

1 Introduction
Le recours aux ressources agricoles et forestières pour la synthèse de bio-polymères présente potentiellement d'importants avantages environnementaux. En effet, à l'inverse de leurs homologues fossiles (matières premières issues de sources pétrolières), les matières et ressources renouvelables issues de la biomasse (agriculture ou agro-ressources) sont susceptibles de réduire la consommation d'énergie et les émissions de gaz à effet de serre, diminuer les impacts sur l'air, l'eau ou le sol [1]. Parmi ces ressources naturelles, on trouve le 1,4:3,6-dianhydro-d-sorbitol (ou isosorbide 1). C'est un diol chiral, avec deux groupements hydroxyles non équivalents, l'un en position exo (C-2) et l'autre en position endo (C-5). L'isosorbide, produit en quantités industrielles, est obtenu par une double déshydratation du d-glucitol (Fig. 1), provenant lui-même de l'hydrogénation du d-glucose, extrait de l'amidon de maïs [2–5].

Obtention et configuration stéréochimique de l'isosorbide.
Dans le cadre de la valorisation et de la meilleure utilisation non alimentaire des sous-produits agricoles, nous nous sommes intéressés à la polycondensation de l'isosorbide pour synthétiser de nouveaux polyéthers (PE 4–8) aliphatiques. Nous avons réalisé ces réactions de polycondensation dans des conditions de catalyse par transfert de phase (CTP) sous irradiation microondes (MO) et par chauffage classique (CC) (Fig. 2). Notre objectif est d'effectuer une comparaison entre les rendements des réactions (réactivité), d'une part, et entre les structures des polyéthers obtenus (sélectivité), d'autre part. Nous étudierons aussi l'effet de la longueur de la chaîne alkyle, l'effet du groupe partant ainsi que celui du temps de réaction.
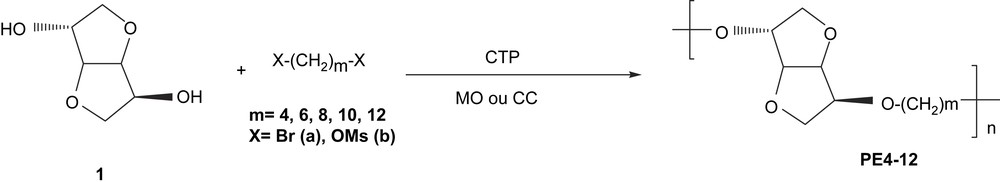
Schéma général de synthèse de polyéthers à base d'isosorbide.
Les différents polyéthers obtenus sont caractérisés par plusieurs techniques physicochimiques complémentaires: spectroscopie IR–TF (infrarouge à transformée de Fourier) et RMN, spectrométrie de masse MALDI–TOF (désorption/ionisation laser assistée par matrice couplée à la spectrométrie de masse à temps de vol), chromatographie d'exclusion stérique (SEC), analyse calorimétrique différentielle (DSC).
2 Résultats et discussions
La catalyse par transfert de phase est une méthode d'activation anionique mettant à profit des échanges d'ions avec des sels d'ammonium quaternaires ou la chélation sélective des cations. Plusieurs exemples de réactions menées dans des conditions de CTP solide–liquide démontrent le succès de cette méthode, en particulier quand elle est couplée avec les microondes. La complémentarité de ces deux méthodes non classiques d'activation s'avère remarquable en ce qui concerne la réactivité, avec de très nettes améliorations en temps et en rendements par rapport aux méthodes classiques [6–8]. Le travail que nous présentons ici s'inscrit dans cette perspective. Il s'agit de synthétiser des polyéthers (PEi) par réaction de polycondensation de l'isosorbide avec des agents dialkylants dibromés (2a–6a) ou dimésylés (2b–6b) en tirant profit des différents avantages que présente la catalyse par transfert de phase. Pendant tout ce travail, nous adopterons cette dénomination pour chaque agent dialkylant et polyéther obtenu (Tableau 1). Ces réactions ont été effectuées sous irradiation microondes dans un réacteur monomodes. Il s'agit d'équipement spécialement dédiés à la synthèse organique, avec un contrôle de tous les paramètres usuels (température, puissance électromagnétique). Les ondes électromagnétiques sont focalisées grâce à un guide d'ondes placé à la sortie du magnétron. Les dimensions du guide d'ondes et l'excitation sont calculées pour ne permettre qu'un seul mode de propagation ou de résonance. Ils permettent d'obtenir une distribution homogène du champ électrique dans le guide d'ondes (faisceau focalisé) et donc dans les produits irradiés. Par conséquent, la puissance énergétique est notablement améliorée. Le réacteur monomode que nous avons utilisé est le Synthewave 402 Prolabo. Ce réacteur offre la possibilité d'opérer à des puissances allant de 15 à 300 W.
Dénomination des différents monomères et polyéthers
| m | Br–(CH2)m–Br | MsO–[CH2]n–OMs | PEi |
| 4 | 2a | 2b | 4 |
| 6 | 3a | 3b | 6 |
| 8 | 4a | 4b | 8 |
| 10 | 5a | 5b | 10 |
| 12 | 6a | 6b | 12 |
En vue de mettre en évidence des effets spécifiques du rayonnement, ces réactions seront aussi réalisées par chauffage classique dans un bain d'huile thermostaté, dans les mêmes conditions de temps et de température. Les polyéthers issus de la réaction de polycondensation effectuée sous irradiation microondes ou par chauffage classique sont isolés en deux temps. À la fin de la réaction, le mélange réactionnel est dissout dans le minimum de chloroforme. Cette solution est versée goutte à goutte, sous forte agitation magnétique, dans 100 ml de méthanol, provoquant ainsi la précipitation d'une fraction de polyéthers insolubles (FPMeOH) : ce sont les polymères les plus lourds (et les plus homogènes: faible indice de polydispersité). Les polymères les plus légers (oligomères) demeurent solubles dans la solution méthanolique. Celle-ci est, par la suite, concentrée et reprise dans 100 ml d'hexane. Nous obtenons ainsi, dans un second temps, la précipitation des polyéthers insolubles dans l'hexane (FPHex) [9].
2.1 Étude d'une réaction modèle
La réaction modèle que nous avons choisi d'étudier est la polycondensation de l'isosorbide (1) et du 1,4-dibromobutane (2a), représentée sur la Fig. 3.
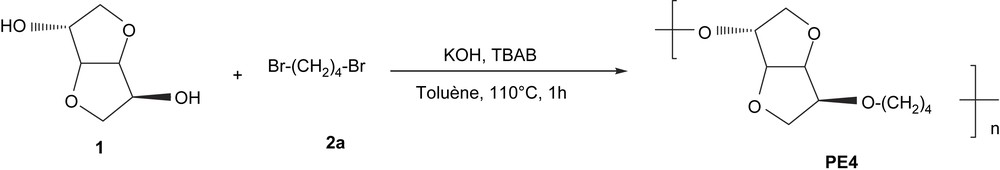
Réaction modèle entre l'isosorbide 1 et le 1,4-dibromobutane 2a à 110 °C pendant 1 h.
En se basant sur des travaux antérieurs dans notre laboratoire [10], nous avons constitué un mélange réactionnel de départ comportant un léger excès de KOH (1,25 équivalent par fonction OH) et une quantité catalytique (10%) de l'agent de transfert de phase (bromure de tétrabutylammonium : TBAB). Nous avons constaté que l'ajout d'une faible quantité de toluène (3 ml pour 5 mmol) permet à la fois un meilleur contrôle de la température et une diminution de la viscosité du milieu [11,12]. Une optimisation, à une température de 110 °C, de deux facteurs intervenant lors de la réaction a été réalisée, afin d'obtenir de bons rendements et des masses molaires élevées [13–15] : ceci est l'effet de l'excès du 1,4-dibromobutane et du temps de réaction.
2.1.1 Effet de l'excès du 1,4-dibromobutane (2a)
L'idée d'ajouter un excès du 1,4-dibromobutane (2a) est intervenue après l'analyse de la composition du mélange réactionnel brut par chromatographie en phase gazeuse (CPG). Au cours de la formation du polyéther (PE4), des réactions secondaires rentrent en compétition avec la réaction souhaitée de substitution (SN2). Il s'agit d'abord de la réaction d'élimination E2, qui donne lieu à des insaturations et, d'autre part, la substitution du brome situé sur la chaîne alkyle par l'ion OH− présent, qui donne une nouvelle fonction éther. La réaction de polycondensation, sous MO ou par CC dans les mêmes conditions, de l'isosorbide et du 1,4-dibromobutane a été menée avec un léger excès du monomère 2a allant de 1 à 5%. Les résultats obtenus ont été traduits graphiquement sur la Fig. 4 (la démarche adaptée pour le calcul des rendements est donnée dans la partie expérimentale).
