

1 Introduction
Nitric oxide (NO) can be reduced catalytically to nitrous oxide (N2O), nitrogen (N2), hydroxylamine (NH2OH) and ammonia (NH3), all of which are relatively stable. Applications in which catalytic NO reduction is involved include the removal of pollutants from exhaust gases, the purification of groundwater and the more efficient production of NH2OH, which is a raw material in nylon production [1]. These applications require selective NO reduction catalysts to either N2 or NH2OH. Since metal catalysts are mainly selective toward N2O and NH3 [2], the search for suitable catalysts continues [3,4].
NO reduction is also an important process in nature. In denitrification respiratory ATP synthesis is coupled to the reduction of nitrate to nitrogen, which occurs via the intermediates NO2−, NO and N2O [5,6]. In this process enzymes such as bacterial nitric oxide reductase [7,8] and cytochrome P450nor [9–12] selectively catalyze the reduction of NO to N2O. Another process in which NO reduction is involved is NO detoxification. An example of a detoxifying NO reductase is flavohemoglobin [13], which also catalyzes the reduction of NO to N2O. Apart from enzymes that reduce NO to N2O there are also some enzymes such as cytochrome c nitrite reductase that can directly reduce NO to NH3 [14,15]. The difference between the enzymes and the metal catalysts is that the enzymes are 100% selective to their desired products. All of these enzymes have in common that a heme group (iron protoporphyrin IX) is present in the active site. This suggests that this group has properties suitable for selective NO reduction. Even though the enzymes are not selective toward the desired products N2 and NH2OH, heme groups might still present an interesting alternative to metal surfaces in the search for selective NO reduction catalysts. Because isolated heme groups are in a distinctly different environment than heme groups in heme proteins their selectivity in NO reduction could differ from that of heme proteins.
Since isolated heme groups aggregate in aqueous environments [16,17], research efforts have focused on characterization of water soluble porphyrins [18,19] and electrochemical NO reduction by immobilized heme groups [20–23] and heme proteins [24–26]. This paper specifically discusses our recent work on electrochemical NO reduction by heme groups immobilized on pyrolitic graphite and shows that, depending on pH and potential, heme groups can be selective catalysts toward N2O or NH2OH [21,22]. The mechanisms for both pathways are also discussed.
2 Electrochemical properties of heme groups immobilized on pyrolitic graphite
Prior to studying electrochemical NO reduction, the heme groups have to be immobilized and characterized. The most commonly employed electrode material is pyrolitic graphite. In contrast to other electrode materials such as gold [27,28] graphite has the advantage that NO reduction can be studied over a wide potential window. The simplest method to immobilize heme groups on pyrolitic graphite is by adsorption from a basic aqueous solution [21,29] or water/ethanol solution [30,31] containing dissolved heme, followed by rinsing with pure water. The electrochemical and spectroscopic properties of heme adsorbed in this way have been studied extensively [21,29,32–35], revealing that the hemes form an ordered structure [29]. The midpoint potential of the FeIII/FeII redox couple is E = −0.40 V versus SCE at pH 7 (E = −0.15 V versus SHE) (Fig. 1). In comparison, the redox potentials at pH 7 of NO reducing enzymes such as cytochrome P450nor and cytochrome c nitrite reductase are −0.31 V and −0.11 V versus SHE, respectively [36,37].

