

1 Introduction
The impressive synergism that has developed in the past few years between biophysical and synthetic inorganic chemists with regard to understanding the spectroscopic signals and functions of the [(SR)zFe(CO)x(CN)y] and [Ni(Cys)4] active sites in [Fe]/[NiFe] hydrogenases has highlighted the chemistry of iron–thiolate cyanocarbonyl and nickel–thiolate complexes [1–12]. The recent report of high-quality single-crystal X-ray structure of [Fe]-only hydrogenase from Desulfovibrio desulfuricans revealed that the active site contains a dinuclear iron(II)–thiolate with mixed CO and CN− coordinated ligands (H cluster) bound to a [4Fe–4S] cubane cluster via cysteinate bridge, and also suggested that the unsaturated Fe center present in the H cluster of the enzyme can act as a binding site for the soft ligand H2 [3]. A more CO/CN−-rich environment was also found for iron in the recently reported X-ray structure of [Fe] hydrogenase isolated from Clostridium pasteurianum (CpI) [4]. A schematic drawing of the active center of [Fe] hydrogenases as deduced from the crystallographic studies is shown in Fig. 1. Moreover, the infrared spectra of [Fe] hydrogenase from Desulfovibrio vulgaris concluded that the weak absorption bands for the CN− ligand range from 2079 to 2106 cm−1, and the broad strong absorption bands (ranging from 2016 to 1894 cm−1) are assigned to the carbonyl stretching frequencies under the various redox states [5].
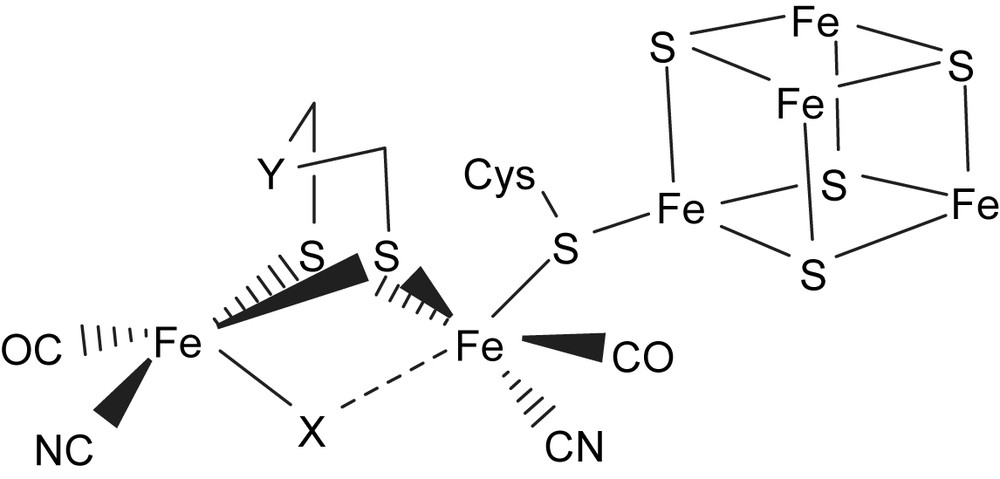
Schematic drawing of the H cluster according to the X-ray crystal structure isolated from D. desulfuricans [Fe] hydrogenase (X = CO, H; Y = NH, CH2) [3,4].
The X-ray crystallographic studies of the active-site structure of [NiFe] hydrogenases isolated from Desulfovibrio gigas, Desulfovibrio vulgaris, Desulfovibrio fructosovorans, and Desulfovibrio desulfuricans ATCC27774, in combination with infrared spectroscopy, revealed that the active site is composed of a heterobimetallic (Scys)2Ni(μ-Scys)2(μ-X)Fe(CO)(CN)2 (X = O2−, HO2−, OH−) cluster (Fig. 2). The bridging ligand X was proposed to be an oxide, hydroxide or hydroperoxide in the oxidized state, and was found to be absent in the reduced state [8–11]. The coordination environment about nickel in the [NiFe] hydrogenases is pseudo-tetrahedral in the reduced state and pseudo-square pyramidal in the oxidized state. Also, the iron site of the [NiFe] hydrogenases isolated from D. gigas has been established as a pyramidal [Fe(CN)2(CO)] unit with the opposite face coordinated to two S-cysteines bridged to a nickel [8–11]. The nickel site has been proposed to be redox active and changes between Ni(III) and Ni(II), while the iron site remains as Fe(II) in all spectrally defined redox states of the enzyme. The active center of [NiFe] hydrogenases exhibits various redox states in the hydrogen catalytic cycle. The EXAFS/EPR studies indicate that the formal oxidation state of the Ni center is paramagnetic Ni(III) in Ni–A, Ni–B and Ni–C states [8–11]. Actually, the active form Ni–C (the paramagnetic Ni–C intermediate) of [NiFe] hydrogenases was proposed to exist as the [(Scys–H)Ni(III)–H–Fe] intermediates after an active state Ni–SIa (silent-active [(Scys–H)Ni(II)(Scys)3]) is passed [8–11]. Ni–R/Ni–SIa states were proposed to exist as [(Scys–H)(Scys)Ni(II)(μ-Scys)2Fe(CO)(CN)2] with a Cys–SH interacting directly with the nickel center (a [Ni⋯H–Scys] interaction). In particular, the recent X-ray absorption spectroscopy shows that the Ni site of the regulatory hydrogenase (RH) in the presence of hydrogen , proposed as Ni–C state, isolated from Ralstonia eutropha is a six-coordinated [NiIII–S2(O/N)3(H)] [12].