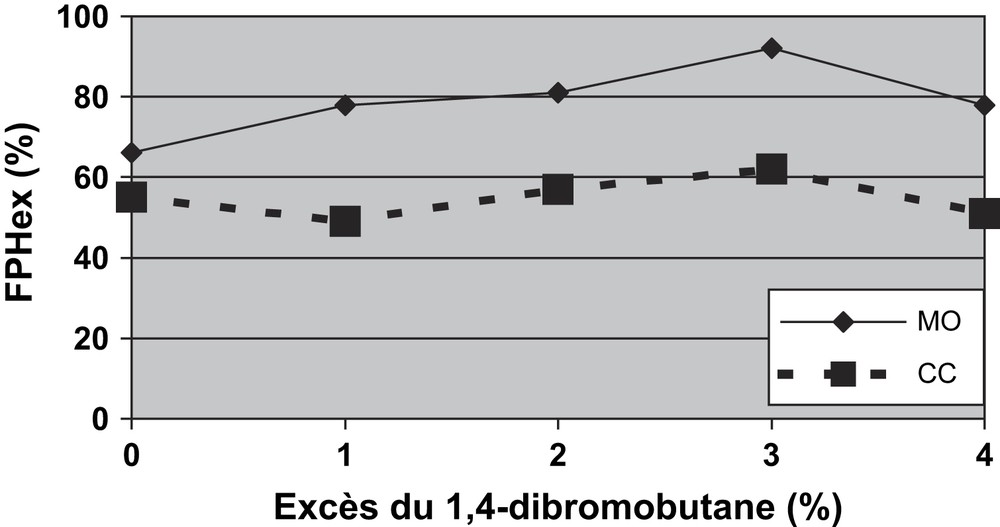
Variation du pourcentage FPHex en fonction de l'excès du monomère 2a.
L'analyse de ce graphique fait apparaître deux résultats :
- (1) quel que soit l'excès utilisé lors de la réaction, nous obtenons des rendements plus grands sous MO par rapport au CC, ce qui met ainsi en évidence des effets spécifiques des microondes;
- (2) pour obtenir un rendement maximal (sous MO: 92% ou CC: 62%), il faut utiliser un excès de 3% du monomère 2a, puisque l'utilisation d'un grand excès (de 5 à 10%) conduit à un abaissement du rendement, indépendamment du mode d'activation.
En conclusion, dans le cas d'un excès en monomère 2a, nous avons dans le mélange réactionnel des quantités non stœchiométriques des groupements fonctionnels (bromure et hydroxyles) et par conséquent la réaction de polycondensation ne peut aller jusqu'à son maximum de conversion. Pour toutes ces raisons, nous avons choisi, pour la suite de ce travail, d'opérer avec un excès de 3% de l'agent dialkylant.
2.1.2 Effets du temps de réaction
Afin d'optimiser davantage les conditions de synthèse de ces polyéthers, nous avons étudié l'effet du temps de la réaction sur les rendements en utilisant toujours un excès de 3% en monomère 2a (Tableau 2). Quel que soit le temps de réaction, nous n'avons pas obtenu de fraction de polyéthers insoluble dans le méthanol (FPMeOH) avec les deux modes de chauffage. En revanche, en ce qui concerne la fraction insoluble dans l'hexane (FPHex), nous constatons clairement que, sous MO et à partir d'une heure de temps de réaction, les rendements sont à leurs maxima. Enfin, nous avons aussi constaté que les rendements obtenus par CC restent, dans tous les cas, plus faibles que ceux obtenus sous MO et qu'il faut attendre 3 h de réaction pour obtenir seulement 66% de rendement.
Effets du temps de la réaction sur les rendements en polyéthers
| Exp. n° | Temps (min) | FPMeOH (%) | FPHex (%) | Rdt total (%) | |||
| MO | Δ | MO | Δ | MO | Δ | ||
| 1 | 30 | 0 | 0 | 70 | 56 | 70 | 56 |
| 2 | 60 | 0 | 0 | 92 | 62 | 92 | 62 |
| 3 | 120 | 0 | 0 | 83 | 66 | 83 | 66 |
| 4 | 180 | 0 | 0 | 85 | 57 | 85 | 57 |
2.1.3 Analyses des polymères
Deux analyses complémentaires ont été effectuées pour caractériser les polymères.
- (1) D'abord, nous avons déterminé les masses moléculaires moyennes en nombre et en poids des polyéthers issus de la polycondensation des monomères 1 et 2a, effectuée sous MO pendant 1, 2 et 3 h (Tableau 3). Les mesures des masses moléculaires ont été effectuées par SEC en utilisant un étalonnage par spectrométrie de masse MALDI–TOF [11–12].
Masses moléculaires moyennes en nombres et en poids des poly(isosorbide-éther)s (PE4) obtenus sous MO
| 1 h | 2 h | 3 h | |
| Mn (g mol−1) | 1250 | 1450 | 1440 |
| Mp (g mol−1) | 1400 | 1650 | 1670 |
| Ip | 1,12 | 1,14 | 1,16 |
Nous avons remarqué que le temps de réaction n'a pas d'effet sur les rendements. En revanche, lorsqu'il a été doublé, nous avons obtenu des polyéthers avec des masses molaires plus élevées (1 h sous MO, Mn = 1250 g mol−1; 2 h sous MO, Mn = 1450 g mol−1). Ceci s'explique par le fait que l'on a laissé suffisamment de temps aux sites actifs pour réagir entre eux. À l'inverse, un temps de réaction de 3 h n'a d'effets, ni sur les rendements, ni sur les masses molaires moyennes.
- (2) Par la suite, nous avons déterminé les structures des polyéthers par spectrométrie de masse (M + Na)+ MALDI–TOF [16–19]. Les Figs. 5 et 6 représentent les spectres de masse MALDI–TOF de la fraction FPHex obtenue lors de la réaction de polycondensation sous MO des deux monomères 1 et 2a, pendant 1 et 2 h, respectivement.

Spectre de masse (M + Na)+ (Da) MALDI–TOF (750–960 Da) du polyéther obtenue lors de la réaction de 1 avec 2a sous MO (Rdt = 92%) pendant 1 h.
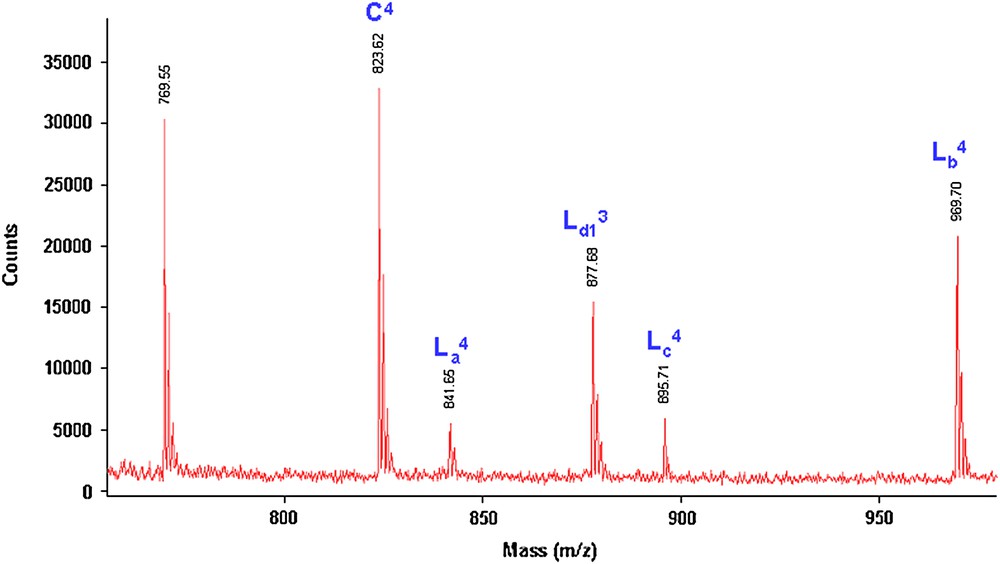
Spectre de masse (M + Na)+ (Da) MALDI–TOF (750–960 Da) du polyéther obtenu lors de la réaction de 1 avec 2a sous MO (Rdt = 92%) pendant 2 h.
Les deux spectres de masse (1 ou 2 h sous MO) montrent les mêmes séries de pics majoritaires, correspondant aux structures C, La, Lb, Lc et Ld1 (M + Na = M + 23)+.
Ces cinq structures, décalées de 200 Da (un maillon de l'unité répétitive de 200 Da), se différencient par leurs bouts de chaînes (Fig. 7).
