

1 Introduction
Irradiated by intense laser pulses, organic molecules can absorb two photons simultaneously; the probability of the two-photon absorption (TPA) phenomenon is proportional to the square of the laser intensity. In organic materials, TPA is a well-known process involved in a lot of different applications [1] for three-dimensional (3D) optical data storage [2], 3D-fluorescence imaging [3], photodynamic cancer therapy [4], optical limiting [5–8], and microfabrication [9–11]. This work concerns molecular engineering for the last two fields.
2 Optical power limiting in the visible range
2.1 General
A lot of investigations focusing on optical limiting materials, for the protection of eyes and optical systems against tunable lasers, have already been reported [12]. The materials involved require high transmission at low irradiance, but a large nonlinear absorption at damageable fluences. Efficient nonlinear absorbers, based on a TPA process, were designed [6,13,14]; this was considered as a very attractive approach due to the instantaneous response and the lack of saturation in highly concentrated materials. Concerning the optical limitation effect on nanosecond time scales, a further level has been reached by the occurrence, in the nonlinear absorption, of an excited-state absorption (ESA) process [13–19]. This three-photon absorption process can be described by two steps during the nanosecond pulse duration: an instantaneous TPA step followed by a very fast relaxation to the lowest excited-state and the ESA process. From the phenomenological point of view, this optical limiting behavior can be described, according to Eq. (1), in which z is the propagation direction and I the beam intensity:(1)
2.2 Experimental section
The TPA experimental set-up has already been presented elsewhere [8,24]. A Nd–YAG laser, pumping an optical parametric oscillator (OPO) and producing 2.6 ns pulses (with 10 Hz repetition rate) in the range of 450–650 nm, was used. TPA excitation spectra were recorded from fluorescence collected at 90° of the incident beam. Calibration of TPA spectra was performed from p-bis(o-methylstyryl)benzene as a standard [25,26] in order to scale two-photon excited fluorescence spectra and the pulse energy was kept low enough to ensure a quadratic dependence of the fluorescence signal on input energy. The ESA cross-section and decay time were measured from Kerr ellipsometry experiments as previously described [22]. The signals which, when observed for “long” delays (500 ps) between pump and probe beams, presented the same absorption profiles as those obtained in the zero delay, could be assigned to S1–Sn absorption. The three-photon coefficient α3 was obtained from nonlinear transmission measurements, as described in Ref. [8], by using relationship (4), where L is the sample thickness:(4)
2.3 Results and discussion
A molecular engineering work to design efficient two-photon absorbers from N4,N4′-bis-(4-methoxyphenyl)-N4,N4′-diphenyl-4,4′-diaminobiphenyl (1) (Fig. 1), for optical limiting in the visible range, was performed [27], by optimization of σTPA values or dynamics of excited-state.
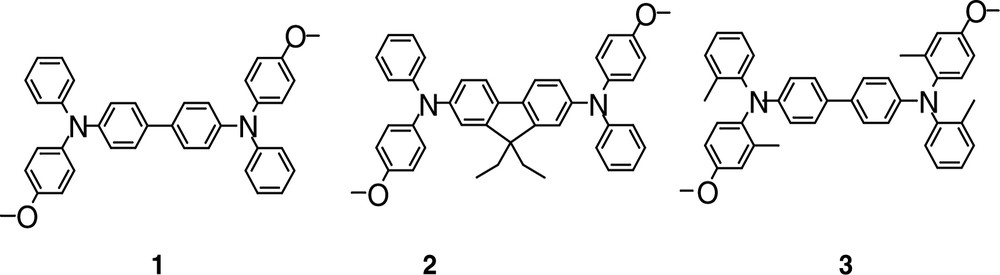
Molecular structures of 1–3.
