

1 Introduction
There has recently been a growing interest toward the design and synthesis of low-coordinate cationic group 13 alkyl species of the types {LX}MR+ and {LX}M(R)(L)+ (L labile) (LX– = monoanionic bidentate ligand, M = Al, Ga, In) for applications in catalysis [1]. Such group 13 cations, readily accessible by reaction of the neutral dialkyl precursors {LX}MR2 with B(C6F5)3 or [Ph3C][B(C6F5)4] [2], incorporate a low coordinated and cationic metal center and are thus extremely Lewis acidic. As such, these highly reactive cationic species are of potential interest in Lewis acid catalysis and some of them have already found applications as epoxide [3], ethylene [2b,c] and dl-lactide [4] polymerization catalysts. However, to date, the obtainment of reasonably stable cationic derivatives often remains tricky, which can be ascribed to their intrinsic high reactivity. Thorough studies on {LX}MR+ and {LX}M(R)(L)+ species have shown that the steric and electronic properties of the chelating ligand LX– were crucial for the stability of the resulting cations [3,5].
Bidentate O,N aminophenolate ligands of type A (Scheme 1) have proven to be versatile ligands in the stabilization of transition metal complexes in various oxidation states [6]. Therefrom, the stabilizing properties of this class of ligands has appeared appealing for their use as LX– ligands for the design and synthesis of stable {LX}MR2, {LX}MR+ and {LX}M(R)(L)+ group 13 complexes. In particular, given the excellent affinity of oxygen and nitrogen for group 13 metals, such bidentate O,N ligands are expected to form stable, while still flexible, six-membered metallacycles. In addition, the steric properties of these O,N ligands are easily tunable via, for example, the introduction of ortho-substituent (R in Scheme 1) on the phenol ring. The nature of the sterics associated with the aminophenolate ligand is of crucial importance since, as will be seen, it largely determines the stability, structure and the reactivity of the derived group 13 complexes. Finally, one should mention that the synthesis of ligands of type A is quite straightforward, involving, in most cases, a one-pot Mannich reaction between the appropriate phenol, formaldehyde and the desired amine [7].
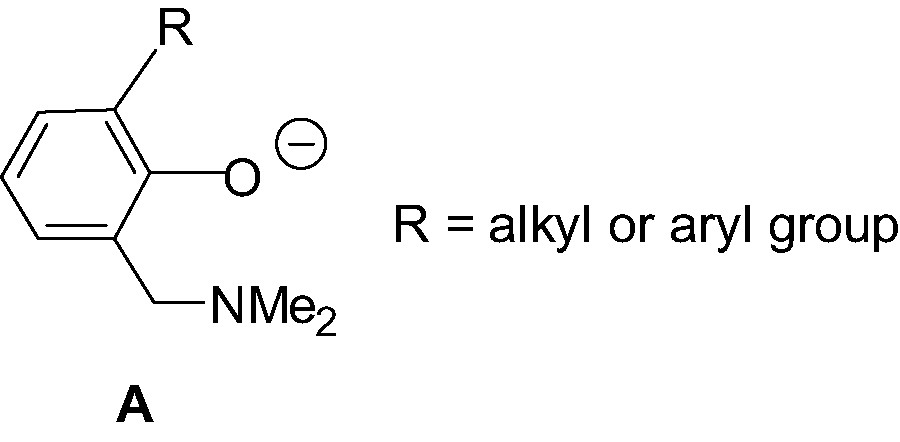
In the present contribution, we first review the synthesis and structures of neutral aminophenolate dialkyl and diphenyl group 13 complexes of the type {LX}MR2; we then present the synthesis, structural characterization and reactivity of the cationic complexes derived from.
2 Neutral mono-aminophenolate dialkyl group 13 complexes
2.1 Synthesis
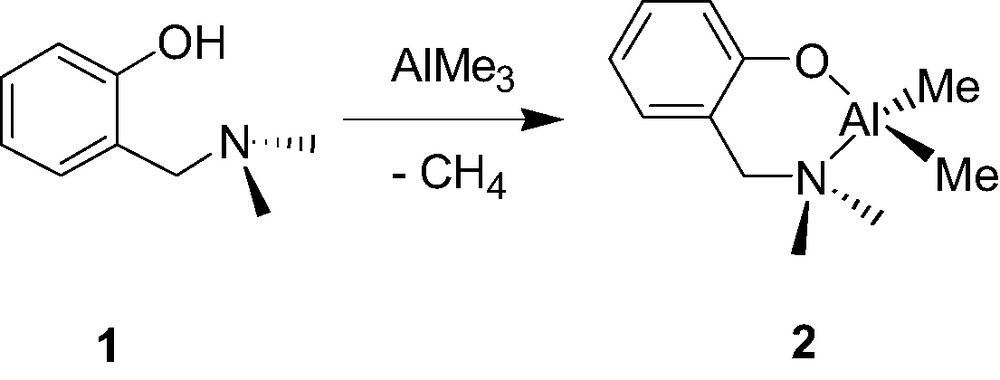
There are a few examples of well-characterized mono-aminophenolate dialkyl group 13 species and those have been quite recently reported. In search for phenolate ligands with potentially intramolecular coordinating ortho-substituents, Hogerheide et al. [8] reported about 10 years ago that the simple aminophenol 2-(CH2NMe2)C6H4OH (1) smoothly reacts with AlMe3 via a classical methane elimination reaction to yield the quantitative formation corresponding aminophenolate chelate Al complex {2-(CH2NMe2)C6H4O}AlMe2 (2, Scheme 2). Although compound 2 was assumed to be monomeric, its structure was not unambiguously established whether in solution or in the solid state.
