

1 Introduction
o-(Diphenylphosphino)benzaldehyde, PPh2(o-C6H4CHO), can be easily prepared by the Hoots et al. [1] modification of the Schiemenz and Kaack [2] original synthesis. Since its preparation it has been used as ligand to several transition metals. It can behave as P-monodentate, as P,O-chelate or as P,C-chelate as in acylhydride complexes formed by the C–H activation of the CHO functionality. It has also been found to stabilize intermediates in the reactions of the aldehyde group with metal atoms.
Hemilabile ligands, with ‘soft’ phosphorus and ‘hard’ nitrogen donor atoms, are of particular interest in catalytic processes. One half of the ligand is capable of dissociating from an otherwise stable chelate complex, generating a vacant site on the metal ion for potential substrate binding. o-(Diphenylphosphino)benzaldehyde has been widely used to prepare hemilabile ligands, by condensation of the aldehyde group with primary amines to give imines. Potentially bidentate PN, terdentate PNN or tetradentate PNNP ligands have been prepared and by using asymmetric primary amines, optically active ligands have been obtained. Their late transition metal complexes, isolated or prepared “in situ”, have been used as catalysts in several organic transformations. PPh2(o-C6H4CHO) has been found to stabilize hemiaminals, which are intermediates in the condensation reaction of aldehydes with amines.
In this account, the behavior of PPh2(o-C6H4CHO) as ligand towards transition metals, its ability to generate hemilabile ligands containing one or two phosphine and one or two imine functionalities and their transition metal compounds will be discussed.
2 Behavior as ligand to transition metals
Four different coordination modes have been reported for PPh2(o-C6H4CHO). It behaves as P-monodentate ligand in complexes of palladium or platinum(II) such as cis-[MCl2(PPh2(o-C6H4CHO))2] [3,4], or [NBu4]2[Pt(C6F5)3(PPh2(o-C6H4CHO))] [5], obtained from halide or benzonitrile compounds by substitution reactions; in the iridium(III) complex [IrCl(PPh2(o-C6H4CHO))(PPh2(o-C6H4CH=NEt))(tetrachloro-o-catecholato)], obtained by oxidative addition of tetrachloro-o-quinone to [Ir(PPh2(o-C6H4CH=NEt))2]+ followed by hydrolysis of one imino group to give a formyl group [6]; or in carbonylated complexes of tungsten(0) [W(CO)5(PPh2(o-C6H4CHO))] [7] or rhodium(I), [Rh(CO)(PPh2(o-C6H4CHO))(μ-pz)]2 (pz = pyrazolato ligand) [8] or trans-[RhCl(CO)(PPh2(o-C6H4CHO))2] [9] obtained by carbon monoxide displacement by PPh2(o-C6H4CHO).
A frequent reaction pattern observed for this ligand 1, is the chelate-assisted oxidative addition to several late transition metals in low oxidation states to yield acylhydride derivatives such as 2–7 in Scheme 1. This scheme shows the products obtained when using (i) platinum(0) [10,11], (ii) cobalt(I) [12] (iii) iridium(I) [13] or (iv–vi) rhodium(I) [9,14–17] complexes as starting materials. These reactions are believed to occur by P-coordination of phosphine followed by oxidative addition of aldehyde to give hydrides containing acylphosphine P,C-chelates.

Oxidative addition reactions of PPh2(o-C6H4CHO).
PPh2(o-C6H4CHO) is one of the simplest bidentate P,O chelating agents. It behaves as chelating phosphine-aldehyde with the aldehyde portion bonded to the metal in a σ-fashion, through oxygen, or in a π-fashion, through both oxygen and carbon. The coordination mode of the aldehyde depends on the transition metal, the oxidation state, the other ligands present in the complex and the formal charge of the complex. Chart 1 shows some examples of both types of coordination modes.

Chelating coordination modes of PPh2(o-C6H4CHO).
