90.380(5) |
| Volume (Å3) | 2169.40(9) | 3947.19(14) |
| Z | 2 | 4 |
| R1 a, wR2 b [I > 2σ(I)] | 0.0710, 0.1745 | 0.0602, 0.1523 |
a R1 = Σ||Fo| – |Fc||/Σ|Fo|.
b wR2 = {Σ[w(Fo2 – Fc2)2]/Σ[w(Fo2)2]}1/2.
4.3 Physical property measurements
Temperature dependence of the electrical resistivity of 1–2 was measured by the standard four-probe method. Gold wires (10 μm diameter) were attached to a single crystal with carbon paste. Magnetic measurements on polycrystalline samples of 2 and 3 in the temperature range 1.9–205 K were carried out with a Quantum Design SQUID susceptometer under an applied magnetic field of 0.1 T. The susceptibilities were corrected for the diamagnetic contribution of the constituent atoms by using the Pascal constants [24]. The infrared absorption spectra of powdered compounds dispersed in KBr pellets with a typical concentration about 1:1000 were measured in the frequency range 400–7000 cm–1 at room temperature, using a FT-IR Perkin–Elmer 1725 spectrometer. The FT-NIR spectra were obtained using a Bruker IFS 66 spectrometer with FRA 106 Raman attachment which operates with Nd:YAG laser with a wavelength 1064 nm. The studied material was ground with KBr and slightly pressed to pellets in order to reduce the problem of sample overheating. This enabled us to increase the laser power and Raman signal without any risk of sample decomposition.
Supporting information available
Tables of reported IR and Raman bands are given as supporting information. CCDC-226286 and 226288 (for 1 and 2, respectively) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Center, 12, Union Road, Cambridge CB2 1EZ, UK; fax: (internat.) +44 1223 336 033; E-mail: deposit@ccdc.cam.ac.uk].