Molecule 1 belongs to the large tetraphenyldiamine (TPD) family, which is currently used in organic luminescent diodes as hole injection layers [28,29] and is generally considered to be thermally stable. The molecular structure of 1 presents a D–π–D structure (in which D is a donor group) with moderate charge transfer character (for an efficiency in the visible range) due to the non-planarity of the biphenyl. Two strategies for 1 were considered in order to optimize α3 (Eq. (2)), allowing in each case, the use of an aminobiphenyl type substituent as the donor group while keeping transparent molecules (λmax between 349 and 374 nm): (a) the optimization of the charge transfer by increasing the system planarity (molecule 2, in where the biphenyl was replaced by a fluorene) in order to boost the TPA cross-section σTPA (from 40 × 10–50 cm4 s–1 photon per molecule—or 40 G.M. for Göppert–Mayer—for 1 to 100 G.M. for 2, see coefficients Table 1); leading, according to relationship (2), to better nonlinear absorption properties (α3 = 7950 cm3 GW–2 at 500 nm for 2 vs. 3900 cm3 GW–2 at 550 nm for 1 as reported in Table 1); (b) the optimization of the excited-state dynamics in order to slow down the relaxation processes and increase the value of τ01 which is also a relevant parameter in the expression of the three-photon absorption coefficient α3 (Eq. (2)).
Experimental TPA, ESA S1 → Sn and nonlinear absorption properties of molecules 1–3
| Compound | 1 | 2 | 3 |
| λmax/nm | 350 | 374 | 349 |
| σTPA/10–50 cm–4 s per photon per molecule | 40 | 100, 120 | 30, 35 |
| (λTPA/nm) | (550) | (550, 640) | (550, 585) |
| σESA/10–16 cm–2 | 1.8 | 1.4 | 3.8 |
| (λESA/nm) | (545) | (585) | (545) |
| τ01/ps | 360 | 320 | 420 |
| α3/cm3 GW–2 | 3900, 2000 | 7950, 2500 | 9000, 6000 |
| (λ3/nm) | (550, 610) | (500, 625) | (535, 585) |
Molecule 3 (Fig. 1), which presents identical charge transfer properties as molecule 1, leads to a similar TPA efficiency. However, when molecule 3 possesses methyl substituents on the peripheral rings which hinder their free rotation, nonradiative de-excitation processes decrease and the lifetime of the lowest excited-state increases compared to that of 1 (τ01 = 360 ps for 1 and 420 ps for 3 as reported in Table 1); moreover, the ESA cross-section (σESA = 1.8 × 10–16 cm2 for 1 and 3.8 × 10-16 cm2 for 3) induces a net increase in the nonlinear absorption properties (α3 = 9000 cm3 GW–2 at 535 nm for 3 and α3 = 3900 cm3 GW–2 at 550 nm for 1), with similar σTPA. A new approach involving unsubstituted oligomers, such as Stilbene 3 (a bistilbene) [17] or polyfluorenes derivatives [8,24], appears very promising in terms of TPA cross-sections in the visible range. In the light of the encouraging preliminary results from Stilbene 3, which presents α3 values larger than 10,000 cm3 GW–2, a study of N-poly(9,9-dihexyl-fluorene-2,7-diyl) (N-PDHF, as shown in Fig. 2) selected for their high photochemical stability was performed in solution as a function of the molecule length (monodispersed [8,24,30] systems and polydispersed for the longest molecules).
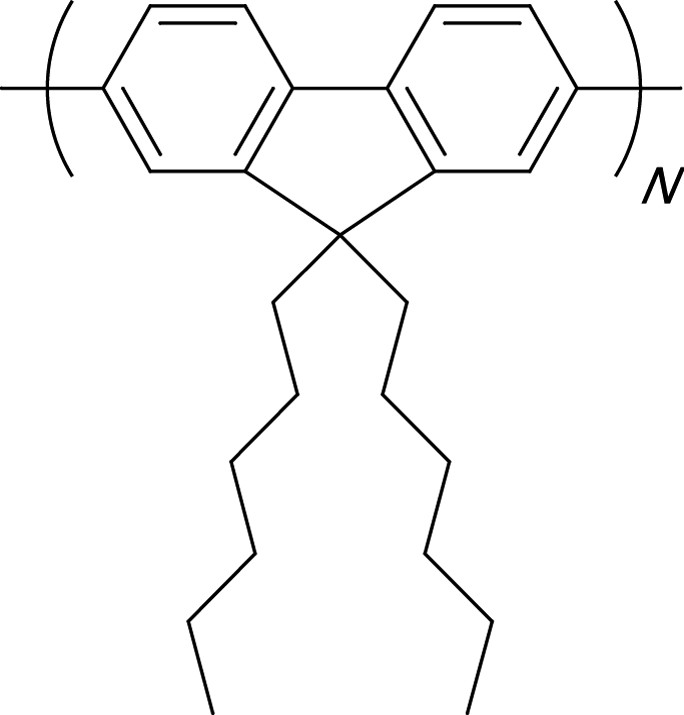
Chemical structures of N-PDHF oligomers.