The P,σ-aldehyde mode (η2-) has been observed in trans,cis-[RuCl2(η2-PPh2(o-C6H4CHO))2] [3], fac-[ReX(CO)3(η2-PPh2(o-C6H4CHO))] (X = Cl, Br) [18] and also in neutral 8 and cationic 9 rhodium(III) complexes shown in Chart 1. Complex 8 has been prepared by reacting [RhCl(Cod)]2 (Cod = 1,5-cyclooctadiene) with PPh2(o-C6H4CHO). The oxidative addition of one PPh2(o-C6H4CHO) and the P,σ-aldehyde coordination of a second molecule occur with concomitant diolefin displacement [9]. In contrast with this, the reaction of [IrCl(Cod)]2 with PPh2(o-C6H4CHO) gives the thermally unstable [IrH(PPh2(o-C6H4CO))Cl(Cod)]. This complex contains coordinated hydride, acyl and olefin and has been proposed as model for an intermediate in the metal catalyzed hydroacylation of olefins [3]. Complex 9 has been prepared by the reaction of [Rh(Cod)2]+ with PPh2(o-C6H4CHO), most likely via an acylhydride olefin intermediate such as [RhH(PPh2(o-C6H4CO))(PPh2(o-C6H4CHO))(Cod)]+, followed by C=C insertion into the Rh–H bond and rearrangement to give the thermodynamically stable η3-allyl derivative. Complex 9 is a catalyst precursor in the catalytic hydroformylation of olefins. It reacts with [Rh(Cod)(PR3)2]+ complexes (R = aryl) to afford stable unsaturated 16-e– rhodium(III) complexes 10 (Eq. (1)). The formation of 10 requires the opening of the P,σ-aldehyde chelate in 9, followed by the oxidative addition of aldehyde to the rhodium(I) complex prior to phosphine exchange [19].

The P,π-aldehyde mode (η3-) of PPh2(o-C6H4CHO) has been observed in cobalt(I) [20] and tungsten(0) [21] complexes 11 and 12 shown in Chart 1. Complex 12 contains also P-monodentate PPh2(o-C6H4CHO) and is transformed into [W(CO)2(η3-PPh2(o-C6H4CHO))2] 13, with two P,π-aldehyde chelates, by a decarbonylation reaction. Further treatment of 13 with PPh2(o-C6H4CHO) results in carbon–carbon coupling of two PPh2(o-C6H4CHO) ligands to yield the dioxyl derivative 14 [W(CO)(η3-PPh2(o-C6H4CHO))(Ph2PC6H4CH(O–)CH(O–)C6H4PPh2], containing a PO2-terdentate ligand, as shown in Eq. (2).
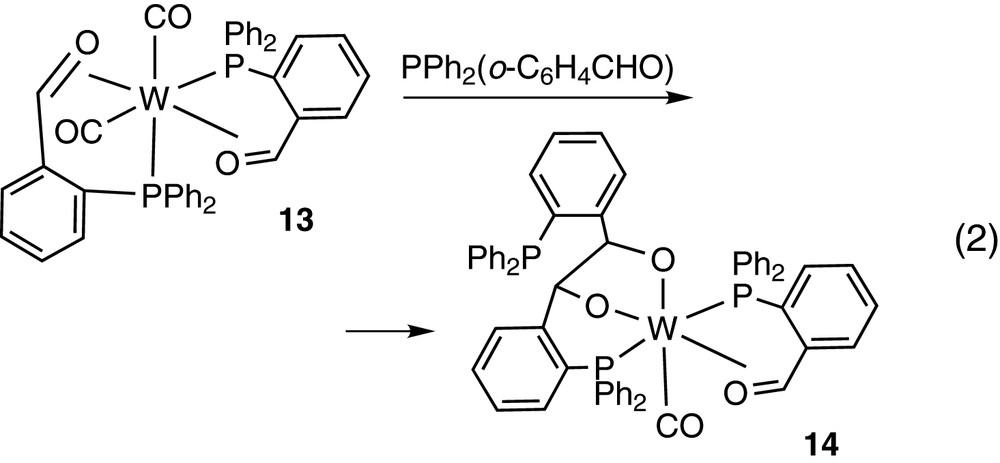
A related technetium complex [TcCl2(η3-PPh2(o-C6H4CHO))(Ph2PC6H4CH(O–)CH(O–)C6H4PPh2], containing the similarly obtained P2O2-tetradentate ligand has also been reported [22].