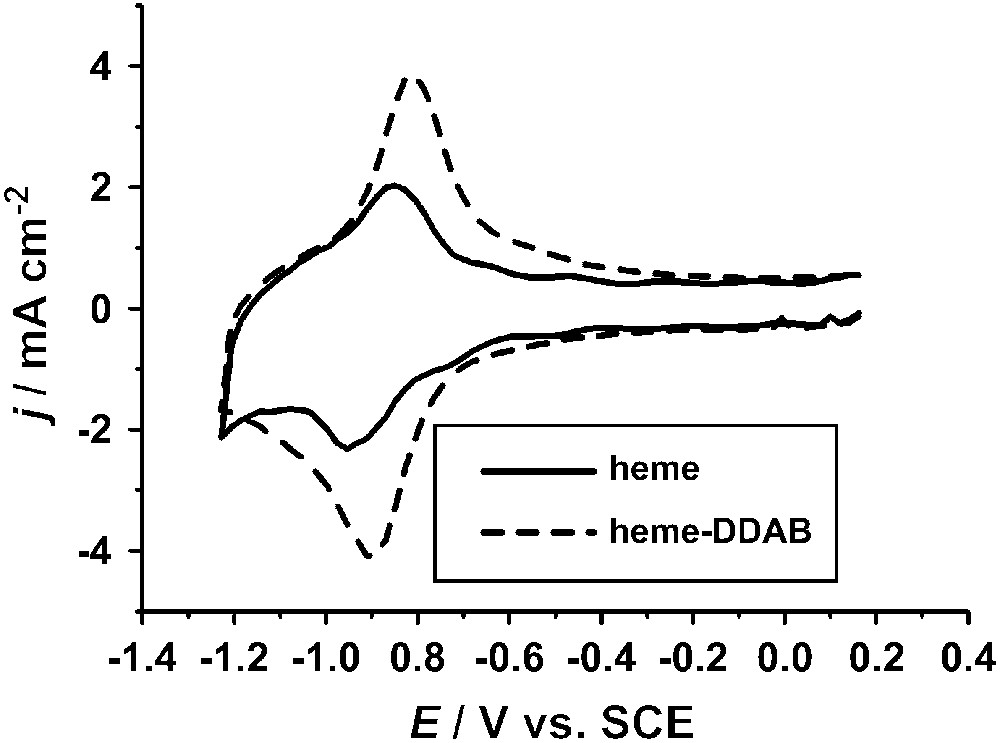
Cyclic voltammograms of a pyrolitic graphite electrode with heme adsorbed from a pH 10 solution (—) and of a film cast from a solution of heme–DDAB in 0.1 M sodium acetate, pH 5 (- - -). Voltammograms were measured in 0.1 M phosphate, pH 7.0, at a scan rate of 500 mV/s. Experimental procedures are described in the literature [21,22].
Heme groups can also be immobilized in other ways and this can distinctly affect their properties. An example is the incorporation in films of surfactants or polymers [20,22,23,38]. Since hemes are relatively hydrophobic they are readily incorporated in vesicles formed by surfactants in aqueous solution. By evaporating a drop of such a solution on an electrode surface a stable film can be formed. Fig. 1 shows the voltammetric response of heme incorporated in a film of didodecyldimethylammonium bromide (DDAB). There are distinct differences compared to heme adsorbed directly on pyrolitic graphite. The FeIII/FeII redox couple is found at E = −0.20 V versus SCE at pH 7 (E = 0.05 V versus SHE), which is 0.2 V more positive than for adsorbed heme. The redox couple is also partly irreversible, with some small cathodic peaks at lower potentials. This has been suggested to be related to a potential-induced phase transition occurring in the DDAB film [22,39]. Additionally, a redox couple is found at −0.80 V versus SHE. This couple has been tentatively assigned to an FeII/FeI transition of the heme group [40]. However, it is still unclear why such a peak is not observed for adsorbed heme.
3 Activity and selectivity in NO reduction
Typical voltammograms for NO reduction catalyzed by adsorbed heme and heme–DDAB films at different pHs are given in Fig. 2a and b. They show that depending on pH and immobilization method the reduction of NO starts between −0.6 V and −0.8 V versus SCE and rapidly increases in rate with decreasing potential. This increase stops when the reduction rate becomes limited by NO mass transport. From the shapes of the voltammograms at different pHs it can be deduced that there are at least two NO reduction pathways for both adsorbed heme and heme–DDAB, one pathway being independent of pH (I) and another pathway shifting to lower potentials with increasing pH (II) [22]. The presence of the two pathways is observed most clearly for the heme–DDAB films, especially from the measurement at pH 10.2, which shows the presence of two reduction waves. For adsorbed heme the presence of the two pathways is less clear. A clear ‘shoulder’ indicative of a pH-independent pathway can be discerned only at pH 11.9. We did not observe the presence of this pathway previously, since measurements on adsorbed heme were restricted to the pH 4–10 range [21].