A red shift of the linear absorption maxima of N-PDHF (in chloroform) is observed as a function of the length of the oligomer (Table 2); nevertheless, all molecules are transparent in the visible range.
Optical properties of N-PDHF
| N | λmax (nm) | λTPA (nm) | σTPA (10–50 cm–4 s photon per molecule) | α3 (cm3 GW–2) |
| 2 | 330 | 540 | 55 | 5000 |
| 4 | 364 | 602 | 600 | / |
| 16 | 373 | 620 | 4000 | 10 000 |
| 25 | 382 | 620 | 9000 | 30 000 |
| 60 | 388 | 620 | 20 000 | 100 000 |
The three-photon absorption coefficient α3 presents high values at resonance for the longest oligomers [31] (α3 = 100,000 cm3 GW–2 for N = 60 at a concentration of 200 g l–1) compared to other organic molecules such as symmetrically substituted molecules [22] which exhibit α3 values weaker than 10,000 cm3 GW–2 measured in the same exact conditions; this lead to efficient optical power limiting in the whole visible range (the maximum transmitted energy being lower than 10 μJ for an input energy of up to 350 μJ in a F/5 optical geometry).
TPA wavelength maxima and cross-sections are reported in Table 2. The TPA cross-sections of small oligomers increase with the length squared. This trend is rationalized in terms of excitonic coupling between monomers within the range of the effective length of the oligomer. Large TPA values, when compared to those of the susbstituted biphenyls 1–3 (see Table 1) optimized for the visible range, were obtained for the longest oligomers, which present a TPA efficiency of the order of 100 × 10–50 cm–4 s photon per molecule (100 G.M.). However, the most relevant parameter allowing to rationalize the TPA efficiency of oligomers is the cross-section density (TPA/N) which permits to overcome concentration effects due to the increase of the number of monomer units and to really determine the influence of the length of the oligomer. The values of this parameter for various N are reported in Fig. 3 and can be readily analyzed from the excitonic interaction model proposed to interpret high TPA cross-sections of this oligomer family [32,33].
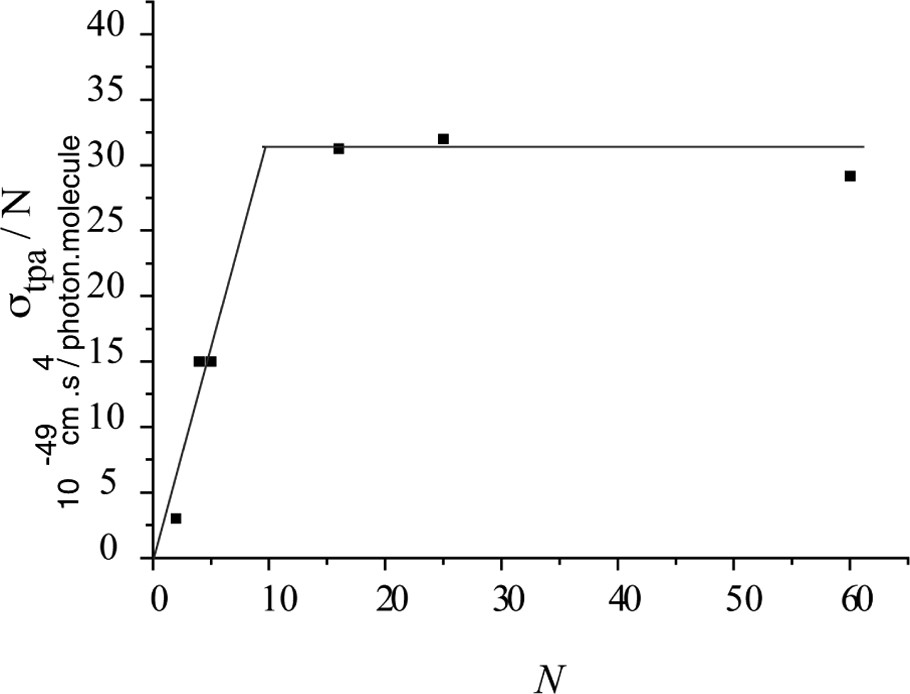
TPA cross-section density of N-PDHF with the length of the oligomer.