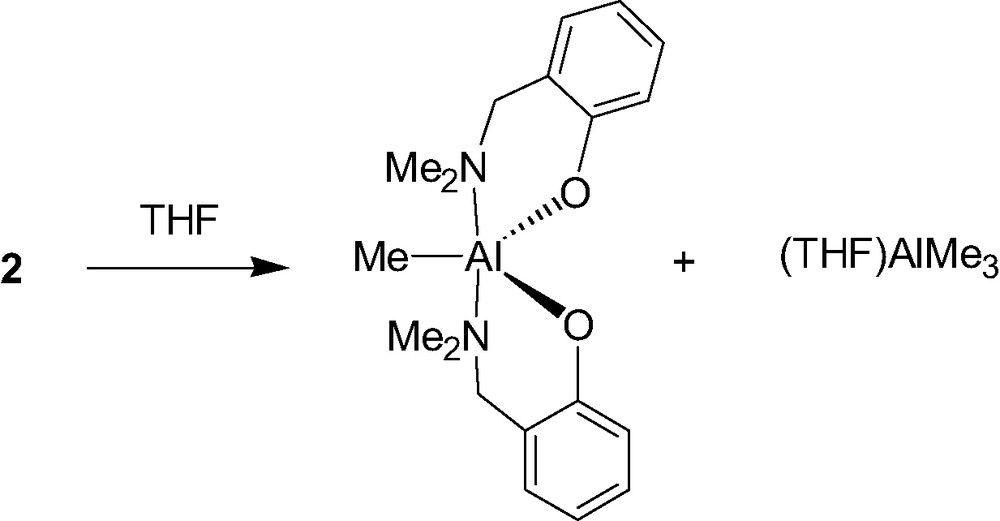
The mono-aminophenolate aluminum complex 2 is unstable in the presence of a Lewis base such as THF since it disproportionates to afford the corresponding bis(aminophenolate) Al species and Al(Me)3(THF) (Scheme 3). For this exchange of ligands to take place, a transition state has to be formed in which the phenolate ligands bridge between two aluminum centers (see Scheme 4). As shown in Scheme 4, the authors propose that the role of the Lewis acid in the exchange reaction of 2 is to make it possible for an amino substituent to dissociate from one aluminum center and, after rotation about the C–O bond of the ligand, to attack on the other aluminum center. This causes a methyl shift and expels Al(Me)3(L) from the newly formed bis(phenolate) Al species [8]. The mechanism for this disproportionation reaction clearly implies a dimeric nature of complex 2, at least in solution.

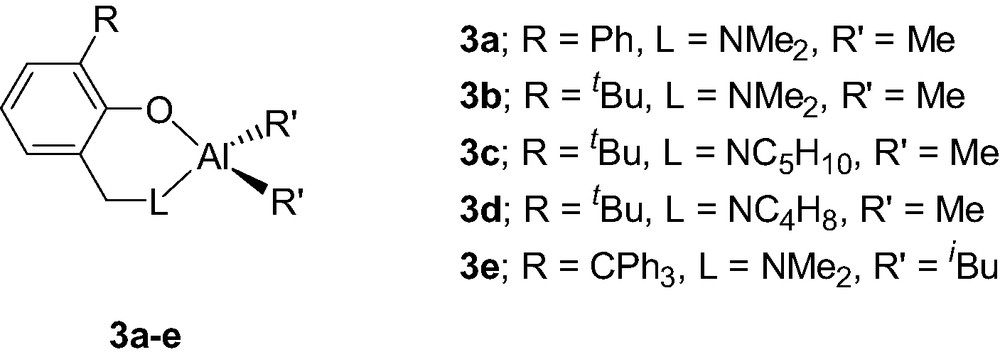
The use of more sterically demanding aminophenol ligands, via the introduction of a second ortho-substituent on the phenol ring, dramatically increases the stability of the formed Al chelates. Thus, the Al complexes incorporating a bulkier aminophenolate {2-(CH2L)-6-R-C6H4O}AlMe2 (3a–e, Scheme 5), also synthesized by a methane elimination reaction between AlMe3 and the desired aminophenol, are stable in THF up to 80 °C, showing no sign of disproportionation reaction [9]. In the present case, it is likely that the steric bulk of the chelating ligand prevents the formation of a dimeric Al species, thus precluding a ligand exchange reaction.
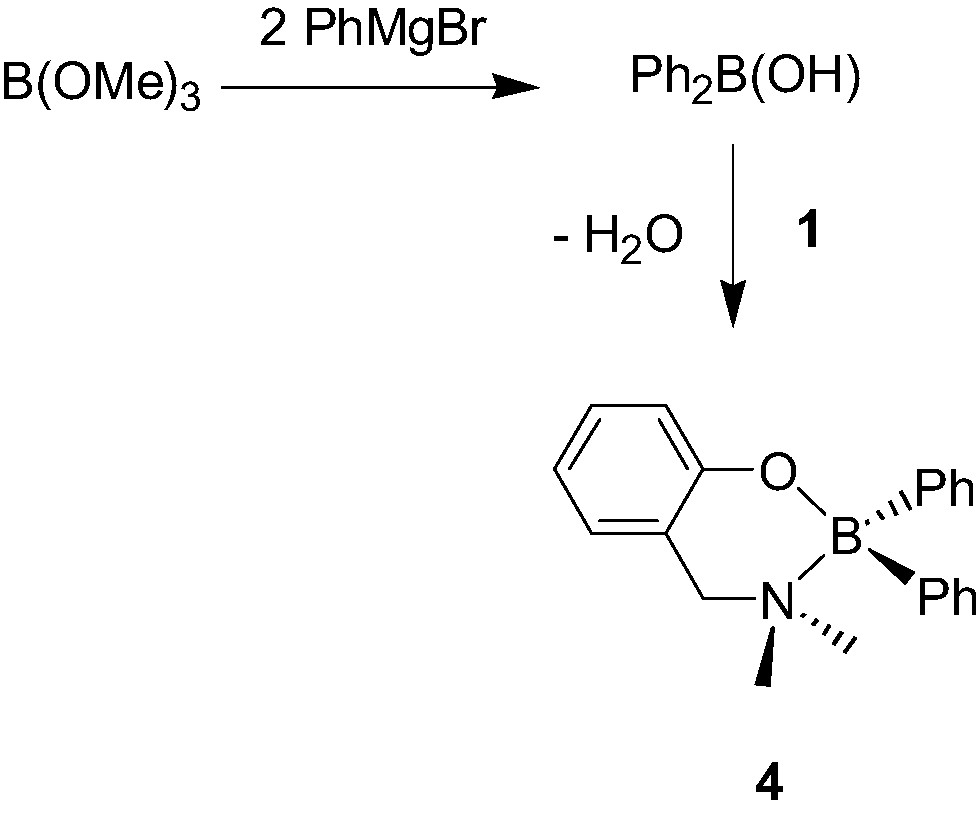
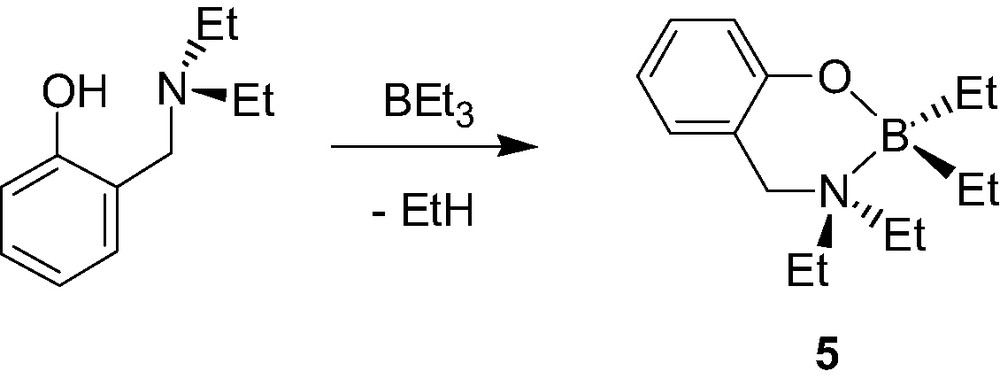
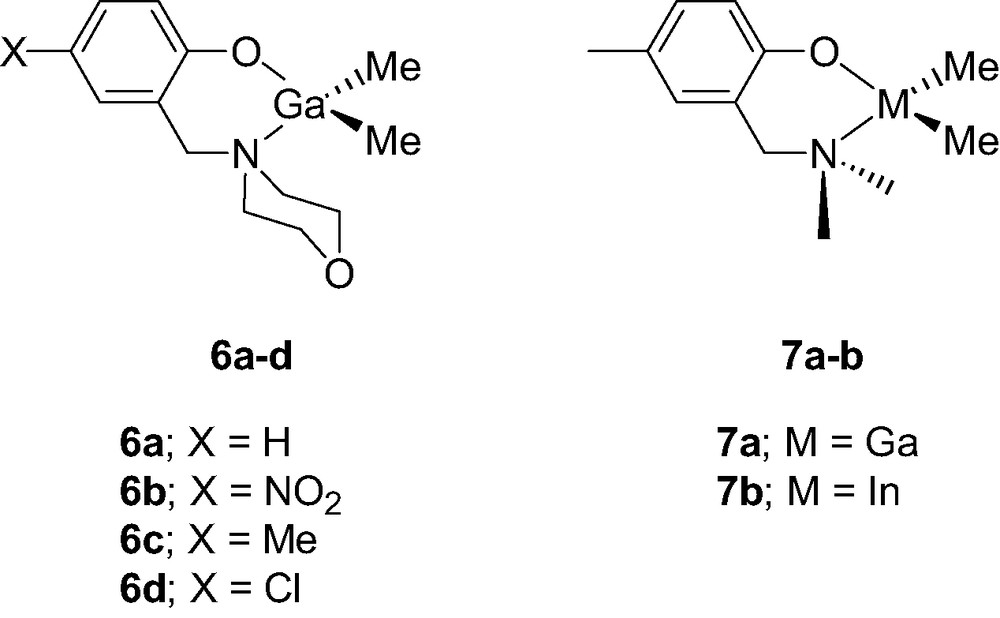