The insertion of aldehydes into transition metal hydrides plays a role in industrial processes such as the reduction of aldehydes to alcohols. PPh2(o-C6H4CHO) has been used to stabilize the proposed intermediate alkoxide or hydroxyalkyl late transition metal complexes. So, chelate assistance makes PPh2(o-C6H4CHO) to insert into the M–H bond of HMn(CO)5 to give the hydroxyalkyl derivative Mn(CO)4(PPh2(o-C6H4CHOH)) [23] as shown in Eq. (3),

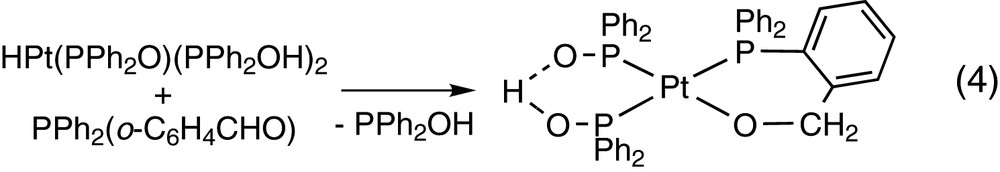
while the platinum derivative HPt(PPh2O)(PPh2OH)2 gives a cyclic platinum alkoxide Pt(PPh2O)(PPh2OH)(PPh2(o-C6H4CH2O)) [24] (see Eq. (4)).
The reactions in Eq. (5) show that P-coordination of PPh2(o-C6H4CHO) occurs prior to the reaction of the aldehyde. The rhodium(I) 2,2′-bipyridine complex undergoes first the oxidative addition of PPh2(o-C6H4CHO) to give the acylhydride 15 that also contains a P-monodentate PPh2(o-C6H4CHO). The free aldehyde inserts then into de Rh–H bond to give the final product 16 [16].

In the reaction of the related dihydrazone derivatives [RhCl(Cod)(H2NN=C(R)C(R)=NNH2)] (R = Me, H) the free aldehyde undergoes two competitive reactions: (i) insertion into Rh–H to give the hydroxyalkyl complex [Rh(PPh2(o-C6H4CO))(PPh2(o-C6H4CHOH))(H2NN=C(R)C(R)=NNH2)]+, similar to 16 and (ii) addition of a free amino group of the dihydrazone to give a stable hemiaminal group in the corresponding complex (vide infra) [9,15].
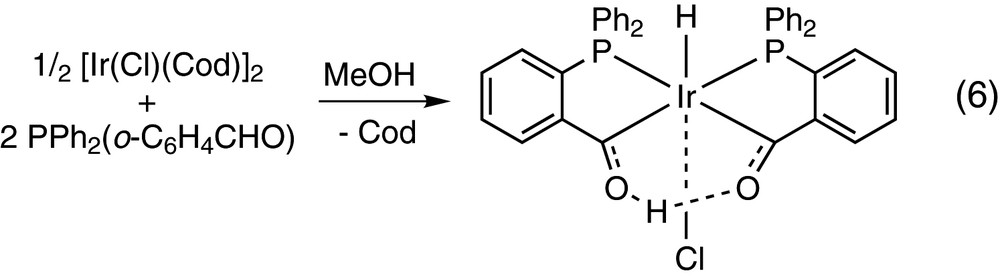
The iridium(I) toluidine complex [IrCl(CO)2(p-NH2C6H4CH3)] also undergoes the oxidative addition of PPh2(o-C6H4CHO) to afford the acylhydride [IrH(PPh2(o-C6H4CO))Cl(CO)(PPh2(o-C6H4CHO))] containing also a P-monodentate PPh2(o-C6H4CHO) and trans P atoms, but in this case no further reaction of the aldehyde has been reported [13]. [IrCl(Cod)]2 reacts with PPh2(o-C6H4CHO) in methanol to yield the hydridoirida-β-diketone complex [IrH{(PPh2(o-C6H4CO))2H}Cl] shown in Eq. (6). This reaction is believed to proceed by two consecutive oxidative additions of PPh2(o-C6H4CHO) to give a diacyldihydride Ir(V) species, which undergoes Ir-to-O proton transfer to give the final product [25].