This coherent coupling of transition dipole moments between monomers (similar to that observed for J-aggregates [34]), induces a power law variation between transition dipole moment of the oligomer N and that of the monomer for the i → j transition, following Eq. (5).(5)
However, as illustrated by Fig. 4 for 2-PDHF, TPA and ESA resonances do not overlap efficiently for this family of compounds [8]; this observation allows to assume that further optimization of α3 properties could be expected.
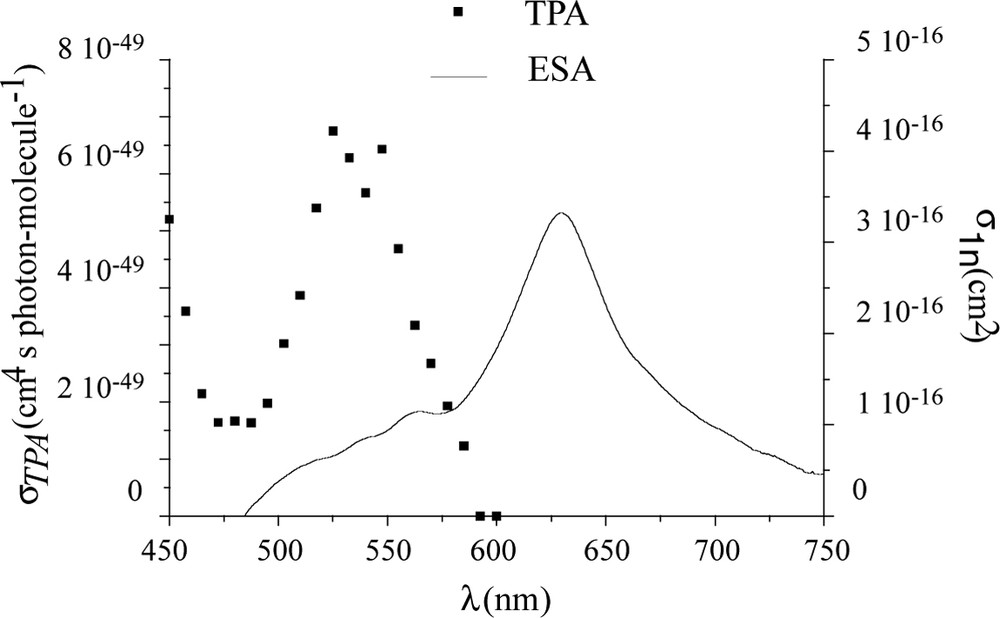
TPA-ESA overlap for 2-PDHF.
Further work is needed to find derivatives showing an improved overlap and therefore a larger optimized nonlinear response; in that perspective, 2D and/or 3D dendrimers of fluorenes could to be of great interest in the design of molecules in order to increase the excited-state level density and enhance TPA cross-sections (i.e. efficient optical power limiting properties).
3 3D microfabrication
3.1 General
TPA induced photopolymerization is a very promising method for the fabrication of 3D microstructures [9,11,36–40]. The chromophore acting as the photoinitiator is excited by the simultaneous absorption of two photons leading to chemical polymerization reactions; due to a tight confinement of the excitation volume around the focal point, this method can produce micrometer sized and highly resolved objects. This TPA based technique led recently to the production of optical devices [41], optical storage [10], photonic bandgaps [42,43] and micromachines [44]. However, high laser intensities and long exposure durations were required due to the use of commercial UV photoinitiators with weak TPA efficiencies. These works were performed using onerous femtosecond Ti: sapphire lasers at intensities close to the damage thresholds of the materials. These operating conditions are a major drawback to the widespread utilization of this promising technique of fabrication. In this context, a molecular engineering work to improve TPA photoinitiators was needed in order to use cheaper lasers with lower peak powers. The 3D microfabrication by TPA polymerization employing a new efficient photoinitiator and using a low cost microlaser has recently been demonstrated [45]. The symmetrically substituted fluorene 2 was used to initiate TPA polymerization of acrylates in the visible range at 532 nm; molecule 2 presents a high TPA efficiency in the visible, as demonstrated above, with substituents able to produce free radicals, as required by the initiation step as shown in Fig. 5.
