

1 Introduction
During the past 20 years, tremendous achievements have been reported in the design and application of organometallic complexes as catalysts for olefin polymerization. These developments have been made possible thanks to a better understanding of the factors that control the stability, reactivity and selectivity of the catalytically active species. In this regard, the cocatalysts, typically represented by methylaluminoxane (MAO), were demonstrated to play a key role in affording highly active and long-lived catalytic systems. Group 4 metallocenes I have clearly been at the forefront of these well-defined single-site catalysts. Changes to the ligand skeleton have also demonstrated their beneficial effects for controlling all of the macromolecular parameters of the polyolefin products. Such improvements are nicely illustrated by the capacity of ansa-metallocenes II and constrained geometry catalysts (CGC) III to polymerize propene with high stereocontrol and to copolymerize ethylene and α-olefins, respectively (Fig. 1) [1].
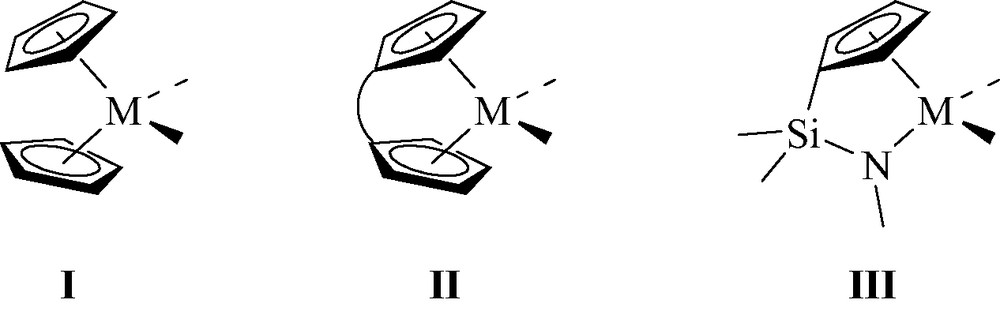
General representation of metallocenes I, ansa-metallocenes II and CGC III.
Over the last 10 years, numerous studies have been devoted to the incorporation of heteroelements, such as boron and phosphorus, in these prototype catalysts I–III. Such changes not only aim at tuning the stability and activity of the complexes by slight variations of their geometric and electronic properties, but also at affording a chemical active site that may participate in the stabilization and/or activation of the transition metal centers. This review will focus on the different approaches reported in this field. The incorporation of boron and phosphorous within the bridging units for ansa-metallocenes as well as constrained geometry complexes will be successively presented. Only group 4 metals catalysts have been considered. The synthesis, structure and catalytic activity of these heteroatom-containing analogs of Cp-based catalysts II–III will be described and whenever possible, comparisons will be drawn between the different systems.
2 Boron analogs
Ansa-metallocenes with single atom bridges have been extensively investigated notably those featuring carbon or silicon units bonded to the two η5-coordinated ligands [2]. Such short bridges typically restrict rotation of the cyclopentadienyl rings, enforce the bending of the rings and widen the area of the ‘equatorial wedge’. In order to further modify the electronic and steric properties of the bridging unit, short boron-based bridges have also been studied over the last 5 years [3]. The propensity of such Lewis acidic bridging units to coordinate Lewis bases should alter the geometry and therefore the reactivity of the complexes. One might also envisage a direct participation of such Lewis acidic bridges during the olefin polymerization, the ultimate goal in this area being the development of self-activating zwitterionic catalysts [4]. In this section, the synthesis, structure and catalytic activity of ansa-metallocenes featuring tetracoordinate neutral, anionic as well as zwitterionic boron-bridges are first discussed. The related complexes featuring neutral tricoordinate boron-bridges are then presented.
2.1 Tetracoordinate neutral boron-bridged metallocenes
The synthesis of the first boron-bridged ansa-metallocene was reported in 1997 by Shapiro and co-workers [5]. The required dicyclopentadienyl borane ligand 2-Ph was first prepared by condensing 2 equiv. of silylated-stannylated cyclopentadiene Me3Sn(Cp)SiMe3 1 with dichlorophenylborane [5a]. Surprisingly, the coordination of 2-Ph could not be achieved with ZrCl4, ZrCl4(thf), ZrCl4(tht) (tht = tetrahydrothiophene) even at elevated temperature. Apparently, the π-acceptor bridging-unit of 2-Ph deactivates the silyl substituent of the Cp rings toward electrophilic substitution by zirconium. Accordingly only the non-oligomeric ZrCl4(SMe2)2 proved to be reactive toward 2-Ph, and the desired zirconium complex was obtained in 33% yield (Scheme 1). The dimethyl sulfide not only stabilize the resulting complex as a Lewis base adduct 3-SMe2 but also probably assists in its formation by coordination to the boron atom of 2-Ph, thereby masking the π-acceptor character of the bridge and facilitating the substitution of the silyl groups for zirconium.
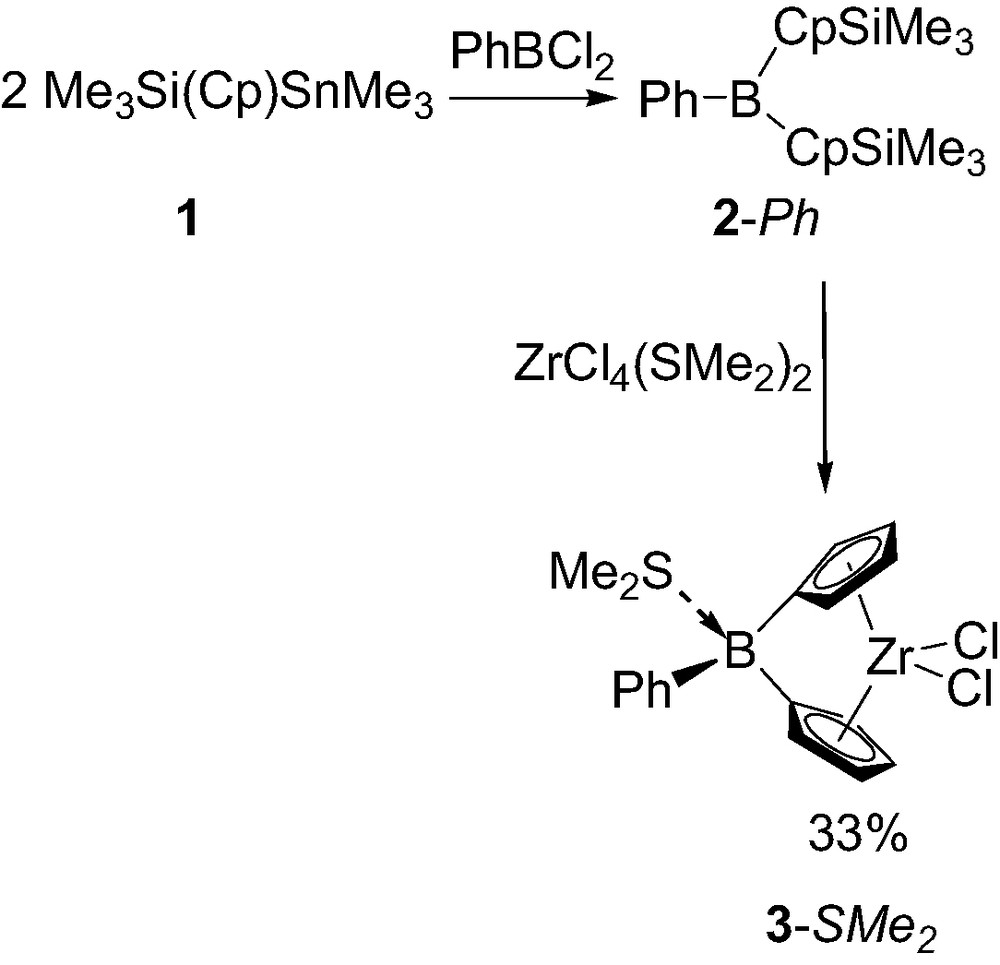
Synthesis of the first boron-bridged ansa-metallocene 3-SMe2.
Complex 3-SMe2 has been fully characterized in solution by multinuclear NMR and its molecular structure has been confirmed by an X-ray analysis [5b]. Table 1 lists the most significant features (bite angle of the bridge Cp–X–Cp, centroid–metal–centroid angle and dihedral angle between the ring planes) for 3-SMe2 compared to those reported for the related C- and Si-bridged zirconium complexes. Of particular interest, the bridge angle Cp–B–Cp (101.1°) is rather similar to that found in the carbon-bridged complex, but significantly wider than that measured in the corresponding silicon-bridged derivative. This value is quite narrow for a tetracoordinate boron atom, suggesting that bridging induces significant strain. As far as the centroid–metal–centroid and Cp–Cp dihedral angles are concerned, the values measured for 3-SMe2 are expectedly at halfway between those of the carbon and silicon-containing ansa-zirconocenes.
Selected angles (°) for C-, B- and Si-bridged zirconium complexes
| Complex | X | Cp–X–Cp | Centroid–M–centroid | Cp–Cp dihedral angle | References |
| Me2C | 99.8 | 116.6 | 71.4 | [6] | |
| Ph(Me2S)B | 101.1 | 121.3 | 65.9 | [5] | |
| Ph(Me3P)B | 100.1 | 121.1 | 68.5 | [5] | |
| Me2Si | 93.2 | 125.4 | 56.8 | [7] |
The structural analysis also revealed important features regarding the boron-bridge itself. Indeed, the rather long boron–sulfur distance (2.01 compared 1.92 Å for the sum of the covalent radii) suggests only a weak dative interaction between the two atoms. This has been confirmed by easy labialisation of the dimethyl sulfide in solution, as deduced from variable-temperature 1H NMR experiments [5b]. The dimethylsulfide was also found to be readily displaced by stronger Lewis bases such as trimethylphosphine (Scheme 2). The new metallocene 3-PMe3 was characterized in solution and in the solid state by X-ray. No noticeable effect on the overall geometry of the complex was observed (Table 1). The only significant change resulting from this Lewis base substitution is the shorter distance between the boron atom and the Lewis base (P–B distance of 1.99 Å for 3-PMe3 compared to S–B distance of 2.01 Å for 3-SMe2) despite the larger covalent radius of phosphorus relative to sulfur (1.10 compared to 1.02 Å). This feature clearly indicates that the boron bridge is much strongly bonded by this Lewis base, and accordingly the phosphine proved to be non-labile in solution on the NMR time scale up to 340 K [5b]. The stronger coordination of the trimethylphosphine was also clearly apparent when the replacement of the chloride atoms on zirconium by more reactive alkyl groups was investigated. Indeed, attempts to alkylate 3-SMe2 only led to decomposition, presumably due to the lability of dimethyl sulfide and subsequent nucleophilic attack at the boron center inducing the cleavage of the boron-bridge. In contrast, the more robust trimethylphosphine adduct 3-PMe3 could be successfully alkylated by various RLi (R = CH2SiMe3, Me) and RMgCl (R = CH2C6H5) reagents [5b].
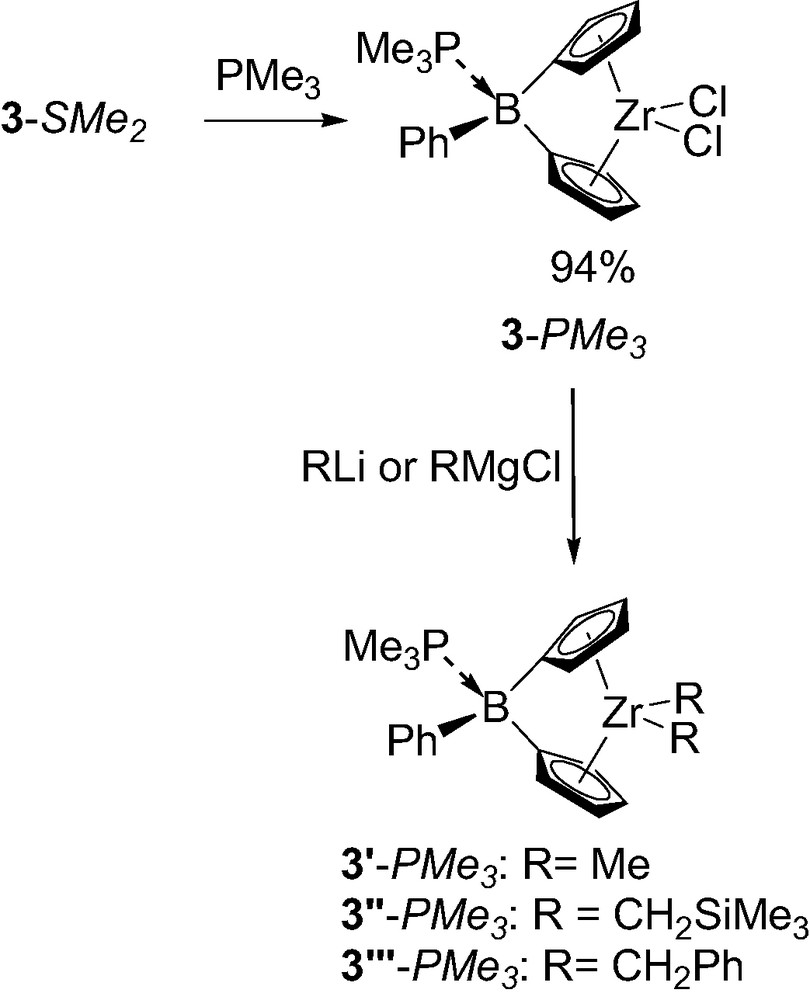
Synthesis of the boron-bridged metallocenes as trimethylphosphine adducts 3-PMe3.
Notably, variation of the B-substituent of the ligand proved to be feasible and to strongly influence the strength of the Lewis base coordination. Indeed, subsequent treatment of 1 with BBr3, LiC6F5 and ZrCl4(SMe2)2 afforded complex 3′-SMe2 featuring a pentafluorophenyl group at boron (Scheme 3), and due to the increased electrophilicity of the boron, this dimethyl sulfide adduct was found to be non-labile on the NMR time scale [3a].
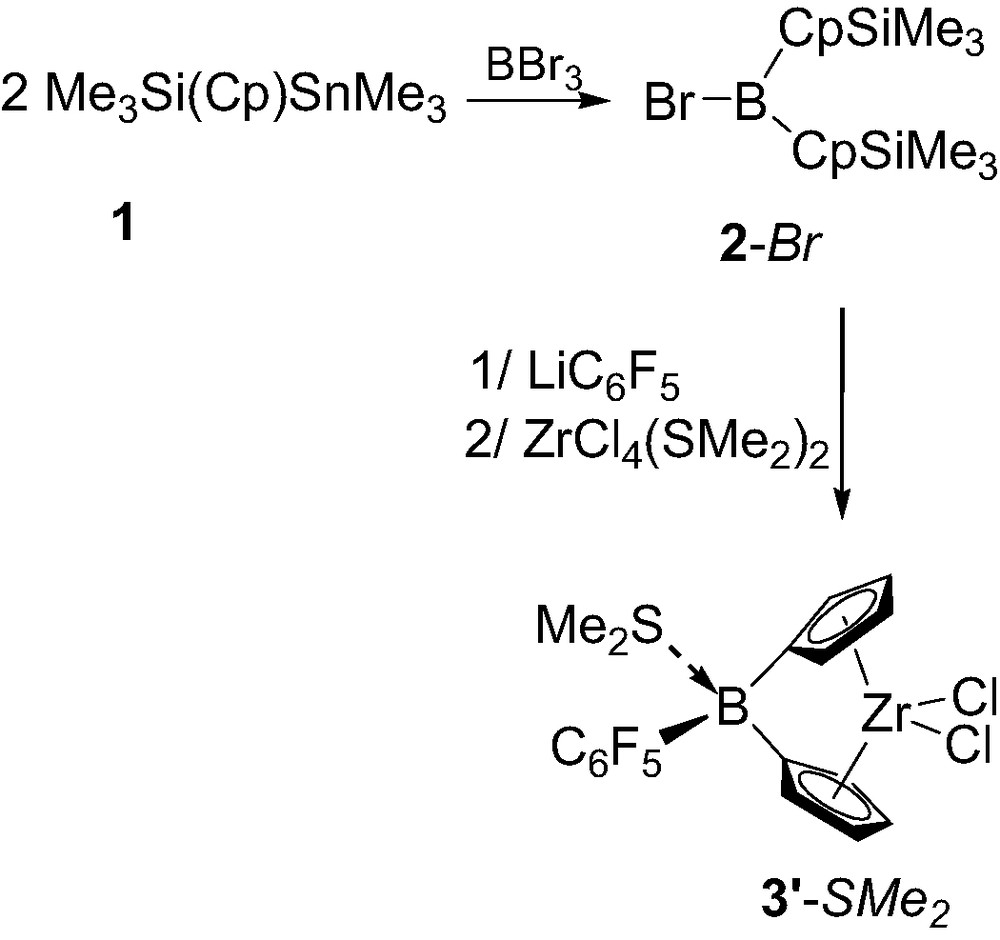
Synthesis of the ansa-metallocene 3′-SMe2 featuring a pentafluorophenyl group at boron.
