

1 Introduction
When describing electronic structures of transition metal complexes one goes a different way from the point of view of quantum chemistry and experimental spectroscopy. Experimentalists make use of ligand field parameterized (effective) Hamiltonians, or of the spin-Hamiltonian when interpreting optical or ESR spectra, or they apply the Heisenberg exchange operator in the case of magnetic exchange coupling. Empirical models of that kind have been therefore tools for description, rather than tools for prediction of ligand field properties. A quantum chemist solves more or less rigorously numerically the Schrödinger equation (ab initio) or the Kohn–Sham equations (density functional theory, DFT) and is able to make predictions as well. However, numerical results are sometimes not easy to interpret or analyze and the bridge between the ab-initio approach and chemical intuition is not always transparent. DFT became increasingly popular in recent time. As manifested by the groups of Baerends, Ziegler [1,2] and Daul [3] it is able to predict both ground and excited states of TM complexes. Recently, a new approach has been developed in our group [4]. It is based on a multi-determinant description of the multiplet structure originating from the well defined dn configurations of a TM in the surrounding of coordinating ligands by combining the CI and the DFT approaches. In doing so, both dynamical (via the DFT exchange-correlation potential) and non-dynamical (via CI) correlation is introduced, the latter accounting for the rather localized character of the d-electron wavefunction. The key feature of this approach is the explicit treatment of near degeneracy effects (long-range correlation) using ad hoc configuration interaction (CI) within the active space of Kohn–Sham (KS) orbitals with dominant d-character. The calculation of the CI-matrices is based on a symmetry decomposition and/or on a ligand field analysis of the energies of all single determinants (SD, micro-states) calculated according to DFT for frozen KS-orbitals corresponding to the averaged configuration, eventually with fractional occupations of the d-orbitals. This procedure yields multiplet energies with an accuracy within 2000 cm–1. Currently, the procedure has been extended to spin-orbit coupling [5] and allows to also treat Zero-Field Splitting (ZFS) [6], Zeeman interactions and Hyper-Fine Splitting (HFS) [7] and magnetic exchange coupling [8] as well.
In this account a more rigorous analysis of our approach utilizing Löwdin's energy partitioning [9] and effective Hamiltonians theory [10,11] is used, providing explicit context for its applicability and allowing to set up more rigorous criteria for its limitations. We will then briefly sketch the mathematical procedure behind our approach. An extension to spin-orbit coupling will be also given. Selected applications cover tetrahedral CrX4 (X = Cl, Br) and FeO42– and octahedral CrX63–(X = F–, Cl–, Br–) complexes. In addition the spin-orbit coupling constant of Cr(acac)3 deduced from a DFT-ZORA calculation will be applied to calculate the ground and excited state zero-field splitting.
A generalization of the LFDFT theory to dimers of transition metals allows to calculate exchange coupling integrals in reasonable agreement with experiment and with comparable success to the broken symmetry approach; in addition they allow to judge ferromagnetic contributions to exchange coupling integrals, the latter are usually being ignored. An illustration of how the procedure works will be given for hydroxo-dimers of Cu2+.
2 Partitioning technique, effective Hamiltonians and the ligand field approach
Let us consider a system consisting of transition metals and ligands, which can be bridging or terminal (Fig. 1). In electronic structure calculations, one usually immediately recognizes antibonding molecular orbitals (MO's), as being dominated by metal |nd〉 functions, which are partly filled and bonding orbitals dominated by ligand AO's which are fully occupied. Following Löwdin [9] we can write down the Schrödinger equation H ψ = E ψ in a discrete representation based on the use of a complete orthonormal set Φ = {Φk} and introduce the Hamiltonian matrix H = and the column vector c = using the relations:(1)

Partioning scheme for LFDFT approach of a pair of transition metals joined by bridging ligands.
Let us subdivide the system into two parts, one build of from metal nd-orbitals (d) and another composed of valence metal (n + 1) s and (n + 1) p and ligand functions (v).