3 Hemilabile phophine-imine ligand precursor
o-(Diphenylphosphino)benzaldehyde has been extensively used to obtain hemilabile bi- tetra- or terdentate ligands, such as those shown in Chart 2, by its reaction with amines.

Hemilabile phosphine-imine ligands prepared from PPh2(o-C6H4CHO).
A variety of bidentate iminophosphines PPh2(o-C6H4CH=N–R) (L1), abbreviated as PN-R, with R = alkyl [26–28], aryl [26,29] or amine [30,31] have been prepared. In most of the corresponding transition metal complexes, collected in Table 1, they behave as chelating ligands.
Transition metal complexes containing bidentate iminophosphines PPh2(o-C6H4CH=NR) (PN-R)
| Complex | R | References |
| [Mo(CO)4(PN-R)] | p-MeOC6H4; CH2=CH–CH2 | [26] |
| Me; Et; Pri; But; Me–NH | [32] | |
| [Pd(PN-R)2]2+ | But | [29] |
| [Pd(CXN)(PN-R)]+ | Me; Et; Prn; Pri; But; C6H5; Me–NH | [30] |
| [Pd(η2-olefin)(PN-R)]+ | p-MeOC6H4; Me; But; Bornyl | [28] |
| [Pd(η2-olefin)(PN-R)2]+ | p-MeOC6H4; But | [28] |
| [Pd(η3-allyl)(PN-R)]+ | p-MeOC6H4; Me; But; Bornyl | [33] |
| C6H5(CH3)CH; Cy(CH3)CH; Naftyl(CH3)CH | [34] | |
| [Rh(PN-R)2]+ | Et; Prn; Pri; But | [27] |
| C6H5(CH3)CH | [35] | |
| [Rh(Cp*)Cl2(PN-R)] | Piperidine | [31] |
| [Ir(PN-R)2]+ | Et; Pri; But | [36] |
The cyclometallated palladium complexes [Pd(CXN)(PN-R)]+ (CXN = azobenzene, 2-phenylpyridine) show relative trans configuration between the phosphorus atom and the nitrogen atom of the ortho-metallated ligand [30]. Olefin palladium(0) complexes may contain bidentate or P-monodentate PN-R ligands in [Pd(η2-olefin)(PN-R)] or [Pd(η2-olefin)(PN-R)2] complexes, respectively [28]. The allyl derivatives [Pd(η3-allyl)(PN-R)]+ are fluxional and show a very fast conformational change of the P–N chelate ring, which moves above and below the P–Pd–N coordination plane. These complexes react with secondary amines to give allyl amines and the amination rates are higher than those of related [Pd(η3-allyl)(α-diimine)]+ derivatives [33].
Palladium complexes in the presence of PN-R ligands (R = ortho-sustituted aryl groups) catalyze the oligomerization of ethene. Increasing the steric bulk at the nitrogen donor results in higher molecular masses [37]. These systems have also been tested in the palladium catalyzed reductive carbonylation of nitrobenzene [29]. The PN-(cyclohexyl) ligand has proved very useful for the palladium catalyzed carbostannylation of alkynes [38]. Several optically active PN-R ligands, prepared by Schiff base condensation with optically active amines, have been used in the palladium catalyzed enantioselective allylic alkylation [34,39] and chiral phosphine-hydrazone ligands have been found to be very effective [40].
Rhodium [27] or iridium [36] complexes [M(PN-R)2]+ (R = alkyl) have been isolated and their reactions with oxygen or carbon monoxide have been studied. The steric crowding of the PN-R ligands and therefore the dimensions of the resulting cavity can account for the different reactivity of the complexes. The rhodium derivatives are efficacious oxygen carriers provided the alkyl group is not too bulky. The phosphine-hydrazone ligand with R = piperidine, behaves as a P-monodentate ligand towards rhodium(III) affording the [Rh(Cp*)Cl2(PN-R)] complex [31].