A few examples of mono-aminophenolate boron, gallium and indium dialkyl and diphenyl complexes have also been reported. The chelate boron complex {2-(CH2NMe2)C6H4O}BPh2 (4, Scheme 6) was recently synthesized in two steps starting from B(OMe)3, although in an overall poor yield [10]. In contrast, aminophenolate dialkylboron derivatives {2-(CH2NEt2)-C6H4O}BEt2 (5, Scheme 7) were shown to be accessible in a straightforward manner, and in excellent yields, via a simple alkane elimination reaction [11]. Similarly, the Ga morpholino dimethyl complexes 6a–d [12], the Ga and In diethyl complexes 7a–b [13] were also obtained via alkane elimination reactions in good yield (Scheme 8).
2.2 Solution and solid-state structural features
The solid-state structures of compounds 3b, 3d and 4–6 as determined by X-ray crystallographic analysis, unambiguously established them as η2(O,N)-bonded aminophenolate dialkyl (or phenyl) group 13 species. For an example, the molecular structure of the Al derivative 3b is illustrated in Fig. 1 [9]. In such complexes, the group 13 metal center adopts a slightly distorted tetrahedral structure. The bite angle of the η2(O,N)-bonded aminophenolate, ranging from 93° to 98°, is compensated for by the opening of the C–M–C, O–M–C and N–M–C bond angles. The main structural feature of these molecules is a puckering of the six-membered chelate metallacycle with, in general, the amine-AlMe2 moiety considerably out of the nearly planar remaining metallacycle backbone, as illustrated in Fig. 2 in the case of 3b.
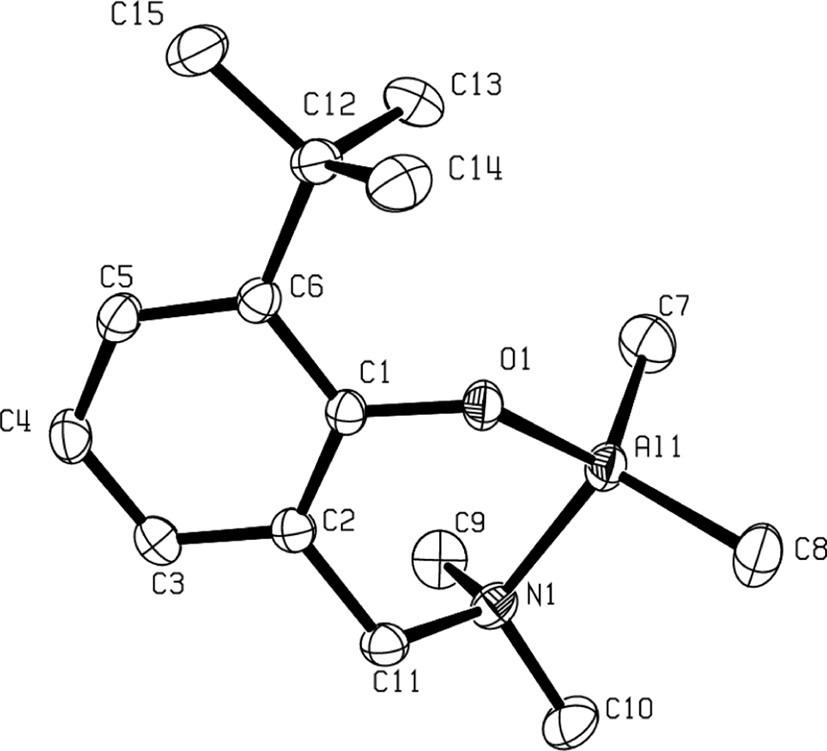
Molecular structure of a mono-aminophenolate dimethyl Al complex: 3b (reproduced with permission of the ACS copyright office).
