

1 Introduction
Proteins containing dinuclear copper centers play paramount roles in biology. Many features include dioxygen transport and/or activation, electron transfer, reduction of nitrogen oxides and hydrolytic chemistry. Many of them have oxidase or oxygenase activities. The structural aspects of the active sites are directly correlated to activity [1]. They are of interest in the field of biomimetic chemistry to provide an improved understanding of the function of biological sites and as potential catalysts for substrate oxidations. We have focused this minireview on the dinuclear dicopper enzyme catechol oxidase [2] with the main point of view: how biomimetic models can help the elucidation of the mechanistic pathway involved in the catalytic cycle of the enzyme?
2 Catechol oxidase: a brief reminder
Catechol oxidases (CO) are ubiquitous plant enzymes which catalyze the oxidation of a broad range of o-diphenols to o-quinones in presence of dioxygen [3,4]. These enzymes belong to the type 3 copper protein family, characterized by an EPR-silent dinuclear copper active site in the native met CuII–CuII form, due to a strong antiferromagnetic coupling between the bridged copper(II) ions. The two other known type 3 copper proteins, hemocyanins (Hc) and tyrosinases (Tyr), have also been extensively studied. Hemocyanins reversibly bind molecular oxygen and tyrosinases catalyze both the hydroxylation of phenols to o-diphenols (monophenolase activity) and the two-electron oxidation of o-diphenols to o-quinones (catecholase activity).
The crystal structure of catechol oxidase from sweet potatoes (Ipomoea batatas, ibCO) has been reported in numerous states: the oxidized CuII–CuII met form, the reduced deoxy CuI–CuI form and the species with the bound inhibitor phenylthiourea (ptu) [5]. In the met state, both metal binding sites involve three histidine ligands. The two cupric ions, likely bridged by a hydroxide ion, are 2.87 Å apart in a trigonal pyramidal coordination sphere. An interesting feature of the dinuclear metal center is a covalent thioether bond formed between a carbon atom of His109 (one of the copper (CuA) ligands) and the sulfur atom of Cys92 (Fig. 1). In the dimetallic center of the enzyme–ptu complex, the hydroxo bridge is replaced by a sulfur bridge from ptu, increasing the CuII–CuII separation to 4.2 Å compared to the met form. The amide nitrogen weakly interacts with CuB leading to still more unsymmetrical coordination spheres for the two copper centers.
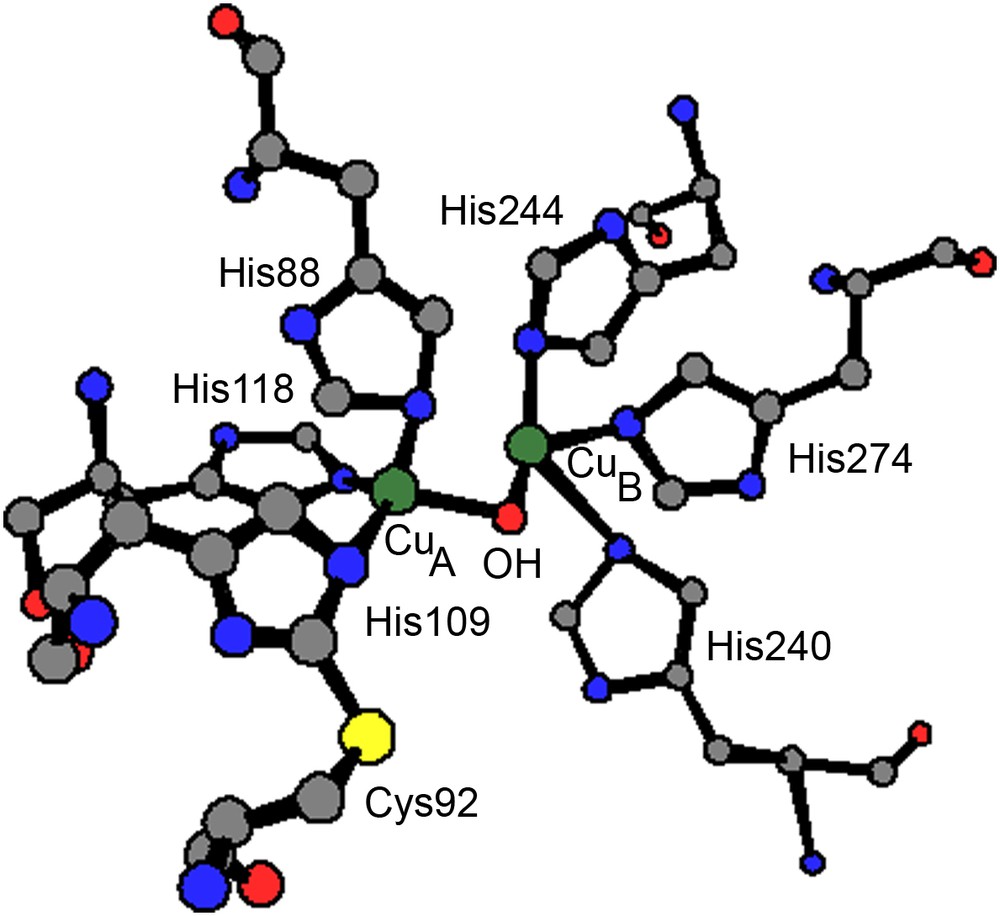
Coordination sphere of the dinuclear copper(II) center of catechol oxidase in the met state.
In the reduced (deoxy) state the copper centers are not bridged (CuI⋯CuI = 4.4 Å) and a water molecule is coordinated to CuA. The coordination sphere is a distorted trigonal pyramidal whereas the coordination of CuB (three histidine ligands) can be described as a square planar with a vacant coordination site.
These structural data evidence the flexibility of the enzyme active center during the catalytic cycle. Furthermore these data combined with biochemical and spectroscopic studies [5,6] as well as with the tyrosinase related cycle [1], allow new insights on the mechanism and a catalytic cycle for catechol oxidase activity (Fig. 2) has been proposed [7]. It begins with the native met form. The monodentate binding of the diphenolic substrate to the CuB is suggested from the crystal structure of the ibCO–ptu complex [5]. Nevertheless, a didentate bridging of catechol is also proposed [2,3]. A stoichiometric amount of the quinone product is formed immediately upon addition of catechol, even in absence of dioxygen. This excludes the oxy form at the starting situation of the catalytic cycle. This form is supposed to be derived from the deoxy form (in the presence of dioxygen) and has been experimentally observed in vitro upon addition of H2O2 to the met form [6,8]. The Cu2O2 species shows strong UV–vis absorptions at 343 and 585 nm assigned to peroxo-to-copper charge transfer transitions and Raman vibrations at ∼750 cm−1 for the intra-peroxide ν(O–O) stretching [6]. These features are characteristic of a μ-η2:η2 dioxygen binding mode as observed in oxy-Hc [9]. In the proposed catalytic cycles, catecholate binds mono- or bidentally to the oxy form, for a two-electron transfer leading to the release of the quinone product and to the initial met form.