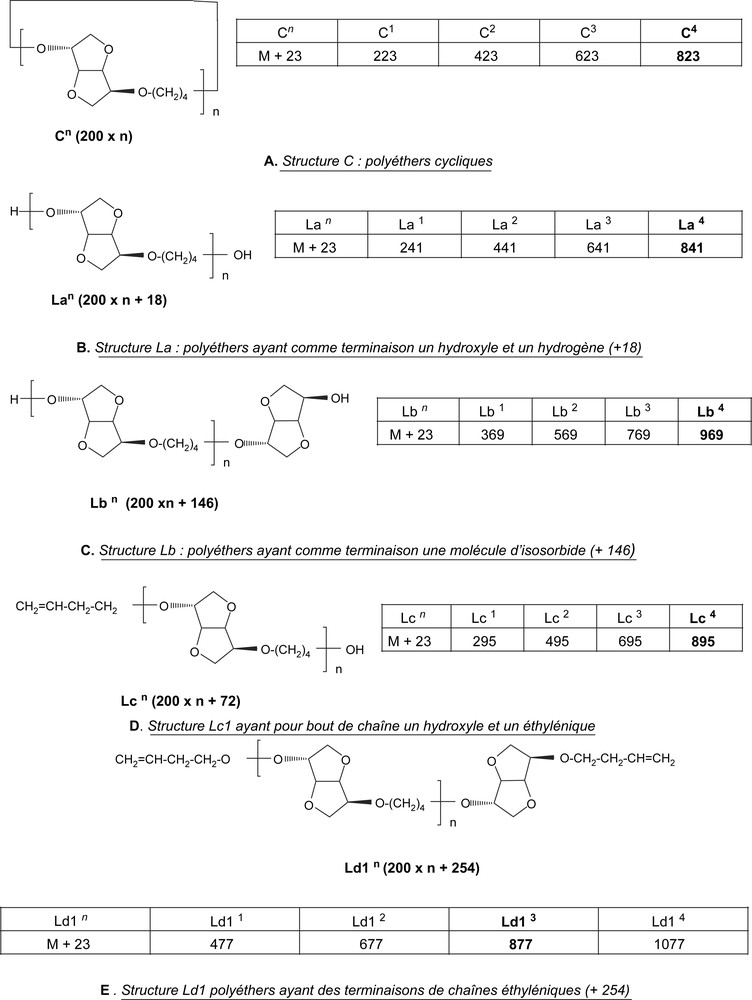
Différentes structures (A, B, C, D et E) obtenues pour les polyéthers PE4 déduites des spectres de masse MALDI–TOF de la Fig. 5 ou de la Fig. 6.
L'étude par spectrométrie de masse MALDI–TOF nous a permis de déterminer les structures exactes des différentes espèces présentes dans le mélange réactionnel (unités répétitives et bout de chaînes) et surtout la présence des structures cycliques C. Cette technique est la seule méthode qui permette de mettre en évidence un polymère cyclique.
Nous avons également constaté une abondance plus importante des polyéthers cycliques lorsque la réaction est effectuée pendant 2 h sous MO. De plus, le spectre de masse du polyéther obtenu après 2 h est plus propre, tous les pics présents dans un motif répétitif ayant pu être attribués (cas du n = 4 par exemple).
En conclusion, après l'étude de cette réaction modèle (polycondensation de l'isosorbide et du 1,4-dibromobutane) et l'analyse du polyéther correspondant par différentes techniques complémentaires, les conditions opératoires que nous allons adopter dans ce travail sont les suivantes: un excès de 3% en agent dialkylant (dibromé ou dimésylé) et un temps de réaction de 2 h.
2.1.4 Effets de la longueur de la chaîne alkyle
Nous avons étudié par la suite l'effet de la longueur de la chaîne alkyle en utilisant un groupe partant dibromé (2a–6a). Dans le Tableau 4 sont rassemblés les pourcentages en polyéthers FPMeOH et FPHex, ainsi que les rendements totaux (la somme des deux fractions) obtenus sous MO ou par CC dans les mêmes conditions.
Polycondensation de l'isosorbide et divers agents dibromés sous MO ou par CC pendant 2 h à 110 °C
| Exp. n° | Br–(CH2)m–Br | FPMeOH (%) | FPHex (%) | Rdt total (%) | |||
| MO | Δ | MO | Δ | MO | Δ | ||
| 1 | 2a | 0 | 0 | 83 | 66 | 83 | 66 |
| 2 | 3a | 0 | 0 | 83 | 53 | 83 | 53 |
| 3 | 4a | 37 | 16 | 59 | 79 | 96 | 95 |
| 4 | 5a | 56 | 34 | 40 | 58 | 96 | 92 |
| 5 | 6a | 74 | 50 | 15 | 31 | 89 | 81 |
Il ressort de ces résultats que le pourcentage en fraction FPMeOH (fraction des polyéthers les plus lourds et les plus homogènes) augmente avec la longueur de la chaîne. Ceci était prévisible, du fait que la masse du motif répétitif augmente et que, par conséquent, les masses molaires moyennes des polyéthers correspondants vont dans le même sens. Dans ce même cas, nous avons mis en évidence des effets spécifiques des microondes. Nous avons obtenu des pourcentages en fraction FPMeOH plus élevés sous MO que par CC.
À titre d'exemple, dans le cas de la polycondensation des monomères 1 et 6a, la fraction FPMeOH en polyéthers (PE12) obtenue sous MO est de 74%, alors que celle obtenue par CC est seulement de 50%.
La détermination des masses molaires moyennes en nombre (Mn) et en poids (Mp) de ces deux fractions a montré que, non seulement les fractions en FPMeOH sont plus grandes, mais qu'aussi les masses molaires sont plus élevées sous MO (Tableau 5).
Masses moléculaires moyennes en nombres et en poids des fractions FPMeOH obtenues sous MO ou par CC (cas du composé 6a)
| MO | CC | |
| Mn (g mol−1) | 1960 | 1460 |
| Mp (g mol−1) | 2540 | 1820 |
| Ip | 1,29 | 1,25 |
La spectrométrie de masse MALDI–TOF nous a permis de déterminer et de comparer les structures des polyéthers obtenus sous MO ou par CC. Les Figs. 8 et 9 représentent les spectres de masse des fractions FPMeOH obtenues sous MO et par CC, respectivement. Ces deux fractions ont été obtenues par polycondensation des deux monomères 1 et 6a pendant 2 h à 110 °C.
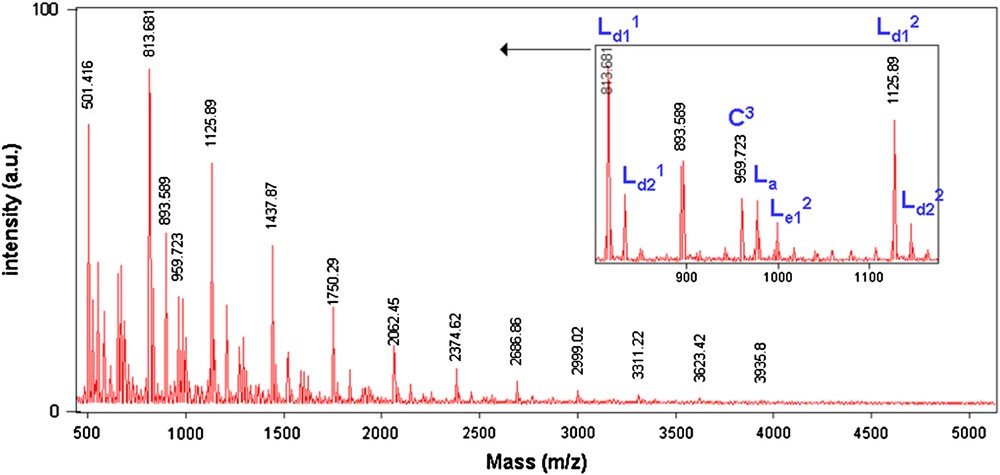
Spectre de masse (M + Na)+ (Da) MALDI–TOF de la fraction FPMeOH du polyéther (PE12) obtenu lors de la réaction de 1 avec 6a sous MO (74%).
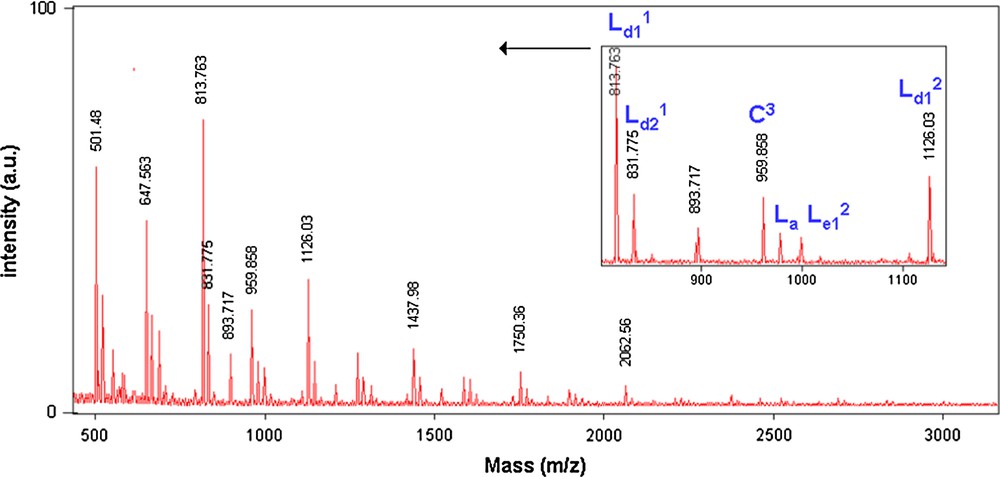
Spectre de masse (M + Na)+ (Da) MALDI–TOF de la fraction FPMeOH du polyéther (PE12) obtenu lors de la réaction de 1 avec 6a par CC (50%).