Cyclic voltammograms of adsorbed heme (a) and of a heme–DDAB film (b) in 0.5 M acetate, pH 5.0 (—); 0.5 M phosphate, pH 7.0 (—); 0.5 M borate, pH 10.2 (—); 0.5 M phosphate, pH 11.9 (—). I indicates the pH-independent pathway and II indicates the pH-dependent pathway. Measurements were performed in saturated NO solution at a scan rate of 50 mV/s and a rotation rate of 16 rps. Experimental procedures are described in the literature [21,22].
The products of the different NO reduction pathways have been determined employing online electrochemical mass spectroscopy (OLEMS) [41], which can detect the formation of N2O and N2, and the rotating ring-disk electrode (RRDE) [42], which can detect the formation of NH2OH. For heme–DDAB films the products of both pathways have been determined to be N2O for the pH-independent pathway (I) and NH2OH for the pH-dependent pathway (II) [22]. For adsorbed heme NH2OH was determined to be the main product at pH 7. At this pH reduction occurs mainly via the pH-dependent mechanism [21]. In that study selectivity measurements at other pHs were not performed. In order to detect the product of the pH-independent pathway, we present some OLEMS measurements at higher pHs in this paper. Fig. 3 shows an increasing N2O formation with increasing pH. The increase in N2O formation is in line with the increase in the ‘shoulder’ we observe in voltammetry (Fig. 2a) and indicates that N2O is the product of the pH-independent pathway (I) for adsorbed heme.
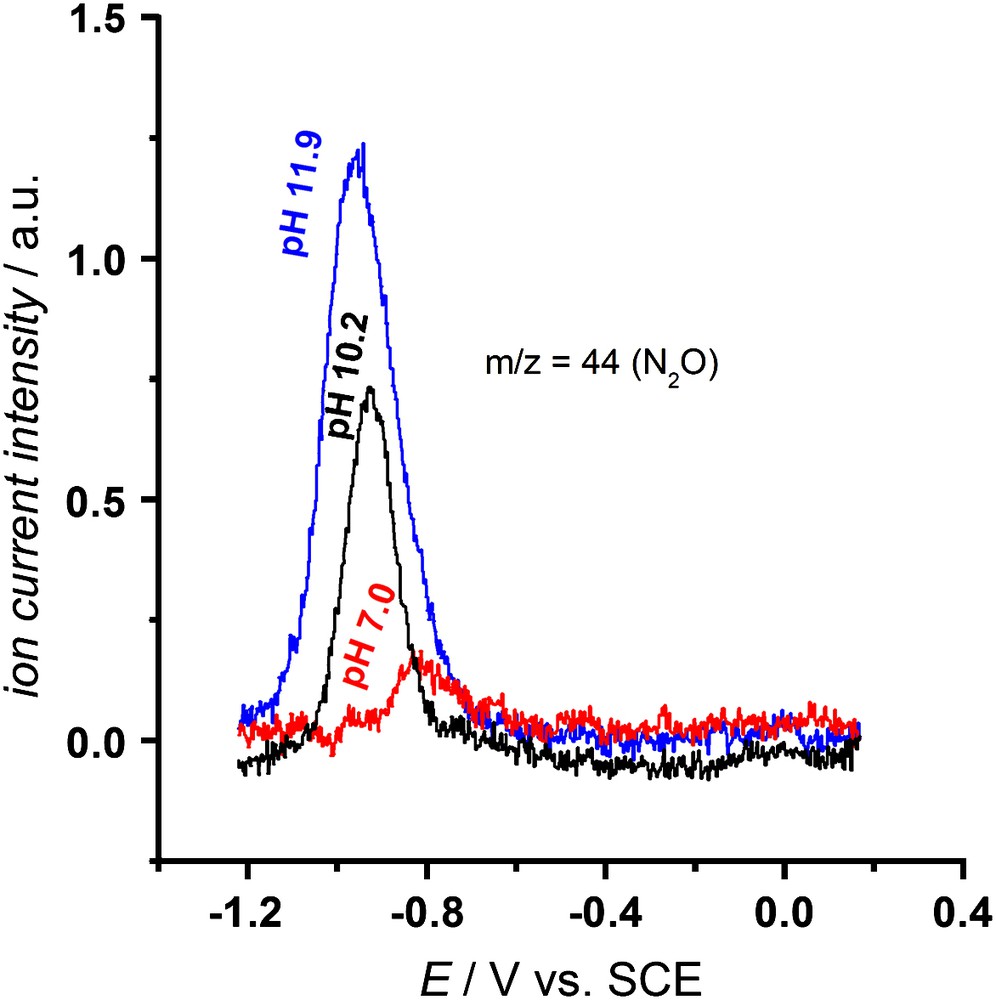
OLEMS measurements on adsorbed heme in saturated NO solution at 2 mV/s. The ion current intensity for m/z = 44 (N2O) in the cathodic scan is plotted as a function of potential. Measurements were performed in 0.5 M phosphate, pH 7.0 (—), 0.5 M borate, pH 10.2 (—) and 0.5 M phosphate, pH 11.9 (—). NO was continuously bubbled through the solution. Experimental procedures are described in the literature [21].
Other pathways, resulting in either N2 or NH3 formation seem to be absent for both adsorbed heme and heme–DDAB films. The absence of N2 formation was proven by OLEMS measurements. NH3 formation was ruled out based on NH2OH reduction experiments, which show that the rate at which NH2OH can be reduced to NH3 is negligible compared to the rate at which NH2OH is formed in NO reduction [21,22]. Moreover, a comparison of NO reduction on adsorbed heme to NO reduction on platinum indicated that the selectivity of adsorbed heme toward NH2OH is 100% at pH 7 [21].