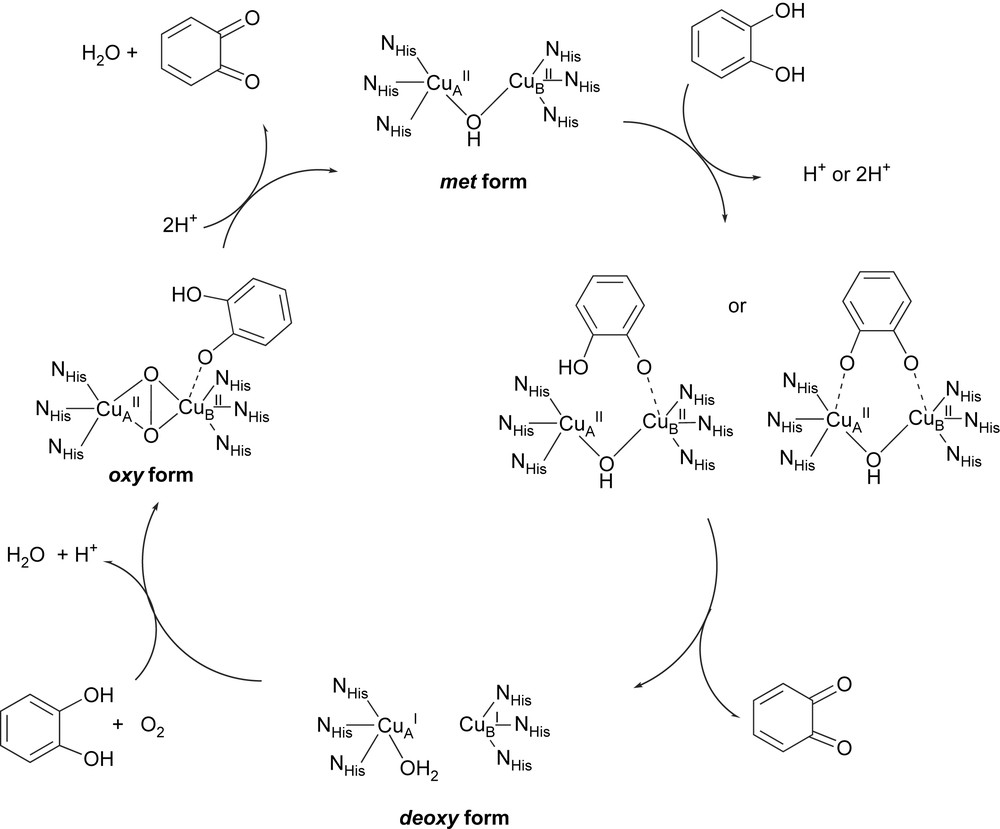
Proposed catalytic cycle for catechol oxidase activity. Redraw after Krebs [7] and Solomon [1].
Structural information were available for hemocyanins [10–14] and very recently for a recombinant tyrosinase [15]. Although comparison of metal coordination region from these structural data evidences high similarities, the functional differences between Tyr, CO and Hc could not be rationalized to date.
However, important questions remain unanswered concerning the catalytic mechanisms (Fig. 2). They mainly concern (i) the control of the catalytic activity (pH, electronic properties), (ii) the coordination mode of the substrate: does it consist of a bridging catecholate between the two copper centers rather than a coordination centered to one metal ion (bridging didentate or monodentate coordination)? (iii) the reactive intermediate involved in the dioxygen activation.
3 Tools for studying catalytic activity
3.1 Models to study the pH dependence for the catecholase activity
The activity of the enzyme [8] is observed in a 5–8 pH range (optimum at pH = 8), with an irreversible loss of activity below pH 4 and above pH 10. Biomimetic studies can enlighten this pH dependence. The pH-induced changes in the coordination spheres of the dicopper(II) complex of the known ligand (Fig. 3): 2,6-bis[(bis(2-pyridylmethyl)amino)methyl]-4-methylphenol, have been studied [16].
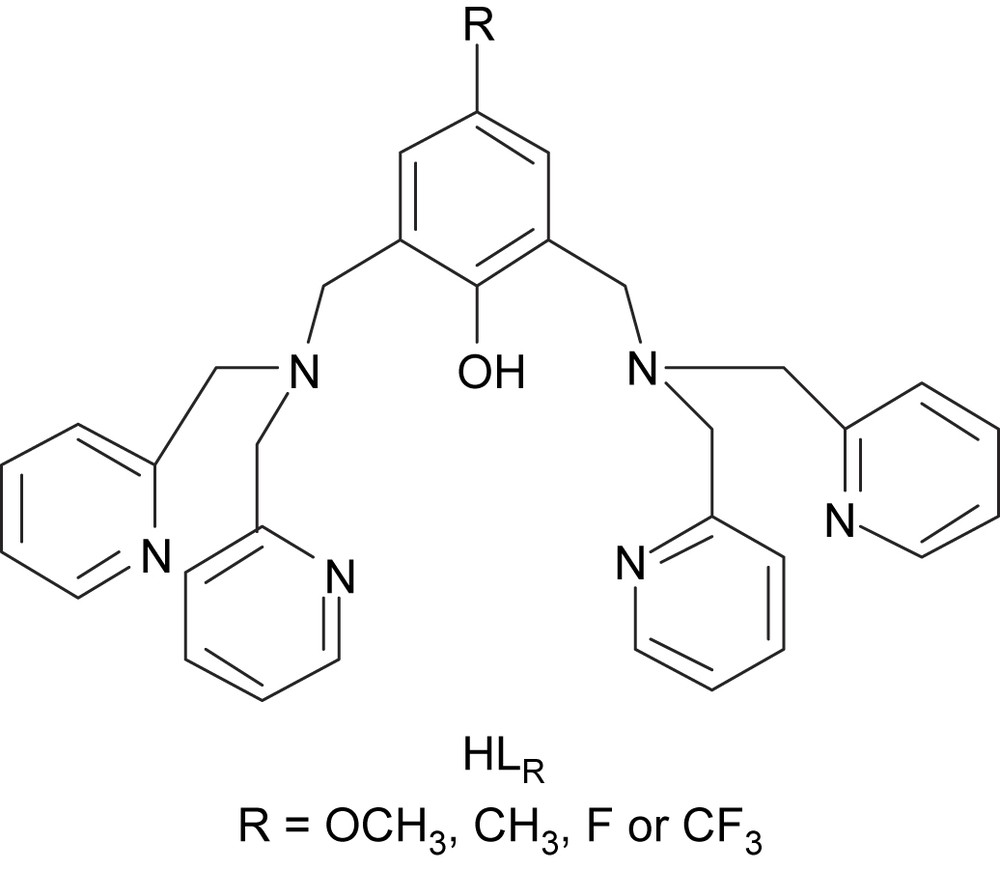
Dinucleating ligands HLR employed to prepare the copper(II) complexes.
From , three complexes are described in solution and two of them in solid state. Their pH-driven interconversions and redox properties have been studied. An X-ray structural view of μ-hydroxo dicationic complex is shown in Fig. 4A. The four Cu1, Cu2, O1 and O2 atoms are in the same plane with a Cu1⋯Cu2 distance of 2.966(1) Å. The complex is EPR-silent in accordance with an antiferromagnetic coupling between the two copper atoms. The X-ray structure of the tricationic complex is shown in Fig. 4B. It is worth noting that in contrast to , the two pyridine groups are trans to each other. The presence of the unique phenoxo bridge leads to an increased Cu⋯Cu′ distance (4.139(1) Å). The EPR spectrum of in frozen solution (H2O (pH = 3)/DMSO, 1/1) reveals a ΔMS = ±2 transition at 160 mT and a broad ΔMS = ±1 signal from 200 to 440 mT. This spectrum is typical for an S = 1 state of two coupled copper(II) ions. The electrochemical behaviors of and have been studied in CH3CN by cyclic voltammetry (CV) and rotating disc electrode (RDE) voltammetry [16].