As far as ethylene polymerization is concerned, complex 3-SMe2 proved to be inactive in the presence of MAO. In marked contrast, low molecular weight PE (Mw = 10,200 g/mol) were obtained with 3-PMe3 in the same conditions (MAO/Zr ratio of 200, room temperature and 1 bar of ethylene pressure) [5b], and a rather high activity of 1664 kg PE/mol Zr per bar per hour was even observed in more drastic conditions (140 °C in toluene under 40 bar of ethylene) [8].
Related boron-bridged bis(indenyl)metallocenes have also been prepared and evaluated in olefin polymerization [9]. Treatment of indenyllithium with PhBCl2 followed by PiPr3-induced diene isomerization first led to the neutral ligand 2′-Ph. Subsequent deprotonation with 2 equiv. of lithium hexamethyldisilazane (LiHMDS) followed by coordination with ZrCl4 in toluene/ether afforded the corresponding complex as a diethyl ether adduct 4-Et2O and a 1:1 mixture of meso/rac isomers (Scheme 4). Pure rac-4-Et2O could be isolated by crystallization in 12% yield and converted into rac-4-THF and rac-4-PMe3 complexes by simple treatment with a THF/CH2Cl2 mixture and PMe3, respectively. Derivative rac-4-Et2O has only characterized by multinuclear NMR in solution, while the molecular structures for rac-4-THF and rac-4-PMe3 have been confirmed by X-ray diffraction studies. The centroid–metal–centroid angles in the two complexes are very similar (122.3° for rac-4-THF and 121.5° for rac-4-PMe3) and stand between those for Me2C- [10] and Me2Si-bridged [11] bis (indenyl) dichlorozirconocenes (118.2° and 127.8°, respectively).

Synthesis of the boron-bridged bis(indenyl)metallocenes rac-4.
These complexes have been evaluated as precatalysts toward ethylene and propene polymerization after activation with MAO for 5 min in toluene [9]. The diethyl ether adduct rac-4-Et2O was found to be a fairly active catalyst for ethylene polymerization (200–2600 kg PE/mol Zr per hour) producing high molecular weight PE (Mw = 700,000 g/mol). However, much lower activities (only 10–75 kg PP/mol Zr per hour using a Al/Zr ratio of 1000 at room temperature) were observed toward propene polymerization and rac-4-Et2O only led to atactic oily PP (Mw = 20,000 g/mol). Notably, the nature of the donor ligand coordinated to the boron center was found to strongly influence the activity and stereoselectivity of the catalytic system. Indeed, the phosphine adduct rac-4-PMe3 proved remarkably active and stereoselective leading to high molecular weights (up to 315,000 g/mol) and high degrees of isotacticities (up to 96%, as deduced from the proportion of mmmm-pentads measured by 13C NMR spectroscopy) (Table 2). Although it is quite clear that the stronger coordination of the phosphine prevents decomposition of the catalyst via MAO-induced labialisation of the Lewis base, the precise reason for the differences observed between the two complexes rac-4-Et2O and rac-4-PMe3 remains to be determined. Anyhow, complex rac-4-PMe3 appears as a promising precatalyst for propene polymerization, competing to some extent with the Me2Si-bridged ansa-zirconocenes [11b].
Propene polymerization with rac-4-Et2O and rac-4-PMe3 (after activation with MAO for 5 min in toluene, under 2 bar of propene pressure)
| Complex | T (°C) | Al/Zr | Activity (kg PP/mol Zr per hour) | Mw (g/mol) | Tacticity (% mmmm-pentads) |
| rac-4-Et2O | 20 | 1000 | 10–75 | 20 000 | – |
| rac-4-PMe3 | 20 | 1000 | 326 | 315 000 | 96 |
| rac-4-PMe3 | 40 | 220 | 783 | 161 000 | 93 |
| rac-4-PMe3 | 40 | 1000 | 1053 | 129 000 | 90 |
| rac-4-PMe3 | 40 | 5000 | 1232 | 97 000 | 93 |
| rac-4-PMe3 | 60 | 1000 | 174 | 62 000 | 85 |
2.2 Tetracoordinate borate-bridged metallocenes
Despite their assumed instability, a few borate-bridged metallocenes have been isolated. The first representative of this new family of boron-containing ansa-zirconocene 5-Me was obtained by taking advantage of the dimethyl sulfide lability in 3-SMe2 (Scheme 5) [8]. Although 2 equiv. of Cp*2AlMe were required to complete the reaction, presumably due to a competitive equilibrium with the liberated dimethyl sulfide, the complete methyl transfer from aluminum to boron was confirmed by 27Al NMR spectroscopy. The related complex 5-Cl was obtained in a similar manner, using bis(triphenylphosphoranylidene)ammonium chloride [PPN]Cl as a chloride source [8]. X-ray diffraction studies confirmed the ion pair structure of both 5-Me and 5-Cl, with Cp*2Al+ and [PPN]+ as respective countercations. Interestingly, the Cp–B–Cp angles of the borate-bridged complexes (97.2° for 5-Me and 99.4° for 5-Cl) are very similar to each other and only slightly narrower than those of their neutral analogs 3-SMe2 (101.1°) and 3-PMe3 (100.1°).
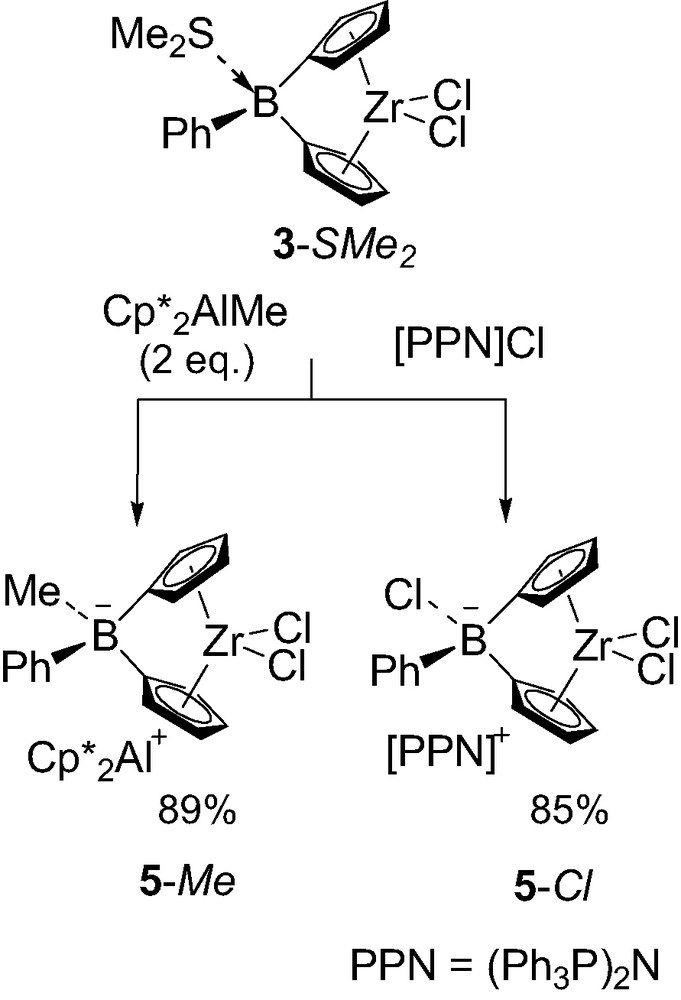
Synthesis of the first borate-bridged metallocenes 5-Me and 5-Cl.
In the presence of excess MAO, both complexes 5-Me and 5-Cl do induce ethylene polymerization, but with significantly lower activities than their neutral analog 3-PMe3 (902 for 5-Me and 574 for 5-Cl compared to 1664 kg PE/mol Zr per bar per hour for 3-PMe3 at 140 °C in toluene under 40 bar of ethylene pressure).
Given the greater stability of tetrakis(pentafluorophenyl)borate compared to tetrakis(phenyl)borate anions toward ligand transfer, Lancaster and Bochmann investigated related borate-bridged complexes featuring C6F5 groups [12]. Notably, pentafluorophenyl lithium was found to preferentially alkylate the zirconium center of 3-SMe2 despite the lability of dimethyl sulfide. Accordingly, alkylation of the boron-bridge required 3 equiv. of LiC6F5 and led to the borate-bridged complex 5-C6F5 that was isolated in 67% yield as a Li(OEt2)x salt (Scheme 6). The countercation of 5-C6F5 can be readily exchanged for NEt4+ or PPh4+ to give complexes 5′-C6F5 and 5″-C6F5, respectively. The replacement of the two C6F5 substituents at zirconium for methyl groups also proved feasible using 10 equiv. of AlMe3, although the corresponding complex 6-C6F5 has only been spectroscopically characterized and has not been isolated so far.
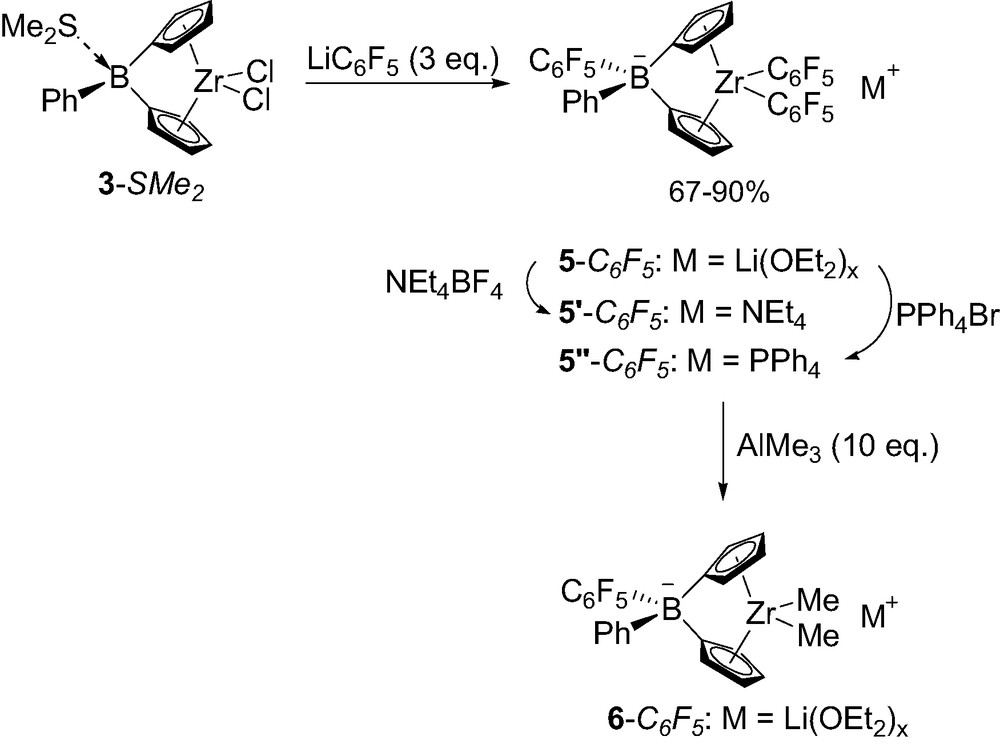
Synthesis of borate-bridged complexes 5-C6F5 featuring one C6F5 group.
Incorporation of two C6F5 groups at the borate bridge has also been investigated. The chloroborane-bridged precursor 3″-SMe2 was first prepared in a classical way by condensing 2 equiv. of silylated-stannylated cyclopentadiene Me3Sn(Cp)SiMe3 1 with trichloroborane followed by reaction ZrCl4(SMe2)2 [12]. Herealso pentafluorophenyl lithium proved to preferentially displace the chlorides at zirconium, and a fivefold excess was required to convert 3″-SMe2 into the desired borate-bridged zirconocene that was isolated in 28% yield as a Li(OEt2)x salt 5-(C6F5)2 (Scheme 7). Notably, the use of 5 equiv. of C6F5MgBr followed by countercation exchange with NEt4Br afforded the related tetraethylammonium ion pair 5′-(C6F5)2 in 40% yield. The replacement of the two C6F5 substituents at zirconium for methyl groups also proved feasible for both derivatives using 2 equiv. of MeLi, the corresponding complexes 6-(C6F5)2 and 6′-(C6F5)2 being obtained with purities higher than 90% according to 1H NMR. All of these complexes have been characterized in solution by multinuclear NMR, the presence of the borate bridge being typically indicated by 11B NMR chemical shifts around –12 ppm [12].
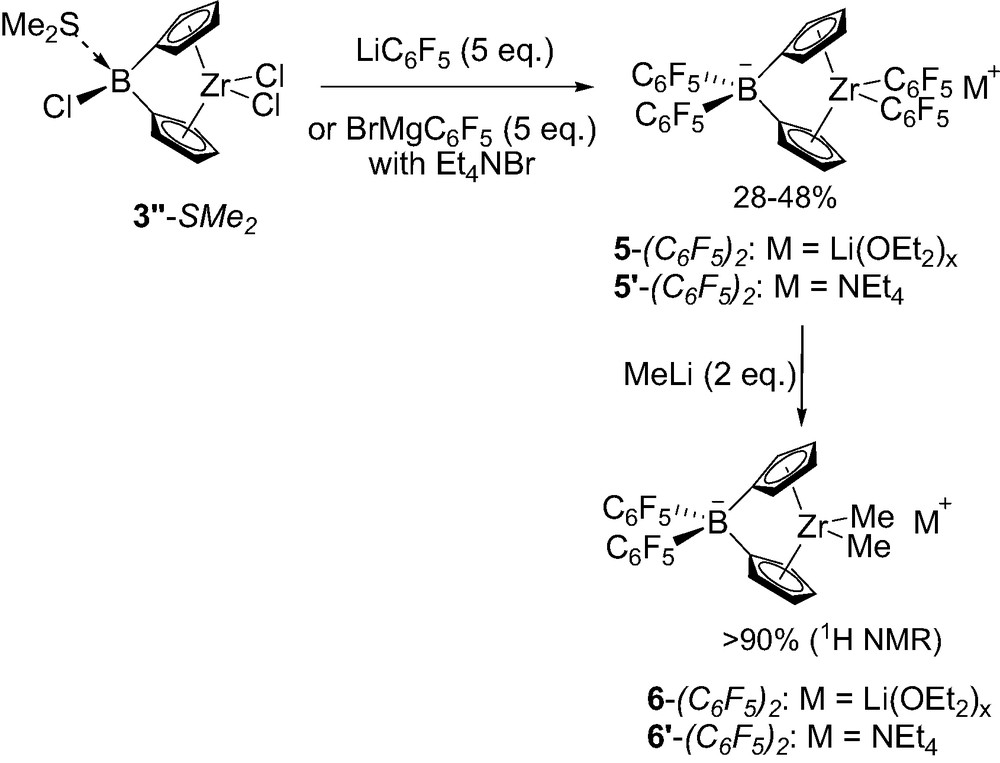
Synthesis of borate-bridged complexes 5-(C6F5)2 featuring two C6F5 groups.