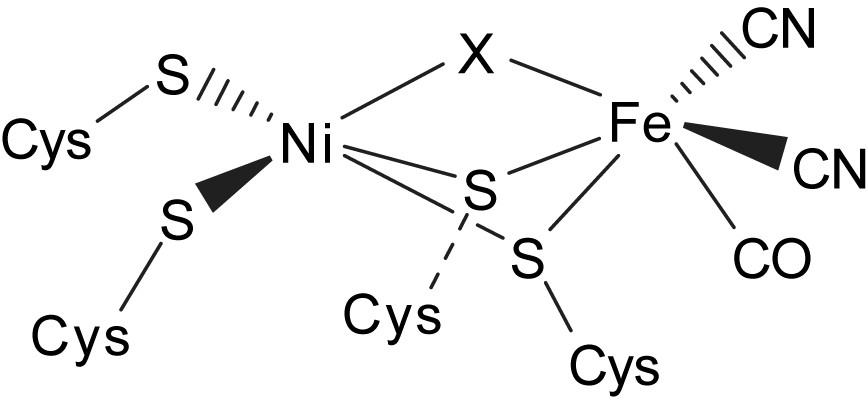
Schematic drawing of the active site of [NiFe] hydrogenases as deduced from crystallographic studies [8–10].
2 Iron–thiolate complexes with mixed CO/CN− ligands
2.1 Mimicking the iron part of [NiFe]/[Fe] hydrogenases
Applications of anionic iron(0)–cyanocarbonyl [Fe(CO)4(CN)]− for preparation of iron(II)–thiolate cyanocarbonyl proved a successful approach in the synthesis of iron–thiolate cyanocarbonyls. When di(2-aminophenyl) disulfide was reacted with the equal molar [PPN][Fe(CO)4(CN)] in THF at room temperature, the air-stable, pentacoordinated, 16-electron Fe(II) complex [PPN][Fe(CO)2(CN)(S,NH–C6H4)] (1) was isolated via oxidative addition of disulfide to the [Fe(CO)4(CN)] motif and subsequent oxygen oxidation accompanied by the concomitant deprotonation of the amine proton (Scheme 1a) [13–15]. The IR spectrum of complex 1 in CH3CN reveals a weak absorption band for the CN− ligand at 2099 cm−1 while the two strong absorption bands (1997 s, 1933 s cm−1) are assigned to the carbonyl stretching frequencies. When a THF solution of complex 1 is purged with 13CO, the IR νCO peaks at 1992 and 1928 cm−1 immediately shift to 1946 and 1885 cm−1 while the IR νCN stretching frequency remains unchanged. The magnitude ∼45 cm−1 of the isotopic shift (ΔνCO) is consistent with the calculated position, based only on the difference in masses between 12CO and 13CO. The shorter Fe–S bond length of 2.226(2) Å and Fe–N bond length of 1.855(4) Å were attributed to the strong π-donating ability of the bidentate [S,NH–C6H4]2− ligand which stabilized the unsaturated Fe(II) complex 1.
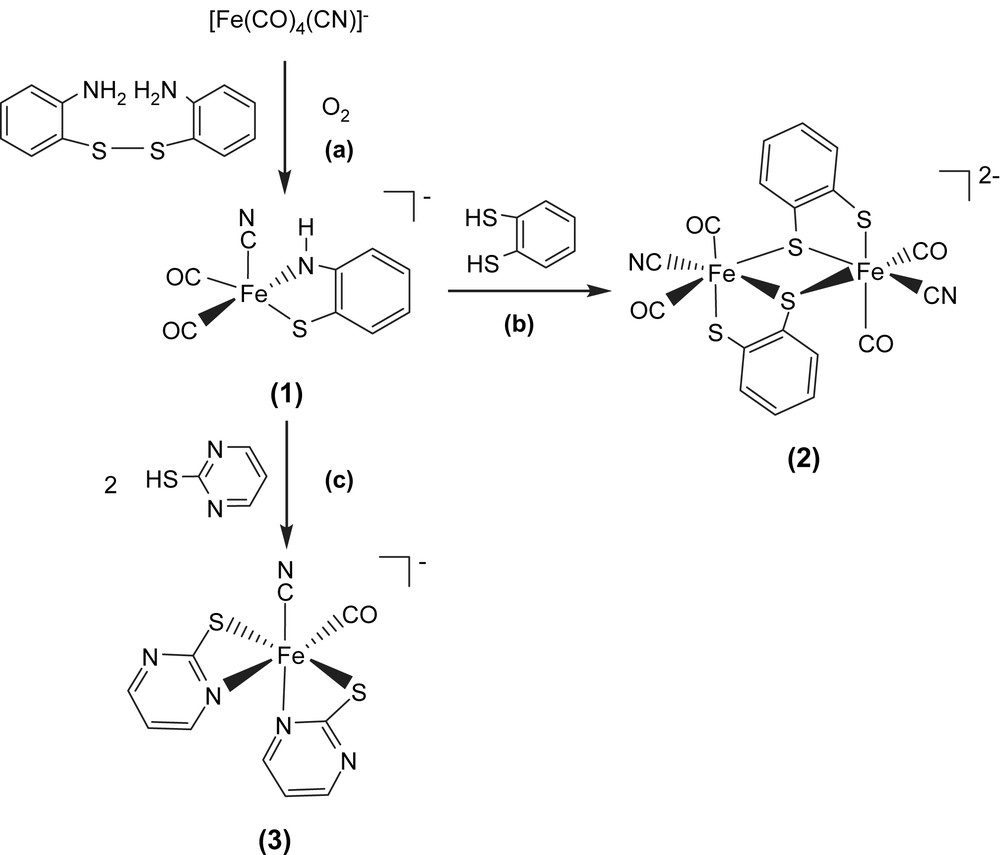
The dinuclear Fe(II)–thiolate cyanocarbonyl complex [PPN]2[(CN)(CO)2Fe(μ-S,S–C6H4)]2 (2) was prepared in a one-step synthesis by treating 1,2-benzenedithiol with complex 1 in THF under N2 atmosphere at −10 °C (Scheme 1b). Complex 2 was isolated as an air-stable brown solid at −10 °C. The dinuclear [(CN)(CO)2Fe(μ-S,S–C6H4)]22− is electronically dianionic and therefore both irons should be divalent. Each iron atom is surrounded pseudo-octahedrally by two bridging thiolates, one terminal thiolate, two terminal carbonyl groups, and one cyanide ligand. The Fe(II)⋯Fe(II) distance of 3.472 Å in complex 2 is longer than