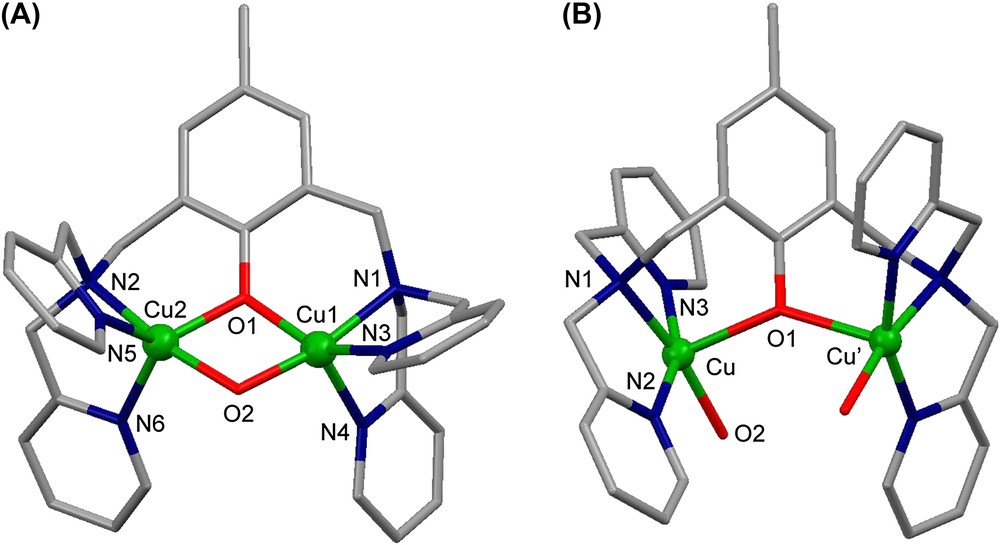
X-ray crystal structures of (A) the dicationic complex and (B) of the tricationic complex .
For in acetonitrile or water solutions, the main UV–vis spectral features are at 410 nm (ɛ = 480 M−1 cm−1) attributed to the ligand (phenoxo) to metal charge transfer transition (LMCT) and at 785 nm (ɛ = 280 M−1 cm−1) to CuII d–d transitions. For the observed absorptions are shifted at 460 nm (ɛ = 710 M−1 cm−1) and 700 nm (ɛ = 180 M−1 cm−1), respectively. UV–vis titration (monitoring the LMCT band) of with perchloric acid (3.8 < pH < 6.6) leads quantitatively to . An isosbestic point observed at 392 nm indicates the presence of only two absorbing species. An intermediate species such as an aquahydroxo dicopper(II) complex is not detected. The reaction is fully reversible by addition of sodium hydroxide to the solution of . A plot of the absorbance vs pH indicates that the deprotonation process has an apparent pK1 value of 4.95 ± 0.05 at 25 °C. Another species is reversibly formed in strong basic medium (pK2 = 12.0 ± 0.1) revealed by UV–vis spectroscopy (which could not be isolated in solid state). The equilibrium between , and the new species observed in basic medium has also been studied by EPR spectroscopy in H2O/DMSO (1/1) at 100 K and by 1H NMR in D2O (DMSO) at 298 K. According to spectroscopic results, a dihydroxo structure (Fig. 5) is proposed. Strong acidification (pH < 3) of leads to the release of free copper ions.
pH-driven interconversions between , and .
It has to be emphasized that a small pH change can induce noticeable variations in the coordination spheres around the metal centers, and consequently in the spectroscopic properties and catalytic reactivities (vide infra).
The 3,5-di-tert-butylcatechol (3,5-dtbc) has been employed as substrate, and the activities and reaction rates have been determined by monitoring the time dependence of absorption of the released quinone (3,5-dtbq) at 410 nm (maximum absorption in acetonitrile). Only exhibits catecholase activity (Table 1). Addition of a small amount of acid or base stopped the reaction which was fully restored when the pH was adjusted to 6–8, i.e. in the range where is the only species present in solution. These results indicate the dependence of changes in metal ion geometry on the catalytic ability.
Selected properties of [Cu2(LR)(μ-OH)]2+ and [Cu2(LR)(H2O)2]3+ complexes (R = OCH3, CH3, F or CF3) [16,17], and catalytic activities
| Compound | d Cu–Cu (Å)a | Electrochemical data (reduction)b | Vmaxe (10−6 M s−1) | KMe (mM) | Turnover number (90 min) | |
| 2.980 | −0.9c | −1.40d | 2.2 | 0.25 | 32 | |
| 2.966 | −0.94c | −1.24d | 1.1 | 1.49 | 16 | |
| [Cu2(LF)(μ-OH)]2+ | 2.969 | −0.90c | −1.30d | 0.27 | 8.8 | 8 |
| f | −0.86c | −1.24d | 0 | – | 0 | |
| f | −0.35c | −1.08d | 0 | – | 0 | |
| 4.139 | −0.33c | −0.86d | 0 | – | 0 | |
| [Cu2(LF)(H2O)2]3+ | 4.084 | −0.32c | −1.14d | 0 | – | 0 |
| 4.222 | −0.78c | −1.18d | 0 | – | 0 |
a From X-ray studies [16,17].
b Measured by CV at 0.1 V s−1, E vs Ag/AgNO3, 10 mM + CH3CN + TBAP 0.1 M.
c E1/2/V (reversible process).
d Ep/V (irreversible process).
e Determined from Lineweaver–Burk plot with 3,5-dtbc as substrate.
f Not determined.
The reaction rate in acetonitrile was obtained from the slope of the trace at 410 nm and Lineweaver–Burk treatment gave: Vmax = 1.1 × 10−6 M s−1 and KM = 1.49 mM. Owing to the fact that is more easily reduced than , the redox potential of the dicopper complexes are not decisively related to the observed catechol oxidase activities. As observed in the structure of , the metallic site is easily accessible and the short Cu–Cu distance (2.96 Å in vs 4.13 Å in ) would allow a bridging catechol coordination compatible with the distance between the two o-diphenol oxygen atoms. It has to be emphasized that (i) this intermetallic distance is close to the value observed for the met form in the enzyme (2.9 Å) which also possesses a μ-hydroxo dicopper center, (ii) the change of coordination sphere from to (from trigonal bipyramid to square pyramid) increases the steric bulk around the copper atoms, preventing the approach of the substrate, and (iii) the pH domain of the μ-hydroxo complex existence corresponds exactly to the pH range of the enzyme activity [8].
3.2 Models to study the influence of electronic effects
For a better understanding of the various parameters that dictate the structural properties and the reactivity, we have prepared and studied dicopper complexes of new ligands derived from , bearing, respectively, strong electron withdrawing or donating groups in the 4-position of the phenol moiety (Fig. 3) [17].
Dinuclear CuII complexes have been synthesized and characterized by spectroscopic (UV–vis, EPR, 1H NMR) as well as electrochemical techniques and in some cases, by single-crystal X-ray diffraction: , , and . The Cu–Cu distance in the two μ-hydroxo complexes (2.980 Å (R = OCH3) and 2.969 Å (R = F)) remains significantly shorter compared to the two bis(aqua) complexes (4.084 Å (R = F) and 4.222 Å (R = CF3)).