The reaction of PPh2(o-C6H4CHO) with primary diamines may give tetradentate PNNP ligands. When using m- or p-phenylenediamines PNNP binucleating agents are formed and the corresponding molybdenum complexes [Mo2(CO)8(PNNP)] contain one imine functionality and one phosphine moiety bonded to each metal atom [26].
Potentially chelating diamines may afford tetradentate chelating PNNP ligands, L2–L5 depicted in Chart 2. N,N′-bis[o-(diphenylphosphino)benzylidene]ethylenediamine (L2H,H) is a most versatile ligand, being capable of coordinating to transition metals as a tetra- (k4-) or terdentate (k3-) ligand in mononuclear complexes and also as bridging ligand, depending on the metal and/or the reaction conditions. The k4-PNNP donor mode has been observed in tetrahedral [M(L2H,H)]+ (M = Cu, Ag), square-planar [Ni(L2H,H)]2+ [41], pentacoordinated [M(L2H,H)L]+n (M = Ag(I), L = ButNC; M = Ni(II), Co(II) L = Br, I) [41,42] and octahedral [RuX2(L2H,H)] (X = OAc, Cl) or [Fe(L2H,H)(CH3CN)2]2+ [43] complexes. The terdentate coordination, in the k3-PNP mode with one of the imine groups remaining uncoordinated, has been reported for tetrahedral [Cu(L2H,H)(ButNC)]+ [41] and the k3-PNN mode with one of the phosphine groups remaining uncoordinated, has been observed in octahedral [M(Cp)(L2H,H)]+ (M = Fe, Ru) [44] or [M(CO)3(L2H,H)] (M = Cr, Mo) [45]. [Ru(Cp)(CH3CN)]2(L2H,H) [44] and [M(CO)4]2(L2H,H) (M = Cr, Mo) [45] are dimer compounds containing the bridging-L2H,H ligand.
PNNP ligands with a biaryl bridging unit such as L3 afford cationic square-planar mononuclear [M(L3Me,Me)]+ (M = Rh, Ir) [46] or [M(L3H,H)]2+ (M = Pd, Ni) [47] complexes. The related N,N′-bis[o-(diphenylphosphino)benzylidene]-2,2′-diimino-1,1′-binaphtyl (L4), more rigid than the ethylenediamine derivative, affords the tricoordinated [Ag(L4)]+ complex that contains a k3-PNP ligand [48] and allows the isolation of the octahedral [RuCl2(L4)] compound and of the stable pentacoordinated [RuCl(L4)]+ complex with k4-PNNP ligand [49]. Other asymmetric PNNP ligands such as L5 [50], L2Me,H [51] and L2Ph,Ph [52] have been prepared. The octahedral [RuCl2L2Me,H] [51] and [RuCl2L5] complexes [53] and the square-planar [Rh(L5)]+ compound [54] have been used in the asymmetric transfer hydrogenation of ketones. The chiral Ru-cluster-based catalyst system generated ‘in situ’ from Ru3(CO)12 and L5 or L2Ph,Ph is extremely effective in this process [52]. The octahedral compound [RuCl2(L5)] is useful in the enantioselective epoxidation of styrene [49].
The potentially terdentate PNN ligand, Ph2P(o-C6H4)CH=N(CH2)2(o-C5H4N) (L6 in Chart 2) has been prepared by the reaction of 2-(2-aminoethyl)pyridine with PPh2(o-C6H4CHO) [55]. This ligand contains three donor atoms, i.e. a diphenylphosphine, an imino and a pyridyl group. Its flexibility facilitates its behavior either as a P-monodentate, P-imino bidentate with dangling pyridine functionality or PNN terdentate ligand towards palladium [55] and as P-imino bidentate or PNN terdentate towards group 6 metals [56]. The corresponding [Pd(k3-PNN)(η1-allyl)]+ complexes are very active catalysts in allylic alkylation reactions [55] and have been used in the 1,3-butadiene telomerization reaction with methanol [57]. L6 reacts with triosmium carbonyl clusters to afford complexes containing chelate and bridging PNN ligands that may undergo methylene or imino C–H activation [58].