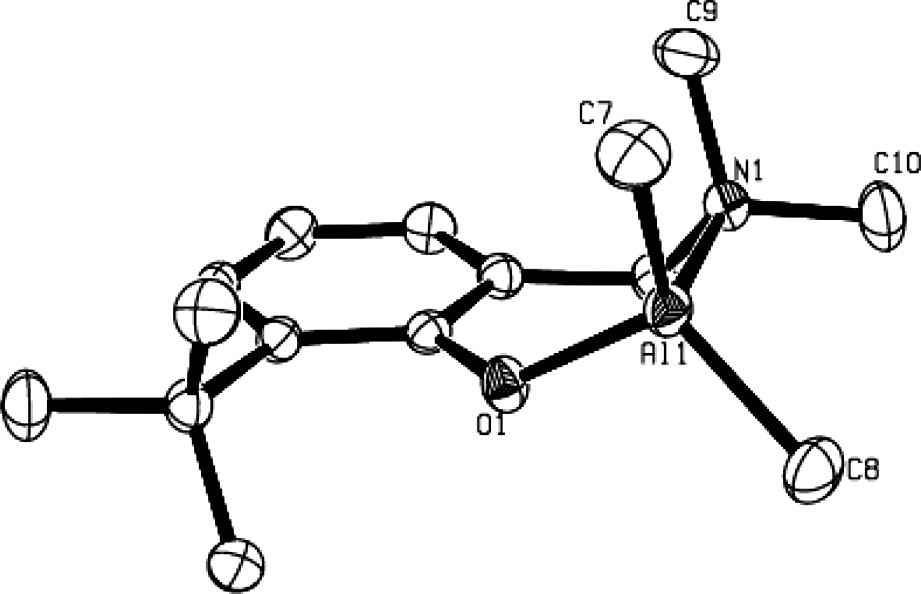
The significant puckering of the Al metallacycle in 3b.
The N,O bidentate chelating structure of mono-aminophenolate dialkyl group 13 complexes observed in the solid state is clearly retained in solution as deducted from NMR data. These data also show an effective Cs symmetry for this class of compounds on the NMR time scale at room temperature, which is due to a fast conformation change, via a ring flip, of the six-membered N,O metallacycle chelate. These fast conformational changes, illustrating the flexible nature of the metallacycle backbone, were studied in details in the case of the boron derivative {2-(CH2NMe2)C6H4O}BPh2, where two different conformers could be observed at –60 °C in d8-toluene [10].
3 Dinuclear cationic aminophenolate alkyl aluminum complexes from the dialkyl precursors
3.1 Synthesis and structure of aminophenolate dinuclear aluminum cations
As stated in Section 1, η2(O,N)-bonded aminophenolate group 13 dialkyl species may be used as neutral precursors for the generation of highly Lewis acidic group 13 cations, via an alkyl abstraction at the group 13 metal center. This approach has been so far applied to Al aminophenolate derivatives 3a–e.
The Al dimethyl complexes {2-(CH2L)-6-R-C6H4O}AlMe2 (3a–d) were found to cleanly react with B(C6F5)3 to afford the dinuclear cations {2-(CH2L)-6-R-C6H4O}AlMe.{2-(CH2L)-6-R-C6H4O}AlMe2+ (9a–d+/9a′–d′+) as a mixture of two diastereomers and as MeB(C6F5)3– salts (Scheme 9) [9]. All these cations are stable for days under an inert atmosphere at room temperature whether in a pure form or in solution, thus showing the suitability of the aminophenolate ligand for the stabilization of Al alkyl cations.
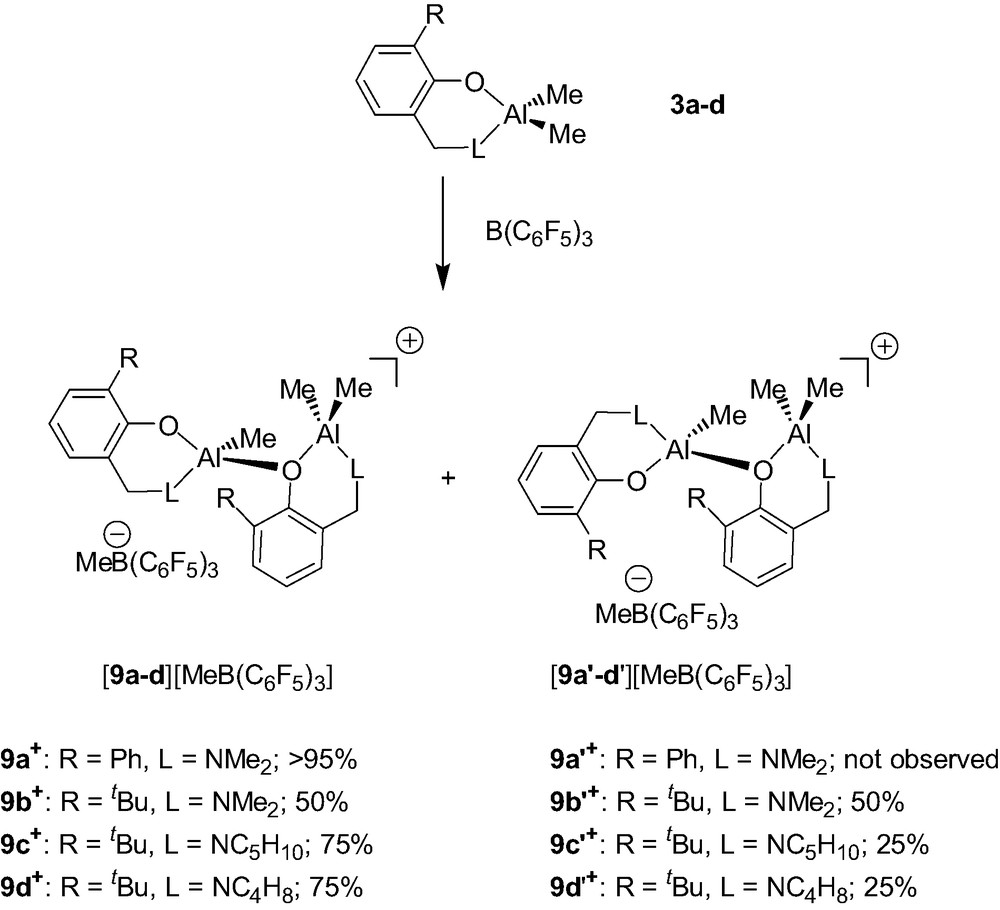
In these cationic systems, the presence of two diastereomers (9a–d+ and 9a′–d′+, Scheme 9) is due to the fact that each dinuclear cation contains two elements of chirality: i.e. a stereogenic tetrahedral Al center and a second element of chirality resulting from the configurational stability of the AlMe2 chelate ring. The latter element of chirality is proposed on the basis of solid-state structural studies and of the lack of dynamic behavior in solution of 9a–d+/9a′–d′+ (vide infra). As could be anticipated, different diastereomeric ratios are observed in the 9a–d+/9a′–d′+ cationic systems (Scheme 9). These different ratios (9a+ vs. 9b+/9b′+ and 9b+/9b′+ vs. 9c–d+/9c–d′+) show that both the phenol and the amino groups influence the diastereoselectivity of these reactions.