Our studies show that for both adsorbed heme and heme–DDAB the products of the pH-independent pathway (I) and the pH-dependent pathway (II) are N2O and NH2OH, respectively. This suggests the presence of two general NO reduction pathways for immobilized heme that are independent of the way in which the heme is immobilized. Nevertheless, the immobilization method has a distinct influence on the potentials at which both pathways occur. The pH-independent pathway starts at potentials about 0.12 V more negative for adsorbed heme compared to heme–DDAB, whereas the pH-dependent pathway starts about 0.05 V more positive. An explanation for these differences in potential will be given in Section 4.
A number of papers have reported NO reduction on myoglobin/DDAB and other protein/DDAB films [24–26]. Recently, we provided strong evidence that heme release occurs in myoglobin/DDAB films on graphite, however [22,43]. Therefore, these films should probably be regarded as equivalent to heme–DDAB films and the results obtained for NO reduction on these films can be directly compared to the results on heme–DDAB films. Mimica et al. also observed the presence of two NO reduction pathways, but assumed that N2O was the product of both pathways [26]. Others argued that there was only one NO reduction pathway toward N2O [24,25]. However, differences between these papers and our heme–DDAB paper are not so much in the observed data, but mainly in the interpretation of the data. The data presented do not contradict our hypothesis that two NO reduction pathways occur.
4 Reaction mechanisms and reaction intermediates
The fact that the same NO reduction products are observed for both adsorbed heme and heme–DDAB films, suggests that the NO reduction mechanisms are independent of the way in which heme is immobilized. More mechanistic information on the pH-dependent and pH-independent pathways can be obtained from the analysis of Tafel slopes, the determination of exact pH dependencies and by electrochemical isolation of intermediates.
For the pH-dependent pathway leading to the formation of NH2OH, Tafel slopes have been determined to be approximately −60 mV/dec for both adsorbed heme and heme–DDAB films [21,22]. This Tafel slope suggests the presence of an electrochemical equilibrium prior to the rate-determining step (EC mechanism) [44]. This electrochemical couple could be a redox couple involving a nitrosyl (NO) to nitroxyl (NO−) transformation. Since a pH dependence of −42 mV/pH has been determined for adsorbed heme, the subsequent rate-determining step could be a proton transfer (see Fig. 4, left-hand side). However, we cannot exclude the possibility that the proton transfer is concerted with the electron transfer and that another step, of which we do not know the nature, is rate determining.