Then the eigenvalue problem (3) can be represented in a form given by:(4)
Collecting terms with same dimension, we get:(5.1)
One can easily express the column vector in terms of (Eq. (6)), and after substitution into Eq. (5.1), one obtains a Hamiltonian completely restricted to the d-subspace:(6)
We thus arrive at a pseudo-eigenvalue equation (Eq. (8)), with the explicit form of given by Eq. (9). No approximation is inherent in Eq. (9). The representation given by Eq. (9) provides:(8)
The one-electron representation of Eq. (9) can easily be extended to systems with more than one TM. The matrix for such cases contains terms that account for d-electron delocalization (via the second term in Eq. (9)) from one metal to another – indirect (via the bridging ligands) or direct (via corresponding off-diagonal terms of Hdd´) [15]. It gives rise to magnetic exchange coupling. This will be the topic of Section 4.
Finally, the one-electron scheme given by Eq. (9) can be extended to the many electron states resulting from the redistribution of all the d-electrons within the active d-orbital subspace. Their treatment requires two-electron repulsion integrals in addition to the one-electron ones. The general scheme we describe in Section 3 allows to also deduce these intergrals from DFT.
3 The ligand-field density functional theory (LFDFT)
The current DFT software includes functionals at the Local Density Approximation (LDA) and Generalized Gradient Approximation (GGA) levels. The former approximation is well adapted for molecular structure calculation: M–L bond lengths are usually accurate to ±0.02 Å but bond energies are too large. The latter approximation, however, is roughly twice as expensive in computer time and yields M–L bond energies accurate to ±5 kcal mol–1. Recently a new generation of functionals called meta-GGA emerged. These functionals are more accurate but also more expensive and their implementation in computer codes is not yet generalized. It is generally accepted that all these functionals (LDA and GGA) describe well the so-called dynamical correlation, however, none of them includes near degeneracy correlation. In the method described next we address this problem specifically and include CI of valence electrons on the d-orbitals. The calculation scheme we developed includes three steps as described next.
We assume that we know the molecular geometry, either from a first principle geometry optimization or from X-ray data. Moreover, for the sake of simplicity, let us focus the following description to open d-shells: the extension to open f-shells is similar. The first step consists in a spin-restricted, i.e. same orbitals for same spin, Self-Consistent Field (SCF) DFT calculation of the average of the dn configuration (AOC), providing an equal occupation n/5 on each MO dominated by the d-orbitals. The KS-orbitals which we construct using this AOC are best suited for a treatment in which, interelectronic repulsion is – as is done in LF theory, approximated by atomic-like Racah parameters B and C. The next step consists in a spin-unrestricted calculation of the manifold of all Slater Determinants (SD) originating from the dn shell, i.e. 45, 120, 210 and 252 SD for d2,8, d3,7, d4,6, and d5 Transition Metal (TM) ions, respectively. These SD-energies are used in the third step to extract the parameters of the one-electron 5 × 5 LF matrix as well as Racah's parameters B and C in a procedure, which we describe below. Finally, we introduce these parameters as input for a LF program allowing to calculate all the multiplets using CI of the full LF-manifold utilizing the symmetry as much as possible. We should note that in classical LF theory, it is only the LF matrix which carries information about the symmetry and the actual bonding in the complex, thus providing useful chemical information.
In order to establish a link between ligand field theory and the energy of each SD mentioned earlier we need to introduce an effective LF-Hamiltonian together with its five eigenfunctions φi which are in general linear combination of the five d-orbitals:(10)
The SD are labeled with the subscript k = 1, …, and where the superscript φ does refer to eigenfunctions of the ligand-field Hamiltonian hLF. The summation i ∈ k of ligand-field splitting matrix elements specify the occupation of the level φi, while and denote Coulomb and exchange integrals; σi are spin functions and E0 represents the gauge origin of energy. This expression does only involve the diagonal matrix elements of . In order to obtain , the full matrix representation of , we make use of the general observation that the KS-orbitals and the set of SD considered in Eq. (11) convey all the information needed to setup the LF matrix. In Ref. [4], we give a justification for this.