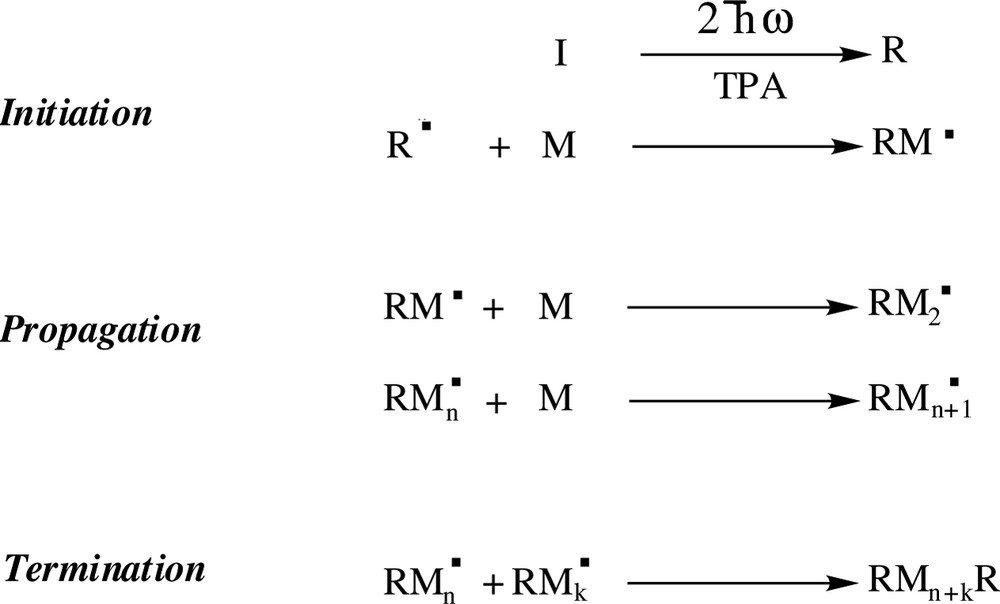
Different steps in a radicalar photopolymerization reaction. I represents an initiator molecule, R is a radical and M a monomer.
New conjugated ketones 5–71 (Fig. 6) with a molecular structure similar to that of the Michler ketone [46] 4, were presented for the IR photoinitiation at 1064 nm.
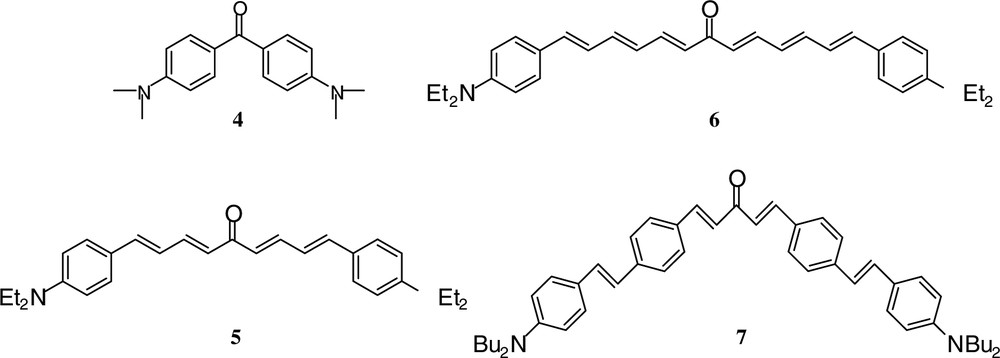
Molecular structures of 4–7.
The oxidation potential, reported in Table 3, is also a relevant parameter for the characterization of the photopolymerization process since a monoelectronic transfer from the initiator to the acrylate monomer is required; the low values obtained for these electron rich photoinitiators, show the ability for both molecules to give an electron from the donor group and create a radical.