The related Ph2P(o-C6H4)CH=NCH2(o-C5H4N) ligand behaves as terdentate in [Mo(CO)3{Ph2P(o-C6H4)CH=NCH2(o-C5H4N)}] [26] and Ph2P(o-C6H4)CH=NNH(o-C5H4N) shows a strong tendency to act as terdentate towards palladium and nickel [59].
The imination of PPh2(o-C6H4CHO) with o-aminophenyldiphenylphosphine, affords 2-(diphenylphosphino)-N-[2-(diphenylphosphino)benzylidene]aniline, a PNP ligand that can behave as PNP-terdentate, PN- or PP-bidentate, P-monodentate or bridging ligand towards chromium, molybdenum and tungsten, depending on the metal and/or the reaction conditions [60].
The imination of aldehydes occurs via the formation of hemiaminal >C(OH)NHR intermediates followed by hydrogen transfer from N to O, as shown in Eq. (7). These intermediates have been rarely isolated or even observed because the second step is usually very easy. It needs both H and OH being on the same side of the N–C bond to allow water elimination [61] and stable hemiaminals such as >C(OH)NH–C=O are known in cyclic molecules [62].
| RC(O)H + R′–NH2 → RCH(OH)−NHR′ → RCH = NR′ + H2O | (7) |
The reaction with rhodium(I) complexes [RhCl(Cod)(NN)], containing bidentate diimine ligands of the dihydrazone type (NN: H2NN=C(R)C(R′)=NNH2; R,R′: H or Me), allows the isolation of rhodium(III) complexes, [RhH(Ph2P(o-C6H4CO)){Ph2P(o-C6H4CHOH–HNN=C(R)C(R′)=NNH2)}]+ 17, containing terdentate PNN ligands forming a seven-member phosphine-imino and a five-member diimino chelate ring. The PN metallocycle includes also a very stable, uncoordinated ‘HN–CH(OH)’ hemiaminal group. The hemiaminals in 17 are reluctant to undergo the condensation reaction shown in Eq. (8), though the imination can be accelerated by the presence of acids, via protonation of the hydroxy group, to give terdentate PNN ligands containing azine groups in 18 [15]. The reaction shown in Eq. (8) is favored when R′ = CH3. The presence of the methyl group makes the seven-member ring to adopt a more suitable conformation to favor the hydrogen transfer from N to O and hence the condensation reaction [9].

The stability of hemiaminals in 17 has been attributed to the fact that they belong to a seven-member metallocycle. Other rhodium(I) complexes [RhCl(Cod)(NN)] containing o-phenylenediamine or 8-aminoquinoline react with o-(diphenylphosphino)benzaldehyde to yield rhodium(III) complexes 19 and 20, respectively, shown in Chart 3. In both cases the imination of the aldehyde goes to completion and the ligands obtained contain terdentate PNN ligands, phosphine-imino-amino, in 19, or phosphine-imino-quinoline, in 20, functionalities. The different behavior, when compared to dihydrazone derivatives, can be due to the different size of the metallocycle formed, six-member in 19 and 20 and seven-member in 17 and/or to the amino group being bonded in the precursor of 19 and 20 and pendant in the precursor of 17 [17].

Coordinated hemilabile phosphine-imine ligands from coordinated ligands.
Other examples of coordinated hemilabile phosphine-imine ligands obtained from coordinated ligands are shown in Chart 3. Complex 21 is obtained by reacting [PdCl2(PPh2(o-C6H4CHO))2], containing P-monodentate PCHO, with [Zn(dien)Cl2] (dien = diethylentriamine) following an imination reaction going to completion [4]. The template synthesis of the heptadentate ligand, [(o-Ph2PC6H4)CH=NCH2CH2]3N coordinated to silver in complex 22 and containing imino functionalities, has been achieved by the reaction of N(CH2CH2NH2)3 with PPh2(o-C6H4CHO) in the presence of Ag(CF3SO3) [63]. In both cases 21 and 22, the metallocycles involving the phosphine groups are six-member, as in 19 and 20.