Après analyse des deux spectres de masse des fractions FPMeOH obtenus sous MO et par CC, nous avons constaté une répartition plus large des pics sous MO, c'est-à-dire que, par activation sous MO, nous obtenons des polyéthers (PE12) de masses molaires plus élevées que par CC. Ce résultat confirme la différence entre les masses molaires obtenues sous MO (Mn = 1960 g mol−1) et par CC (Mn = 1460 g mol−1). En revanche, sous les deux modes d'activation, nous avons obtenu les mêmes structures (C, La, Ld1 et Le1) (M + Na = M + 23)+ (Fig. 10).
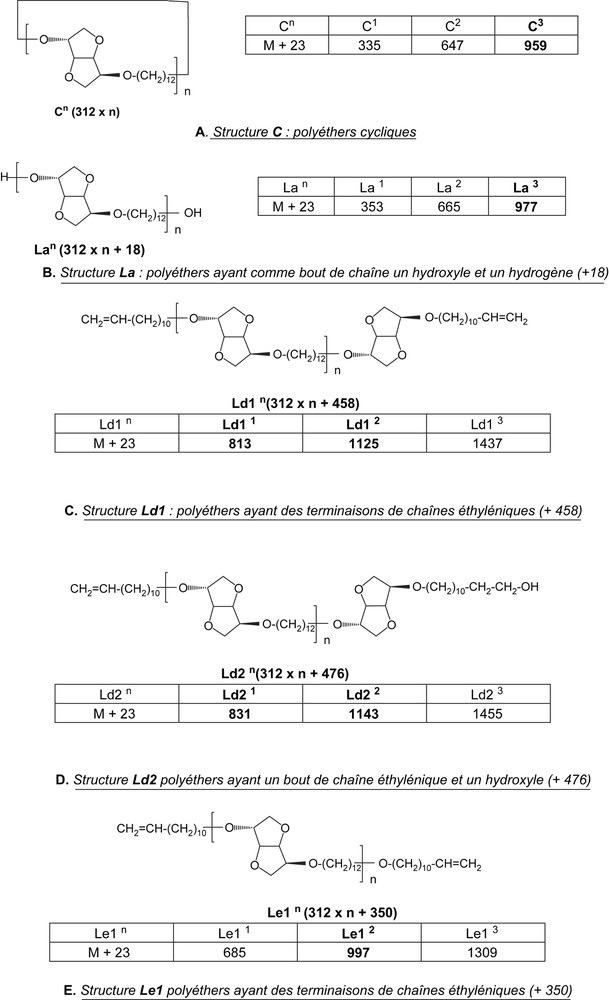
Structures (A, B, C, D et E) des polyéthers PE12.
Les structures C, La, Ld1 et Ld2 résultent d'une croissance normale de la chaîne macromoléculaire par réaction de polycondensation entre l'alcoolate à base d'isosorbide et le 1,12-dibromododécane. En revanche, la structure Le1 est obtenue par un chemin différent, dans lequel il y a eu auparavant une réaction d'hydrolyse des groupements bromure en bout de chaînes, suivie d'une réaction de β-élimination selon le schéma de la Fig. 11.

Schéma des réactions secondaires.
2.1.5 Effets du groupe partant
Après avoir étudié et optimisé la réaction de polycondensation de l'isosorbide avec divers agents dialkylants aliphatiques dibromés (produits commerciaux), nous avons examiné, dans cette partie, l'effet de l'utilisation d'un groupe partant mésylé sur la fraction en FPMeOH, les masses molaires moyennes et les structures des polyéthers.
Nous avons montré, lors de travaux récents dans notre laboratoire [20,21], que la formation de la fonction éther à base d'isosorbide a lieu plus facilement avec un groupe partant mésyle. Les résultats qui ont été obtenus lors de la polycondensation des monomères 1 et 2b–6b sous MO ou par CC pendant 2 h à 110 °C sont rassemblés dans le Tableau 6.
Polycondensation de l'isosorbide et divers agents dimésylés sous MO ou par CC pendant 2 h à 110 °C
| Exp. n° | MsO–[CH2]n–OMs | FPMeOH (%) | FPHex (%) | Rdt total (%) | |||
| MO | Δ | MO | Δ | MO | Δ | ||
| 1 | 2b | 17 | 0 | 61 | 81 | 78 | 81 |
| 2 | 3b | 21 | 0 | 51 | 75 | 76 | 75 |
| 3 | 4b | 40 | 29 | 52 | 53 | 92 | 82 |
| 4 | 5b | 77 | 55 | 20 | 38 | 97 | 93 |
| 5 | 6b | 88 | 67 | 8 | 23 | 93 | 91 |
D'après ces résultats, l'utilisation d'un groupe partant mésylé présente plusieurs avantages, qui sont exposés sur la Fig. 12 :
- (1) nous sommes parvenus à augmenter la fraction FPMeOH obtenue sous MO ou par CC;
- (2) nous avons pu obtenir une quantité de l'ordre de 20% de fraction FPMeOH, même avec les courtes chaînes aliphatiques, en utilisant les MO;
- (3) des effets spécifiques (non purement thermiques) des microondes ont été mis en évidence en comparant les fractions FPMeOH obtenues sous MO et CC.
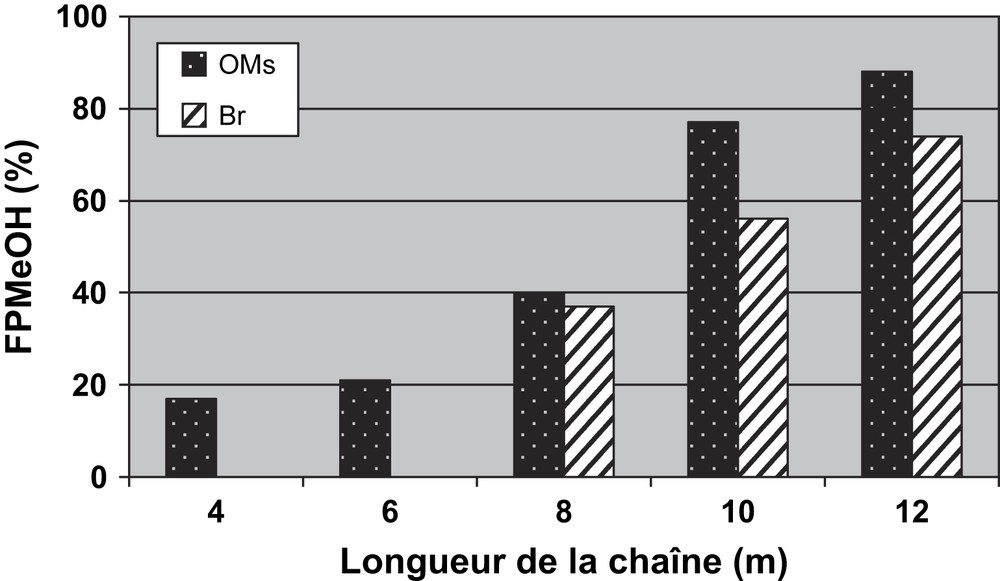
Effets du groupe partant sur la fraction FPMeOH obtenue sous MO.
À titre de comparaison, nous avons déterminé les masses molaires moyennes en nombre et en poids du polyéther (PE12) issu de la polycondensation entre les monomères 1 et 6b réalisée sous MO ou par CC (Tableau 7).
Masses moléculaires moyennes en nombres et en poids des fractions FPMeOH obtenues sous MO ou par CC (cas du composé 6b)
| MO | CC | |
| Mn (g mol−1) | 1950 | 1590 |
| Mp (g mol−1) | 2530 | 1970 |
| Ip | 1,29 | 1,24 |
La comparaison des résultats concernant les fractions en FPMeOH obtenues pour les deux groupes partants par activation MO avec ceux obtenus par CC nous permet aisément de mettre en évidence les effets spécifiques des MO.
Nous avons étudié, par spectrométrie de masse MALDI–TOF, les structures des fractions FPMeOH obtenues sous MO ou par CC des différents polyéthers (PE4–12). Sur la Fig. 13, nous donnons le spectre de masse (750–1100 Da) de la fraction FPMeOH (17%) du polyéther PE4 obtenue sous MO.
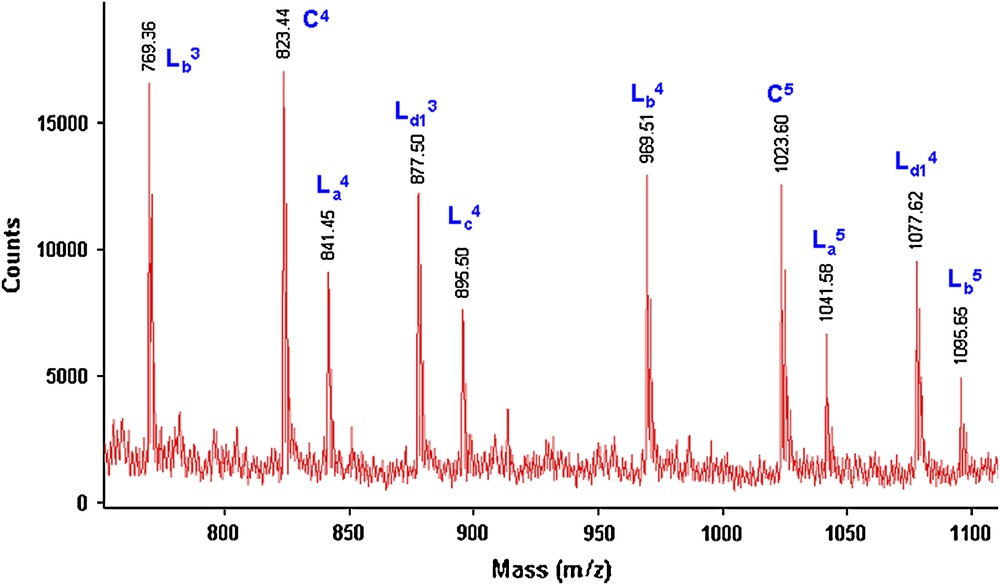
Spectre de masse (M + Na)+ (Da) MALDI–TOF (750–1100 Da) de la fraction FPMeOH (17%) du polyéther PE4 obtenu lors de la réaction de 1 avec 2b sous MO.