Among all of these derivatives, complexes 5′-C6F5, 5-(C6F5)2, 6-(C6F5)2 and 6′-(C6F5)2 have been investigated as precatalysts toward ethylene polymerization (Table 6) [12]. The structural modifications (replacement of the phenyl substituent at boron for a second pentafluorophenyl ring, substitution of the C6F5 rings at zirconium for methyl groups, exchange of the Li(OEt2)x countercation for an ammonium salt) did not induce dramatic variations, as indicated by the similar activities observed for all of these complexes. According to these preliminary studies, rather high activities were observed (around 200–300 and 700–800 kg PE/mol Zr per bar per hour at 20 and 60 °C, respectively, under 1 bar of ethylene pressure using a large excess of MAO), but these results are hardly comparable with those obtained with the other borate-bridged complexes 5-Me and 5-Cl since the polymerization conditions are rather different. Notably a somewhat higher productivity was observed when the precatalyst 5-(C6F5)2 was premixed with MAO for 1 h, suggesting that the active species are slowly generated in these conditions. The mode of activation of the precatalyst also proved to be critical so that higher productivities up to 2130 kg PE/mol Zr per bar per hour at 60 °C were achieved with 5-(C6F5)2 in the presence of [Ph3C+,B(C6F5)4–] and AliBu3. However, such an increase in the catalytic activity was not observed for the dimethyl derivative 6-(C6F5)2 (Table 3).
Selected angles (°) for aminoborylidene-bridged zirconium complexes
| Complex | R2 | CxHy | Cx′Hy′ | Cp–B–Cp | Cp–Cp dihedral angle | References |
| 13a-Zr | Me2 | Cp | Cp | 105.9 | [19b] | |
| 13b-Zr | (iPr)2 | Cp | Cp | 103.9 | 65.5 | [18] |
| 13′b-Zr | (iPr)2 | Cp | Flu | 104.7 | 68.1 | [20] |
| rac-14b-Zr | (iPr)2 | Ind | Ind | 104.9 | 66.8 | [18] |
| 13a-Hf | Me2 | Cp | Cp | 105.2 | 64.7 | [19d] |
| 13e-Hf | –(CH2)5– | Cp | Cp | 105.5 | 64.9 | [19d] |
Ethylene polymerization using the borate-bridged complexes featuring one or two C6F5 groups (under 1 bar of ethylene pressure)
| Precatalyst | T (°C) | Cocatalyst (relative amount to Zr) | Time (s) | Activity (kg PE/mol Zr per bar per hour) |
| 5′-C6F5 | 20 | MAO (500) | 600 | 280 |
| 5′-C6F2 | 60 | MAO (500) | 240 | 800 |
| 5-(C6F5)2 | 20 | MAO (1000) | 1200 | 190 |
| 5-(C6F5)2 | 60 | MAO (1000) | 600 | 670 |
| 5-(C6F5)2a | 60 | MAO (600) | 600 | 1100 |
| 5-(C6F5)2 | 20 | [Ph3C+,B(C6F5)4–] (2) | 180 | 900 |
| AliBu3 (500) | ||||
| 5-(C6F5)2 | 60 | [Ph3C+,B(C6F5)4–] (2) | 180 | 2130 |
| AliBu3 (500) | ||||
| 6-(C6F5)2 | 20 | MAO (2000) | 300 | 230 |
| 6-(C6F5)2 | 60 | MAO (2000) | 300 | 690 |
| 6-(C6F5)2 | 20 | [Ph3C+,B(C6F5)4–] (1) | 300 | 290 |
| AliBu3 (500) | ||||
| 6-(C6F5)2 | 60 | [Ph3C+,B(C6F5)4–] (1) | 300 | 370 |
| AliBu3 (500) | ||||
| 6′-(C6F5)2 | 20 | [Ph3C+,B(C6F5)4–] (2) | 300 | 610 |
| AliBu3 (500) | ||||
| 6-(C6F5)2 | 60 | [Ph3C+,B(C6F5)4–] (2) | 300 | 390 |
| AliBu3 (500) |
a MAO and 5-(C6F5)2 premixed 1 h before polymerization.
2.3 Tetracoordinate zwitterionic borate-bridged metallocenes
In order to protect the boron-bridge from nucleophilic attack, metallocenes featuring zwitterionic tetracoordinate boron-bridge have been recently investigated [13]. Thanks to the high affinity of phosphorus ylides for Lewis acidic boron centers [14], ylides such as Ph3P=CH2 replace Me2S from the boron-bridge in 3-SMe2 giving the zwitterionic complex 7 (Scheme 8). Complex 7 has been isolated in 75% yield and fully characterized in solution and in the solid state. The P–C and C–B bond lengths within the boron bridge (1.79 and 1.67 Å, respectively) are in the range of P–C(sp3) and C–B(sp3) bonds and similar to those of Ph3PCH2B(C6F5)3 (1.79 and 1.67 Å, respectively) [14c]. The centroid–Zr–centroid angle (120.2°) and the dihedral angle Cp–Cp (66°) are within the range of those observed in the tetracoordinated boron neutral bridged complexes (121.3°/65.9° for 3-SMe2 and 121.1°/68.5° for 3-PMe3). The Cp–B–Cp angle (97.1°) is very similar to that of 5-Me (97.2°) but narrower than that of 5-Cl (99.4°) and of the neutral complex 3-PMe3 (100.1°). These data indicates that the Lewis basicity of the ylide is comparable to that of methyl anion.
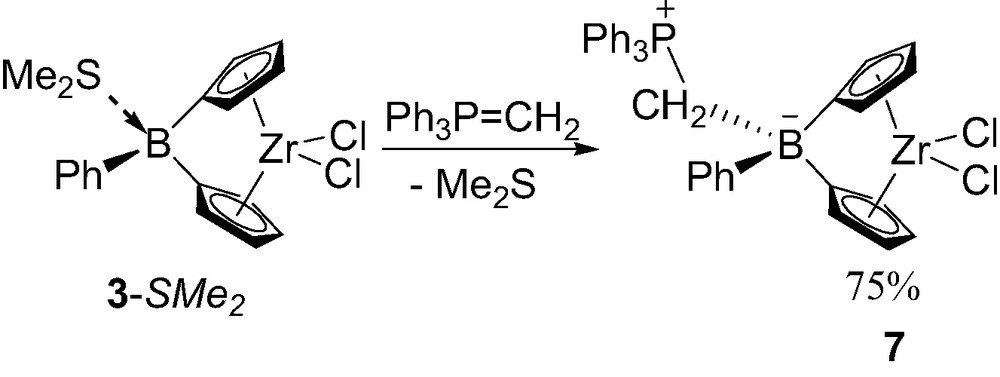
Synthesis of the zwitterionic tetracoordinate borane-bridged complex 7.
When activated with 1000 equiv. of MAO, complex 7 exhibited remarkable activity toward ethylene polymerization that is an order of magnitude greater than that of the commercial precatalyst [Cp′SiMe2-NtBu]TiCl2 (Table 4). In the presence of oct-1-ene, little, if any, α-olefin was incorporated, but complex 7 was found to be rather active in propene polymerization, with similar efficiency than [Me2SiCp2]ZrCl2.
Ethylene/oct-1-ene copolymerization and propene polymerization using precatalyst 7
| Precatalyst | Alkene | Efficiency (gP/gM) |
| 7′ | Ethylene/oct-1-enea | 6 835 920 |
| Propeneb | 27 186 | |
| [Cp′-SiMe2-NtBu]TiCl2 | Ethylene/oct-1-enea | 441 729 |
| [Me2SiCp2]ZrCl2 | Ethylene/oct-1-enea | 93 725 |
| Propeneb | 34 421 |
a 140 °C under 3.4 MPa of ethylene.
b 70 °C for 150 g of propene.
The bis(indenyl)analogs of 8-Ph were prepared following the same strategy than that used for the neutral indenylboron complexes 4 [13]. The diene isomerization was occurred without PiPr3 and ZrCl4(SMe)2 was used instead of ZrCl4 for the complexation (Scheme 9). The poor solubility and stability of the new complex 8-Ph complicated its purification. Furthermore, the related complex 8-Me featuring the Me3PCH2B bridge was found to be even more sensitive than 8-Ph.
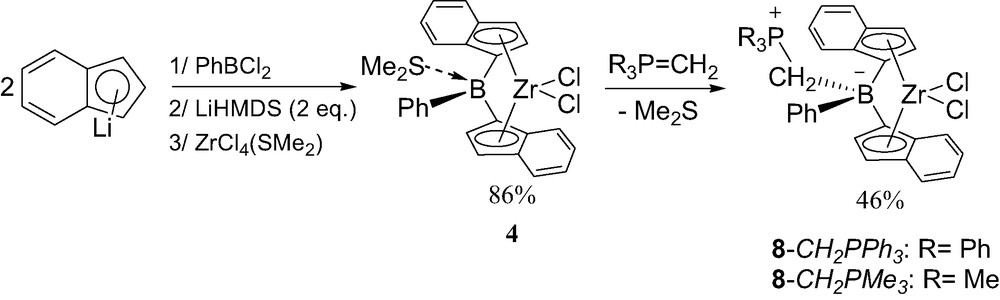
Synthesis of the neutral zwitterionic borate-bridged metallocenes 8 featuring indenyl rings.
Preliminary screenings of polymerization activities of 8-Ph and 8-Me toward propene were rather promising, 8-Ph featuring the Ph3PCH2B bridge being by far the best candidate [13]. Aiming at improving the solubility of such neutral zwitterionic borate-bridged metallocenes, complex 9 featuring 2-methyl-4-phenylindenyl rings was prepared [13]. The increased solubility of 9 allowed for its purification, crystallization and spectroscopic characterization in solution (Fig. 2).
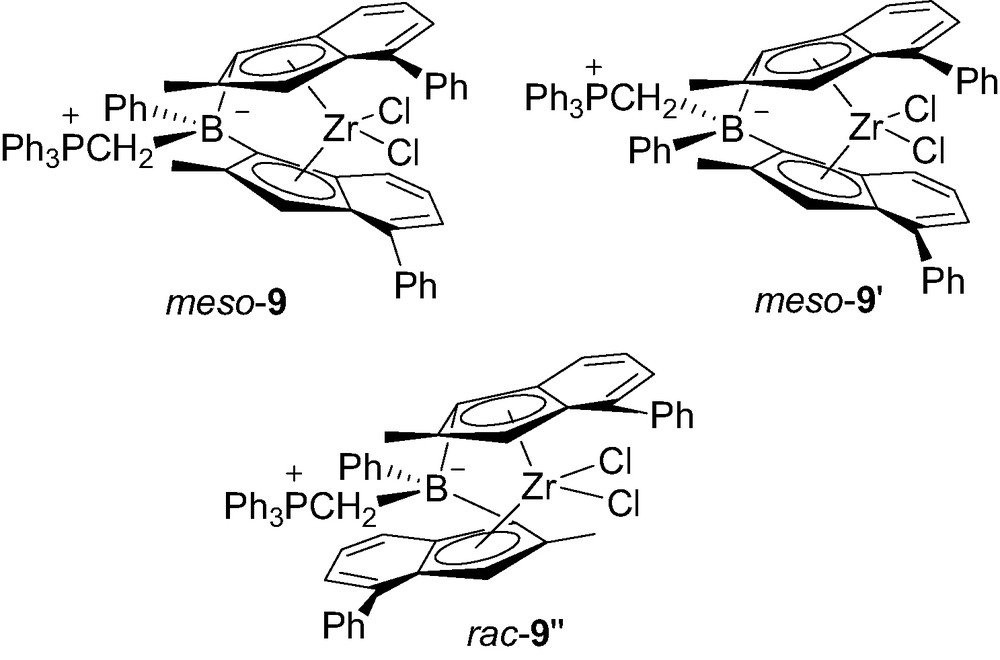
Structure of the three isomers of 9.
The 31P spectrum showed three distinct resonances, one for each isomer meso-9, meso-9′ and rac-9″. The meso/rac ratio was estimated to 2:1, on the basis of the 2-Me signals for the indenyl rings groups assigned in the 1H NMR spectrum. The molecular structures of rac-9″ and meso-9 were determined by X-ray diffraction studies. The P–C and C–B bond lengths within the boron bridge of rac-9″ (1.81 and 1.66 Å, respectively) and meso-9 (1.79 and 1.69 Å, respectively) are very similar to those of 7 (1.79 and 1.67 Å, respectively). Notably, the bite angle at boron in rac-9″ (97.6°) is smaller than in the related rac-4-THF (101.5°) and rac-4-PMe3 (99.4°). But this has little effect on the geometry of the indenyl rings, as indicated by the similar values observed for the centroid–Zr–centroid angles (123.2° in rac-9″ versus 122.3° in rac-4-THF and 121.5° in rac-4-PMe3). The molecular structure of meso-9 is one of the very few reported structures of meso ansa-bis(indenyl)zirconocene complexes [15]. The influence of the span of the bridge on the tilt of the indenyl rings is evident by comparing the centroid–Zr–centroid and dihedral angles of meso-9 (123.2° and 68.3°, respectively, with those of the corresponding ethandiyl-bridged meso-C2H4[4,4′-(2,7-dimethylindenyl)2]ZrCl2 (131.8° and 52.9°, respectively) [1d].
The mixture of meso and rac diastereomers 9 was evaluated toward propene polymerization under MAO activation and compared to the related silicon-bridged complex rac-(2-Me,4-Ph-Ind)2Zr(1,4-Ph2-butadiene) 10 [16]. The activity of 9 is more than twice that of 10 at 70 °C and 85 °C but this trend is inverted at temperature ≥ 100 °C (Table 5). The molecular weights of the produced polymers are higher with 9 than with 10, but quickly drop with increasing temperature for both catalytic systems. The relatively narrow polydispersity indexes (around 2) and high melting temperature of the PP (~155 °C) prepared with 9 suggest that rac-9″ is the most active species in the mixture of isomers. Accordingly, 13C NMR analysis of the polymer microstructures revealed a 63.6/26.5/9.8 ratio of mm/mr/rr triads and a mmmm pentad composition of 52.4%.
Propene polymerization using the zwitterionic tetracoordinate boron-bridged complex 9 and the related silicon-bridged derivative 10
| Precatalyst | Tp(°C) | Efficiency (g P/g Zr) | MW (g/mol) [PDI] | Tm (°C) |
| 9 | 70 | 1 081 024 | 373 000 [1.96] | 156.2 |
| 9 | 85 | 1 020 311 | 196 000 [2.24] | 155.3 |
| 9 | 100 | 713 375 | 132 000 [2.14] | 154.5 |
| 9 | 115 | 333 920 | 31 900 [2.89] | 152.7 |
| 10 | 70 | 450 108 | 513 000 [1.80] | 158.8 |
| 10 | 85 | 502 987 | 309 000 [2.00] | 156.8 |
| 10 | 100 | 834 442 | 184 400 [3.30] | 153.7 |
| 10 | 115 | 900 218 | 79 800 [2.81] | 153.7 |
2.4 Tricoordinate boron-bridged metallocenes
As mentioned before, bridging via a Lewis acidic neutral tricoordinate boron atom offers the possibility not only to finely tune the steric and electronic properties of the complexes, but also to develop self-activating zwitterionic catalysts. Such borylidene-bridged metallocenes were first reported by Rufanov et al. [17] with phenyl, methyl and tris(trimethylsilyl)methyl substituents at boron. However, these results have been highly controversial [3a] since the complexes were only partially characterized. Attempts to abstract the labile dimethyl sulfide from 3-SMe2 have also been reported [3a], but the corresponding phenylborylidene-bridged metallocene proved to be more electrophilic than expected so that only partial abstraction of the Lewis base was achieved, even with an equimolar amount of B(C6F5)3. So far, definitive proof for borylidene-bridged metallocene structures have only been gained with amino groups at boron [3b]. Compared to ansa-complexes based on tetracoordinate boron bridges, these new complexes can be reasonably anticipated to display original properties, especially (i) higher geometry constraint due to the sp2 hybridization of the boron-bridge and (ii) somewhat higher electrophilicity of the metal center, despite the expected π-donation of the amino substituent to the bridging boron atom.