The μ-hydroxo and bis(aqua) complexes are reversibly interconverted upon acid/base titration. In basic medium, new species are reversibly formed and identified as the bis-hydroxo complexes except for the complex from which is irreversibly transformed near pH = 10. For the fluorinated complexes, the changes in the coordination sphere around the copper centers are evidenced by the fluorine chemical shift changes (19F NMR, see Section 5). For all the complexes described here, investigations of the catecholase activities showed the pH dependence for the catalytic abilities of the complexes, related with changes in the coordination sphere of the metal centers: only the μ-hydroxo complexes from , HLF and exhibit catecholase activity (Table 1). In addition, modification on R-substituent induces a drastic effect on the catecholase activity: the presence of an electron donating group on the ligand increases this activity, the reverse effect is observed with an electron withdrawing group. These results evidenced that the catalytic properties of the complexes depend on the electronic effects reflected by the redox potential values determined for the μ-OH dicopper(II) complexes, even if it is clear that electronic effects do not control solely the reactivity. In particular, as previously shown for the complexes from , the redox potential of the dicopper complexes are not decisively related to the observed activities, since bis(aqua) complexes are more easily reduced than the μ-hydroxo complexes. It should be stressed that substitution by the strongly electron withdrawing trifluoromethyl group completely inhibits the activity of the corresponding μ-OH complex, although possesses similar structural features than the other μ-OH active complexes. Catechol binding studies investigated by UV–vis spectroscopy with tetrachlorocatechol (tcc, a non-easily oxidizable catechol) or with 3,5-dtbc in anaerobic conditions, show that complex does not bind catechols. This demonstrates that the formation of an intermediate between the catechol and the CuII complex prior to electron transfer is a key step. Applying the Michaelis–Menten approach, the KM parameters have been determinated. The values (Table 1) are associated to the ability of the complex to form an adduct with the substrate and are correlated with the electronic effects from R-groups. Several parameters are involved in the catalytic efficiency of these complexes, especially intermetallic distances, geometric features around the copper atoms and redox potentials, modulated herein by the pH and the electronic features of the R-substituent on the ligand to allow an optimal interaction between the copper(II) complexes and the substrate.
4 Substrate binding studies using biomimetic models as tools
With dinuclear complexes, only few structurally characterized complex/substrate adducts are known. The first, described by Karlin et al. [18], was prepared by the ‘oxidative addition’ reaction of a phenoxo-bridged dicopper(I) complex to tetrachloro-o-benzoquinone (tcbq). The complex obtained presents a bridging tetrachlorocatechol ligand (tcc) between the two copper(II) ions with a metal–metal separation of 3.248 Å. More recently, different adducts with tcc have been isolated and characterized by X-ray crystallography [19,20]. These studies exemplify various coordination modes of catechol substrates: monodentate or didentate (bridging or not).
4.1 Tcc and 3,5-dtbc binding studies
Comparative binding experiments with the two o-diphenols substrates, were carried out with the active μ-OH and the inactive bis(aqua) dicopper(II) complexes. Our series of closely related dinuclear complexes offers a unique opportunity to perform mechanistic investigations in order to study the first step of the catalytic cycle, i.e. the coordination of the substrate to the copper centers before the intervening of dioxygen in the mechanistic pathway [21]. From the catecholase activities previously described (Table 1), it appears that the affinity of the substrate (revealed by the KM value) is the main factor tuned by the R group. During the progressive addition of the substrate, the fixation is evidenced by UV–vis and EPR changes. As the oxidation of tcc is highly much more difficult compared to 3,5-dtbc (the difference in redox potentials is 0.56 V), the former has been used in aerobic experiments and the latter in anaerobic conditions.
The changes observed in the UV–vis and EPR spectra of in acetone upon progressive addition of tcc are depicted in Fig. 6.
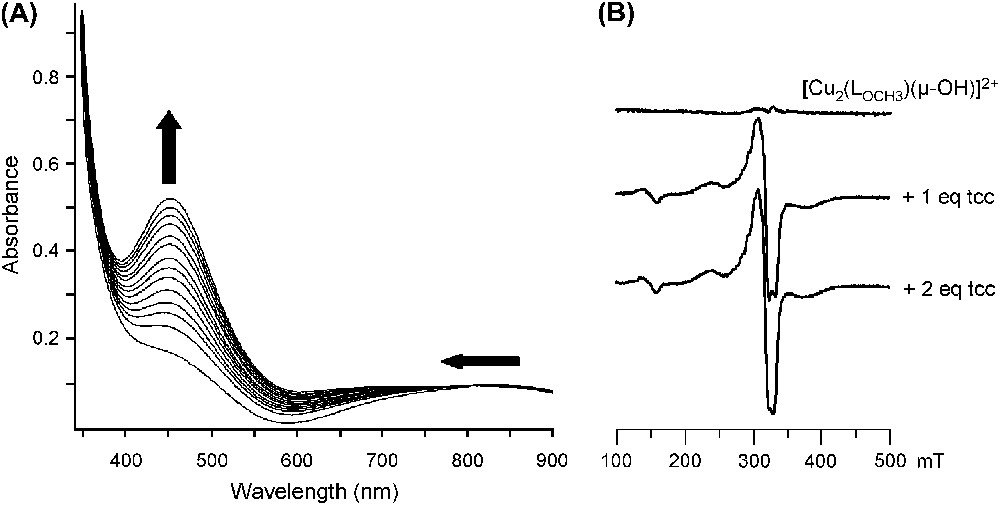
Changes in (A) the UV–vis spectrum (25 °C, acetone) and in (B) the EPR spectrum (77 K, acetone) of upon addition of tcc (from 0 to 2 equivalents).
The LMCT band at 440 nm is shifted up to 450 nm and ɛ is raised from 560 to 1050 M−1 cm−1 (1 equivalent of tcc added) and then to 2100 M−1 cm−1 (2 equivalents of tcc added) (Fig. 6A). The d–d transitions are also affected, especially during the addition of the second equivalent of substrate. These features indicate that a first substrate binding occurs followed by a second one. Further addition of tcc does not induce additional changes in the spectrum.
From the EPR-silent OH-bridged starting complex, addition of 1 equivalent of tcc leads to drastic changes with a ΔMS = ±2 signal at 150 mT and a ΔMS = ±1 signal from 200 to 440 mT (Fig. 6B). These EPR features, similar to that obtained with the corresponding free complex [17], indicate two copper(II) ions in a moderate interaction, thus suggesting the cleavage of the hydroxo bridge after binding of one tcc. Minor changes are then observed upon addition of the second equivalent of tcc. The binding of 2 equivalents of substrate on the bis(aqua) complexes has been revealed only by UV–vis since no noticeable changes appeared on the EPR spectrum.
Owing to the fact that the fixation of tcc is fast, experiments with the complex have been run using stopped-flow techniques. Two successive steps have been evidenced when the complex is mixed with 2 equivalents of tcc. The fixation of the first substrate is still too fast to allow the determination of a kinetic constant but the slower fixation of the second substrate can be monitored at 450 nm. The corresponding curve (described by the following equation in which Ae is the absorbance at the equilibrium and At the absorbance at the given time t) leads to kobs = 1.3 s−1.
While two successive steps have been observed for the fixation of two tcc to complex (fast for the first, slow for the second), the fixation of two tcc to complex is too fast to be distinguished.
The influence of added tcc or 3,5-dtbc on the electrochemical behavior of the dicopper(II) complexes of HLR type ligand was studied by CV and RDE voltammetry in CH3CN vs Ag/AgNO3 [21]. The electrochemical behavior of is deeply affected by the addition of tcc or 3,5-dtbc in the electrolytic solution despite that, as judged from RDE experiments, no change in the redox state of the metal center occurs; the copper centers remain at +2 redox state.