σTPA, Epa and Imin values for molecules 2, 5–7
| Photoinitiator | Weight (%) | σTPA (cm–4 s) λirradiation (nm) | Epa (mV) vs. EFc+/Fc | Imin (W m–2) for texpo = 10 ms (laser wavelength in nm) |
| 2 | 3 | 80 × 10–50 (532) | / | 1.4 × 1014 (532) |
| 2 | 0.5 | 80 × 10–50 (532) | +250 | 2.8 × 1014 (532) |
| 5 | 0.5 | 50 × 10–50 (1064) | +260 | 8.7 × 1014 (1064) |
| 6 | 0.5 | 100 10–50 (1064) | +170 | 5.1 × 1014 (1064) |
| 7 | 0.5 | 65 × 10–50 (1064) | +170 | 3.9 × 1014 (1064) |
3.2 Experimental section
As described above, TPA spectra of compounds 5–7 were obtained (Table 3), in the 780–1120 nm range; TPA cross-section at 1064 nm were determined using Rhodamine B as a standard [47] in order to scale the two-photon excited fluorescence spectra and are reported in Table 3. All TPA induced photopolymerization experiments at 532 nm were performed using a frequency doubled Nd–YAG Nanolase microlaser from JDS Uniphase (λ = 532 nm, 0.5 ns pulse duration, 4 μJ maximum pulse energy, 6.5 kHz repetition rate) focused through a ×5 objective lens; for 1064 nm experiments, the Nd–YAG Nanolase microlaser produced pulses with a duration of 0.64 ns, a maximum energy of 5.4 μJ, and a repetition rate of 13.6 kHz focused through a ×40 objective lens with a numerical aperture (NA) of 0.75. The films were placed on a three axis piezoelectric stage allowing their translation relative to the laser focal point. The resins consisted in 0.5–3% photoinitiator, 70% monomer (tris(2-hydroxyethyl) isocyanurate triacrylate with a radical inhibitor level of 100 ppm) and 27–30% polymer binder (poly(styrene-co-acrylonitrile) (75/25)). Films with a thickness of between 20 and 50 μm were obtained by solvent evaporation from a chloroform solution.
The initiation efficiencies of these systems were evaluated [48] by the determination of the threshold intensities for a given exposure duration. The incident intensity distribution, which is assumed to be Gaussian, determines the shape of the polymerized volume. Using the microlasers at 532 or 1064 nm, several studs of polymer were fabricated in the resin. Fig. 7 presents an image of a typical stud from the scanning electron microscope; the studs, which can be compared to a rotation ellipsoid with their long axis corresponding to the focal axis of the laser beam, lie down on the glass substrate during the dissolution of polymerized areas in acetone.

Scanning electron microscope image of a stud lying on the glass substrate after dissolution with acetone.
The threshold intensity inducing polymerization Imin, can be assessed from the intensity associated to the edge of the polymerized volume (Fig. 8).
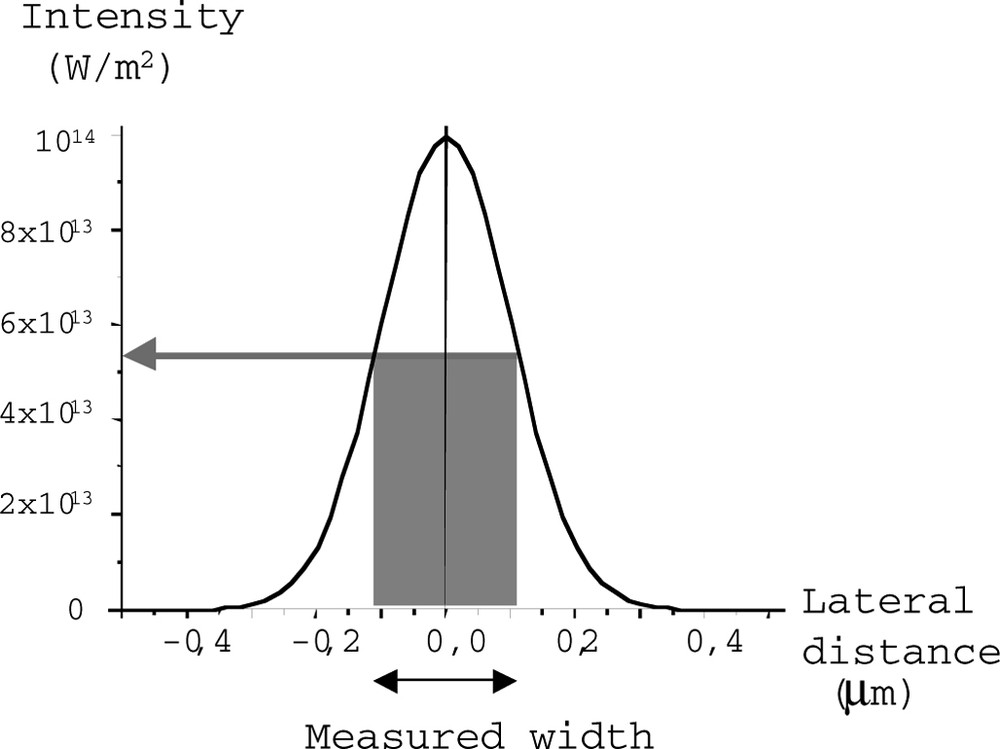
Determination of the polymerization threshold intensity.