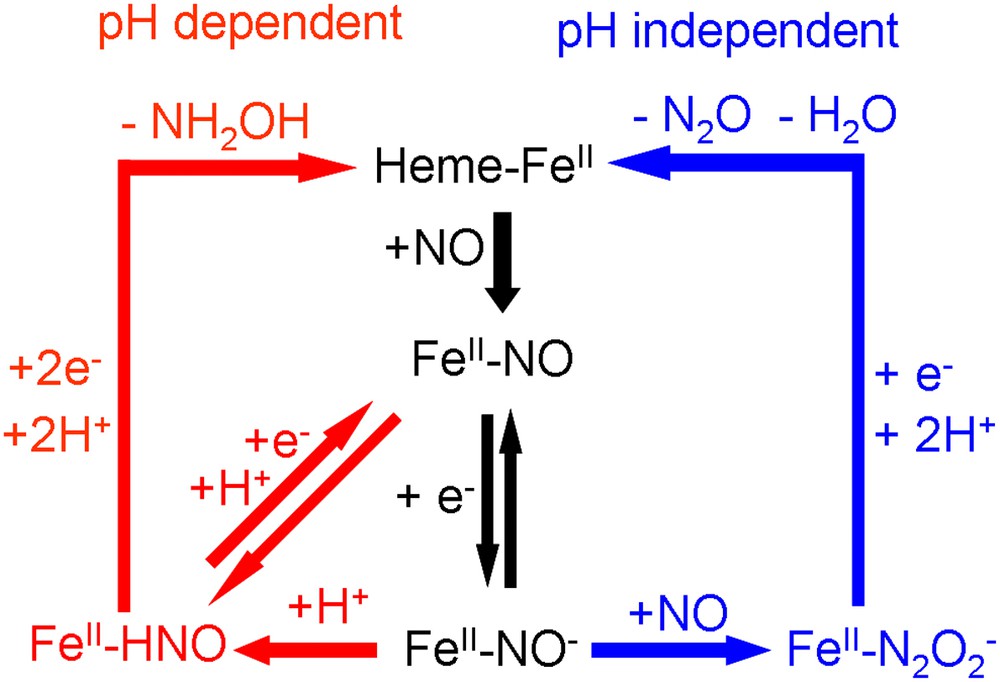
Mechanisms of the two pathways for NO reduction observed for immobilized heme on pyrolitic graphite.
On the basis of this mechanism, one would expect to be able to observe an FeII–NO/FeII–NO− or FeII–NO/FeII–HNO redox couple at high scan rates. Fig. 5 shows that such a couple is indeed observable for both adsorbed heme and heme–DDAB. It has a potential of approximately −0.86 V versus SCE for heme–DDAB and −0.90 V versus SCE for adsorbed heme and is independent of pH [22]. However, the couple is only observable at high pHs (>10) and not at low pHs. Even at scan rates up to 1000 V/s a redox couple cannot be observed around pH 7. This suggests that the rate-determining step is very fast at lower pH values, which is supported by the fact that the onset of the pH-dependent pathway is about 0.15 V more positive than the position of the redox couple at pH 7. This strong pH dependence suggests that a proton transfer is indeed the rate-determining step. In a previous study on myoglobin/DDAB films a pH-independent FeII–NO/FeII–NO− redox couple has also been observed at E = −0.87 V versus SCE [24]. In line with our results, it was reported that the lifetime of this couple decreased with decreasing pH.
Cyclic voltammograms of adsorbed heme (—) and a heme–DDAB film (---) in 3 M KOH, showing a FeII–NO/FeII–NO− redox couple. Measurements were performed in saturated NO solution at a scan rate of 10 V/s. Experimental procedures are described in the literature [21,22].