Several X-ray determined molecular structures of this new family of dinuclear cations, clearly established the adduct nature of these cationic species, as illustrated in Fig. 3 in the case of 9a+ [9]. Thus, as shown in Fig. 3, cation 9a+ exhibits an overall C1 symmetry and can be seen as an adduct of the three-coordinate cation {2-(CH2NMe2)-6-Ph-C6H4O}AlMe+ and the neutral four-coordinate Al complex {2-(CH2NMe2)-6-Ph-C6H4O}AlMe2, in which the two Al centers are linked by a μ2-O bridging aminophenolate ligand through O(2). Interestingly, the six-membered-ring Al(2) metallacycle containing the bridging aminophenolate adopts a boat like conformation, which further illustrates the excellent flexibility of the chelating ligand. Such a conformation can be rationalized by the geometrical constraints imposed on the aminophenolated ligand by both the coordination of O(2) to Al(1) and the tetrahedral geometry preferred by Al(2).
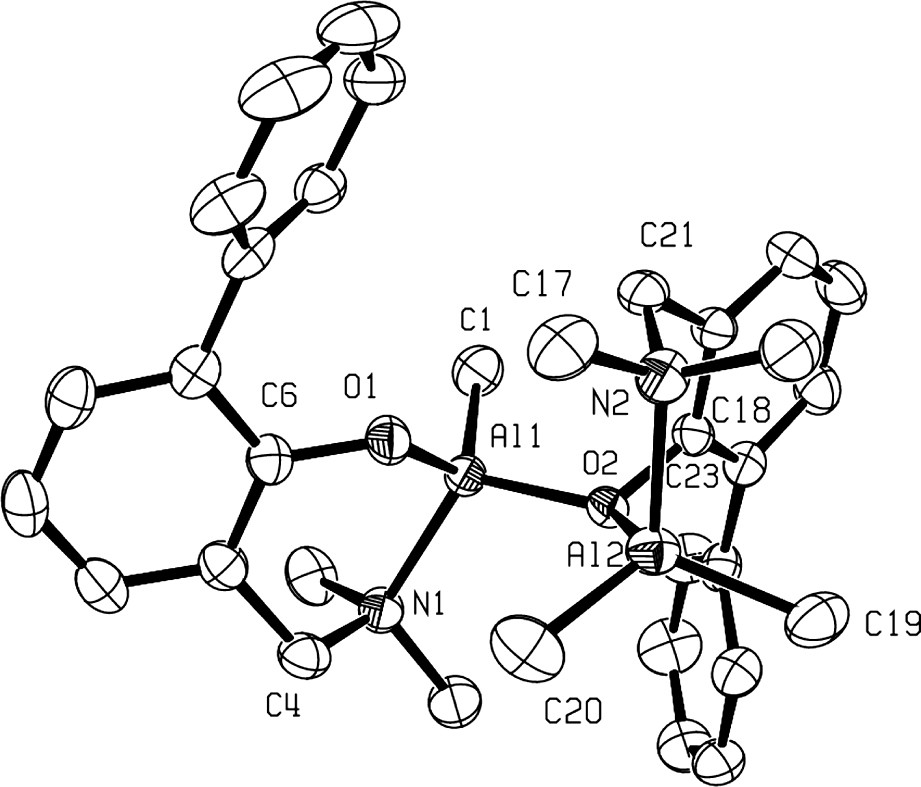
Molecular structure of the dinuclear Al cation 3a+ (reproduced with permission of the ACS copyright office).
Detailed 1D and 2D NMR studies of all Al cations are consistent with their solid-state structures being retained in solution up to 80 °C in aromatic and halogenated solvents and no dynamic behavior was observed in solution from room temperature to 80 °C, indicating the robustness of these derivatives. In addition, no close cation–anion interactions were observed in solution under the studied conditions.
From the above studies, one can conclude that the presence of a Ph or a tBu ortho-substituent on the phenol ring of the aminophenolate ligand does not provide enough steric shielding to prevent the formation of aggregates: i.e. in the present case, the formation of dinuclear Al cationic species. As observed in other Al cationic {LX}AlMe+ systems [5], the tendency to generate polynuclear species, a common feature in group 13 (III) chemistry, seems to be enhanced when going from neutral to cationic derivatives.