All of the aminoborylidene-bridged metallocenes were synthetized following the same multistep one-pot procedure [18–20]. Accordingly, an aminodihalogeno borane R2NBX2 11a–c (R = Me, iPr or SiMe3) was first reacted with 2 equiv. of metallated cyclopentadienyl (Cp), indenyl (Ind) or fluorenyl (Flu) rings. The corresponding neutral boron-bridged ligands 12a–c were eventually obtained as mixtures of double-bond isomers. Coordination to titanium was then typically achieved by deprotonation (with LDA or BuLi) followed by reaction with TiCl3(thf)3 and subsequent oxidation of the intermediate Ti(III) species with PbCl2 (Scheme 10). The related zirconium and hafnium complexes 13-Zr and 13-Hf could be prepared by direct reaction of the dilithiated ligands with the appropriate metal chloride MCl4(thf)x. This synthetic strategy allowed for the preparation of complexes featuring identical as well as different rings (Cp–Cp and Ind–Ind derivatives as well as mixed Cp–Ind and Cp–Flu combinations) [19b,d,20].
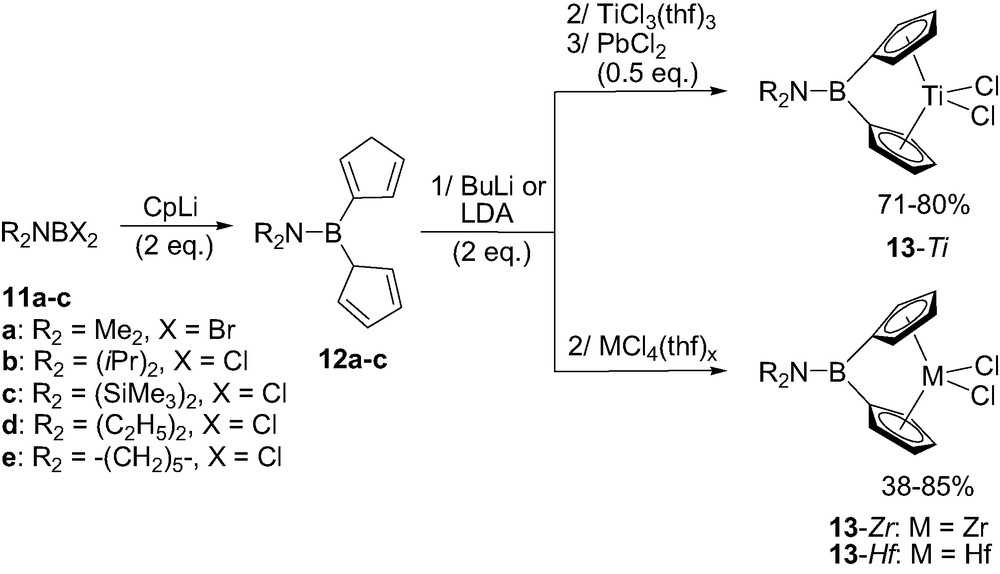
General synthesis of aminoborylidene-bridged ansa-metallocenes 13-M (M = Zr, Hf).
Following this multistep synthesis, the diisopropylaminoborylidene bis(indenyl)zirconium complex was obtained as a 60:40 mixture of rac/meso isomers. This ratio increased to 87:13 when the coordination to zirconium was achieved in two steps using subsequently Zr(NMe2)4 and Me3SiCl (for the amido/chloride exchange) and the desired isomer rac-14b-Zr could be isolated in 33% yield by recrystallization in toluene (Fig. 3) [18]. A more favorable situation was encountered for the related zirconium and titanium complexes featuring a bis(trimethylsilyl)aminoborylidene bridge, the general synthetic procedure leading exclusively to the rac-isomer, as deduced from the single resonance observed in NMR for the methyl groups of the dimethyl derivative rac-14′c-Zr [19b].
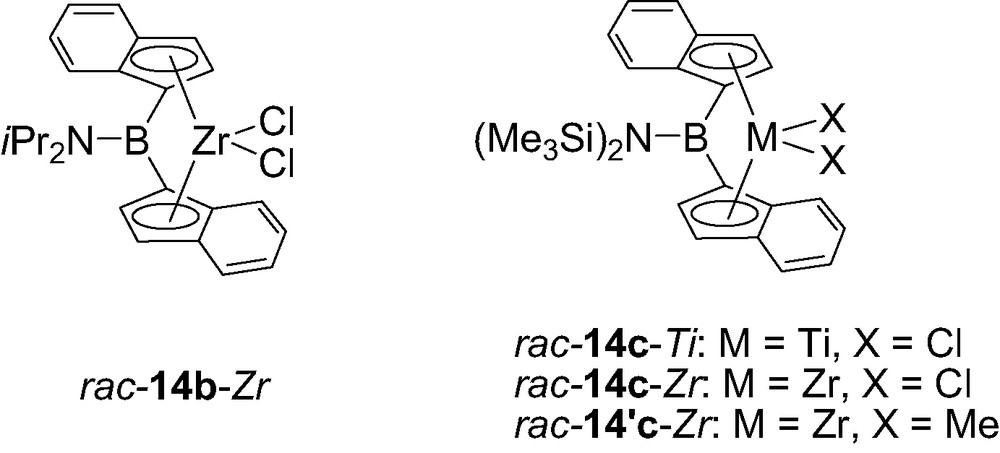
Structure of the aminoborylidene bis(indenyl) complexes 14-M (M = Zr, Hf).
The structures of all aminoborylidene-bridged metallocenes have been assessed in solution by multinuclear NMR spectroscopy. In particular, a resonance was typically observed at about δ = 44 ppm in 11B NMR for the tricoordinate boron atom. Moreover, up to 200 °C, two sets of signals were observed in 1H NMR for the two isopropyl groups of the mixed complex iPr2NB(Cp)(Flu)ZrCl2. Accordingly, the rotational barrier about the B–N bond was estimated to be higher than 23.8 kcal/mol, suggesting a strong π-bonding between the two atoms [20]. Four zirconium and two hafnium complexes have also been characterized by X-ray diffraction studies, and very similar features were observed, reflecting the similar size of zirconium and hafnium atoms (Table 6). The bridging boron atom always adopts a perfectly planar geometry and the B–N bond length is very short (1.38 Å), confirming a strong B–N π-bonding. Despite the sp2-hybridization of the bridging atom, the bite angles CBC in the borylidene-bridged complexes (around 104°) are only marginally wider than in the sp3-hybridized derivatives 3-SMe2 and 3-PMe3 (100–101°) and significantly narrower than in the related unbridged derivative PhB(CpTiCl3)2 (127°) [5a]. This clearly evidences the considerable strain induced by the tricoordinate boron-bridge. As expected, the dihedral angles between the Cp rings in these borylidene-bridged complexes (65–68°) are also similar to those observed in 3-SMe2 and 3-PMe3 (66–68°), and significantly wider than in the Me2Si-bridged zirconocene (62°). In a first approximation, this reflects the smaller size of the boron-based bridges and suggests higher accessibility of the metal center.
According to preliminary investigations, aminoborylidene-bridged complexes are rather efficient precatalysts toward Ziegler–Natta polymerization under MAO activation. As far as ethylene polymerization is concerned, catalytic activities of 8905 and 5258 kg PE/mol catalyst per hour were reported for complexes Me2NB(Cp)2ZrCl2 13a-Zr and (Me3Si)2NB(Ind)2TiCl2 rac-14c-Ti, respectively [19b], but these values can be hardly compared to those obtained with other complexes since the polymerization conditions were not precisely defined. As already reported for other Zr- and Hf-based systems, the activity of the hafnium complexes in ethylene polymerization (260 kg PE/mol catalyst per hour, in toluene at 60 °C, under 2 bar of ethylene pressure and for a Al/Hf ration of 4500) is significantly lower than that of the zirconium derivatives [19d].
Ethylene/hex-1-ene copolymerization and propene polymerization have also been assessed with various diisopropylaminoborylidene-bridged complexes (Tables 7 and 8). Accordingly, the nature of the rings (Cp/Cp, Cp/Flu and Ind/Ind) proved to strongly influence the catalytic activity and selectivity of the precatalysts. In both cases, the following order of activity was observed: bis(cyclopentadienyl) 13b-Zr < mixed (cyclopentadienyl)(fluorenyl) 13′b-Zr < bis(indenyl) rac-14b-Zr. As far as the tacticity of the PP is concerned, 13′b-Zr resulted in modest syndiotacticity whereas rac-14b-Zr-induced moderate isotacticity. Although these results cannot be quantitatively compared with those obtained under somewhat different conditions with C- and Si-bridged ansa-(cyclopentadienyl)(fluorenyl) zirconium complexes, the following order of syndioatacticity could be qualitatively established: SiMe2 < C2H4 ≈ BNiPr2 < CMe2 [20].
Ethylene/hex-1-ene copolymerization using the diisopropylaminoborylidene-bridged complexes iPr2NB(CxHy)(Cx′Hy′)ZrCl2 (under 34 bar of ethylene pressure at 140 °C with Al/Zr = 1000)
| Precatalyst | (CxHy)/(Cx′Hy′) | Activity (kg PP/mol Zr per bar) |
| 13b-Zr | Cp/Cp | 700 |
| 13′b-Zr | Cp/Flu | 3900a |
| rac-14b-Zr | Ind/Ind | 17,000 |
a According to its density (0.9 g/ml), the resulting polymer has incorporated about 5.7 mol % of oct-1-ene.
Propene polymerization using the diisopropylaminoborylidene-bridged complexes iPr2NB(CxHy)(Cx′Hy′)ZrCl2 (under 23.3 bar at 70 °C)
| Precatalyst | (CxHy)/(Cx′Hy′) | Activity (kg PP/mol Zr per bar) | Triad ratio (mm/mr/rr) |
| 13b-Zr | Cp/Cp | 64 | |
| 13′b-Zr | Cp/Flu | 770 | 4:15:81 |
| rac-14b-Zr | Ind/Ind | 2200 | 82:12:6 |
Taking into account that metallocenes bridged by two carbon atoms usually give polymers of higher molecular weight than those bridged with one carbon [1d], Braunschweig et al. [21] recently investigated diboronbridged-metallocene. The required ligand 15 was obtained in quantitative yield as a mixture of isomers by reaction of the 1,2-bis(dimethylamino)-1,2-dibromoborane with NaCp (Scheme 11). Deprotonation of 15 with nBuLi at –80 °C followed by addition of ZrCl4(thf)2 or HfCl4 gave 16-Zr and 16-Hf, respectively. The two complexes exhibit very similar spectroscopic data. Due to hindered rotation around the B–N bonds, two singlets are observed for the methyl groups in 1H NMR, and the protons of the Cp rings resonate as two pseudo-triplets. The 11B NMR chemical shift for both complexes 16-M (42.9 ppm) is very similar to that of the ligand precursor 15 (46 ppm).

Synthesis of the diboron-bridged metallocenes 16-M (M = Zr, Hf).
The molecular structure of 16-Hf was confirmed by X-ray diffraction and compared to that of its boron-bridged analog Me2NBCp2HfCl2 16′-Hf (Table 9). The B–N bond lengths are very short (1.38 Å), confirming a strong B–N π-bonding. The diboron bridge induces much less, if any, constrain than the single-atom bridge, as evidenced by comparing the centroid–hafnium–centroid angles (130.7° in 16-Hf versus 121.4° in 16′-Hf) and the tilt angles between the Cp rings (51.8° in 16-Hf versus 64.7° in 16′-Hf). In fact, the structural data for 16-Hf are very similar to those of the unbridged complex Cp2HfCl2 (centroid–hafnium–centroid angle 129.2° and tilt angle of 53.5°).
Selected angles (°) for (di)boron-bridged and unbridged hafnium complexes
| Complex | Centroid–hafnium–centroid | Cp–Cp tilt |
| 16-Hf | 130.7 | 51.8 |
| Me2NBCp2HfCl2 16′-Hf | 121.4 | 64.7 |
| Cp2HfCl2 | 129.4 | 53.5 |
Preliminary experiments showed that both complexes 16-Zr and 16-Hf efficiently polymerize ethylene under MAO activation with activities of about 27000 g (PE)/mmol. Polymers with very high molecular weights were obtained (MV = 896 000 for 16-Zr and 1600,000 g/mol for 16-Hf). Given the structural similarity between 16-Hf and Cp2HfCl2, the catalytic performance of 16-Zr and 16-Hf was attributed to the electronic rather than the geometric influence of the diboron bridge.
Lastly it should be mentioned that boron-bridged constrained geometry complexes have been recently prepared. Subsequent treatment of the dichloroborane 11b with CpNa and PhNHLi first led the neutral ligand 17. Aminolysis of 17 with Ti(NMe2)4 then afforded the corresponding titanium complex 18-Ti. Exchange of the amido groups at titanium for chlorides was then readily achieved with Me3SiCl to give 18′-Ti (Scheme 12) [22]. The X-ray diffraction analysis carried out on 18-Ti revealed herealso the typical geometry for a strong BN π-bonding with planar environments for both atoms, negligible torsion angle (2°) and short bond distance (1.41 Å). According to preliminary experiments, 18′-Ti was expectedly found more active than 18-Ti toward ethylene polymerization, with an activity of 500 kg PE/mol Ti per hour (in toluene at 60 °C, under 5 bar of ethylene pressure and for a Al/Ti ratio of 1000) but further studies are clearly necessary to determine the precise influence of a Lewis acidic boron bridge on the activity of such constrained geometry complexes.
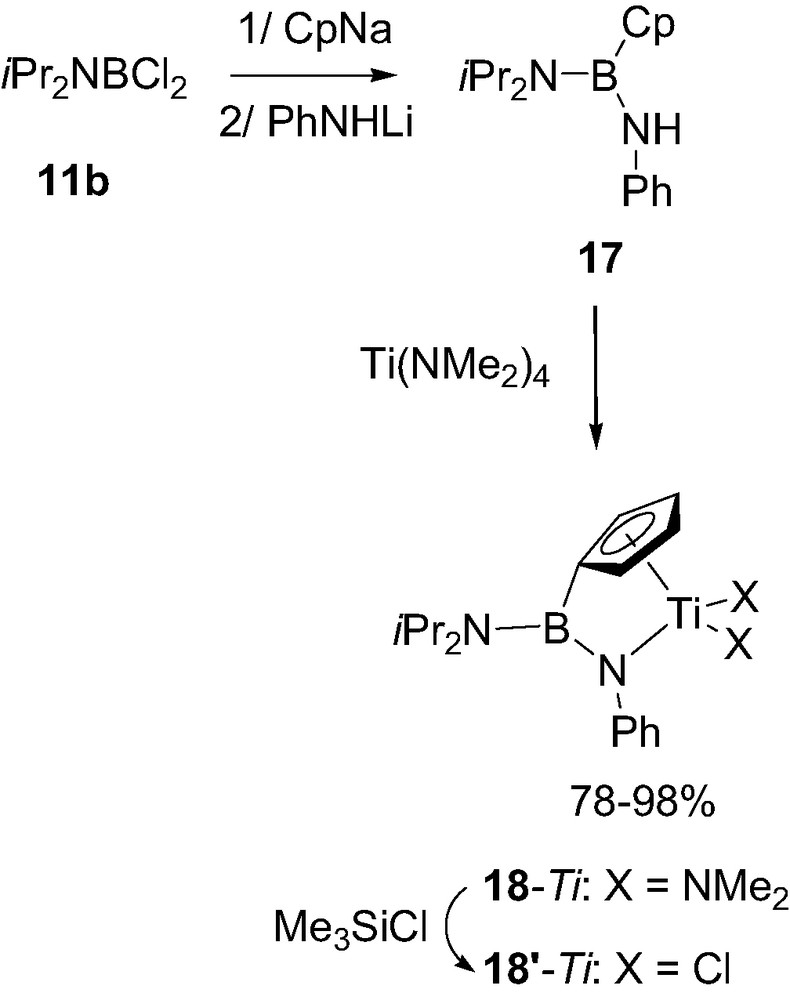
Structure of the boron-bridged constrained geometry complexes 18-Ti.