The main conclusion which can be drawn from these results is that is made more easily oxidizable (by 0.46 V or 0.09 V) and reducible (by 0.53 V or 0.41 V) in the presence of tcc or 3,5-dtbc, respectively. Its stability domain is decreased from 1.55 V down to 0.56 V or 1.05 V in the presence of tcc and 3,5-dtbc. The electrochemical reactivity of the substrate/complex species compared to the free complex is thus improved towards further electron transfer and suggests deep modifications in the coordination sphere around the metal center. Interaction with the substrate strongly affects the electronic density around the metal center. Furthermore, the electrochemical wave due to the irreversible oxidation of the OH-bridge progressively vanishes in the presence of substrate (tcc or 3,5 dtbc), indicating that substrate fixation causes (in accordance with EPR results) the cleavage of the OH-bridge.
On the contrary, the electrochemical behavior of is weakly affected by the addition of tcc or 3,5-dtbc in the electrolytic solution. In particular, no significant changes are observed in the characteristic potentials of the anodic and cathodic responses centered on the complex [21].
4.2 Substrate specificity
A particular point of interest is the substrate specificity observed on the enzyme itself: only o-diphenols are oxidized into quinone [22]. Studies on catechol oxidase from Lycopus europaeus reported substrate specificity for different phenolic substrates.
Comparative binding experiments with different diphenol substrates (m- and p-) were carried out with the catalytically active species. The interactions with 2,3-dimethylhydroquinone (2,3-dhq) and 4-ethylresorcinol (4-er) have been evidenced by changes in UV–vis and EPR data. In addition, binding studies with ptu, a known inhibitor of the enzyme [22] is reported. For each m- and p-diphenols, no oxidation occurs whereas the binding of the substrate on the dicopper(II) center is observed from UV–vis spectra [21]. The observed modifications on the EPR spectrum (77 K, acetone) have been also reported [21]. After addition of 1 equivalent of 2,3-dhq (or 4-er), the spectrum remains silent, indicating no important changes in geometric and magnetic properties around the copper centers and suggesting that the hydroxo bridge have been preserved in the 1:1 complex/substrate adduct.
Owing to the fact that a didentate coordination on one copper atom or a bridging didentate coordination on the two copper centers is not possible with m- and p-diphenols, these results reveal a monodentate coordination of one phenolate to one copper center in the case of 2,3-dhq and 4-er with the preserved hydroxo bridge.
The binding of ptu on complex has been also studied by UV–vis. However, after addition of 1 equivalent of ptu, no changes are observed on the d–d transitions and the EPR spectrum remains silent. These observations indicate no important changes around the dimetallic center which remains in strong interaction. When the catalytic 3,5-dtbc oxidation is studied in the presence of ptu, strong inhibition effects are observed (as for the enzyme [22]). Attempts to characterize adduct between ptu and a μ-OH dicopper complex by single-crystal X-ray structure studies have unfortunately been unsuccessful so far.
We have observed with our model complexes that dioxygen was needed even for the oxidation of the first o-diphenol substrate. Although the enzyme is able to oxidize stoichiometrically 1 equivalent of catechol under strict anaerobiosis [7], the series of model compounds described herein are good tools for binding studies on the first part of catalytic cycle and give new lights regarding the substrate binding. The fixation of the substrate occurs with the concomitant cleavage of the hydroxo bridge. The resulting monodentate hydroxyl serves to assist the complete deprotonation of the monodentate bound catechol and leads to a bridging catecholate prior to the electron transfer. Our suggested mechanism (Fig. 7) reconciles Krebs's [7] and Solomon's [1] proposals.
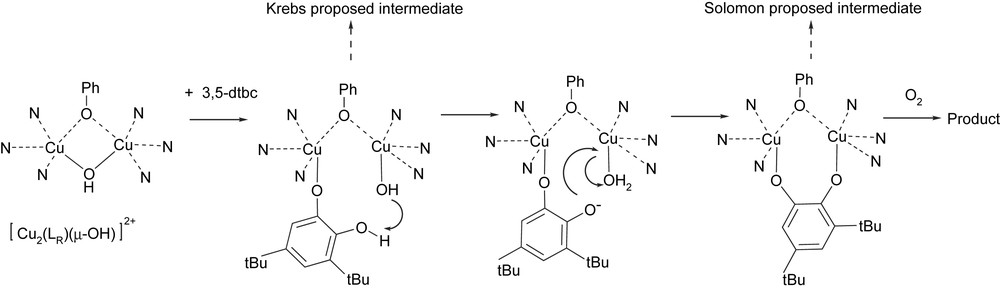
Proposed mechanism for the interaction between the series of dinuclear copper(II) μ-OH complexes and the 3,5-dtbc substrate.
All together this demonstrates: (i) how the pH dependence of the catalytic abilities of the complexes is related to change in the coordination sphere of the metal centers, (ii) how the modification on R-substituent induces a drastic effect on the catecholase activity: the presence of an electron donating group on the ligand increases this activity, the reverse effect is observed with an electron withdrawing group, and (iii) the importance of the OH-bridge cleavage during the fixation of the catecholate and underlines the key role of the resulting pendant hydroxo ligand.
5 19F-labeling, an efficient tool for studying well-suited catechol oxidase model complexes by 19F NMR
The 100% natural abundance of 19F combined with (i) its 1/2 nuclear spin, (ii) the wide range of fluorine chemical shifts and (iii) the relatively slow exchange compared to 1H, justify the use of 19F NMR for mechanistic studies.
Moreover, even if 19F NMR has rarely been applied to paramagnetic copper complexes, it constitutes a powerful tool and justifies in our opinion a section in this paper.
5.1 3,5-dtbc Binding studies (anaerobic conditions) using 19F NMR
When the fluorine labeled derivatives are available, modifications in the coordination sphere around bridged dicopper(II) centers are easily evidenced by changes in fluorine chemical shifts on 19F NMR spectra [17,23]. The UV–vis measurements show that all studied complexes herein are able to form 2:1 substrate/complex adduct.
The evolution of the 19F NMR spectra of the [Cu2(LF)(μ-OH)]2+ and [Cu2(LF)(H2O)2]3+ complexes, upon progressive addition of 3,5-dtbc in acetone-d6 is depicted in Fig. 8. For [Cu2(LF)(μ-OH)]2+ (Fig. 8A), the resulting spectra exhibit well-resolved resonances indicating that the equilibrium processes are slow compared to the 19F NMR time scale, allowing the observation of a mixture of two species simultaneously present in the medium. Spectra on Fig. 8A show that two products are successively formed upon addition of substrate (1 and 2 equivalents), characterized by chemical shifts at 44.9 and 49.2 ppm, respectively. The 19F NMR chemical shifts observed for each species are slightly modified due the presence of other compounds in solution. With the bis(aqua) complex [Cu2(LF)(H2O)2]3+, a different behavior is observed (Fig. 8B). From the relatively broad signal of the starting material, addition of 1 and 2 equivalents of 3,5-dtbc leads to the formation of two different adducts characterized by signals located at 51.2 and 52.8 ppm, respectively. In the presence of 0.5 and 1.5 equivalents, the peak broadening could also be due to exchange processes faster than the 19F NMR time scale in addition to the paramagnetic properties of the isolated species.