Thus, let us denote KS-orbitals dominated by d-functions which result from an AOC dn DFT-SCF calculation with a column vectors . From the components of the eigenvector matrix build up from such columns one takes only the components corresponding to the d functions. Let us denote the square matrix composed of these column vectors by U and introduce the overlap matrix S:(12)
Since U is in general not orthogonal, we use Löwdin's symmetric orthogonalization scheme to obtain an equivalent set of orthogonal eigenvectors (C):(13)
We identify now these vectors as the eigenfunctions of the effective LF-Hamiltonian , we seek as(14)
Thus, the fitting procedure described below will enable us to estimate and hence the full representation matrix of as(15)
Having obtained energy expressions for each : , B, C and E0 are estimated using a least-square procedure. Using matrix notation, we obtain an overdetermined system of linear Eq. (19), with the unknown parameters stored in and given by:(19)
It is worthwhile to note that the KS-eigenvalues of the orbitals with dominant d character are almost equal to the ligand-field parameters obtained in the fitting procedure, i.e.:(21)
Thus, we conclude this section with the statement that the separation between KS-eigenvalues of orbitals with dominant d-character are good approximations for the ligand field splitting parameters.
For cases where the fine-structure is sought it is now easy to include spin–orbit coupling and calculate:(22)
From the splitting (due to the combined effect of spin-orbit splitting and perturbations of symmetry lower than Oh), say of the 3A2 and 4A2 of hexa-coordinate ground states of Ni(II), d8 and Cr(III), d3, it is possible to obtain the ZFS D-tensor using a conventional spin-Hamiltonian approach:(24)
4 LFDFT and magnetic exchange coupling
The approach of Section 3 has been extended to the calculation of the exchange coupling constant of Heisenberg Hamiltonian (Eq. (25)) in the case of pairs of TM joined by bridging ligands [8]. Taking a TM pair with S1 = S2 = 1/2 spins, the singlet–triplet separation is given by Eq. (26), implying a positive and negative values of J12 for ferromagnetic and antiferromagnetic constants, respectively:(25)
Six micro-states or SD are possible. Two are doubly occupied , and four are singly occupied , , , . The doubly occupied SD correspond to closed shells and are spin singlets, whereas the singly occupied SD correspond to a singlet and to a triplet. The two SD with MS = 0: and , are mixed states belonging to a singlet and to a triplet. The energies of all these determinants can be easily calculated from DFT. Let us denote their energies, respectively, by:(28)
To obtain these energies, a two-step calculation scheme is applied. First a spin-restricted calculation with a so-called average of configuration (AOC) occupation (…a1b1) is carried out yielding a corresponding set of MOs {… a, b, …}. In a second step, these Kohn–Sham orbitals are kept frozen in order to evaluate the four SD-energies E1, E2, E3 and E4 (spin-polarized DFT) without further SCF iterations. We note that the E4 – E3 difference equals the exchange integral [ab|ab] which is also the quantity accounting for the mixing (1:1 in the limit of a full localization) between the a+a– and b+b– functions. This leads to matrix (Eq. (29)) which after diagonalization yields the eigenvalues E– and E+ and the singlet-triplet energy separation E– – E3, i.e. J12:(29)
Within the Anderson exchange model [17], dl1 and dl2 are singly occupied in the ground state giving rise to a triplet and a singlet with wave functions ψT and ψS (Eqs. (30, 31)). There are two further singlet states and arising when either of the two magnetic electrons is transferred to the other magnetic orbital, i.e.:(30)
U is again a positive and large quantity (typically 5–8 eV). The interaction matrix element between ψS and (Eq. (34)) reflects the delocalization of the magnetic electrons due to orbital overlap, the quantity t12 being referred to as the transfer (hopping) integral between the two sites, i.e.:(34)
Calculations show that T12 = t12 in a very good approximations, differences being generally less than 0.002 eV. This term tends to lower the singlet over the triplet-energy and is intrinsically connected with the gain of kinetic energy (kinetic exchange). The interaction matrix (Eq. (35)) describes the combined effect of these two opposite interactions:(35)
Perturbation theory yields Eq. (36) for the (ES – ET)P energy separation, i.e. J12 . This allows us to decompose J12 into a ferromagnetic () and an anti-ferromagnetic () part.(36)
As has been pointed out already in [18], the parameters K12, U and T12 can be expressed in terms of the Coulomb integrals (Jaa, Jbb and Jab), exchange integral Kab and of ɛ(b) – ɛ(a), the KS-orbital energy difference. Eqs. (37–39) below, resume these relations:(37)
We like to point out that these expressions are furthermore related with the energies of the SD |a+a–|, |b+b–|, |a+b+|, |a+b–| (i.e. E1, E2, E3 and E4, respectively). In deriving these expressions we made use of Eqs. (40–43).(40)
Thus, Eqs. (37–39) allow us to obtain K12, U and T12 directly from DFT data and to compare them with the corresponding empirical values checking the consistency of the current functionals. Such empirical estimates of K12, U and T12 can be deduced by a fit to magnetic and spectroscopic data (valence-bond CI approach (VBCI), Sawatzky [19,20], Solomon [21]). We get therefore a model of localized magnetic orbitals, whose parameters are readily obtained from the DFT SD-energies E1, E2, E3 and E4 of the dimmer. An application of the approach for calculation exchange integrals in bis-hydroxo bridged Cu(II) dimers is given in Section 6.4.