3 Phosphorus analogs
3.1 Ansa-metallocenes
Although most of the ansa-metallocenes feature carbon-, silicon- or boron-bridges, P-bridged derivatives have also been investigated. The main interests for the incorporation of phosphorus are probably the opportunity (i) to tune the electronic properties of the complexes by varying the oxidation state and charge of the bridging unit (from neutral tricoordinate to neutral as well as cationic tetracoordinate) and (ii) to modify the size of the bridge (from a single phosphorus atom to a C2PC2 skeleton) with eventually a direct participation of the phosphorus via coordination to the metal center. All of these possibilities have been recently illustrated by the preparation of complexes 19–22 (Fig. 4) [23]. The synthesis, structure and properties of these P-bridged metallocenes will be discussed, including their catalytic activity toward olefin polymerization.
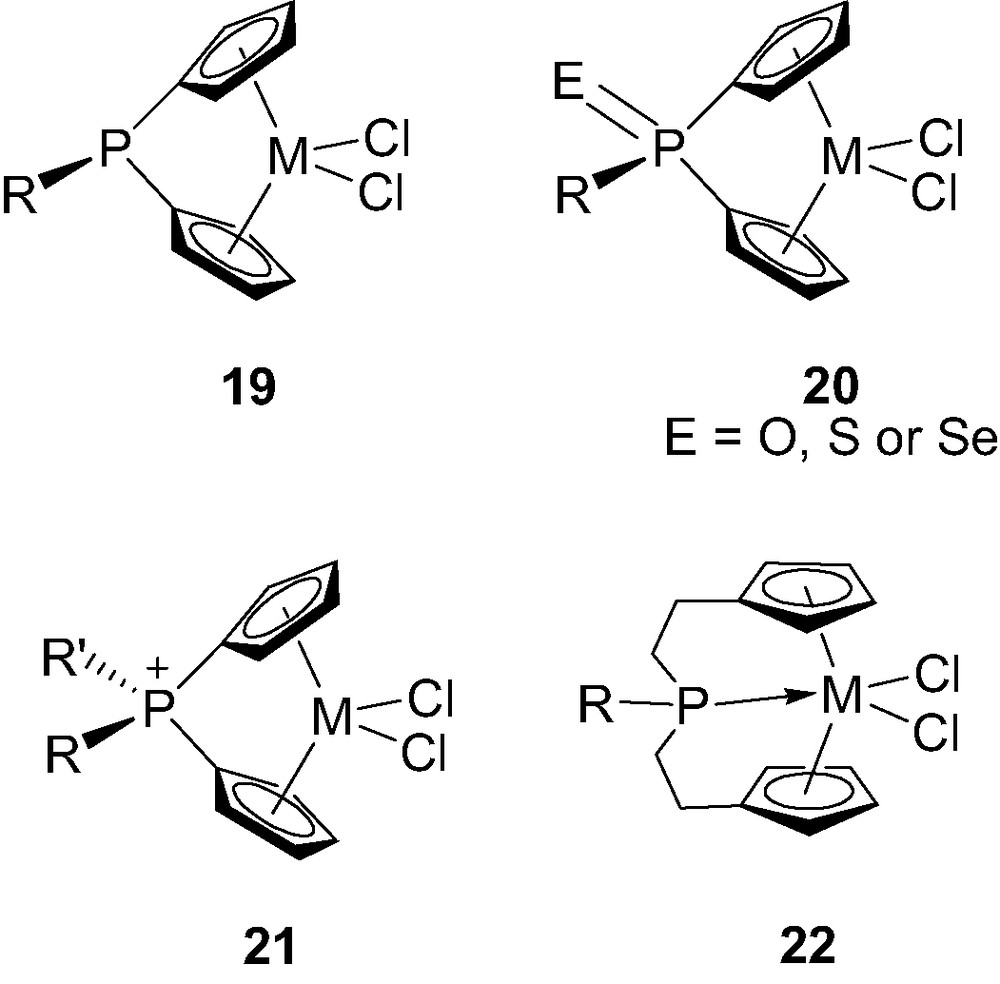
Structure of the phosphorus-bridged metallocenes 19–22-M (M = Ti, Zr, Hf).
3.1.1 Synthesis of phosphorus-bridged metallocenes
Although phosphorus-bridged ferrocenes are usually obtained by reacting the dilithioferrocene/TMEDA adduct with a dichlorophosphine, with group-4 metals, the coordination follows the preparation of the ansa-ligands. Indeed, a dichlorophosphine RPCl2 23 (R = Ph, Me, Et, iPr, tBu) was first treated with 2 equiv. of metallated cyclopentadiene CpM′. The resulting neutral derivatives 24a were then doubly deprotonated and subsequently reacted with the appropriate metal chlorides to give the desired P-bridged bis(cyclopentadienyl) metallocenes 19a-M (M = Ti, Zr) (Scheme 13) [24,25]. This strategy is very similar to that developed for boron-bridged ansa-metallocenes, excepted that (i) even the titanium complexes could be directly obtained using TiCl4 and (ii) thallium reagents CpTl and TlOEt might be preferred to avoid base-induced cleavage of the neutral ansa-ligands 24a [26].

Synthesis of the P-bridged bis(cyclopentadienyl) metallocenes 19a-M (M = Ti, Zr).
Anyhow this procedure could be easily extrapolated to tetramethylcyclopentadiene (Cp′) as well as fluorenylidene (Flu) rings, leading to the corresponding complexes 19b-M (M = Zr, Ti and Hf) [27] and 19c-Zr [28], respectively. C2-symmetric complexes featuring two indenyl (Ind) or two 2-methyl-4-phenylindenyl (Ind′) rings have also been prepared under the same conditions. Accordingly, PhP(Ind)2ZrCl2 was obtained as a 1:1 mixture of rac/meso isomers from which the desired rac-19d-Zr could be isolated by crystallization, while PhP(Ind′)2ZrCl2 19e-Zr was obtained as an unseparable 1:2 mixture of rac/meso isomers (Fig. 5) [29].
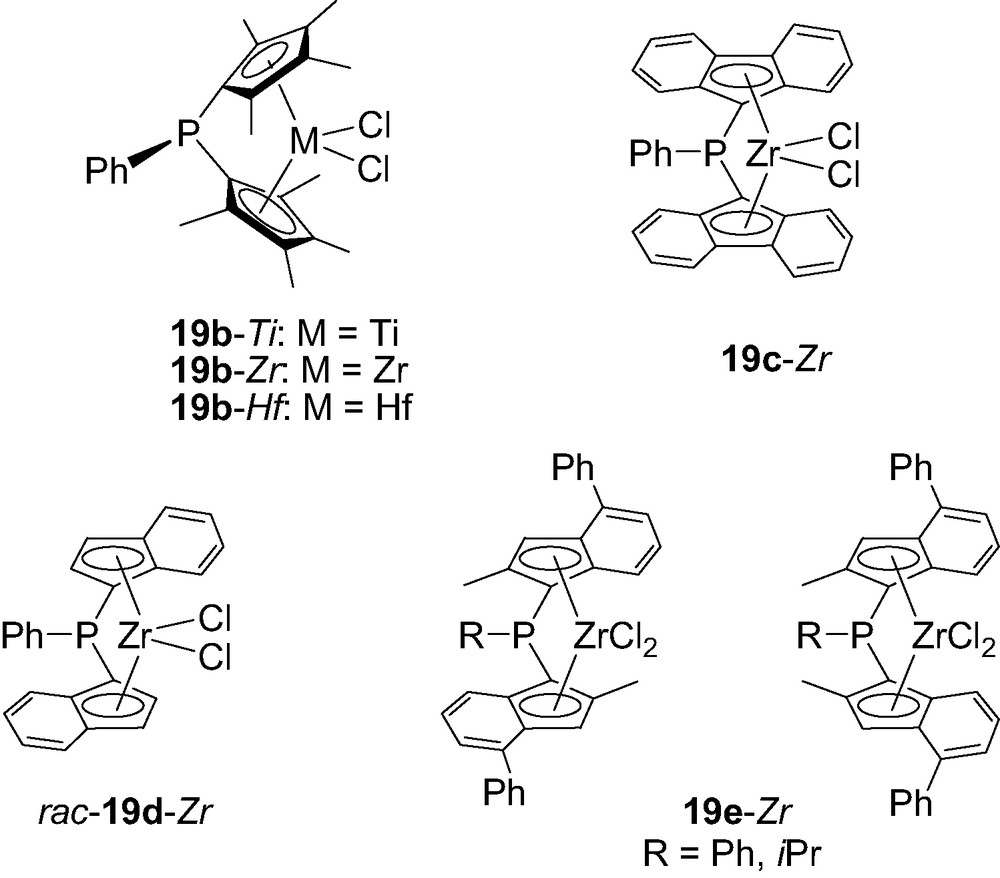
Structure of the P-bridged metallocenes 19b–e-M featuring Cp’, Flu, Ind or Ind’ rings.
Starting from a chloroethoxyphosphine 23′ instead of a dichlorophosphine and sequential addition of fluorenyl- and cyclopentadienyl-lithium FluLi and CpLi, a mixed complex PhP(Cp)(Flu)ZrCl2 19f-Zr could also be prepared in a similar way (Scheme 14) [29].
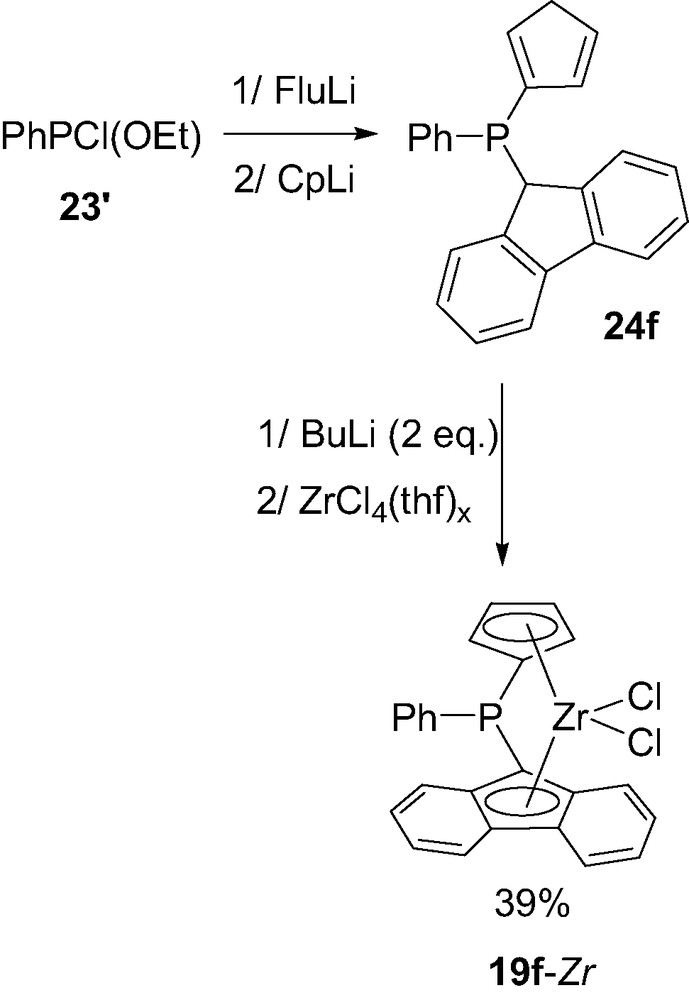
Synthesis of the mixed (cyclopentadienyl)(fluorenyl) P-bridged zirconocene 19f-Zr.
Since the RPCl2 route only affords (3)-P susbstituted indenyl ligands, the preparation of (2)-P-substituted indenyl ansa-complexes required an alternative route based on palladium catalyzed phosphination [30]. Reaction of 2 equiv. of (2)-bromoindene with primary phosphines RPH2 (R = Ph, tBu) 23″ in the presence of tetrakis(triphenylphosphine)palladium afforded the ansa-ligands [di(1H-inden-2-yl)]phosphines 24g (Scheme 15). Deprotonation of 24g followed by reaction of ZrCl4(thf)2 led to the corresponding complexes 19g. Note that due to the presence of the RP bridge in position 2 of the indenyl rings, complexes 19g present Cs symmetry.
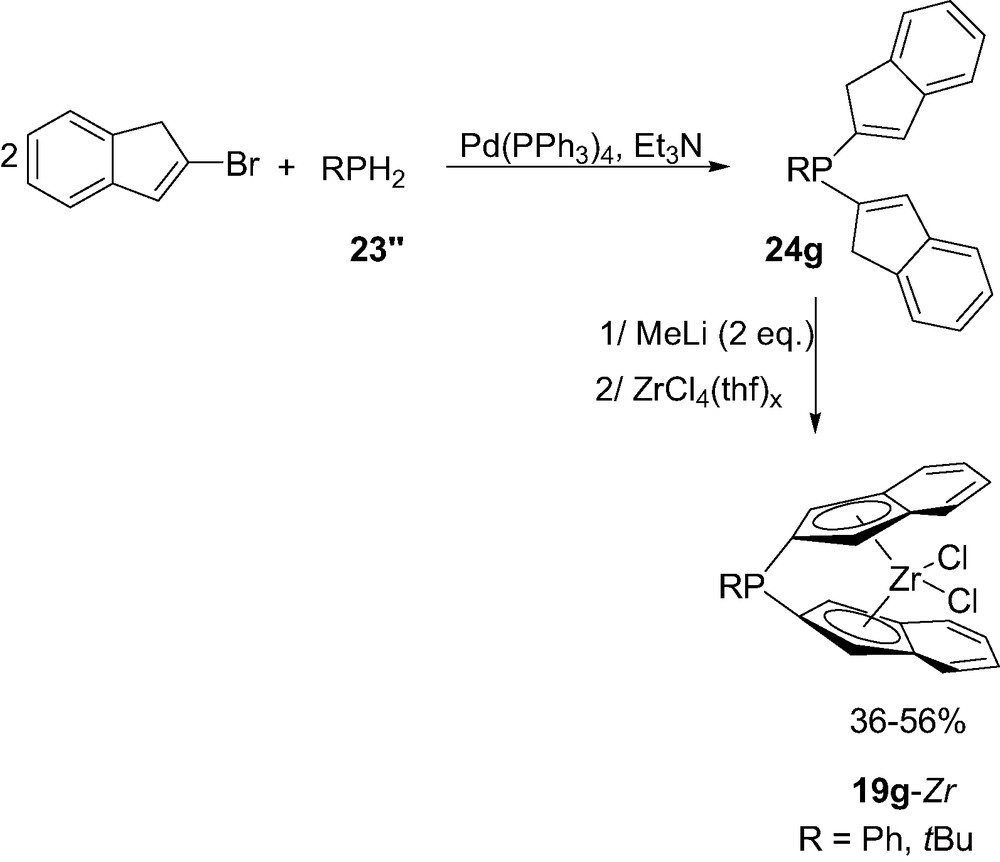
Synthesis of the indenyl P-bridged zirconocene 19g-Zr.
The preparation of bimetallic complexes such as (LnM)RP(Cp)2ZrCl2 demonstrated that the lone pair at the tricoordinate bridging-phosphorus remains active in RP(Cp)2ZrCl2 19a-Zr [26]. Taking advantage of this feature, Shin et al. [27] oxidized 19b-Zr with O2, S8 or Sex to synthesize the related ansa-metallocenes 20b-Zr featuring neutral tetracoordinate phosphorus-bridges (Scheme 16).