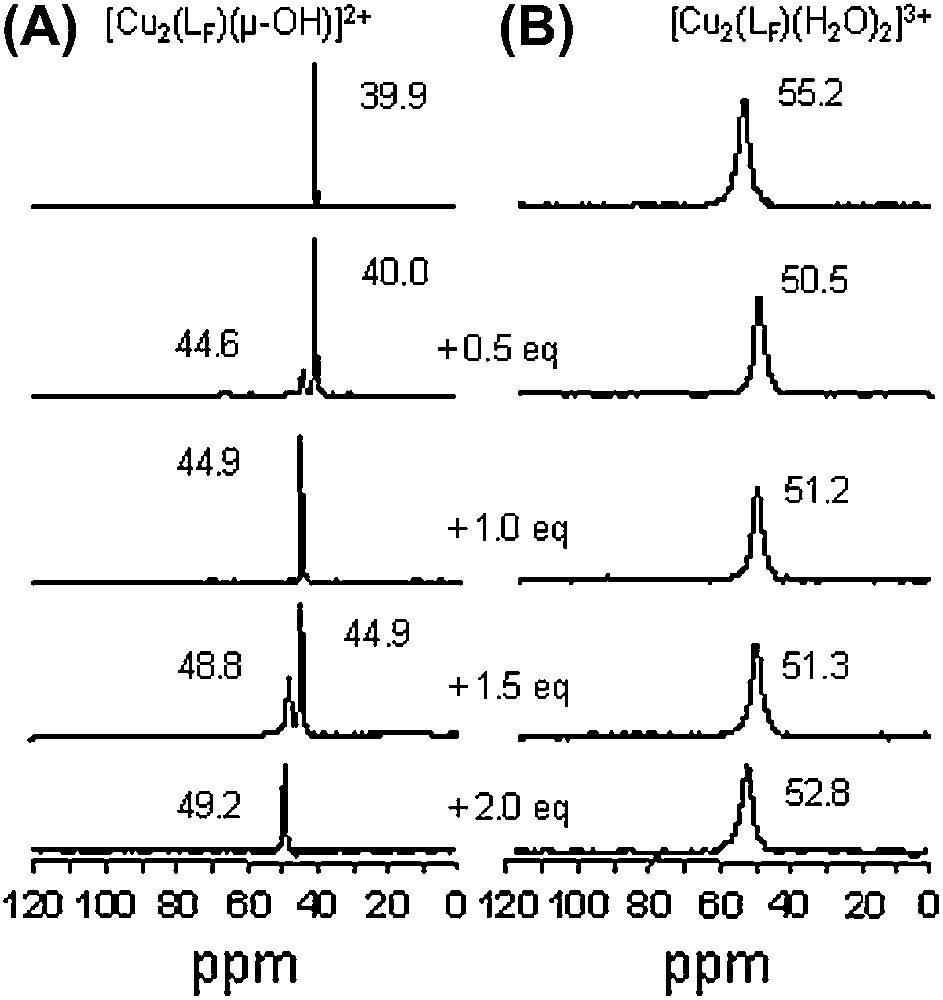
19F NMR (ppm vs C6F6) in acetone-d6 for (A) [Cu2(LF)(μ-OH)]2+ and (B) [Cu2(LF)(H2O)2]3+ and upon progressive addition of 3,5-dtbc (from 0 to 2 equivalents, anaerobic conditions).
These observations underline the interest of 19F NMR as a probe in mechanistic studies to complete UV–vis and EPR investigations (vide supra). Different binding processes of 3,5-dtbc have been evidenced herein. Both [Cu2(LF)(μ-OH)]2+ and [Cu2(LF)(H2O)2]3+ species can bind one or two substrates, but different adducts are formed. From the hydroxo bridged species, the substrate binding occurs in two successive steps; on the contrary, from bis(aqua) complex, no successive steps can be clearly evidenced [21]. Taken all together, these observations indicate that the observed 19F signals are very sensitive to the local environment (structural and magnetic) of the dimetallic center, despite the fact that the fluorine atom is far from the metal centers. It has to be emphasized that 1H NMR spectra of the same solutions are non-exploitable.
5.2 Towards model to discriminate mono vs didentate substrate coordination
A new dinucleating phenol-based ligand with two pendant pyridine arms bearing a fluoro group and the corresponding OH-bridged dinuclear complex has been synthesized (Fig. 9). This complex is catalytically active and substrate binding studies are in progress (unpublished results). With this complex, a bridging didentate coordination of substrate would lead to a single fluorine peak on NMR spectrum while a monodentate fixation would be revealed by two non-equivalent fluorine signals.
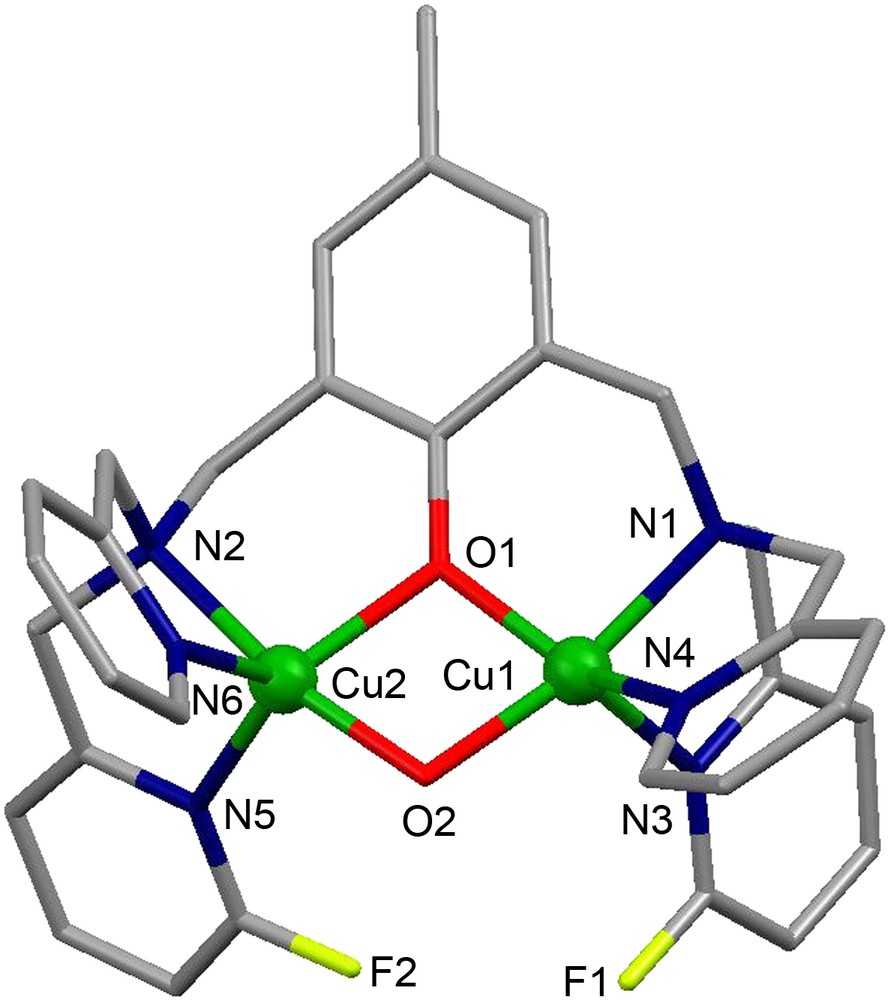
X-ray crystal structure of the dicationic complex .