3.2 Stability and reactivity with THF of aminophenolate dinuclear Al cations
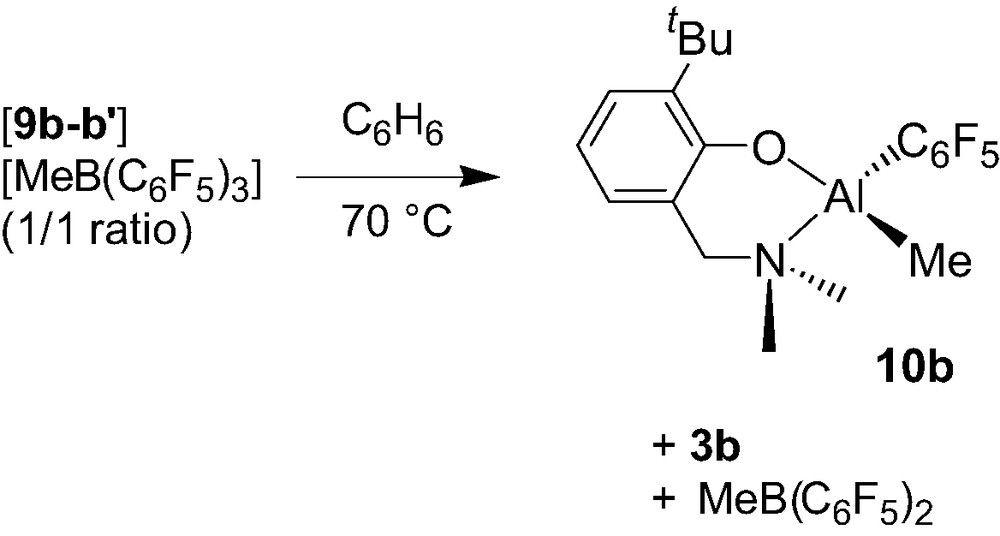
The stability of the dinuclear aminophenolate Al cations was studied in the case of the 9b+/9b′+ diastereomeric mixture (1:1 ratio). Complete decomposition of the salt species [9b/9b′][MeB(C6F5)3] is observed after overnight in benzene (70 °C) giving rise to a 1:1:1 ratio of the neutral monomethyl complex {2-(CH2NMe2)-6-tBu-C6H4O}Al(Me)(C6F5) (10b), complex 3b, and three-coordinated borane MeB(C6F5)2 (Scheme 10). Similar degradation reactions between cationic Al alkyl systems and the MeB(C6F5)3– anion occurring via a C6F5– transfer from the anion to the Al center has previously been observed [14]. However, degradations of this type usually take place at a lower temperature, thus illustrating the excellent stability of the 9b+/9b′+ system.
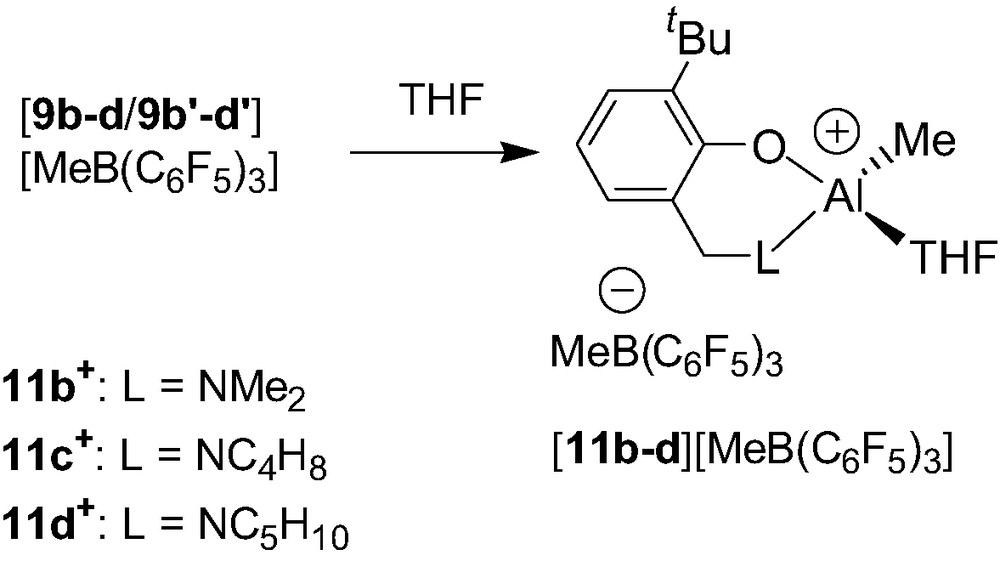
The reactivity of the dinuclear cations 9b–d+/9b′–d′+ has also been investigated with a Lewis base such as THF. Thus, with the notable exception of cation 9a+, cationic species 9b–d+/9b′–d′+ readily react with THF to afford a 1:1 mixture of the cationic THF adduct Al alkyl complexes {2-(CH2L)-6-tBu-C6H4O}Al(Me)(THF)+ (11b–d+) and the neutral precursors 3b–d (Scheme 11). The obtainment of the Al–THF cationic adducts 11b–d+ from the dinuclear species 9b–d+/9b′–d′+ is consistent with the proposed adduct structure of the latter cations. In contrast, the reaction of 9a+ with THF has been reported to afford an untractable mixture, which remains quite perplexing given the apparent structural similarities, in solution and in the solid state, of all the synthesized cations.
3.3 Applications of aminophenolate dinuclear Al alkyl cations in Lewis acid catalysis
As cationic low-coordinate aluminum species, the dinuclear aminophenolate Al cations 9b–d+/9b′–d′+ are expected to be highly Lewis acidic species, thus of interest in catalysis. Therefore, their reactivity has been studied with ethylene and propylene oxide.
Cations 9b–d+/9b′–d′+ are not active in ethylene polymerization, as might be expected by the robustness and the lack of fluxional behavior of these adducts species. These cations, do, however, initiate the polymerization of propylene oxide under mild conditions to afford atatic PPO in reasonable yields and Mw/Mn values (Table 1).
| Cationic complex | Yield in PPO (%) | Mn | Mw/Mn |
| 9b+/9b′+ a | 55 | 2840 | 1.54 |
| 9b+/9b′+ b | 70 | 9022 | 1.73 |
| 9c+/9c′+ | 51 | 2560 | 1.61 |
| 9d+/9d′+ | 61 | 3076 | 1.50 |
a Reaction conditions: 200 equiv of PO, toluene, room temperature, 15 min.
b Reaction conditions: 200 equiv of PO, toluene, 0 °C, 15 min.
As suggested by the data in Table 1, lowering the temperature increases the yield in PPO as well as the molecular weight of the obtained PPO. Overall, the catalytic activity of cations 9b–d+/9b′–d′+ for PO polymerization compares with that of Schiff base–AlEt2Cl systems [15] but is lower than that of porphyrinato-aluminum chloride complexes [16].
4 Extremely bulky cationic aminophenolate alkyl Al complexes
The obtention of mononuclear cationic group-13 complexes of the type {LX}AlMe+ is, in general, more interesting than that of dinuclear analogues, because mononuclear species usually exhibit a higher reactivity toward Lewis bases and are often easier to structurally characterize. However, a dramatic steric crowding around the group-13 metal center, possible via the incorporation of an extremely bulky LX– ligand in {LX}AlMe+, is required for the formation of such cations and thus to prevent aggregation from occurring. Accordingly, the design and synthesis of an extremely bulky aminophenolate LX– seemed appropriate.