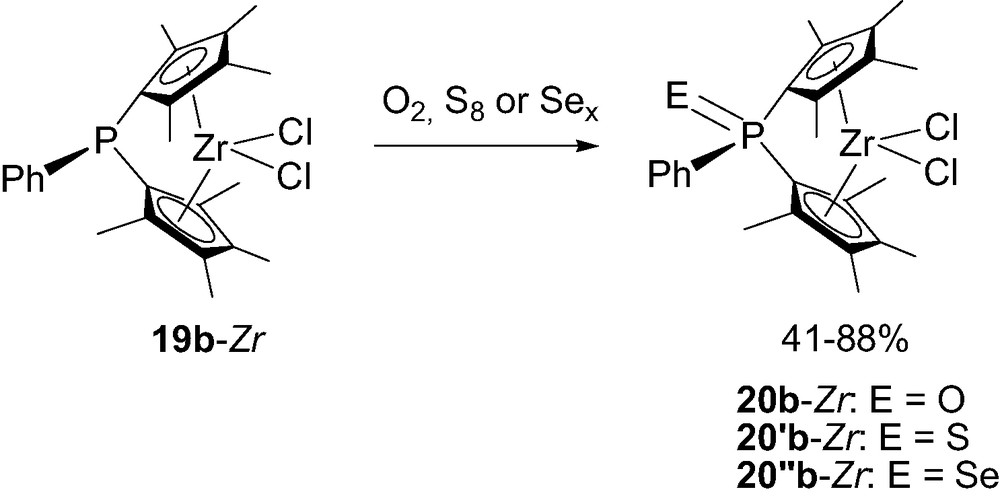
Synthesis of the neutral tetracoordinate phosphorus-bridged metallocenes 20b-Zr.
To complete the series of P-bridged metallocenes, phosphonium-bridged derivatives 21 have also been investigated. Since the R2P+ and R2Si fragments are isoelectronic, these complexes are cationic analogs of the well-studied silylene-bridged metallocenes. In marked contrast with the RP(=E)-bridged complexes 20, cationic P-bridged metallocenes 21 have not been prepared from the corresponding neutral tricoordinate phosphorus-bridged derivatives 19 but rather by coordination of pre-assembled ansa-ligands [31,32]. The required cationic ligands 25 were first obtained by treatment of a dichlorophosphine 23 with 2 equiv. of 2-methyl-4-tertiobutylcyclopentadienylithium followed by P-alkylation with an alkyliodide (Scheme 17) [31]. Coordination to zirconium was then achieved classically by reaction with butyllithium followed by zirconium chloride. Due to the substitution pattern of the cyclopentadienyl rings, complexes 21h-Zr were obtained as mixtures of rac/meso isomers than could not be separated [31].

Synthesis of the phosphonium-bridged zirconocenes 21h-Zr.
Following the same strategy, Shin et al. [32] have prepared and isolated three Me2P+-bridged metallocenes 21b-M (M = Zr, Hf) featuring two tetramethylcyclopentadienyl rings (Fig. 6).
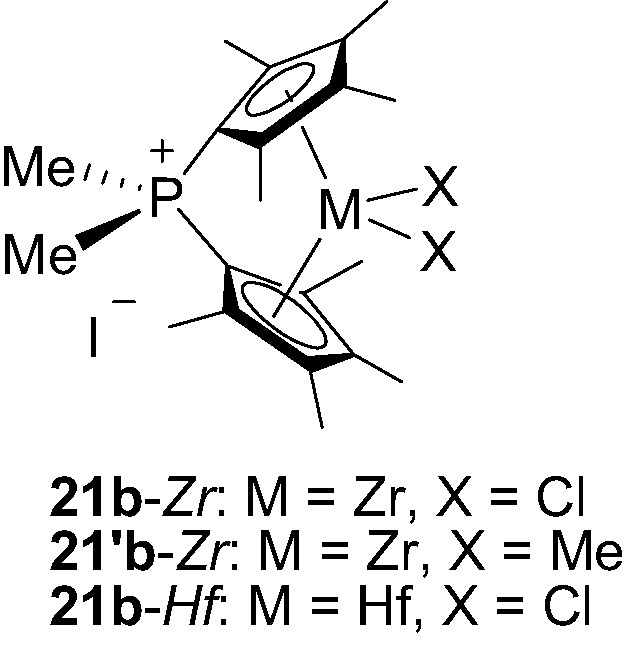
Structure of the phosphonium-bridged metallocenes 21b-M (M = Zr, Hf).
As already mentioned, the incorporation of phosphorus in ansa-metallocenes has also offered the opportunity to modify the size of the bridge, from a single atom to a C2PC2 skeleton. Indeed, Curnow et al. [33] have been interested in tridentate ligands featuring a phosphine tethered to two cyclopentadienyl rings via ethylene spacers. The corresponding zirconocene 22a-Zr has been prepared in 22% yield by addition of dilithiophenylphosphine PhPLi2 to the highly strained spiro [2,4]hepta-4,6-diene, followed by coordination to ZrCl4(thf)2 (Scheme 18). The propensity of the bridging phosphorus to coordinate the zirconium center was indicated in solution and in the solid state by 31P NMR spectroscopy and X-ray analysis, respectively (see below).
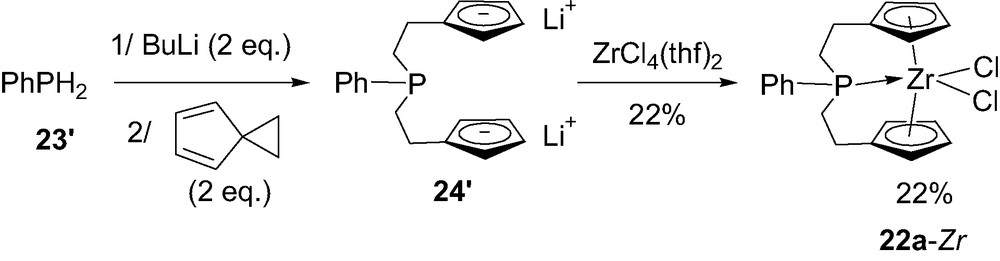
Synthesis of the C2PC2-bridged zirconocene 22a-Zr.
3.1.2 Structure and properties of phosphorus-bridged metallocenes
As expected, 31P NMR spectroscopy is a convenient tool to characterize P-bridged metallocenes (Table 10). Although the chemical shifts associated with tricoordinate bridging phosphorus atoms strongly depend on the substituent, they all appear at a rather high field, significantly more deshielded signals being observed for neutral as well as cationic tetracoordinate bridging phosphorus (δ 5–20 ppm). The influence of the bridging unit was also clearly evidenced by comparing the chemical shifts for PhP(Cp)2ZrCl2 (–28.9 ppm) and PhP(CH2CH2Cp)2ZrCl2 (+ 17.5 ppm). This high deshielding clearly indicates an intramolecular P→Zr coordination that can only take place with the long and flexible C2PC2 bridge.
31P NMR chemical shifts (ppm) and geometric data [bond angles (°) and bond lengths (Å)] for P-bridged metallocenes
| Complex | δ 31P NMR | Centroid–M–centroid | M-centroid | References |
| MeP(C5H4)2ZrCl2 | –53.7 | 122.2 | 2.200 | [25] |
| Me2Si(C5H4)2ZrCl2 | – | 125.4 | 2.197 | [25] |
| Me2C(C5H4)2ZrCl2 | – | 116.6 | 2.192 | [25] |
| MeP(C5Me4)2ZrCl2 | – | 125.8 | 2.226 | [27] |
| PhP(C5Me4)2ZrCl2 | –37.7 | 125.9 | 2.225 | [27] |
| PhP(C5Me4)2TiCl2 | –37.8 | 129.4 | 2.117 | [27] |
| PhP(C5Me4)2HfCl2 | –37.2 | 126.3 | 2.212 | [27] |
| PhP(=S)(C5Me4)2ZrCl2 | 25.8 | 126.3 | 2.234 | [27] |
| PhP(=Se)(C5Me4)2ZrCl2 | 4.7 | 126.3 | 2.302 | [27] |
| [Me2P(C5Me4)2ZrCl2]I | 16.0 | 126.0 | 2.240 | [32] |
| PhP(CH2CH2C5H4)ZrCl2 | 17.5 | 127.2 | 2.252 | [33] |
| 2.234 | ||||
| tBuP[(2)-(C9H7)]2ZrCl2 | –12.0 | 122.7 | 2.225 | [30] |
| 2.219 |
More insight into the precise structure of these P-bridged metallocenes has been gained thanks to X-ray analyses performed on representative complexes (Table 10):
- ● the centroid–M–centroid angles in complexes X(Cp)2ZrCl2 were found to vary in the following order: X = CMe2 (116.6°) < PMe (122.2°) < SiMe2 (125.4°) [25]. A very similar evolution has been predicted theoretically for the model active species [X(Cp)(Flu)ZrH]+: X = CH2 (121.5°) < PH (127.2°) < SiH2 (129.4°) [29]. The wider centroid–M–centroid angles of phosphorus and silicon-bridged complexes versus carbon-bridged derivatives clearly result from the larger size of the bridging atom (Si ≈ P > C). Notably, the following order was also predicted for the CpXCp angles: PH (87.7°) < SiH2 (94.3°) < X = CH2 (99.3°), in agreement with the ability of the bridging atom to accommodate acute bond angles;
- ● the introduction of four methyl substituents at the cyclopentadienyl rings induced as expected a slight widening of the centroid–M–centroid angle (from 122.2° to 125.8°) whereas replacement of the cyclopentadienyl rings by indenyl rings has practically no influence [27,30];
- ● the replacement of the methyl group at phosphorus for a phenyl ring did not induce significant distortions [27];
- ● as expected, the geometries of zirconium and hafnium complexes were very similar, somewhat shorter M-centroid bond lengths and wider centroid–M–centroid angle being observed in the related titanium derivative [27];
- ● the variation of the oxidation state and charge of the bridging phosphorus (from neutral tricoordinate to neutral as well as cationic tetracoordinate) did not induce significant variations [27,32];
- ● an intramolecular P→Zr interaction was found in the solid-state structure of PhP(CH2CH2C5H4)2ZrCl2, with a P–Zr bond length of 2.78 Å and an approximately trigonal bipyramidal environment around zirconium [33]. With this long and flexible C2PC2 skeleton, the centroid–M–centroid angle (127.2°) is wider than in the single phosphorus-bridged complexes (122–126°) and but still more acute than in the related non-bridged metallocene (129.2° for Cp2ZrCl2).
The influence of the phosphorus bridge on the electron ability of the π-rings has been estimated based on the stretching frequencies for the related dicarbonyl zirconocenes (Table 11) [27]. Since the higher the frequency, the lower the M→CO backdonation, the P-bridged ansa-ligand [PhP(C5Me4)2] was found to engender a more electrophilic metal center than both Me2Si(C5Me4)2 and Me2C(C5Me4)2 ligands.
Stretching frequencies for dicarbonyl zirconocenes
| Complex | νCO ( per cm)a |
| (C5Me5)2Zr(CO)2 | 1946, 1853 |
| Me2Si(C5Me4)2Zr(CO)2 | 1956, 1869 |
| PhP(C5Me4)2Zr(CO)2 | 1959, 1874 |
a Asymmetric and symmetric stretching combinations.
3.1.3 Catalytic activity of phosphorus–bridged metallocenes in Ziegler–Natta polymerization
Although only preliminary investigations have been reported so far, P-bridged metallocenes seem to be rather efficient precatalysts toward Ziegler–Natta polymerization under MAO activation. As far as ethylene polymerization is concerned, PhP(=X)(Cp′)2ZrCl2 complexes (X = lone pair, O, S, or Se) were found to be significantly less active than the related Me2C-bridged derivative but almost as active as the Me2Si-bridged complex (Table 12) [27]. Notably, the catalytic activity was not significantly altered by oxidation of the bridging phosphorus with oxygen, sulfur or selenium. This suggests that PhP(Cp′)2ZrCl2 is not deactivated by coordination of the tricoordinate phosphorus to MAO cocatalyst, probably due to the higher steric hindrance around phosphorus.
MAO activated ethylene polymerization at room temperature, in toluene, under 1 bar of ethylene pressure and with an Al/Zr ratio of 460
| Precatalyst | Activity (g PE/mmol Zr per atm per min) |
| (C5Me5)2ZrCl2 | 5.3 |
| Me2Si(C5Me4)2ZrCl2 | 1.9 |
| PhP(C5Me4)2ZrCl2 | 1.7 |
| PhP(=O)(C5Me4)2ZrCl2 | 1.8 |
| PhP(=S)(C5Me4)2ZrCl2 | 1.7 |
| PhP(=Se)(C5Me4)2ZrCl2 | 1.9 |
Several P-bridged zirconocenes have also been evaluated by Schaverien et al. toward propene polymerization. Atactic polypropenes (a-PP) are completely amorphous polymers that are usually obtained in only small amounts with metallocene precatalysts due to enantiomorphic-site and/or chain-end control. In this regard, PhP(Flu)2ZrCl2 appeared rather promising, affording a-PP with similar molecular weight (Mv = 190,000 g/mol) and triad distribution to those obtained with the related Me2Si-bridged complex (Table 13) [29]. However, the P-bridged metallocene was found to be significantly less active.
Aspecific propene polymerization under MAO activation (Al/Zr ratio of 1000) at 50 °C in liquid propene
| Precatalyst | Activity (kg PP/g Zr per hour) | Distribution of mm/mr/rr triads |
| PhP(Flu)2ZrCl2 | 8 | 15.7/49.5/34.8 |
| Me2Si(Flu)2ZrCl2 | 176 | 19/49/32 |
In marked contrast, the mixed complex PhP(Cp)(Flu)ZrCl2 was found to give syndiotactic polypropenes (s-PP) with rather high activities (100 and 155 kg PP/g Zr per hour at 50 and 67 °C, respectively) and 81 mol% of rrrr pentads [29]. These values are slightly lower than those obtained with the related Me2C-bridged derivative but significantly higher than those observed with the corresponding Me2Si-bridged complex (Table 14). Ab initio calculations on the model active species X(Cp)(Flu)ZrH+ (X = CH2, SiH2 and PH) allowed for rationalizing this trend. Indeed, the centroid–M–centroid angles were predicted to vary as follows: X = CH2 < PH < SiH2. Since the smaller this bite angle, the more difficult the inversion at the metal center, the amount of stereoerrors measured with the P-bridged complex is logically higher (respectively, lower) than with the C- (respectively, Si-)bridged derivative [29].
Syndiospecific propene polymerization under MAO activation (Al/Zr ratio of 1000) in liquid propene
| Precatalyst | T (°C) | Activity (kg PP/g Zr per hour) | Tacticity (mol% of rrrr pentads) |
| PhP(Cp)(Flu)ZrCl2 | 50 | 100 | 34 |
| PhP(Cp)(Flu)ZrCl2 | 67 | 155 | Not determined |
| Me2C(Cp)(Flu)ZrCl2 | 67 | 400 | 85 |
| Me2Si(Cp)(Flu)ZrCl2 | 67 | 18 | 51 |
It should be mentioned that the presence of the tetrahedral PhP bridge does not affect the syndiospecificity of PhP(Cp)(Flu)ZrCl2 precatalyst, the local symmetry at the metal center being apparently not affected by this remote perturbation. This stimulated the investigation of C2 symmetric P-bridged zirconocenes as isospecific precatalysts (Table 15). The bis(indenyl)derivative PhP(Ind)2ZrCl2 was found to be only moderately active (24 kg PP/g Zr per hour at 67 °C), affording poorly isotactic polypropene (i-PP). Significantly better results were obtained with the related zirconocenes featuring 2-methyl-4-phenylindenyl rings (Ind′), with activities up to 576 kg PP/g Zr per hour at 67 °C and stereoselectivity up to 97.5–98.0% (according to the mol% of mmmm pentads). Raising the polymerization temperature from 50 to 67 °C resulted in a significant increase of the activity while raising the Al/Zr ratio from 8000 to 37 000 did not bring any improvement. Replacement of the phenyl ring for an isopropyl substituent at the bridging phosphorus resulted in even higher activities (up to 1265 kg PP/g Zr per hour at 67 °C) and similar isotacticities (98%). However, the major limitation of these C2 symmetric P-bridged bis(indenyl)zirconocenes toward isospecific propene polymerization is probably the concomitant obtention of small amounts of a-PP (from 2.0 to 7.8 mol%). The formation of this undesirable atactic polymer has been attributed to the fact that these precatalysts have been obtained as unseparable mixtures of rac/meso isomers.