6 Model to study the role of dioxygen in catechol oxidase activity
In the previously presented model systems, the reduced CuI–CuI states are not stable and consequently do not permit the study of the dioxygen role in catechol oxidation. New insights into the dioxygen fixation on reduced dicopper(I) complex and the reactivity of the (peroxo)-dicopper(II) species towards the substrate were obtained from a monohydroxo-bridged dicopper(II) complex with N-donor macrocyclic ligand, [22]py4pz (Fig. 10) [24,25].
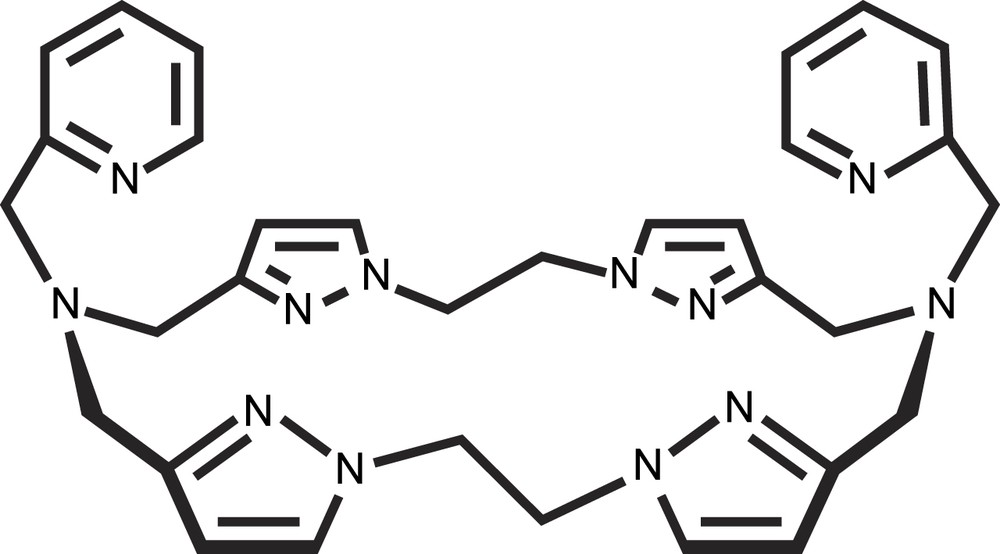
Schematic representation of the ligand [22]py4pz.
The crystal structure of the complex cation [Cu2([22]py4pz)(μ-OH)]3+ is shown in Fig. 11A. The CuII ions have distorted trigonal bipyramidal geometry constituted by N4O atoms. The oxygen atom O23 of the hydroxo group bridges two copper ions, occupying the axial position in their coordination spheres and keeping them at a distance of 3.7587(11) Å.
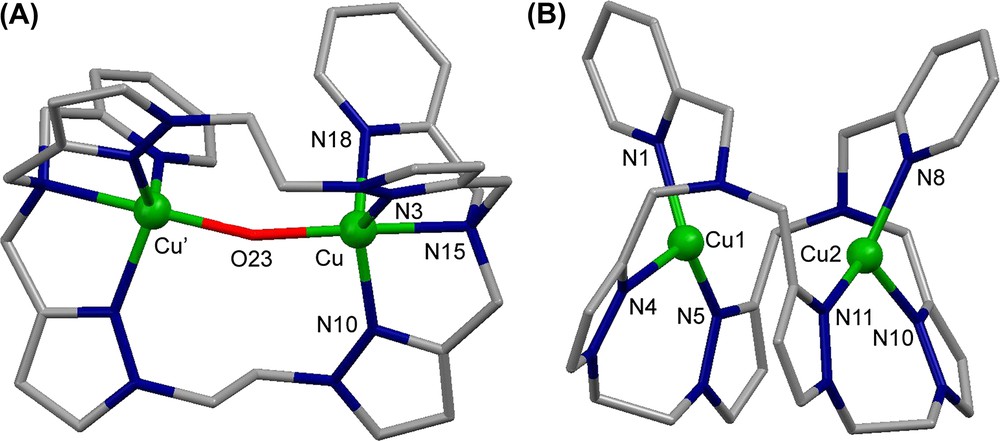
Crystal structure of (A) the complex cation [Cu2([22]py4pz)(μ-OH)]3+ and (B) the complex cation [Cu2([22]py4pz)]2+. Hydrogen atoms are omitted for clarity.
The compound [Cu2([22]py4pz)(μ-OH)]3+ exhibits a very strong antiferromagnetic coupling (2J = −691(35) cm−1) between the copper ions, leading to the EPR-silent complex in both the solid state and in a frozen acetonitrile solution [24]. The UV–vis spectrum of the complex in CH3CN solution shows two absorption bands at 350 nm (ɛ = 7176 M−1 cm−1) corresponding to the LMCT band from the hydroxo bridge to the copper ions, and at 821 nm (ɛ = 336 M−1 cm−1) assigned to the d–d transitions of the CuII ions. The catalytic activity of [Cu2([22]py4pz)(μ-OH)]3+ in the oxidation of 3,5-dtbc was studied in acetonitrile by monitoring the development of the UV–vis band of the produced quinone. Upon treating a 0.25 mM solution of the complex with 100 equivalents of 3,5-dtbc, a turnover number of 36 was determined, and the reaction follows a Michaelis–Menten behavior (Vmax = 1.3 × 10−6 M s−1 and KM = 4.9 mM).
As shown previously by Krebs and co-workers [7], the natural met form of catechol oxidase reacts with 1 equivalent of catechol stoichiometrically in anaerobic conditions, producing 1 equivalent of the corresponding quinone. We have studied the interaction of [Cu2([22]py4pz)(μ-OH)]3+ with 3,5-dtbc in acetonitrile in anaerobic conditions, and 1 equivalent of the corresponding quinone was produced along with the reduced species. This can be evidenced from the disappearance of the LMCT band and the d–d band of the CuII ions, and the appearance of a new band at 400 nm, characteristic of the formed quinone (Fig. 12).
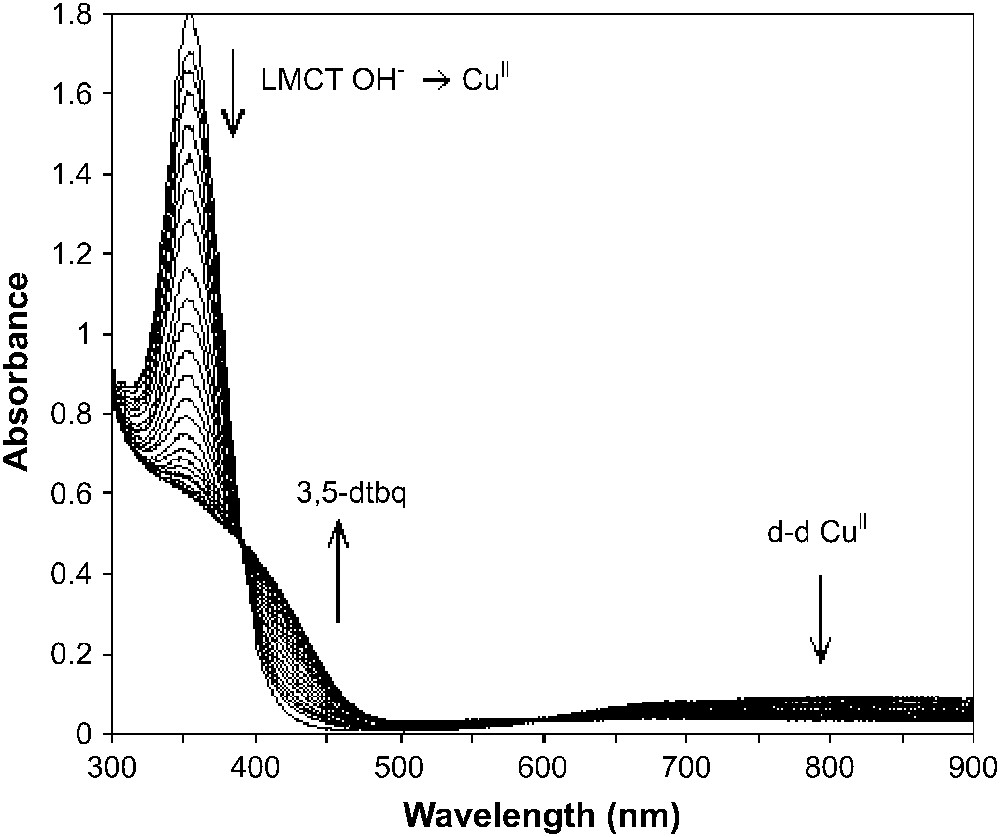
Changes in the UV–vis spectra of [Cu2([22]py4pz)(μ-OH)]3+ (CH3CN, anaerobic conditions) upon addition of 3 equivalents of 3,5-dtbc.