4.1 Synthesis and structure
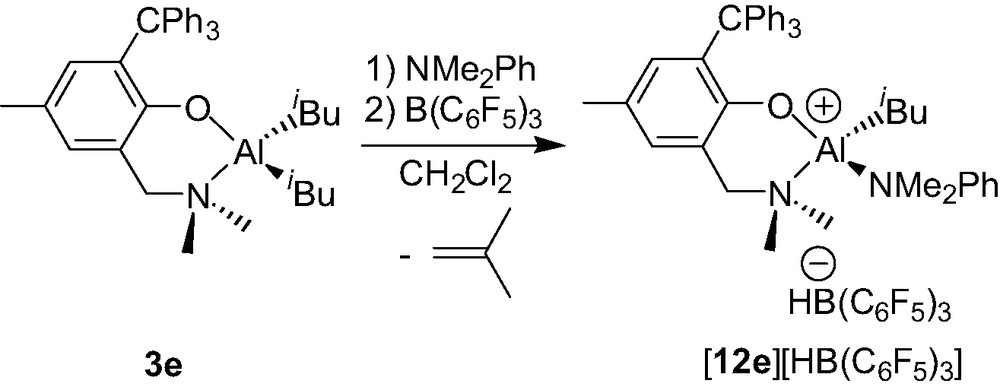
While the ionization chemistry of the dimethyl Al complexes 3a–d yields the clean formation of dinuclear cationic species 9b–d+/9b′–d′+, that of the extremely bulky aminophenolate Al diisobutyl derivative {2-(CH2NMe2)-4-Me-6-CPh3-C6H2O}AliBu2 (3e, Scheme 12), appears more complicated according to preliminary results: a mixture of Al species seems to be obtained and their characterization is under study [17]. However, in the presence of a Lewis base such as NMe2Ph, the ionization of compound 3e quantitatively affords the mononuclear Al cation {2-(CH2NMe2)-4-Me-6-CPh3-C6H2O}Al(iBu)(NMe2Ph)+ (12e+) as a HB(C6F5)3– salt (Scheme 12) [18].
The salt species [12e][HB(C6F5)3], whose molecular structure was determined by X-ray crystallography (Fig. 4), is rather unusual in the sense that it incorporates a Lewis acidic Al cation associated with a Lewis base entity such the HB(C6F5)3– anion, a hydride source. The excellent stability of this salt species may be ascribed to the significant steric hindrance around the Al center in 12e+ preventing an attack of the borohydride anion HB(C6F5)3–. The structural data for [12e][HB(C6F5)3] deduced from solid state and solution structural studies are all consistent with no close cation–anion interactions.
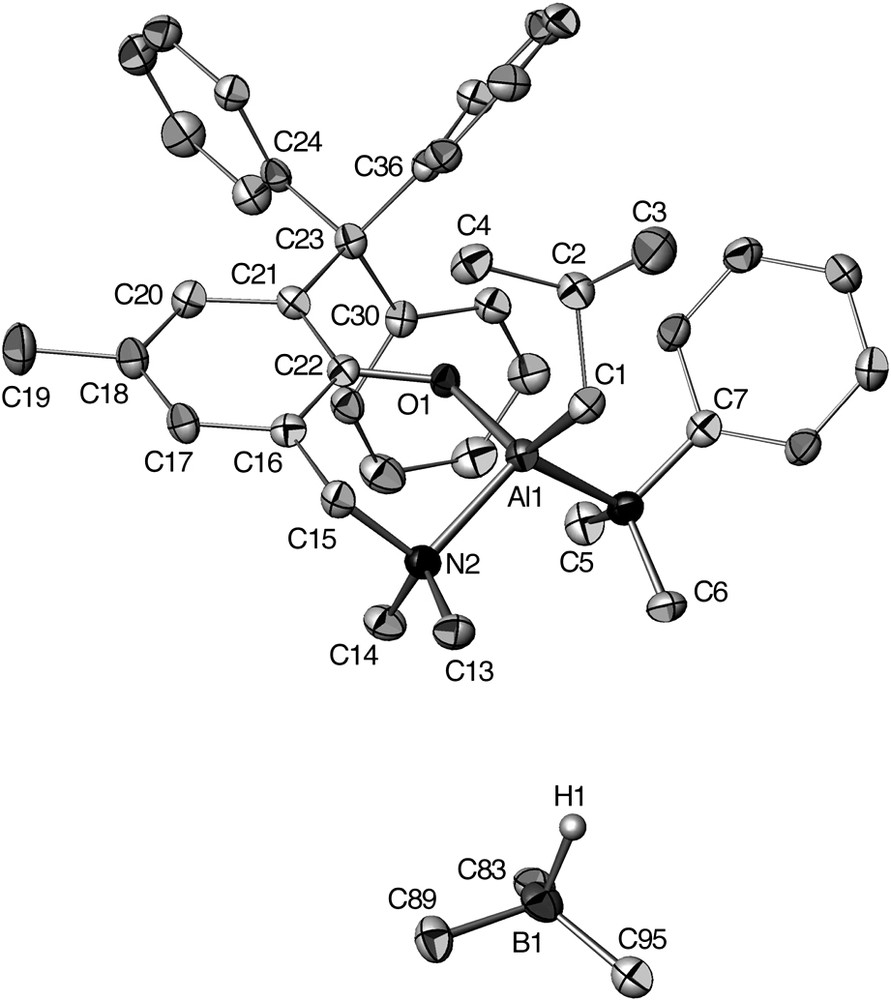
Molecular structure of [12e][HB(C6F5)3]. The C6F5 rings of the HB(C6F5)3– anion are omitted for clarity (reproduced with permission of the ACS copyright office).
4.2 Implications of an aminophenolate Al cation in a B(C6F5)3-catalyzed hydroalumination of Ph2CO and PhCHO
The reactivity of the Al cation/HB(C6F5)3– salt compound [12e][HB(C6F5)3] as a Lewis acid and base system has been studied with carbonyl substrates. Such substrates appeared attractive since they may act as Lewis bases toward the formed Al cation while being susceptible to hydride attack from HB(C6F5)3–.