5 Computational details
All DFT calculations have been performed using the ADF program package [22–25] (program release ADF2003.01). The approximate SCF KS one-electron equations are solved by employing an expansion of the molecular orbitals in a basis set of Slater-type orbitals (STO). All atoms were described through triple-ς STO basis sets given in the program data base (basis set TZP) and the core-orbitals up to 3p for the TM and up to 1s (for O, N), 2p (Cl) and 3d (Br) were kept frozen. We used the local density approximation (LDA), where exchange-correlation potential and energies have been computed according to the Vosko, Wilk and Nusair's (VWN) [26] parameterization of the electron gas data.
6 Applications
6.1 Tetrahedral d2 CrX4 (X = Cl–, Br–)
Tetrahedral d2 complexes possess a 3A2(e2) ground state as well as 3A2 → 3T2 and 3A2 → 3T1, e → t2 singly excited states. They give rise to broad d–d transitions in the optical spectra. In addition, spin-flip transitions within the e2 configuration lead to sharp line excitations. Multiplet energies from LDA agree within a few hundred cm–1 with experimental data. In particular the 3A2 → 3T2 transition energy and thus 10Dq nicely agrees with experiment as is seen from inspection of Table 1. Experimental transition energies for CrCl4 and CrBr4 as well as values of B, C and 10Dq deduced from a fit to experiment for CrCl4 are also listed.
Electronic transition energies of CrX4, X = Cl and Br, with geometries optimized using LDA functional and calculated using values of B, C and 10 Dq from least square fit to DFT energies of the Slater determinants according to the method described in Section 2
| Cr Cl4 | CrBr4 | ||||
| Term | This work | LF-fit | Exp.a | This work | Exp.a |
| 3A2(e2) | 0 | 0 | 0 | 0 | 0 |
| 1E(e2) | 6542 | 6089 | – | 6373 | 6666 |
| 1A1(e2) | 11 114 | 10 586 | – | 10 698 | 10 869 |
| 3T2(e1t2 1) | 7008 | 7010 | 7250 | 6163 | – |
| 3T1(e1t2 1) | 10 316 | 10 440 | 10 000 | 9269 | – |
| 1T2(e1t2 1) | 13 454 | 12 991 | 12 000 | 12 434 | – |
| 1T1(e1t2 1) | 15 074 | 14 718 | – | 14 037 | – |
| 1A1(t22) | 32 099 | 30 599 | – | 30 120 | – |
| 1E(t22) | 21 121 | 20 716 | – | 19 271 | – |
| 3T1(t22) | 16 033 | 16 229 | 16,666 | 14 424 | 13 258 |
| 1T2(t22) | 21 217 | 20 822 | – | 19 373 | – |
| R(M–X) | 2.104 | – | – | 2.264 | – |
| B | 355 | 376 | – | 347 | – |
| C | 1903 | 1579 | – | 1855 | – |
| 10 Dq | 7008 | 7250 | – | 6162 | – |
| S.D. | 0.030 | – | – | 0.030 | – |
a P. Studer, Thesis, University of Fribourg, 1975.