Le spectre de masse MALDI–TOF de la fraction FPMeOH (cas du composé 2b) obtenu pour le groupe partant OMs laisse apparaître quatre structures (C, La, Lb, Lc et Ld1), qui sont identiques à celles obtenues avec le groupe partant Br. Ce spectre de masse traduit une abondance plus importante des polyéthers vers les hauts poids moléculaires. Ceci est évident, car cette fraction (avec OMs) est insoluble dans le méthanol (polyéthers plus homogènes et plus lourds), alors que la fraction (avec Br) est insoluble dans l'hexane (polyéthers en phase de croissance, donc des oligomères).
Enfin, nous avons constaté, après analyse des différents spectres de masse MALDI–TOF des polyéthers (PE4–12), qu'il y a une forte présence de polyéthers cycliques en utilisant des courtes chaînes aliphatiques (cas des composés 2a–b et 3a–b), alors que l'utilisation des longues chaînes aliphatiques (cas des composés 4a–b, 5a–b et 6a–b) donne lieu à des polyéthers avec une forte abondance en chaînes linéaires. Ceci pourrait s'expliquer par le fait que les réactions réalisées en l'absence de solvants défavorisent le libre mouvement des longues chaînes alkyles et empêchent la réaction entre les bouts de chaînes réactifs d'une même molécule et, par conséquent, le phénomène de cyclisation. En revanche, les oligomères demeurent aptes à la cyclisation, du fait de leur courte chaîne.
2.1.6 Effets du temps de la réaction par CC
Ces réactions de polycondensation ont été effectuées entre l'isosorbide et les agents dialkylants dimésylés (+3% d'excès) à 110 °C pendant 2 h, 24 h ou 7 j. Cette étude a pour objectif de suivre l'évolution de la fraction FPMeOH, des masses molaires moyennes et des structures des polyéthers au cours du temps par CC. Dans un premier temps, nous avons commencé par déterminer la fraction FPMeOH pour chaque agent dimésylé (Tableau 8).
Pourcentage de la fraction FPMeOH obtenu par CC pendant 2 h, 24 h ou 7 j
| Exp. n° | MsO–[CH2]n–OMs | FPMeOH (%) | ||
| 2 h | 24 h | 7 j | ||
| 1 | 2b | 0 | 49 | 43 |
| 2 | 3b | 0 | 42 | 42 |
| 3 | 4b | 29 | 47 | 45 |
| 4 | 5b | 55 | 57 | 63 |
| 5 | 6b | 67 | 71 | 83 |
Au bout de 2 h de réaction sous CC, nous n'avons pas obtenu de fractions FPMeOH pour les courtes chaînes (2b et 3b). En revanche, nous avons pu obtenir des fractions FPMeOH acceptables (allant de 29% à 67%) pour les longues chaînes (4b, 5b et 6b). L'influence du temps de la réaction par CC est importante: après 24 h, nous avons obtenu des fractions FPMeOH allant de 42% à 49% pour les courtes chaînes. Pour les longues chaînes, nous n'avons pas eu de modifications significatives au niveau des fractions FPMeOH. Nous avons aussi constaté qu'un temps de réaction aussi long que 7 j n'engendrait pas d'effet remarquable sur les fractions FPMeOH, et ceci pour toutes les chaînes alkyle dimésylées.
Au cours de cette étude, nous avons mis en évidence l'intérêt de travailler sous irradiation MO. En effet, il est intéressant de signaler que la fraction FPMeOH obtenue après 2 h de réaction sous MO est très voisine de ce qui résulte, en termes de rendement de FPMeOH, d'une réaction menée pendant 24 h sous CC (Fig. 14).
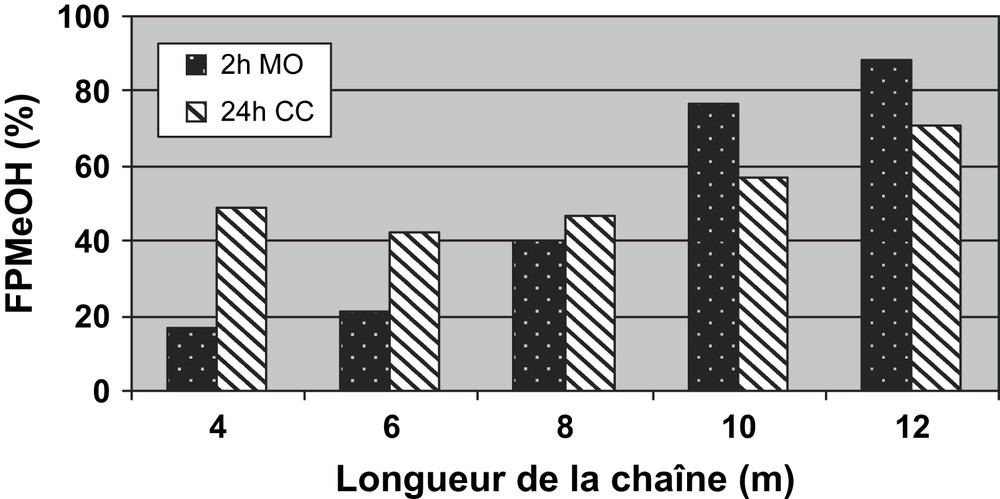
Comparaison des fractions FPMeOH obtenues sous MO (2 h) et CC (24 h).
Après avoir suivi l'évolution des fractions FPMeOH obtenues par CC (2 h, 24 h et 7 j), nous avons déterminé les masses molaires moyennes de chaque fraction obtenue, soit sous MO (2 h) soit par CC (24 h) à titre de comparaison, et ceci pour chaque chaîne alkyle (Tableau 9). Nous constatons une augmentation croissante des masses molaires moyennes en allant vers les chaînes les plus longues sous les deux modes de chauffage. Les masses molaires les plus élevées sont obtenues pour les fractions en FPMeOH les plus grandes.
Comparaison des masses molaires moyennes en nombre des fractions FPMeOH obtenues sous MO (2 h) et par CC (24 h)
| Exp. n° | MsO–[CH2]n–OMs | Mn (g mol−1) | Ip = Mn/Mp | ||
| MO (2 h) | CC (24 h) | MO (2 h) | CC (24 h) | ||
| 1 | 2b | 1350 | 1410 | 1,14 | 1,17 |
| 2 | 3b | 1520 | 1670 | 1,12 | 1,20 |
| 3 | 4b | 1680 | 1770 | 1,10 | 1,23 |
| 4 | 5b | 1830 | 1920 | 1,13 | 1,19 |
| 5 | 6b | 1980 | 2100 | 1,14 | 1,27 |
À titre de comparaison, les Figs. 15 et 16 donnent les deux spectres de masse MALDI–TOF relatifs aux fractions FPMeOH obtenues, respectivement, sous MO (2 h) et par CC (24 h) par polycondensation des deux monomères 1 et 3b.
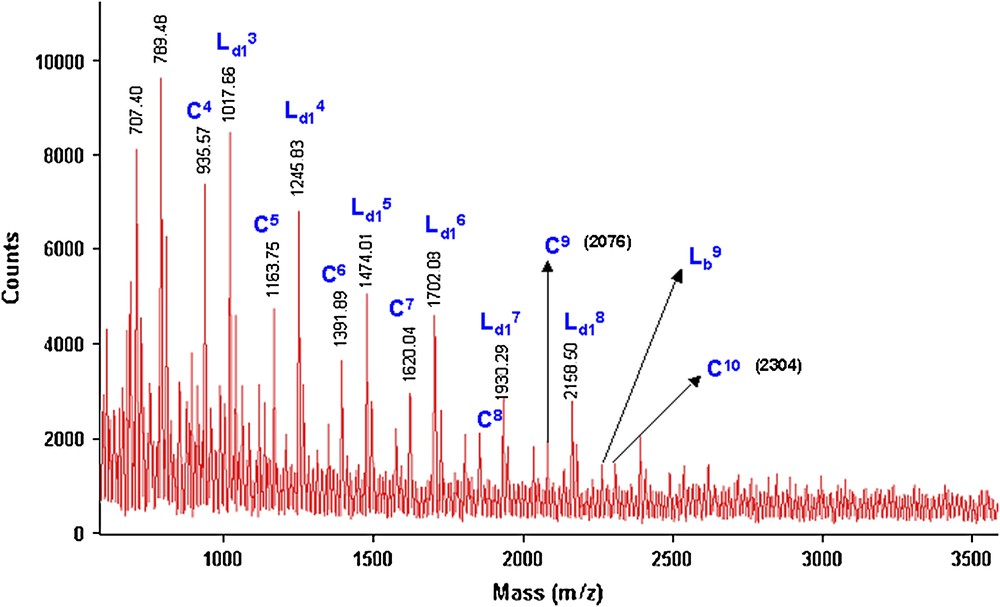
Spectre de masse (M + Na)+ (Da) MALDI–TOF de la fraction FPMeOH (21%) du polyéther PE6 obtenu lors de la réaction de 1 avec 3b sous MO pendant 2 h.