The reduced species was prepared in dry glove box from [Cu(CH3CN)4]ClO4 and the free [22]py4pz ligand. The molecular structure of [Cu2([22]py4pz)]2+ is depicted in Fig. 11B. The complex cation encloses two copper(I) ions at a distance of 3.3922(7) Å, which is shorter than the intermetallic distance in [Cu2([22]py4pz)(μ-OH)]3+. The Cu1 and Cu2 cations are surrounded by the nitrogen atoms of the pyridine rings (N1 and N8, respectively) and the nitrogen atoms of two pyrazole rings (N4, N5 and N10, N11, respectively).
Upon dioxygen addition to a solution of [Cu2([22]py4pz)]2+ in acetonitrile at −40 °C, the UV–vis spectrum displays two absorption bands at 523 nm (ɛ = 4320 M−1 cm−1) and 617 nm (ɛ = 2212 M−1 cm−1), ascribed, respectively, to πσ∗ → d and πσ∗ → d peroxo-to-CuII LMCT transitions. The resonance Raman spectrum of the final product is characterized by the vibration at 840 cm−1, corresponding to an intra-peroxide ν(O–O) stretching. These UV–vis and Raman spectroscopic data are typical for (trans-μ-1,2-peroxo)-dicopper species and suggest that dioxygen is bound to both copper(II) ions in a symmetric end-on fashion [26]. The formed (trans-μ-1,2-peroxo)-dicopper complex ([Cu2([22]py4pz)(O–O)]2+) is stable for ca. 3 h in acetonitrile solution at −40 °C, and the dioxygen binding was found to be irreversible.
Relatively few examples of the interaction of (peroxo)-dicopper(II) ((μ-η2:η2-peroxo)- or (bis-μ-oxo)-dicopper complexes) with catechol substrates are described in the literature [27–30]. Only the deprotonation of tetrachlorocatechol upon reaction with (trans-μ-1,2-peroxo)-dicopper species was evidenced by Comba and co-workers [31].
We have studied spectrophotometrically the anaerobic interaction of our (trans-μ-1,2-peroxo)-dicopper complex (in situ) with 3,5-dtbc and deuterated 3,5-dtbc at −40 °C in acetonitrile solution. The changes in the UV–vis spectrum upon addition of 1 equivalent of catechol to a solution of [Cu2([22]py4pz)(O–O)]2+ complex are shown in Fig. 13. The oxidation process clearly proceeds in two steps: the characteristic absorbance of the (peroxo)-dicopper(II) species at 523 nm gradually disappears along with the appearance of the new peak at 342 nm. The isosbestic point at 440 nm indicates the presence of two species. Then, a band around 400 nm develops indicating the formation of 1 equivalent of quinone (dotted line, Fig. 13).

Changes in the UV–vis spectrum upon addition of 1 equivalent of deuterated 3,5-dtbc to a solution of [Cu2([22]py4pz)(O–O)]2+ complex in anaerobic conditions (in situ, CH3CN, −40 °C). Solid lines: the spectra recorded each 0.2 min after deuterated 3,5-dtbc addition. The dotted lines correspond to the gradual formation of 1 equivalent of 3,5-dtbq.
A high kinetic isotope effect (KIE) was observed for the first step ( for deuterated 3,5-dtbc, whereas for non-deuterated 3,5-dtbc the reaction is too fast to allow the determination of the reaction rate constant). For the second process, the reaction rate constant could be determined for both non-deuterated and deuterated substrate. Thus, in the second step of the reaction, no KIE is observed, whereas a proton is obviously transferred in the first step. The bubbling of dioxygen through the solution after completion of the reaction showed that the (trans-μ-1,2)-peroxo core could not be restored. This suggests that the copper(II) ions are not reduced upon interaction with the substrate; thus, the oxidation of the substrate is likely to be performed by the peroxo moiety. Upon addition of the excess of 3,5-dtbc in dioxygen presence to (trans-μ-1,2)-peroxo-dicopper complex, a catalytic oxidation of catechol takes place.
On the basis of the results described above, we propose the mechanism for the catechol oxidation by [Cu2([22]py4pz)(μ-OH)]3+, as depicted in Fig. 14 [25].

Proposed mechanism for the oxidation of 3,5-dtbc by [Cu2([22]py4pz)(μ-OH)]3+.
On the first stage of the reaction, a stoichiometric oxidation of catechol by [Cu2([22]py4pz)(μ-OH)]3+ takes place. This step does not require the presence of dioxygen. At the second stage, the resulted dicopper(I) complex [Cu2([22]py4pz)]2+ reacts with dioxygen to form the (trans-μ-1,2-peroxo)-dicopper(II) species. This intermediate oxidizes a second equivalent of catechol through a two-electron transfer from the catechol to the peroxide moiety. The reaction proceeds in two steps: the proton transfer from the substrate to the nucleophilic peroxo core resulting in the formation of a ternary complex of a formal composition [Cu2([22]py4pz)(OOH)–substrate], and the oxidation of the bound catecholate. After the quinone molecule is released, the complex [Cu2([22]py4pz)(μ-OH)]3+ is regenerated, and the catalytic cycle can continue. Quinone (2 equivalents) is thus generated per catalytic cycle. These results show that trans-μ-1,2-peroxo-dicopper complex is able to oxidize catechols as reported for (μ-η2:η2)-peroxo- or (bis-μ-oxo) dicopper species [28–30].
7 Conclusion
Some years ago, Holm and Solomon wrote in a preface [32] concerning bioinorganic enzymology: “research can now focus more intensely on learning how function relates to structure.” We think that the biomimetic approach can contribute to this goal. In this paper we have described some chemical tools for mechanistic studies related to catechol oxidase activity. These tools have led to new insights concerning structure–activity relationships for such and such step of the catalytic cycle. Iterative feedback from enzymes and model studies are undoubtedly a fruitful strategy for a better understanding, at a molecular level, of the mechanistic pathway involved in metalloenzyme activities.
Acknowledgements
The authors are grateful to Prof. J. Reedijk, Dr. I. Koval and Dr. P. Gamez for their extensive contribution and insights in Section 6 of the present work. It was supported by French Ministry of Research and Foreign Affaires (Egide) and NOW (Van Gogh program).