6.2 Octahedral CrIII d3 complexes
In Table 2 we list the predicted (this work), adjusted (LF fit to exp.) and observed (Exp.) multiplet energies for CrX63– (X = F–, Cl–, Br–) complex ions. We used a LDA functional to calculate the CrIII–X bond lengths and we compare these results with energies from a LF-calculation utilizing values of B, C and 10 Dq obtained from a best fit to spectra from experiment. Bond lengths are too long while values of 10 Dq are too small compared to experiment. The situation improves if instead of optimized, experimental bond lengths are taken for the calculation. Even in this case, spin-forbidden transitions come out by 3000–4000 cm–1 too low in energy compared to experiment. Clearly, in this example of highly charges species, our prediction is much less accurate. In order to improve the quality of the prediction we obviously need to consider the environment of the CrX63– chromophore by adding an appropriate embedding potential to the KS-hamiltonian. Already the use of experimental bond lengths does significantly improve the precision of our calculation as mentioned before. A full analysis of this problem is given in [27].
Electronic transition energies of CrX63–, X = F–, Cl–, Br– with geometries optimized using LDA functionals calculated using values of B, C and 10Dq from least square fit to DFT energies of the Slater determinants and to experiment. The values of (10Dq)orb as deduced from the eg – t2g KS-orbital energy difference taken from the …t2g1.8eg1.2 SCF KS-energies are also listed. Experimental transition energies are also listed
| Term | CrF63– | CrCl63– | CrBr63– | ||||||
| This work | LFT fit to exp. | Exp. | This work | LFT fit to exp. | Exp. | This work | LFT fit to exp. | Exp. | |
| 4A2g(t2g3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 2Eg(t2g3) | 12 497 | 15 802 | 16 300a | 10 756 | 14 426 | 14 430b | 10 333 | 13 900 | 13 900b |
| 2T1g(t2g3) | 13 044 | 16 461 | 16 300a | 11 180 | 14 873 | – | 10 694 | 14 348 | – |
| 2T2g(t2g3) | 18 628 | 23 260 | 23 000a | 15 918 | 21 037 | – | 15 185 | 20 281 | – |
| 4T2g(t2g1eg1) | 13 569 | 15 298 | 15 200a | 10 911 | 12 800 | 12 800b | 9816 | 12 400 | 12 400b |
| 4T1g(t2g1eg1) | 19 443 | 22 262 | 21 800a | 15 618 | 18 198 | 18 200b | 13 992 | 17 700 | 17 700b |
| 2A1g(t2g1eg1) | 24 071 | 28 709 | – | 20 056 | 25 351 | – | 18 709 | 24 459 | – |
| 2T1g(t2g1eg1) | 26 348 | 31 473 | – | 21 878 | 27 421 | – | 20 316 | 26 503 | – |
| 2T2g(t2g1eg1) | 25 959 | 30 970 | – | 21 568 | 27 079 | – | 20 047 | 26 159 | – |
| 2Eg(t2g1eg1) | 27 819 | 33 341 | – | 23 147 | 29 098 | – | 21 530 | 28 126 | – |
| 4T1g(t2g1eg1) | 30 339 | 34 636 | 35 000a | 24 375 | 28 455 | – | 21 861 | 27 643 | – |
| R(M–X) | 1.957 | - | 1.933c | 2.419 | – | 2.335d | 2.588 | – | 2.47e |
| B | 605 | 734 | – | 484 | 550 | – | 427 | 543 | – |
| C | 2694 | 3492 | – | 2403 | 3450 | – | 2395 | 3296 | – |
| 10Dq | 13 598 | 15 297 | – | 10 911 | 12 800 | – | 9816 | 12 400 | – |
| SD | 0.113 | – | – | 0.105 | – | – | 0.113 | – | – |
| (10Dq)orb | 13 928 | – | – | 10 775 | – | – | 9622 | – | – |
a K3CrF6: G.C. Allen, A.M. El-Sharkawy, K.D. Warren, Inorg. Chem. 10 (1971) 2538.