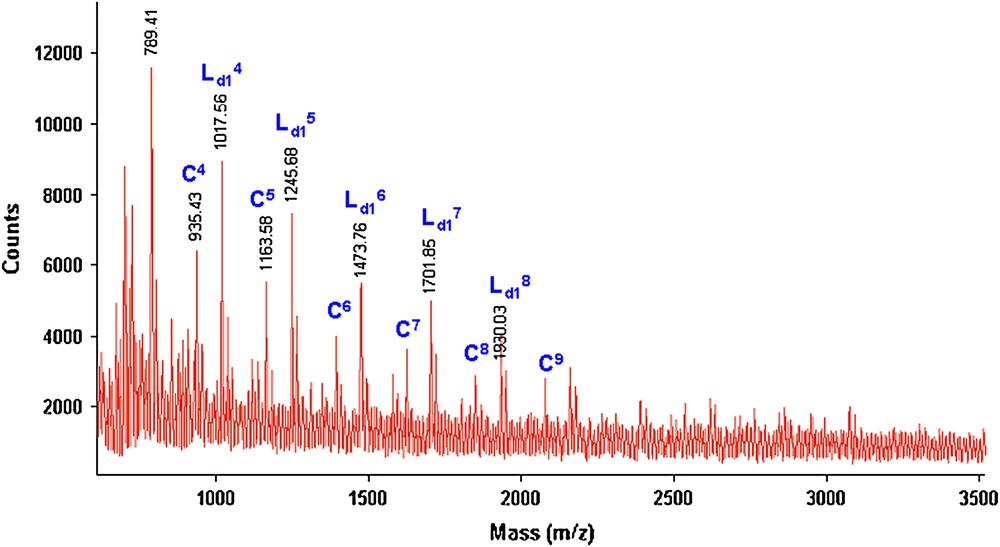
Spectre de masse (M + Na)+ (Da) MALDI–TOF de la fraction FPMeOH (42%) du polyéther PE6 obtenu lors de la réaction de 1 avec 3b par CC pendant 24 h.
Nous remarquons que les deux spectres de masse sont identiques et qu'il y une présence majoritaire de deux séries de pics correspondant aux structures C et Ld1. Quand la réaction est réalisée sous MO, la structure Lb est minoritaire par rapport aux deux autres (C et Ld1). Nous avons aussi constaté que, quel que soit le mode d'activation (2 h sous MO ou 24 h par CC), la structure cyclique est toujours présente d'une manière importante. Ceci confirme notre hypothèse, suggérant que la formation des polyéthers cycliques est favorisée avec les chaînes aliphatiques courtes (m = 4 ou m = 6), et ceci indépendamment du mode d'activation (MO ou CC).
2.2 Étude du comportement thermique par DSC
Le comportement thermique des fractions FPMeOH obtenues lors de la polycondensation de l'isosorbide et des différents agents dialkylants dimésylés sous MO pendant 2 h a été étudié par DSC entre −40 et 110 °C, avec une vitesse linéaire de chauffage de 10 °C min−1 (Tableau 10). L'analyse des thermogrammes des polyéthers à longues chaînes alkyles (PE8, PE10 et PE12) révèle des températures de fusion qui augmentent avec la longueur de la chaîne alkyle.
Répartition des températures de fusion trouvées pour les différents polyéthers synthétisés
| Polyéthers | PE4 | PE6 | PE8 | PE10 | PE12 |
| Tfusion (°C) | – | – | 15 | 19 | 37 |
2.3 Interprétation des effets spécifiques des microondes
D'une façon générale, lors de la polycondensation de l'isosorbide avec les divers agents dialkylants dibromés ou dimésylés, nous avons toujours obtenu des fractions FPHex (Fig. 1) ou FPMeOH (Fig. 17) plus élevées sous irradiations microondes que par chauffage classique.
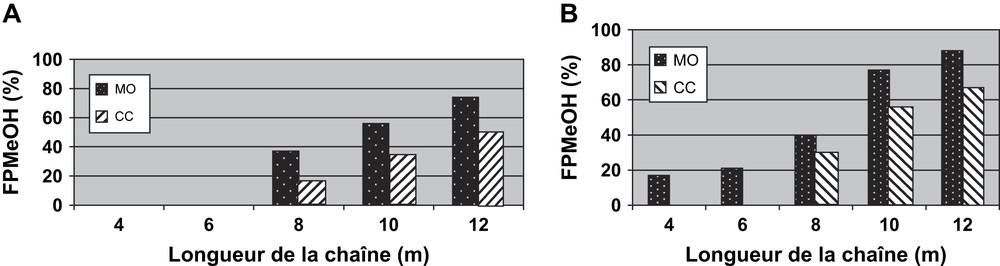
(A) Fractions FPMeOH obtenues sous MO ou par CC en utilisant un groupe partant Br. (B) Fractions FPMeOH obtenues sous MO ou par CC en utilisant un groupe partant OMs.
Nous donnons, dans la Fig. 18, les profils de montée en température sous microondes et par chauffage classique dans le cas de la réaction de polycondensation de l'isosorbide (1) par le 1,4-dimésylate d'octyle (2b).
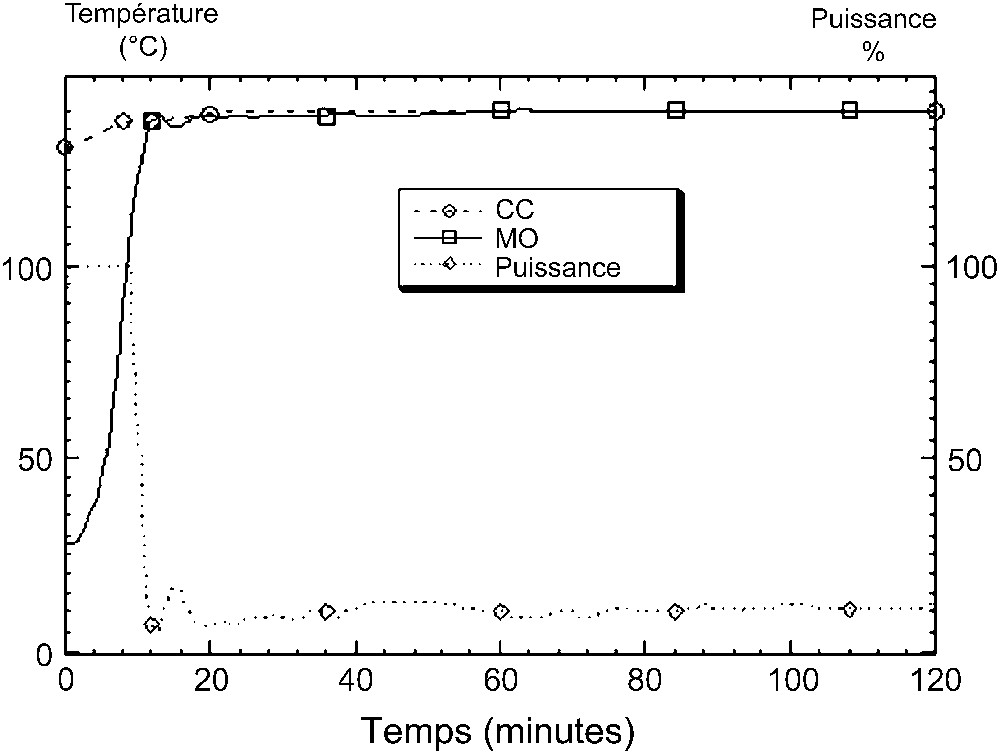
Profils de montée en température sous MO et par CC dans le cas de la réaction de polycondensation de l'isosorbide (1) par le 1,4-dimésylate d'octyle (2b).
Au vu des courbes de montée en température quasiment voisines pour les deux types d'activation, il est évident que l'intervention d'un effet spécifique des microondes, autre que celui purement thermique, est à l'origine des excellents résultats obtenus. Cette réaction de substitution nucléophile (réaction de Williamson) fait intervenir l'anion de l'une des fonctions hydroxyle de l'isosorbide sur un groupe partant bromé ou mésylé. Cette réaction est du premier ordre par rapport à l'alcool et à l'agent dialkylant (X–R–X) et met en jeu un mécanisme de type SN2, selon le schéma de la Fig. 19.

Illustration de l’évolution de la polarité entre l’état initial et l’état de transition.