For the pH-independent pathway leading to the formation of N2O a Tafel slope of −108 mV/dec has been determined for heme–DDAB films. This value suggests that the first electron transfer in the mechanism is rate-determining, which is the reduction of FeII–NO to FeII–NO−. This result seems to be in contradiction with the mechanism suggested for the pH-dependent pathway, in which the FeII–NO/FeII–NO− redox couple is in equilibrium. How can the rate-determining step in one mechanism be in equilibrium in another mechanism? The answer can only be that the electron transfer is only the rate-determining step at high potentials and under conditions where NO mass transport is not rate limiting. At lower potentials, where the high reaction rate causes a very low NO concentration at the electrode, we expect that the second NO transfer to the FeII–NO− adduct becomes rate limiting. A similar change in rate-determining step with NO concentration has also been reported for the enzyme cytochrome P450nor [9].
Based on these mechanisms we can provide a plausible explanation for the differences in NO reduction behavior between adsorbed heme and heme–DDAB. The differences probably originate from the fact that adsorbed heme is directly exposed to the aqueous solution, whereas the heme in a heme–DDAB film is embedded in a hydrophobic environment. The NO saturation concentration in such a hydrophobic environment has been suggested to be almost two orders of magnitude higher than in water [24], which would increase the rate of the pH-independent pathway. On the other hand, the H+ concentration in such an environment is probably significantly lower, thereby decreasing the rate of the pH-dependent pathway.
5 Selectivity of NO reduction by immobilized hemes compared to enzymes and metal catalysts
Our results show that immobilized hemes can reduce NO to N2O or NH2OH. By tuning the conditions (pH, immobilization method, potential and NO concentration) 100% selectivity toward either of these products can be obtained. We now make a comparison to the NO reducing enzymes cytochrome P450nor and cytochrome c nitrite reductase and to metal catalysts.
Similar to cytochrome P450nor NO can be reduced to N2O selectively. The mechanism of NO reduction by cytochrome P450nor resembles the pH-independent pathway in that it occurs via an FeII–NO− or FeII–HNO intermediate that reacts with a second NO molecule. A difference is that cytochrome P450nor does not reduce NO to NH2OH. This is not surprising since its electron donor NADH only provides two electrons and hence further reduction of the FeII–HNO intermediate is impossible in the case of cytochrome P450nor. The transfer of more electrons from a second NADH is unlikely since only one NADH can bind close to the heme [45].
A comparison to the enzyme cytochrome c nitrite reductase suggests that the mechanism by which the enzyme reduces NO to NH3 is similar to the pH-dependent pathway of immobilized heme toward NH2OH. The question therefore arises why immobilized hemes do not reduce NH2OH further to NH3, in a way similar to cytochrome c nitrite reductase. The answer is probably that in contrast to the enzyme, the individual heme is not able to break the N–O bond. Reduction of NO to either N2O or NH2OH does not require the breaking of the N–O bond, whereas reduction to NH3 does. This inability to break the N–O bond is probably related to the way in which NO binds to the catalyst. This occurs in a bended end-on configuration, in which nitrogen binds to the iron (Fig. 6) [46,47]. The overlapping of the anti-bonding orbitals of NO with the d-orbitals of Fe weakens the N–O bond, as is reflected in its lower stretching frequency, which is about 1670–1680 cm−1 compared to 1904 cm−1 in the gas phase [48,49]. However, this weakening of the NO bond is apparently insufficient to enable NO bond breaking. One could argue that in our aqueous system hydrogen bonds with water molecules from solution could further weaken the bond, but vibrational studies in which aqueous and UHV (Ultra High Vacuum) systems have been compared suggest that this is unlikely [50,51].