b Cs2NaYCl[Br]6: Cr3+, R.W. Schwartz, Inorg. Chem. 15 (1976) 2817.
c K. Knox, D.W. Mitchell, J. Inorg. Nucl. Chem. 21 (1961) 253.
d Estimated for Cs2NaCrCl6 and Cs2NaCrBr6, F. Gilardoni, J. Weber, K. Bellafrouh, C. Daul, H.-U. Guedel, J. Chem. Phys. 104 (1996) 7624.
e estimated for Cs2NaCrCl6 and Cs2NaCrBr6, F. Gilardoni, J. Weber, K. Bellafrouh, C. Daul, H.-U. Guedel, J. Chem. Phys. 104 (1996) 7624.
6.3 Application of the LFDFT to the calculation of the zero-field splitting in Cr(acac)3
In octahedral ligand fields the t2g orbital of the TM are purely π-bonding. The π-electrons of the acac-ligand lead to a significant anisotropy; as has been recognized already by Orgel [28], this anisotropy can lower the symmetry of the ligand field from Oh to D3 with clear manifestations in the spectrum. For the acac ligand, the topmost π-orbital which dominates its π-donor functions is characterized by pi-orbitals with the same sign (in-phase), the out of-phase counterparts being much lower in energy. Three such in-phase coupled functions, when combining in a complex of a D3 symmetry give rise to species of e and a2 symmetry. From these only the e-combinations interact with the TM counterpart of the same symmetry, the a1 component of t2-obital having no counterpart from the ligand and being non-bonding in this approximation. For d-orbitals of Cr which are antibonding in Cr(acac)3 this leads to a splitting of the Oh t2g level in D3 with an a1 < e orbital energy sequence even though the geometrical arrangement of the oxygen ligator is very close to octahedral. This qualitative picture has been quantified in terms of phase-coupling ligand field model [29–32], which could explain both the splitting (800 cm–1) of the 4A2–4T2 band in the electronic absorption spectrum and its polarization behavior and the ground 4A2 and excited state 2E zero-field splittings, 1.1–1.2 and 250 cm–1. The latter have been detected in high-resolution optical spectra and further supported by detailed optically detected magnetic resonance (ODMR) studies [32]. We applied the LFDFT method to this system and results are collected in Table 3b. LFDFT values of the spin-orbit coupling constant ς, the parameters B and C (Table 3c) and the 5 × 5 LF matrix (Eq. (33)) have been utilized in standard LF-calculation yielding multiplet energies:(44)
LFDFT ground state fine-structure levels and the energies of spin-forbidden transition of Cr(acac)3 with and without accounting for spin-orbit coupling. Data from experiment, when available, are also listed
| ς = 0 | ς = 211 | Experiment [32] |
| 4A2 0 | Γ56(2 A) 0.0 | 0.0 |
| Γ4(E) 1.16 | 1.20 | |
| 2E 8618 | Γ56(2 A) 8520 | 12 940 |
| Γ4(E) 8713 | 13 200 | |
| 2A2 10,444 | Γ4(E) 10 442 | – |
| 2E 10,676 | Γ4(E) 10 674 | – |
| Γ56(2 A) 10 677 | – | |
| 2A1 16,707 | Γ4(E) 16 701 | – |
| 2E 17,832 | Γ4(E) 17 797 | – |
| Γ56(2 A) 17 890 | – |
LFDFT values of energies of spin-allowed transitions in comparison with experiment (ς = 0)
| LFDFT | Experiment [31] | |
| 4A2 | 0.0 | |
| 4A1 | 21 306 | 17 700 |
| 4E | 22 655 | 18 500 |
| 4A2 | 26 742 | 22 700 |
| 4E | 28 748 |
4A2 ground- and 2E excited state zero-field splittings (in cm–1) – Dg and De, respectively, of Cr(acac)3 from a LFDFT—ZORA calculation of the spin-orbit coupling constant (ς = 211 cm–1) and from experiment