L'état initial (EI) est constitué, d'une part, d'une paire d'ions entre l'alcoolate (formé in situ) et l'ammonium quaternaire de l'agent de transfert de phase et, d'autre part, du bromure ou du mésylate de la chaîne alkyle. L'état de transition (ET) est constitué d'une paire d'ions lâche, dans la mesure où il met en jeu un anion à charge délocalisée. Ceci lui confère une polarité accrue par rapport à l'état initial, de par un accroissement de la dissociation ionique. L'exaltation de la polarité dans l'état de transition permet une meilleure interaction de type dipôle–dipôle avec le champ électromagnétique, en raison du phénomène de polarisation dipolaire. Il s'ensuit alors une stabilisation accrue de l'état de transition (ET) par rapport à l'état initial (EI). La réaction est, de ce fait, accélérée sous microondes par suite d'un abaissement de son énergie d'activation (Fig. 20) [22–24].

Stabilisations relatives de EI et ET sous l'effet des MO par interactions de type dipôle–dipôle (ET est plus polaire que EI).
3 Conclusions
Nous avons étudié, au cours de ce travail, l'influence de plusieurs paramètres sur la synthèse des poly(isosorbide-éther)s et leur cyclisation selon deux modes d'activation (MO et CC).
En ce qui concerne la longueur de la chaîne alkyle, plus elle est courte, plus la proportion des polymères cycliques tend à augmenter. Ceci nous permet de dégager une hypothèse selon laquelle, plus les chaînes s'allongent, plus les sites actifs s'éloignent les uns des autres et ne peuvent plus réagir entre eux pour pouvoir donner lieu à la cyclisation. Par ailleurs, plus les chaînes sont courtes, plus les rendements en FPMeOH sont faibles ou, à la limite, nuls.
Quant à l'influence du temps de la réaction, elle diffère selon le mode d'activation. En effet, l'augmentation du temps de chauffage au-delà de 2 h (pour MO) et de 24 h (par CC) n'a pas d'effet sur le rendement, mais influe, en revanche, sur les masses molaires moyennes en nombre Mn et en poids Mp. Ce paramètre nous a aussi permis de mettre en évidence les effets spécifiques des MO, puisque le rendement d'une réaction de 1 h sous MO est très voisin de celui d'une réaction de 24 h par CC. Ces effets spécifiques résultent de la stabilisation des molécules polaires sous l'effet du champ électromagnétique des MO par polarisation dipolaire et de l'abaissement de l'énergie d'activation. Cependant, nous obtenons les mêmes structures de polyéthers, indépendamment du mode d'activation.
Sur un autre plan, nous avons dégagé l'importance du groupe partant quant au rendement de la réaction, en observant que le groupe partant mésyl (OMs) est meilleur, puisqu'il permet d'accéder à des rendements plus élevés et conduit à l'obtention d'une fraction FPMeOH pour les chaînes courtes, ce qui n'est pas le cas en travaillant avec un groupe partant bromure (Br).
Enfin, nous avons noté, par une étude du comportement thermique, que les températures de fusion augmentent avec l'allongement des chaînes alkyle du monomère de départ. Cependant, le mode d'activation semble ne pas avoir d'influence sur ce paramètre.
4 Partie expérimentale
4.1 Réactifs et équipements
Les produits de départ ont été achetés chez Aldrich ou Acros (Geel, Belgique) et ils ont été utilisés sans purification. L'isosorbide a été offert par la société Roquette Frères (France), et a été recristallisé auparavant dans l'acétone.
Un réacteur monomode à ondes focalisées par un guide d'ondes bien dimensionné, le Synthewave 402 de Prolabo, a été utilisé. Toutes les réactions ont été effectuées dans des matras de tailles variables. Le matra contenant le mélange réactionnel est introduit dans le réacteur monomode. La puissance (15 à 300 W), la température et le temps de réaction sont programmés à l'aide d'un ordinateur. Ce dernier permet l'asservissement de la puissance (ou de la température) et l'enregistrement des courbes (température et puissance en fonction du temps) lors de la réaction. Le mélange réactionnel peut être agité à l'aide d'une pale fixe plongée dans le tube en rotation. Lorsque la température fixée est atteinte, la puissance est alors asservie avec une valeur minime de 15 W afin de contrôler le palier de température. Les températures sont mesurées grâce à un détecteur infrarouge situé sur le fond du réacteur et qui évalue la température de surface.
Les spectres de masse MALDI on été enregistrés avec un spectromètre de masse dernière génération PerSeptive Biosystems Voyager Elite (Framingham MA, USA). Cet instrument est équipé d'un laser azote (337 nm), d'un extracteur et d'un réflecteur. Le potentiel d'accélération est de 20 kV dans les deux modes, linéaire et réflectron. Les spectres de masse enregistrés représentent une moyenne sur 256 impulsions laser (fréquence de répétition: 3 Hz). Des peptides et des porphyrines ont été utilisés pour étalonner l'instrument, avec le programme de calibration 3.07.1 de PerSeptive Biosystems.
Les solutions de polymères ont été préparées dans le THF à une concentration de 2 g l−1. Les fractions SEC ont été évaporées à sec et diluées dans 50 ml de THF. La matrice, l'acide 2,5-dihydroxybenzoïque (2,5-DHB), a été utilisée sans purification supplémentaire. Elle a été aussi dissoute dans le THF (10 g l−1) et 5 μl de solution de polymère ont été mélangés avec 50 μl de solution matrice. Cinq microlitres de solution d'iodure de sodium (5 g l−1 dans le THF) ont été ajoutés, dans le cas de quelques manipulations, pour induire la cationisation. Un microlitre de la solution finale a été déposé sur l'appareil et séché à l'air.
Les mesures SEC ont été réalisées en utilisant un appareil Knauer travaillant à température ambiante, avec le THF comme éluant et un débit de 1 ml min−1, équipé de colonnes PLgel 5–10 μm (8 × 250 mm). Cent microlitres de solution polymère (concentration 1% dans THF) ont été injectés. Un détecteur de l'indice de réfraction a été utilisé et les masses molaires ont été calculées à la base de la courbe de calibration (les masses molaires des échantillons utilisés pour l'étalonnage sont situées entre 400 et 10 000 g mol−1).
Les analyses élémentaires ont été réalisées par le service central de microanalyse de l'ICSN à Gif-sur-Yvette avec un appareil PerkinElmer 2400.
Les spectres RMN 1H ont été enregistrés, à température ambiante, sur un appareil Brüker AC 200 en utilisant le tétraméthylsilane (Me4Si: TMS) comme référence interne. Le solvant employé est le chloroforme deutérié.
4.2 Préparation des dérivés dimésylés aliphatiques (2b–6b)
Dans une solution sous agitation magnétique de diol (80 mmol) et de triéthylamine 12,33 g (122 mmol) dans 240 ml de chlorure de méthylène, maintenue à 0 °C sous argon, on ajoute, à l'aide d'une seringue, du chlorure de méthanesulfonyle 20,4 g (180 mmol), à raison de 2,23 ml min−1. La solution est ensuite agitée pendant 1 h à cette température, puis hydrolysée par 250 ml d'eau. On lave successivement par 200 ml de HCl 1,5 N et 200 ml de NaHCO3 à 5% dans l'eau : la phase organique est alors séchée sur MgSO4 et concentrée sous vide. Le produit brut est cristallisé dans l'éthanol absolu (rendement allant de 87% à 93%, suivant le diol de départ).
4.3 Procédé général de synthèse et de purification des polyéthers
La synthèse de ces polyéthers a été réalisée sous MO ou par CC dans les mêmes conditions. Dans un tube spécialement adapté pour le réacteur microondes, 1,46 g (10 mmol) d'isosorbide sont mélangés avec 1,62 g (25 mmol) de KOH en poudre (contenant 15% d'eau) et 10% de bromure de tétrabutylammonium (TBAB), soit 0,644 g. On introduit ensuite 10 mmol de dérivés dibromés ou de dérivés dimésylés et 3 ml de toluène.
Le mélange réactionnel est maintenu sous agitation mécanique constante pendant 30 min. À la fin de l'irradiation, le mélange est refroidi, puis dilué dans 50 ml de chloroforme pour extraire les produits organiques. Nous filtrons pour éliminer la potasse en excès et les sels minéraux, puis nous concentrons la solution chloroformique et nous précipitons dans le méthanol (75 ml) les fractions de polyéthers à longues chaînes FPMeOH. La deuxième fraction est obtenue, après évaporation du méthanol, par précipitation dans le n-hexane (FPHex). Ces deux fractions de polyéthers sont séchées sous vide poussé jusqu'à l'obtention d'une masse constante. Le pourcentage en fraction de polyéthers insoluble dans le méthanol (FPMeOH) est déterminée comme suit : une fois cette fraction isolée, elle est séchée sous vide jusqu'à obtenir une masse constante; on détermine sa masse et on la divise par la masse théorique (nombre de moles multiplié par la masse du motif répétitif. Par exemple, dans le cas du PE4, la masse théorique est de 0,01 × 200 = 2 g). On effectue la même chose avec la fraction de polyéthers insoluble dans l'hexane (FPHex). Le rendement total est la somme des deux fractions FPMeOH et FPHex.