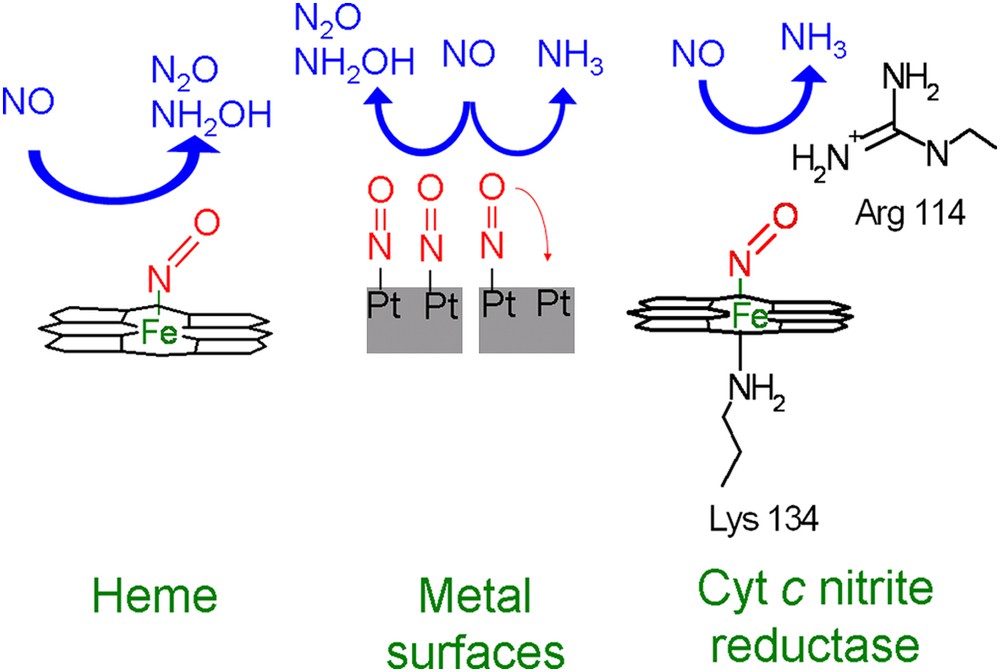
Schematic overview of the selectivity of NO reduction by immobilized heme, metal electrodes, and cytochrome c nitrite reductase.
A related question is how the enzyme cytochrome c nitrite reductase is able to sufficiently weaken the N–O bond in order to reduce NH2OH further to NH3. In this case one should take into account that the heme is surrounded by protein residues, which can interact with NO. An example is Arg114, which can form a hydrogen bond with the oxygen of NO [14]. Additionally, there is a lysine residue trans to NO that also interacts with the heme. It has been shown experimentally and theoretically that such a ligand can weaken the NO bond [52–56]. It is unclear whether immobilized heme groups with bound NO also have such a trans ligand. For adsorbed heme groups this seems unlikely since they are adsorbed almost flat on the surface [29,34]. To what extend the surrounding protein residues and the trans ligand weaken the NO bond in the protein is still unknown, since the stretching frequency of NO bound to cytochrome c nitrite reductase has not yet been determined. However, for other heme proteins NO stretching frequencies of around 1600 cm−1 have been observed [57,58], indicating that these effects can indeed significantly weaken the NO bond. This extra weakening might not only enable reduction of NH2OH to NH3, but could also be one of the reasons why cytochrome c nitrite reductase is able to reduce NO at potentials much more positive (∼500 mV) than immobilized heme [36].
The relation between weakening of the N–O bond and selectivity not only applies to hemes and heme enzymes, but also to metals. It has been suggested in literature that NH3 formation on most metals is only possible if adsorbed NO has space to tilt in such a way that the oxygen of NO can coordinate to a second metal site (Fig. 6) [59]. If this is not possible, e.g. if the surface is fully covered with NO, only N2O and NH2OH are the reaction product analogous to immobilized heme. This suggests that similar to immobilized heme groups NO bond breaking on metals is not possible when coordination is only by a metal–N bond.
6 Conclusions
Hemes immobilized on pyrolytic graphite can reduce NO via two pathways, a pH-independent pathway leading to the formation of N2O and a pH-dependent pathway leading to the formation of NH2OH. Which pathway is favored depends on potential, pH, immobilization method and NO concentration. This ability of immobilized hemes to catalyze different NO reduction pathways is in line with the ability of different heme enzymes to reduce NO to different products. Most interesting is the fact that immobilized hemes cannot reduce NH2OH further to NH3, since the heme–NO interaction apparently is insufficient to break the N–O bond.
Acknowledgment
This work was supported by the National Research School Combination Catalysis (NRSC-C).