| LFDFT-ZORA | Experiment | |
| Dg | 1.16 | 1.20 |
| De | 193 | 260 |
6.4 Exchange splitting in Cu(OH)2Cu dimers
The usual pattern of an exchange coupling between pairs of TM with open shells is anti-ferromagnetic spin-alignment corresponding to a weak delocalization of unpaired spin density from one center to another center (weak covalent bond as described by the term –4 T122/U, Eq. (36)). It out weights the contribution of the first term (2 K12), the latter tending to lower exchange (Pauli) repulsion between electrons with parallel spins. It has been therefore challenging to find systems where the latter effect dominates, leading a ferromagnetic spin-alignment. This is the case if magnetic orbitals are orthogonal to each other or nearly so, a situation encountered in edge sharing square planes or octahedra with M1–X–M2 bridging angles β close to 90° [33]. This is the case in bis bipyridyl-μ-dihydroxo-dicopper (II) nitrate with a Cu–OH–Cu bridging angle of 95.6° and an exchange coupling constant J12 = 0.021 eV [34]. A DFT–LDA geometry optimization using a [(NH3)2CuOH]22+ model cluster has lead to a geometry of the bridging Cu(OH)2Cu2+ moiety very close to the experiment (Fig. 2). Unpaired electrons on Cu2+ are characterized by a dx2 – y2 ground state which is weakly affected by long axial contacts to NO3–, which we neglect here. The LFDFT calculated exchange coupling constant J12 = 0.021 eV (Table 4a) matches perfectly the value from experiment, but deviates from the anti-ferromagnetic one given by the broken symmetry (BS) DFT approach [35] (J12BS = –0.099 eV). In Fig. 3, we compare energies of the four independent Slater determinants as given by our DFT procedure with the state energies after taking the a+a––b+b– configurational mixing into account. The former configuration is stabilized by localization leading to a final singlet function, but it does not cross (as different to usual cases) the triplet term T. Experimental data show [34] that J12 becomes strongly anti-ferromagnetic upon increasing the Cu–O–Cu bridging angle (β) by structural manipulations allowing one to tune this structural parameter. Thus the increase of the value of β to 104.1° in [Cu(tmen)OH]2Br (tmen = N,N,N´,N´-tetramethylethylenediamin) correlates with a large negative reported value of J12 (–0.063 eV [34]). Antiferromagnetism for this geometry is also obtained by LFDFT but the resulting value exceeds now the experimental one by a factor of 2.88 (however the BSDFT value is by 4.61 larger). The reason is that DFT leads to systematically lower values of the energy U to cause an increase of the –4T122/U term, in cases where this terms plays an important role (see [8] for other examples and an analysis).

Bond distances (in Å) and bond angles (in °) from a DFT geometry optimization (spin-unrestricted, S = Ms = 1, LDA–VWN functional, non-relativistic TZP basis Cu-2p, O-1s, N-1s, frozen cores) of a [Cu(NH3)2(OH)]22+ model cluster and experimental parameters as reported from X-ray diffraction study of bis-bipyridil-μ-dihydroxo-dicopper(II) nitrate [Cu(C10H8N2)µ(OH)-NO3]2, R.J. Majeste, E.A. Meyers, J. Phys. Chem. 74 (1970) 3497.