The value of Imin is given by Eq. (5), where W is the stud width (polymerized volume) and ω0 the beam waist.(5)
3.3 Results and discussion
Results were analyzed for exposition time texpo = 10 ms (see Table 3) comparing data obtained for molecule 2 at 532 nm and molecules 5–7 at 1064 nm.
The comparison of the data for molecule 2 at 3.0% or 0.5% shows that, as expected, a lower concentration leads to a lower sensitivity. Although both molecules present similar TPA properties, photoinitiators 5–7 designed for IR are less sensitive than 2 designed for visible light. Comparing to the commercial resins for UV photopolymerization generally involved in TPA, these optimized initiators led to a significant increase of the sensitivity during fabrication.
This work provides a quantitative study of the TPA induced polymerization efficiency and allows us to compare different photoinitiators. However, values of threshold energies represent the global initiation efficiency of molecules: (1) their TPA efficiency, (2) their ability to create radicals which (3) will react with the monomer to induce the polymerization, while value of the TPA cross-section reflects only the efficiency of molecules to induce the first step of the initiation stage.
4 Conclusion
This paper summarizes a wide range of work on engineering and characterization of TPA chromophores, targeting optical limiting in the visible and 3D microfabrication applications. A same molecule, adequately substituted can be used efficiently for both applications.
For optical limiting in the visible range, the oligomer approach based on excitonic interactions opened a new way to enhance the TPA efficiency, leading to promising nonlinear absorption responses. The study of different dendritic systems to optimize this response through ESA properties is in progress [49,50].
3D micro-objects can be prepared by TPA based photopolymerization: a 20 μm diameter Euro coin [45] and a micrometer-sized Archimedes' screw are presented in Fig. 9. The design of efficient TPA initiators at 532 and 1064 nm have allowed to use weak power microlasers making this promising technique viable for industrial applications. From this point of view, the study of 3D micro-rotors controlled by optical tweezers is in progress under various conditions of power laser and rotation speed [51].
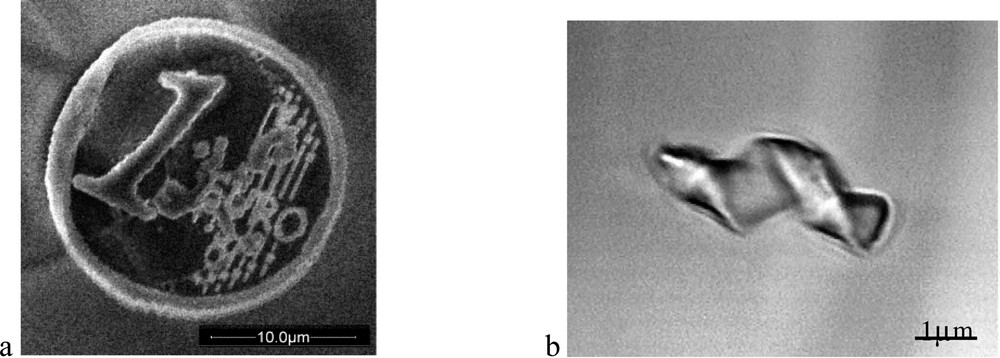
Two fabricated micro-objects: (a) a micro Euro coin and (b) a micrometer sized Archimedes' screw.
Acknowledgments
The authors would like to thank the ‘Délégation générale pour l'armement’ (DGA) and the GDR ‘Matériaux et Fonctions de l'optique non linéaire’ for their financial supports.
1 Synthesis will be published elsewhere.
Vous devez vous connecter pour continuer.
S'authentifier