Isospecific propene polymerization under MAO activation in liquid propene
| Precatalyst | T (°C) | Al/Zr | Activity (kg PP/g Zr per hour) |
| PhP(Ind)2ZrCl2a | 67 | 24 | |
| PhP(Ind′)2ZrCl2b,c | 50 | 37 000 | 200 |
| PhP(Ind′)2ZrCl2b,c | 50 | 8000 | 184 |
| PhP(Ind′)2ZrCl2b,c | 67 | 37 000 | 576 |
| iPrP(Ind′)2ZrCl2b,c | 30 | 8186 | 180 |
| iPrP(Ind′)2ZrCl2b,c | 50 | 8004 | 495 |
| iPrP(Ind′)2ZrCl2b,c | 50 | 37 600 | 1265 |
a Pure rac isomer.
b Mixture of rac/meso isomers.
c Ind′ = 2-methyl-4-phenylindenyl.
According to these preliminary studies, simple variation of the π-donor rings in P-bridged metallocenes allows the aspecific, syndiospecific as well as isospecific polymerization of propene. However, further investigations are clearly deserved, especially for P-bridged metallocenes complexes 21 and 22 that have not been evaluated so far.
Lastly, preliminary studies revealed that complex 19g featuring a PtBu bridge exhibits very high activity toward ethylene polymerization and ethylene/oct-1-ene copolymerization (from 64 050 to 76 300 g PE/mmol Zr per hour per atm at 80 °C under 5 atm of ethylene pressure), producing low molecular weight (co)polymers [30].
3.2 CGC
The spectacular achievements reported over the last 15 years for the CGC have stimulated the investigation of numerous structural analogs of the initial ‘CpSiN’ complexes III. In particular, complexes 26–28 featuring a phosphido group instead of the amido moiety, and bridging units varying from a single atom (silicon or carbon) to a C2 backbone, have been described (Fig. 7). Alternatively, phosphorus has been used to bridge the cyclopentadienyl and nitrogen ligands, such as in the recently studied complexes 29–31.
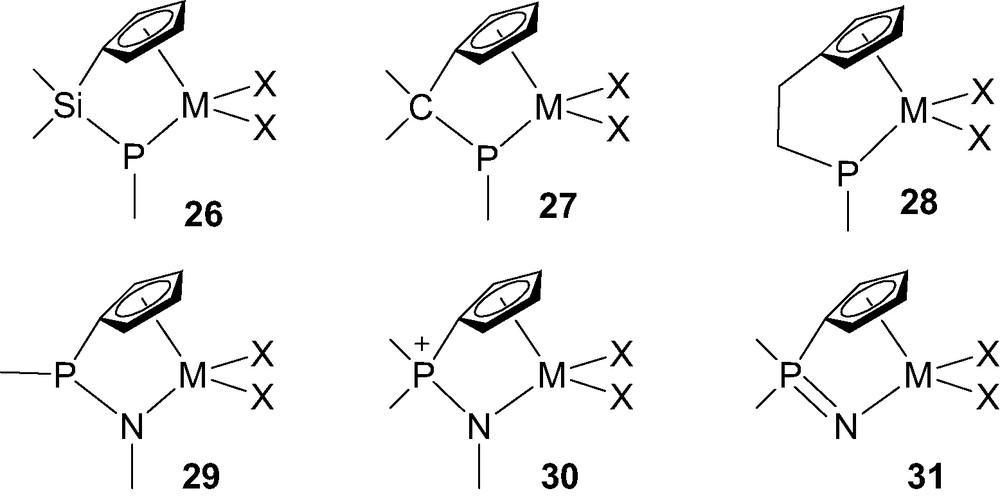
Phosphorus-containing constrained geometry complexes 26–31-M (M = Ti, Zr, Hf).
3.2.1 Mixed (cyclopentadienyl)(phosphido) complexes featuring R2Si or R2C bridges
The synthetic route developed to access ‘CpSiN’ and ‘CpCN’ complexes could be readily extrapolated to the related ‘CpSiP’ and ‘CpCP’ derivatives 26 and 27. Indeed, the required neutral ‘CpSiP’ ligand 32 was isolated in > 80% yield by treatment of the Me2SiCl-functionalized tetramethylcyclopentadiene with CyPHLi. Coordination was then achieved by double deprotonation with butyllithium followed by reaction with the appropriate metal chloride (Scheme 19). In contrast to that usually observed with (cyclopentadienyl)(amido) ligands, this step proved unsuccessful with simple MCl4(thf)x precursors [34] and required diamido MCl2(NR2)2(thf)x reagents [35]. Accordingly, titanium and zirconium ‘CpSiP’ complexes 26-Ti and 26-Zr were obtained in 67% and 59% yield, respectively.
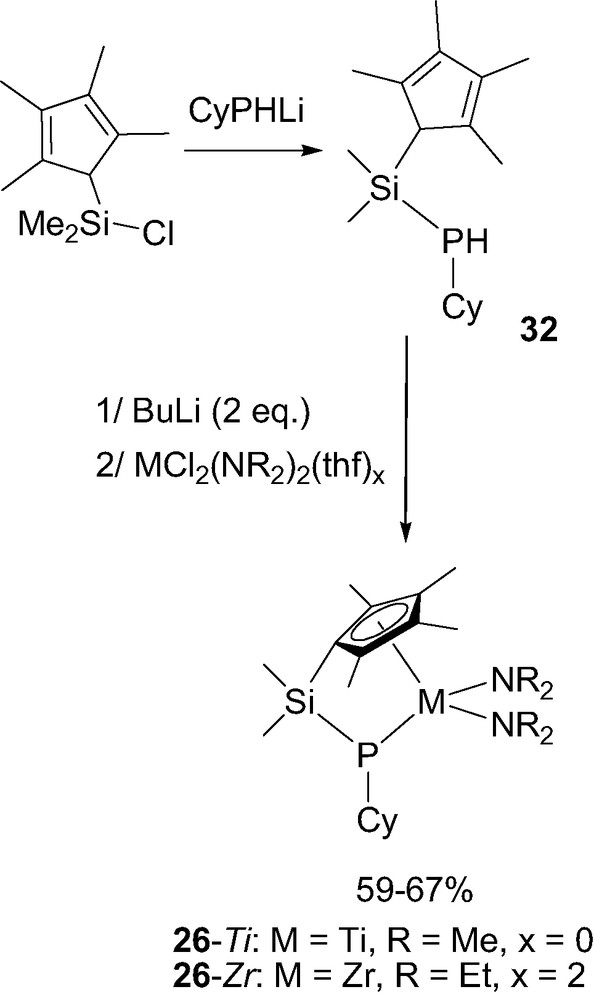
Synthesis of the ‘CpSiP’ complexes 26-M (M = Ti or Zr).
The related ‘CpCP’ framework was readily prepared in its singly deprotonated form 33 by nucleophilic addition of a lithium phosphide RPHLi (R = Cy or Ph) to a fulvene. Further deprotonation with LDA followed by reaction with the diamido MCl2(NR2)2(thf)x reagents finally led to the desired ‘CpCP’ complexes 27 in good yields (Scheme 20) [36]. Following this three-step procedure, the substituents at both the chelating phosphorus and bridging carbon atoms could be easily varied and accordingly, several titanium and zirconium complexes 27a–d-M were prepared. Notably, even the enolizable 6,6-dimethylfulvene could be engaged in this reaction due to the higher nucleophilicity/basicity ratio of lithium phosphides compared to lithium amides.
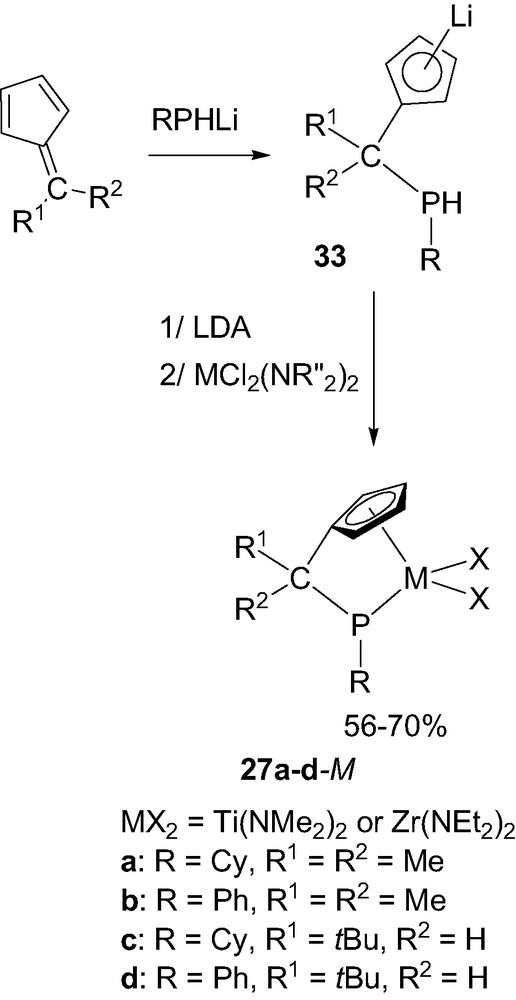
Synthesis of the ‘CpCP’ complexes 27a–d-M (M = Ti or Zr).
X-ray diffractions studies performed on ‘CpSiP’ complexes 26-Ti and 26-Zr revealed the expected constrained geometries although significant distortions were noticed compared to the related ‘CpSiN’ derivatives (Table 16). The steric constraints induced by the ligand framework are clearly evidenced by the narrow endocyclic CipsoSiP angles (ca. 92–94°). These values are very similar to those observed in the related ‘CpSiN’ complexes Me2Si(Cp)(NtBu)Ti(NMe2)2 (CipsoSiN angles of 93.9°) and Me2Si(Cp′)(NtBu)Zr(NMe2)2 (CipsoSiN angles of 94.5°) [37], suggesting that the replacement of nitrogen by the larger phosphorus does not induce the expected relief to the steric constraints. This feature is corroborated by the marginal differences observed between the sum of bond angles at Cipso (356° and 349–352° in the ‘CpSiP’ and ‘CpSiN’ complexes, respectively) and the centroid–M–P(N) angles (98–104° and 100–105° in the ‘CpSiP’ and ‘CpSiN’ complexes, respectively). In fact, it is the phosphorus coordination that makes ‘CpSiP’ complexes 26 so different from their ‘CpSiN’ analogs. Indeed, the sum of the bond angles around phosphorus only amount to ca. 310–320°, indicating that the environment around this atom strongly deviates from the planar geometry typically observed around nitrogen in (Cp)(amido) complexes. This pyramidalization clearly results from the higher inversion barrier of phosphorus versus nitrogen and suggests only partial P–M π-bonding, in agreement with the rather long P–M bond lengths (2.50–2.54 Å for Ti, 2.65 Å for Zr).
Selected angles (°) for ‘CpSiP’ and ‘CpSiN’ complexes
| Complex | Cipso–Si–P(N) | Sum of bond angles at Cipso | Centroid–M–P(N) | Sum of bond angles at P(N) |
| Me2Si(Cp′)(PCy)Ti(NMe2)2 26-Tia,b | 91.8–93.1 | 355.7–356 | 103.9 | 310.1–321.1 |
| Me2Si(Cp′)(PCy)Zr(NEt2)226-Zrb | 94.2 | 356.1 | 98.2 | 321.2 |
| Me2Si(Cp)(NtBu)Ti(NMe2)2 | 93.9 | 349.2 | 105.5 | 359.4 |
| Me2Si(Cp′)(NtBu)Zr(NMe2)2b | 94.5 | 352.2 | 100.2 | 359.6 |
a Two independent enantiomorphic molecules with slightly different geometric data were found in the crystal.
b Cp′ = tetramethylcyclopentadienyl.
Although X-ray data are not available for the related ‘CpCP’ complexes 27, detailed NMR investigations combined with DFT calculations revealed analogous structural features [36b]. In particular, the pyramidal environment around phosphorus was unambiguously evidenced by decoalescence of the C5H4 1H NMR signals at low temperature. Moreover, activation energies ΔG# ranging from 7.5 to 10.0 kcal/mol could be estimated for the P-inversion process in these ‘CpCP’ complexes. These rather low values (compared to ca. 35 kcal/mol typically encountered with phosphines) have been rationalized by computational studies, the stabilization of the corresponding transition states via P–M π-bonding being indicated by phosphorus planarization and shortening of the P–M bond lengths.
All of these (cyclopentadienyl)(phosphido) complexes have been evaluated toward Ziegler–Natta polymerization under MAO activation. As far as ethylene polymerization is concerned, the best results were obtained for the ‘CpCP’ zirconium complexes 27a-Zr and 27b-Zr but in most cases, only moderate activities were observed whatever the bridging unit and the phosphorus substituent (Table 17).
MAO activated ethylene polymerization at 60 °C, in toluene, under 2 bar of ethylene pressure
| Precatalyst | Al/M | Activity (g PE/mmol catalyst per bar per hour) | References |
| Me2Si(Cp′)(PCy)Ti(NMe2)2 26-Tia | 1100 | 40 | [35] |
| Me2Si(Cp′)(PCy)Zr(NEt2)2 26-Zra | 1100 | 66 | [35] |
| Me2C(Cp)(PCy)Ti(NMe2)2 27a-Ti | 825 | 21 | [36b] |
| Me2C(Cp)(PPh)Ti(NMe2)2 27b-Ti | 840 | 56 | [36b] |
| (tBu)HC(Cp)(PCy)Ti(NMe2)2 27c-Ti | 1200 | 18 | [36b] |
| (tBu)HC(Cp)(PPh)Ti(NMe2)2 27d-Ti | 1000 | 9 | [36b] |
| Me2C(Cp)(PCy)Zr(NEt2)2 27a-Zr | 1450 | 193 | [36b] |
| Me2C(Cp)(PPh)Zr(NEt2)2 27b-Zr | 825 | 78 | [36b] |
| (tBu)HC(Cp)(PCy)Zr(NEt2)2 27c-Zr | 1400 | 6 | [36b] |
| (tBu)HC(Cp)(PPh)Zr(NEt2)2 27d-Zr | 950 | 6 | [36b] |
a Cp′ = tetramethylcyclopentadienyl.
Similar trends were observed for ethylene/oct-1-ene copolymerization (Table 18). In all cases a reasonable incorporation of the long-chain 1-alkene was achieved (ethylene/oct-1-ene ratios around 6). Both the metal and bridging unit were found to have a critical influence on the catalytic performances [zirconium > titanium and Me2C > Me2Si ≈ (tBu)HC], the highest activities being herealso obtained with the ‘CpCP’ zirconium complexes 27a-Zr and 27b-Zr.
MAO activated ethylene/oct-1-ene copolymerization at 90 °C, in toluene, under 2 bar of ethylene pressure
| Precatalyst | Al/M | Activity (g copolymer/mol of catalyst per bar per hour) | Ethylene/oct-1-ene | References |
| Me2Si(Cp′)(PCy)Ti(NMe2)2 26-Tia | 1100 | 16 | 6 | [35] |
| Me2Si(Cp′)(PCy)Zr(NEt2)2 26-Zra | 1320 | 9 | 6 | [35] |
| Me2C(Cp)(PCy)Ti(NMe2)2 27a-Ti | 960 | 15 | 6 | [36b] |
| Me2C(Cp)(PPh)Ti(NMe2)2 27b-Ti | 825 | 25 | 5 | [36b] |
| (tBu)HC(Cp)(PCy)Ti(NMe2)2 27c-Ti | 960 | 18 | 6 | [36b] |
| (tBu)HC(Cp)(PPh)Ti(NMe2)2 27d-Ti | 1000 | 5 | 7 | [36b] |
| Me2C(Cp)(PCy)Zr(NEt2)2 27a-Zr | 1650 | 860 | 6 | [36b] |
| Me2C(Cp)(PPh)Zr(NEt2)2 27b-Zr | 1500 | 400 | 6 | [36b] |
| (tBu)HC(Cp)(PCy)Zr(NEt2)2 27c-Zr | 1400 | 40 | 6 | [36b] |
| (tBu)HC(Cp)(PPh)Zr(NEt2)2 27d-Zr | 1500 | 12 | 6 | [36b] |
a Cp′ = tetramethylcyclopentadienyl.