Energies (in eV) of Slater determinants for the geometry-optimized (NH3)2Cuμ(OH)2Cu(NH3)22+, the calculated singlet(S)–triplet(T) splitting ES – ET, the value J12(p) = (ES – ET)P given by perturbation theory (J12(p)f + J12(p)af = 2 K12 – 4 T122/U), by the broken symmetry calculation (ES – ET)BS as well as the one from experiment
| E1(a+a–) | E2(b+b–) | E3(a+b+) | E4(a+b–) |
| –4.434 | –3.798 | –4.692 | –4.238 |
| J12 = ES – ET | J12(p) | (ES – ET)BS | (ES – ET)exp [34] |
| 0.021 | 0.010 | –0.099 | 0.021 |
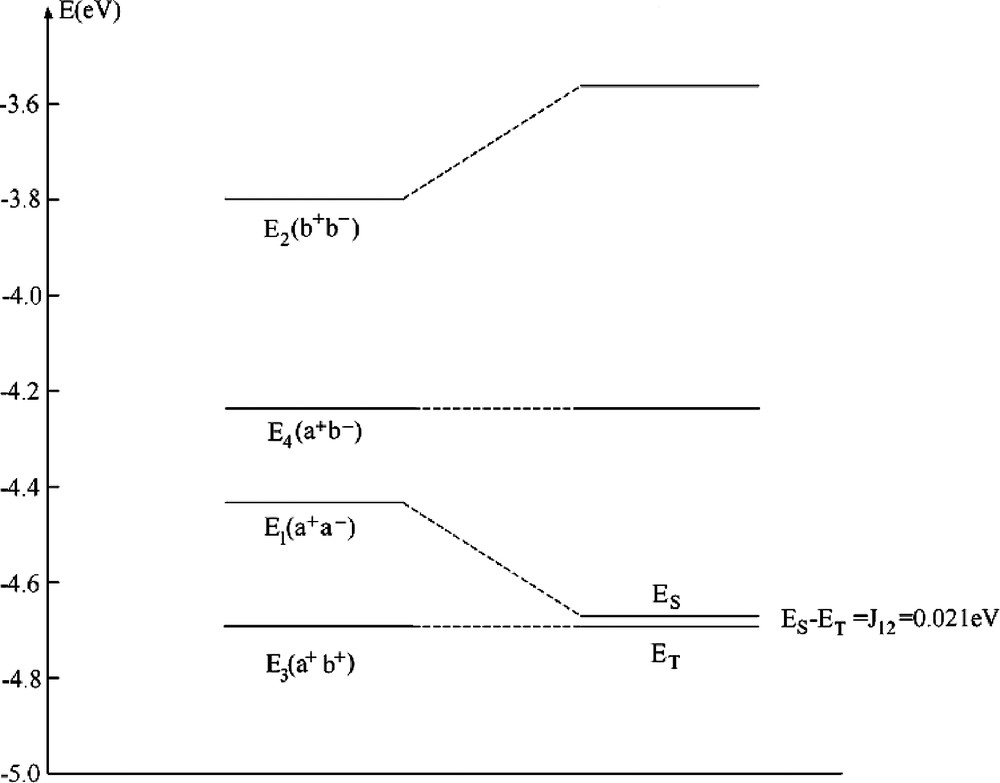
Correlation diagram between the energies of single determinants from DFT and the resulting multiplets of relevance for the magnetic exchange coupling in a [Cu(NH3)2(OH)]22+ model cluster with a ferromagnetic spin alignment.
It is remarkable that ferromagnetic contributions to J12 seem to be realistically described by the LFDFT procedure and our results show that such terms could be indeed rather important (as large as 0.061 eV in the chosen example, see Table 4b). Such terms have been neglected in earlier studies [36] or deemed by physicists to be small [17].
Decomposition of J12(p) into ferro- and anti-ferromagnetic contributions to the exchange along with the transfer(hopping) integral (t12), the Heisenberg exchange integral K12(= J12(p)f/2) the effective charge, transfer energy U are also included. For bond angles and distances characterizing the bridging geometry, see Fig. 2
| J12(p)f | J12(p)af | K12 | T12 | U |
| 0.121 | –0.111 | 0.061 | 0.159 | 0.909 |
7 Conclusion
The model we present here is simple and easy to implement. The quality of the predictions is exceptional in regard of the low computer time consumption. Keeping in mind that time dependent (TD) DFT is restricted to closed shell molecules an is still problematic for TM complexes and difference dedicated CI approaches [37–39] could be applied with success but only to systems of a smaller size, the model presented here is probably unique to address excited states of molecules with open d- and f-shells. Moreover, the concepts used here (LF theory, Racah parameter) are familiar to all chemists and spectroscopists. Thus, the quantities involved in the calculations provide immediate insights and facilitates communication between theorists and experimentalists. On the basis of our results we can conclude that DFT provides a rigorous interpretation of the LF-parameters and leads to a justification of the parametric structure of the classical LF theory. It is remarkable to mention that a theory which was discovered three quarter of a century ago is still modern.
Acknowledgements
This work was supported by the Swiss National Science Foundation.