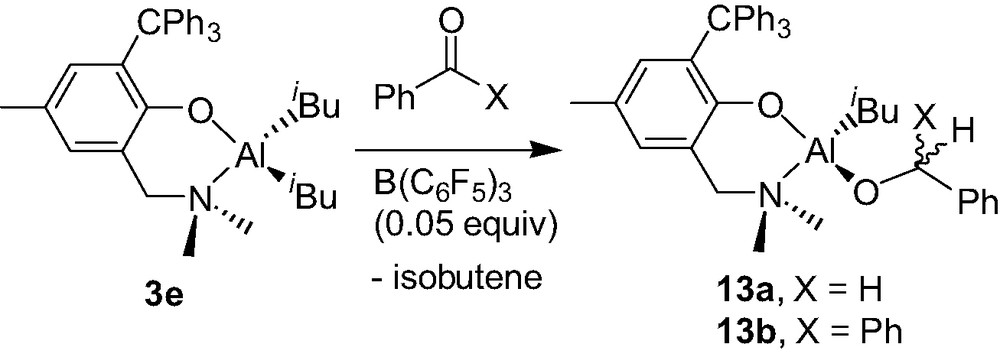
The diisobutyl Al complex 3e, in the presence of benzophenone or benzaldehyde, was found to be cleanly converted to the corresponding neutral monoalkoxy monoisobutyl complex {2-(CH2NMe2)-4-Me-6-CPh3-C6H2O}Al(iBu)(O–C(H)(R)Ph) (13a–b) provided a catalytic amount of B(C6F5)3 is used (Scheme 13) [17]. This B(C6F5)3-catalyzed hydroalumination reaction, which proceeds smoothly at room temperature, most likely involves a mononuclear cation of the type described in Section 4.1, as proposed in Scheme 14.
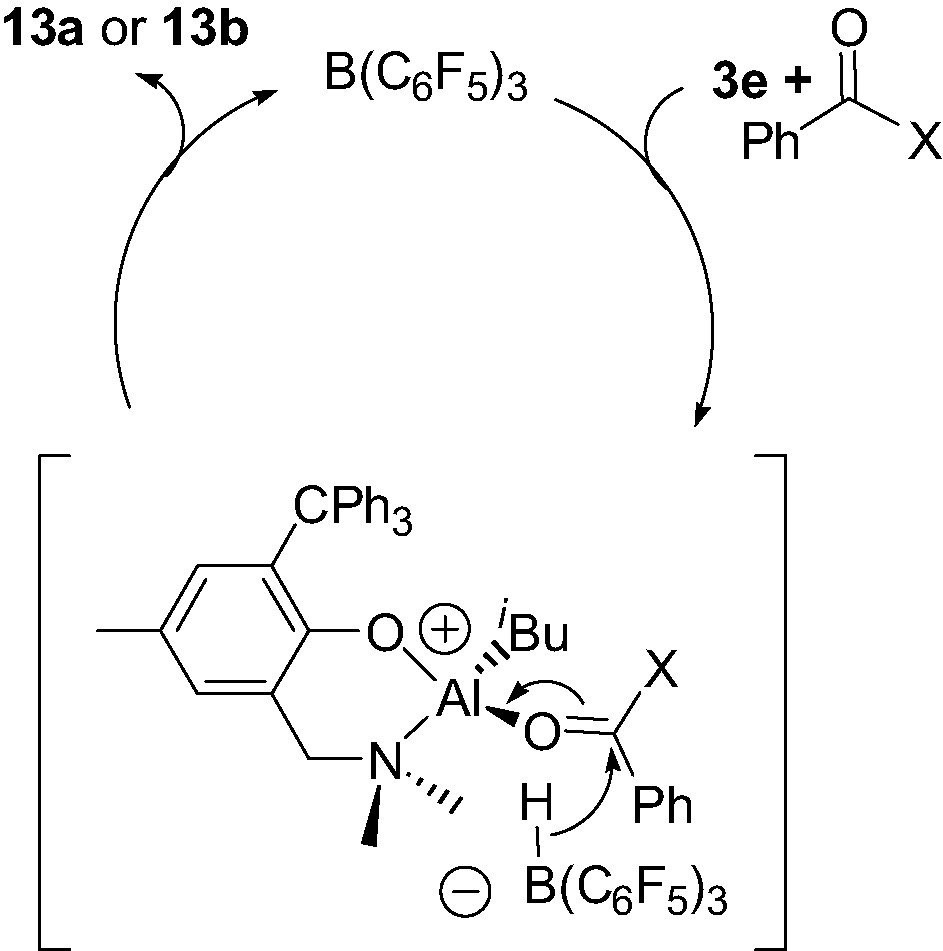
According to Scheme 14, the initial reaction of complex 3e with B(C6F5)3, in the presence of the carbonyl derivative acting as a Lewis base L, probably leads to the formation of a transient species that can be related to 12e+: i.e. a 4-coordinate cationic Al–L adduct 6-(CH2NMe2)-2-CPh3-4-Me-C6H2O}Al(iBu)(L)+ as a HB(C6F5)3– salt, along with release of isobutene. The HB(C6F5)3– borohydride may then transfer back an hydride to the quite electrophilic C=O carbon, thus producing complex 13a or 13b and re-generating B(C6F5)3. The mechanism proposed here: i.e. a B(C6F5)3-mediated hydride abstraction/transfer reaction, is reminiscent to that found in the B(C6F5)3-catalyzed hydrosilation of carbonyl functions, studied by Parks et al. [19].
5 Summary, concluding remarks and perspectives
Bidentate monoanionic O,N aminophenolate ligands are suitable for coordination to group 13 metal centers as shown by the research work performed in this area over the last 10 years. This suitability may be illustrated by the excellent stability of neutral group 13 mono-aminophenolate derivatives, provided there is a sufficient steric protection of the metal center to avoid ligand exchange reactions. In addition, these dialkyl group 13 complexes are easily accessible via classical alkane elimination reactions.
While the ionization of {LX}MR2 group 13 complexes usually yield poorly stable low-coordinated cationic species, stable aminophenolate aluminum have been reported and shown to be active as catalysts for PO polymerization. The higher stability of these aminophenolate cations versus other cationic systems may be related to the flexibility of the chelating aminophenolate backbone, allowing this ligand to adapt to the steric and electronic requirements of the chelated metal center.
In terms of perspectives, the Lewis acidic Al cations presented in this short review may broaden the scope of catalytic applications of this class of species, a result of their higher stability. In addition, as shown here by a B(C6F5)3-catalyzed hydroalumination, these robust species might also be useful for the mediation of unusual transformations. Finally, an extension of this ionization chemistry to gallium and indium derivatives, which are known to exhibit an increased stability vs. Al analogues, may also be of interest.