4.3.1 Polyéther PE4
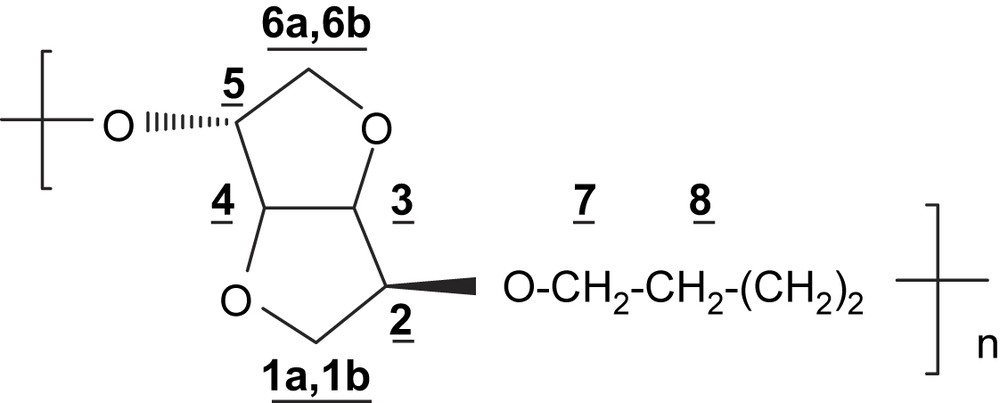
RMN 1H (200 MHz, CDCl3) δ (ppm): 4,61 (dd, 1H, H-4, J3,4 = J4,5 = 4,1 Hz); 4,47 (d, 2H, H-3, J3,4 = 4,1 Hz); 4,08–3,83 (m, 7H, H-2; H-5; H-1a; H-7); 3,71 (m, 1H, H-1b); 3,41–3,29 (m, 2H, H-6); 1,61 (m, 4H, H-8).
IR (cm−1): 3440, 2932, 2858, 1096, 1017, 776, 726.
Analyse élémentaire: (C10H16O4)n; n(200)
| Carbone | Hydrogène | Oxygène | |
| Calculée (%) | 60,00 | 8,00 | 32.00 |
| Trouvée (%) | 59,21 | 7,85 | 32,94 |
4.3.2 Polyéther PE6

RMN 1H (200 MHz, CDCl3) δ (ppm): 4,57 (dd, 1H, H-4, J3,4 = J4,5 = 4,1 Hz); 4,51 (d, 2H, H-3, J3,4 = 4,1 Hz); 4,11–3,79 (m, 7H, H-2; H-5; H-1a; H-7); 3,69 (m, 1H, H-1b); 3,37–3,21 (m, 2H, H-6); 1,65 (m, 4H, H-8); 1,35 (m, 4H, H-9)
IR (cm−1): 3440, 2932, 2858, 1096, 1017, 776, 726.
Analyse élémentaire: (C12H20O4)n; n(228)
| Carbone | Hydrogène | Oxygène | |
| Calculée (%) | 63,15 | 8,77 | 28,07 |
| Trouvée (%) | 62,97 | 8,61 | 28,42 |
4.3.3 Polyéther PE8
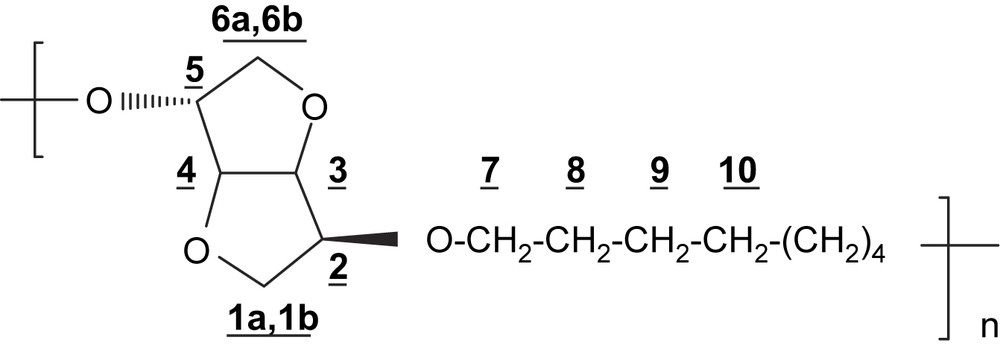
RMN 1H (250 MHz, CDCl3) δ (ppm): 4,60 (dd, 1H, H-4, J3,4 = J4,5 = 4,1 Hz); 4,45 (d, 2H, H-3, J3,4 = 4,1 Hz); 4,10–3,80 (m, 7H, H-2; H-5; H-1a; H-7); 3,62 (m, 1H, H-1b); 3,50–3,38 (m, 2H, H-6); 1,53 (m, 4H, H-8); 1,3 (m, 8H, H-9–H-10).
IR (cm−1): 3440, 2932, 2858, 1096, 1017, 776, 726.
Analyse élémentaire: (C14H24O4)n; n(256)
| Carbone | Hydrogène | Oxygène | |
| Calculée (%) | 65,62 | 9,37 | 25,00 |
| Trouvée (%) | 65,21 | 9,47 | 25,16 |
4.3.4 Polyéther PE10
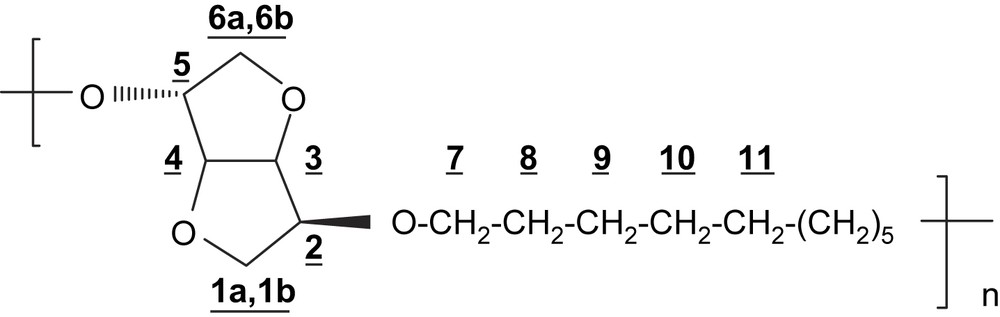
RMN 1H (250 MHz, CDCl3) δ (ppm): 4,65 (dd, 1H, H-4, J3,4 = J4,5 = 4,1 Hz); 4,51 (d, 2H, H-3, J3,4 = 4,1 Hz); 4,01–3,79 (m, 7H, H-2; H-5; H-1a; H-7); 3,52 (m, 1H, H-1b); 3,45–3,33 (m, 2H, H-6); 1,63 (m, 4H, H-8); 1,25 (m, 8H, H-9–H-10–H-11).
IR (cm−1): 3440, 2932, 2858, 1096, 1017, 776, 726.
Analyse élémentaire: (C16H28O4)n; n(284)
| Carbone | Hydrogène | Oxygène | |
| Calculée (%) | 67,60 | 9,85 | 22,53 |
| Trouvée (%) | 67.19 | 9,51 | 23,30 |
4.3.5 Polyéther PE12
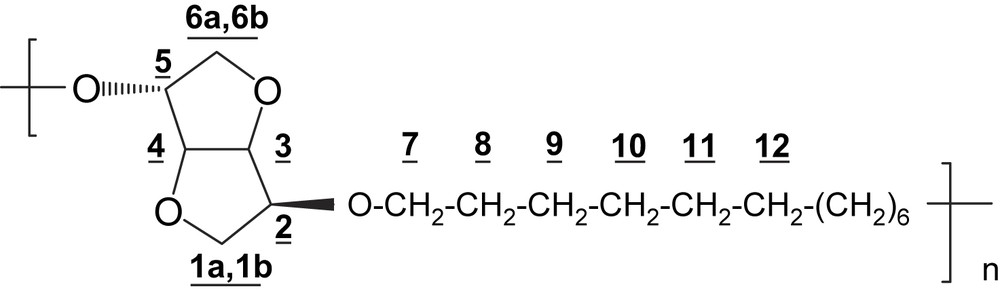
RMN 1H (250 MHz, CDCl3) δ (ppm): 4,63 (dd, 1H, H-4, J3,4 = J4,5 = 4,1 Hz); 4,63 (d, 2H, H-3, J3,4 = 4,1 Hz); 4,01–3,88 (m, 7H, H-2; H-5; H-1a; H-7); 3,61 (m, 1H, H-1b); 3,49–3,36 (m, 2H, H-6); 1,57 (m, 4H, H-8); 1,27 (m, 8H, H-9–H-10–H-11–H-12).
IR (cm−1): 3440, 2932, 2858, 1096, 1017, 776, 726.
Analyse élémentaire: (C18H32O4)n; n(312)
| Carbone | Hydrogène | Oxygène | |
| Calculée (%) | 69,23 | 10,25 | 20,51 |
| Trouvée (%) | 68,91 | 9,93 | 21,16 |
Remerciements
Les auteurs tiennent à remercier M. Jean-Claude Blais (université Pierre-et-Marie-Curie) pour la détermination des spectres de masse MALDI–TOF et M. Darek Bogdal (université de Cracovie, Pologne) pour la détermination des masses molaires moyennes par chromatographie d'exclusion stérique (SEC).