3.2.2 Mixed (cyclopentadienyl)(phosphido) complexes featuring an ethylene bridge
Mixed (cyclopentadienyl)(phosphido) complexes featuring an ethylene bridge 28 have also been reported. The ‘CpC2P’ framework was first prepared in its singly deprotonated form 34 by nucleophilic addition of MesPHLi (Mes = 2,4,6-trimethylphenyl) on the spiro [2,4]hepta-4,6-diene. Coordination to zirconium and hafnium was then achieved by silylation or stannylation of the anionic ‘CpC2P’ ligand 34 followed by reaction with MCl4(tht)2 (Scheme 21). Accordingly, the (cyclopentadienyl)(phosphine) complexes 35-M were obtained as tetrahydrothiophene (tht) adducts in high yields [38a]. Finally, the desired phosphido derivatives were generated by deprotonation of 35-M with NaCPh3 or FluLi and isolated in ca. 40% yield as N-methyl imidazole (NMI) adducts 28a-M [38b]. Alternatively, treatment of 35-M with Grignard reagents allowed for the substitution of the chlorides at the metal by benzyl or allyl groups, subsequent elimination of toluene (upon heating) or propene (spontaneous) affording the di(benzyl) and di(allyl) complexes 28b-M and 28c-M, respectively [38c].

Synthesis of the ‘CpC2P’ complexes 28a–c-M (M = Zr or Hf).
So far, only complex 28a-Zr has been structurally characterized [38b,c]. Of particular interest, the Zr–P bond length (2.60 Å) is shorter than in the related ‘CpSiP’ complex 26-Zr (2.65 Å) and the phosphorus is in a less pyramidal environment (sum of the bond angles around P: 341° in 28a-Zr versus 321° in 26-Zr), indicating stronger P–Zr π-bonding. It may also be anticipated that the longer and more flexible ethylene bridge induce less steric constraints, as indicated by the perfectly planar environment around Cipso (sum of the bond angles around Cipso: 360° in 28a-Zr versus 356° in 26-Zr). Unfortunately, none of these ‘CpC2P’ complexes has been evaluated toward olefin polymerization so that the precise influence of the bridging unit on the catalytic activity of (cyclopentadienyl)(phosphido) complexes remains to be determined.
3.2.3 P-bridged (cyclopentadienyl)(amido) complexes
The preparation of P-bridged (cyclopentadienyl)(amido) complexes 29 was found to be much more difficult than that of (cyclopentadienyl)(phosphido) complexes. Indeed, pre-assembled ‘CpPN’ ligands could not be coordinated to group 4 metals despite numerous attempts. In fact, the synthesis of such a derivative only proved successful when the readily available PCl-functionalized cyclopentadienyl complex 36 was treated with an equimolar amount of tBuNHLi and NEt3 (Scheme 22) [39]. No X-ray data are available for the resulting complex but the pyramidal environment around phosphorus could be established by NMR spectroscopy, 29-Ti being obtained as a mixture of syn/anti diastereomers (relative to the orientation of the tBu groups at the phosphorus atom and Cp ring). According to preliminary investigations, complex 29-Ti proved to be a moderately active precatalyst for ethylene polymerization in the presence of MAO, leading to high molecular weight PE with an activity of 100 kg PE/mol Ti per bar per hour at 80 °C under 3 bar of ethylene pressure and for an Al/Ti ratio of 2000 [39].
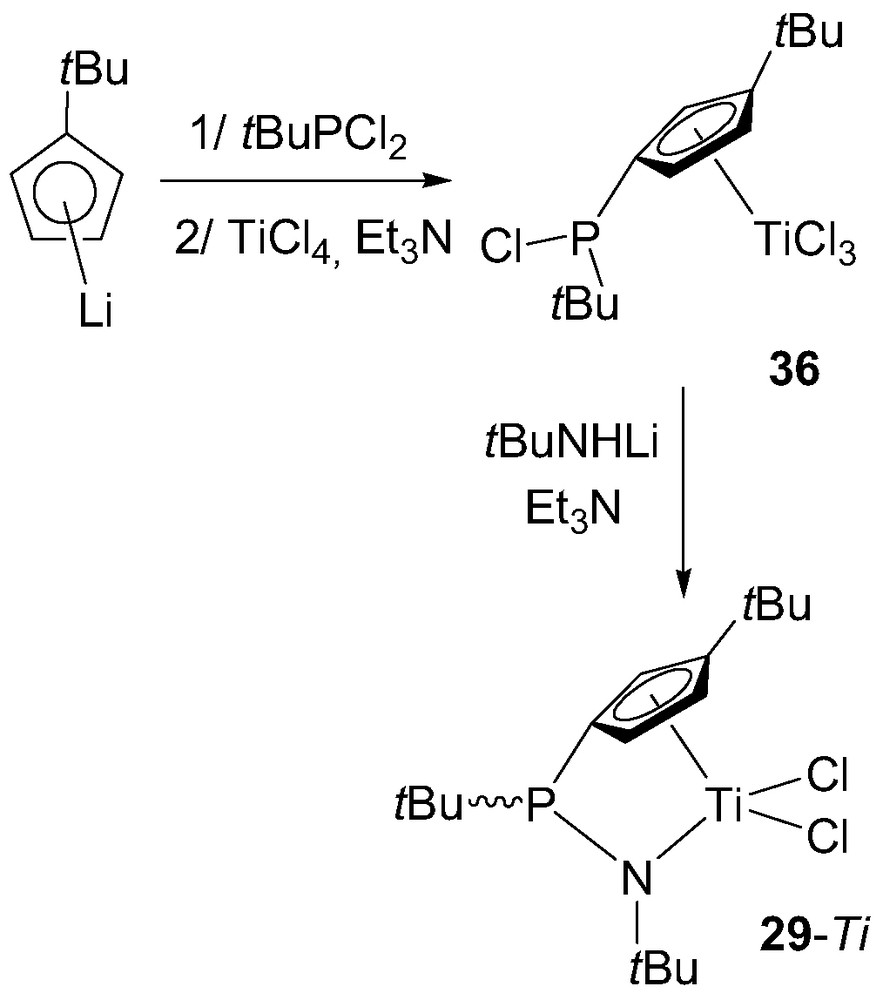
Synthesis of the P-bridged (cyclopentadienyl)(amido) complex 29-Ti.
Lastly, it should be mentioned that DFT calculations have been reported for the related complexes 30 and 31 featuring phosphazene and phosphinimido arms [40]. Accordingly, the positive charge in the model complex H2P(Cp)NHZrCl2+ 30*-Zr was found to mainly develop at the metal center and as a result the metal electrophilicity was predicted to significantly increase by replacing the silylene bridge R2Si for a phosphenium unit R2P+. Moreover, the neutral model complex H2P(Cp)NZrCl2 31*-Zr was found to be geometrically and electronically strongly related to the corresponding ‘CpSiN’ complex H2Si(Cp)N(H)ZrCl2, but markedly more constrained than its isomer HP(Cp)N(H)ZrCl2 29*-Zr. These theoretical predictions clearly deserve experimental confirmations.
4 Mixed boron/phosphorus analogs
Two-center bridges in ansa-metallocenes usually involve non-polar or weakly polar bonds (C–C, Si–C…). Taking advantage of the P/B affinity, Starzewski et al. [41] reported in 1999 a new class of ansa-metallocene catalysts 37 featuring a strongly polar donor/acceptor (D/A) bridge (Fig. 8). In these complexes, one Cp ring bears a pendant donor group (a phosphine unit) while the other bears a Lewis acid moiety (a borane). As a result, the two π ligands are strongly bonded through a dative interaction.
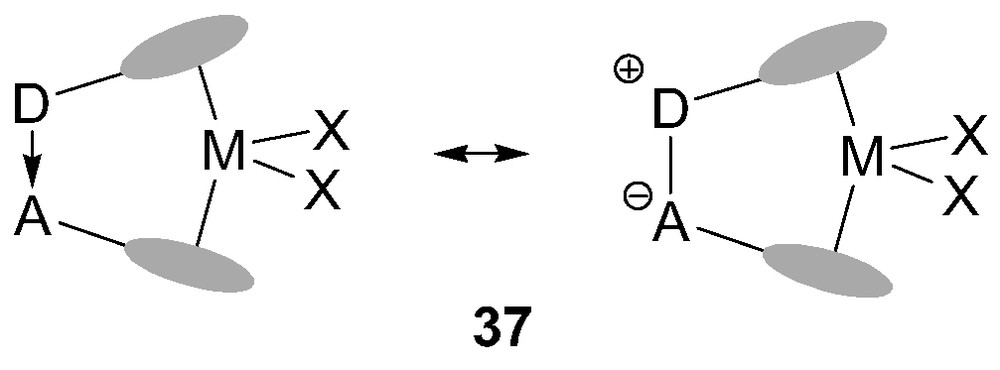
General representations for ansa-metallocenes 37 with donor/acceptor bridge.
Such a donor–acceptor ansa-metallocene 37a has been prepared by reaction of the silylated phosphorus-containing precursor Me2P(Cp)SiMe3 with zirconium tetrachloride, and subsequent reaction of the resulting complex Me2P(Cp)ZrCl3 with the silylated boron-containing unit Cl2B(Cp)SiMe3 (Scheme 23) [41]. Notably, the synthesis of this complex markedly differs from that generally involved for ansa-metallocenes. Indeed, it does not require the pre-formation of a bridging ligand, since the dative P→B interaction spontaneously forms as soon as the two Cp-ligands are coordinated to the metal center.
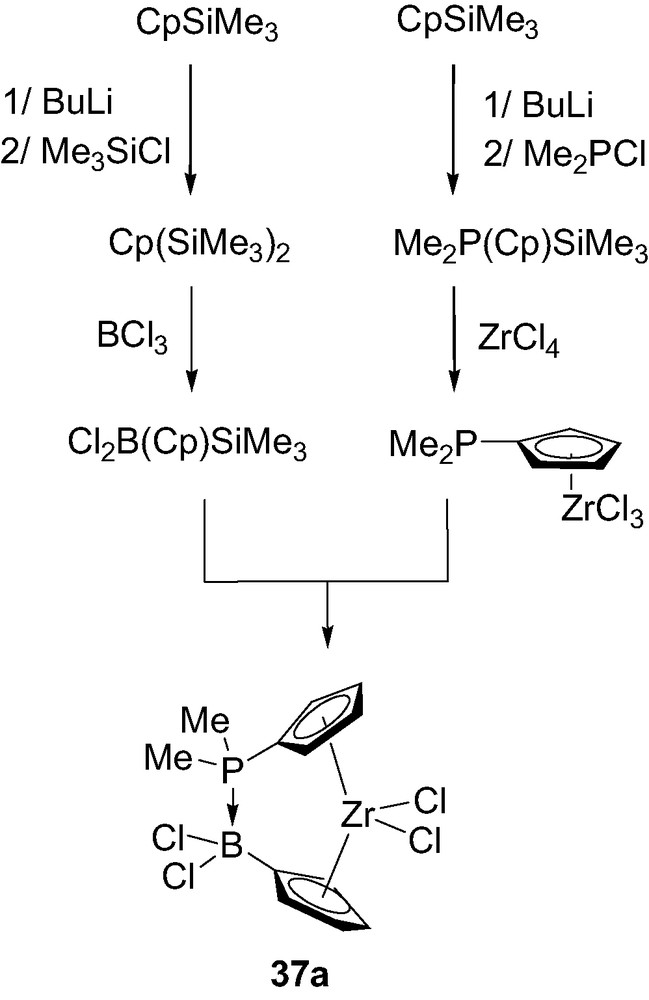
Synthesis of the donor–acceptor ansa-metallocene 37a.
The molecular structure of 37a has been confirmed by an X-ray analysis. Accordingly, the P–B bond length (1.98 Å) is at the upper limit of those observed for P→B dative bonds and the centroid–Zr–centroid angle (127.9°) is very comparable to that observed in the related ansa-metallocene featuring an ethylene bridge (128°) [42]. Multinuclear NMR investigations (δ 31P = 104 ppm, δ 11B = –0.5 ppm and 1JPB = 120 Hz) indicated the presence of the P→B bridge in solution as well. Moreover, this interaction proved to be highly thermally stable, no change being observed in the 1H NMR spectrum up to 100 °C.
Under MAO activation, complex 37a was found to be highly active for ethylene polymerization. Indeed, at 100 °C under 10 bar of ethylene, high molecular weight polyethylene (PE) was obtained (Mv = 106 000 g/mol) with a catalyst activity of about 118 000 kg PE/mol Zr per hour.
Due to the hindered rotation of the Cp rings, ansa-metallocenes are particularly suitable for stereocontrolled polymerization of propene. The racemic catalyst 37b, in which both the donor and acceptor moieties have been introduced on 2-methylindenyl rings, has been investigated in this respect. Activation with triisobutylaluminium/dimethylanilinium-tetrakis(pentafluorophenyl)borate allowed the production of highly isotactic polypropene (i-PP). According to 13C NMR, the selectivity in isotactic mmmm-pentads was higher at room temperature (92% in toluene solution, 94% in bulk) than at 50 °C (82% in bulk) (Table 19). Such a transformation could previously only be achieved with metallocenes featuring synthetically demanding patterns [43].
Polymerization of propene using 37b activated with triisobutylaluminium/dimethylanilinium-tetrakis(pentafluorophenyl)borate under 2 bar
| Solvent | Temperature | mol% of mmmm-pentads (%) | Isotacticity index (%) | Mv (g/mol) |
| Toluene | rt | 92 | 97 | 422 000 |
| Bulk | rt | 94 | 98 | 2 000 000 |
| Bulk | 50 °C | 82 | 92 | 434 000 |
Due to their smaller centroid–metal–centroid angles, ansa-metallocenes are also particularly attractive for the polymerization and copolymerization of higher olefins. In this regard, the use of donor–acceptor catalysts for the incorporation of cycloolefins (such as norbornene) has been patented [44].
Varying the electronic and steric properties of the donor and acceptor bridging units in such ansa-metallocenes clearly offer the possibility to adjust the strength and length of the dative D→A interaction, and thereby to tune the architecture and catalytic activity of the metallocenes. This aspect was first illustrated with the preparation of the related titanocene 37c featuring a significantly shorter N→B bridging unit (1.74 Å) (Fig. 9) [41].
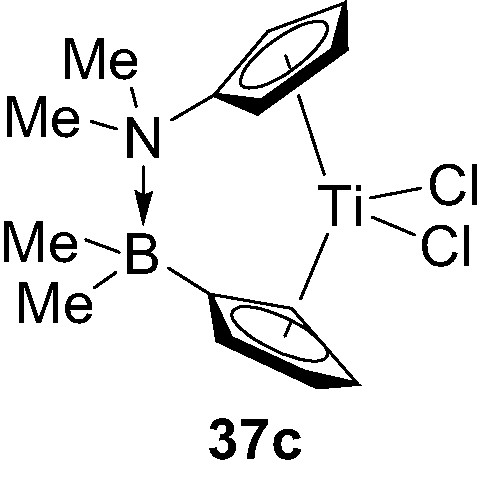
Structure of the donor/acceptor ansa-metallocene 37c.
5 Conclusion and perspectives
From this review, it is clear that the incorporation of boron and phosphorus in metallocene, ansa-metallocene and CGC has been an extremely intense research area over the last 10 years. Most of the synthetic challenges have been successfully addressed and promising results in olefin polymerization have been reported for almost all of the investigated boron- and phosphorus-containing complexes. Further progress in this direction will certainly be achieved in near future but besides the fundamental and synthetic interests of all these studies, the influence of each structural modification will clearly have to be precisely determined if practical applications complementary to those of the classical catalysts are to be found.
Acknowledgments
The ‘Centre national de la recherche scientifique’, the University Paul-Sabatier (France) and the French Ministry of Research and New Technologies (ACI JC4091) are gratefully acknowledged for their support.