those reported for the diiron subcluster active site (2.6 and 2.62 Å) of [Fe] hydrogenases [3,4]. The IR spectrum of complex 2 in CH2Cl2 reveals a weak absorption band for the CN− ligands at 2100 cm−1, and two strong absorption bands for the CO groups at 1960 and 2013 cm−1 [14–16]. When the CH2Cl2 solution of complex 2 was exposed to 13CO at 0 °C, stretching bands ν(13CO) at 1968 and 1915 cm−1 appeared within 10 min. Reappearance of the 2013 and 1960 cm−1 bands on removal of the 13CO and replacement with 12CO atmosphere demonstrated reversibility of CO ligand lability of complex 2.
In contrast, protonation of complex 1 by 2 equiv of 2-mercaptopyrimidine in THF under N2 at ambient temperature yielded hexacoordinated iron(II)–thiolate cyanocarbonyl complex [PPN][Fe(CO)(CN)(S–C4H3N2)2] (3) with two anionic [S–C4H3N2]− ligands bound to the Fe(II) ion in a bidentate manner (S,N-bound) (Scheme 1c). The IR spectrum of complex 3 in CH3CN displayed a weak νCN band at 2090 cm−1 and a broad strong νCO band at 1945 cm−1, well within the range observed for [Fe] hydrogenase isolated from D. vulgaris [5]. The reversibility of the CO ligand lability of complex 3 was demonstrated by exposing the CH2Cl2 solution of complex 3 to 13CO. The IR νCO peak at 1948 cm−1 shifted to a single absorbance at 1904 cm−1 [14]. The vibrational spectroscopies of the Fe(CO)2(CN) and Fe(CO)(CN) fragments (νCN ranges from 2087 to 2106 cm−1, νCO ranges from 1928 to 2013 cm−1) found in complexes 1–3 may be regarded as spectroscopic references of [Fe] hydrogenases in the various enzymatic states (Table 1).
Selected infrared data for complexes 1–3 and hydrogenases
| Compounds | νCN, cm−1 | νCO, cm−1 | Ref. |
| 1 | (CH3CN) 2099 | 1997, 1933 | [14] |
| (THF) 2101 | 1992, 1928 | ||
| (CH2Cl2) 2095 | 2000, 1936 | ||
| 2 | (CH3CN) 2102 | 2011, 1961 | [14] |
| (THF) 2106 | 2008, 1950 | ||
| (CH2Cl2) 2100 | 2013, 1960 | ||
| 3 | (CH3CN) 2090 | 1945 | [14] |
| (THF) 2094 | 1942 | ||
| (CH2Cl2) 2087 | 1948 | ||
| D. vulgaris, [Fe] hydrogenase | (Air) 2106, 2087 | 2007.5, 1983, 1847.5 | [5a] |
| (H2/Ar) 2095, 2079 | 2016, 1972, 1965, 1940 | ||
| (H2/CO) 2096, 2088 | 2016, 1971, 1964 | ||
| (H2) 2079, 2041 | 1965, 1941, 1916, 1894 | ||
| Chromatium. vinosum, [NiFe] hydrogenase | 2093, 2083 | 1945 | [1c] |
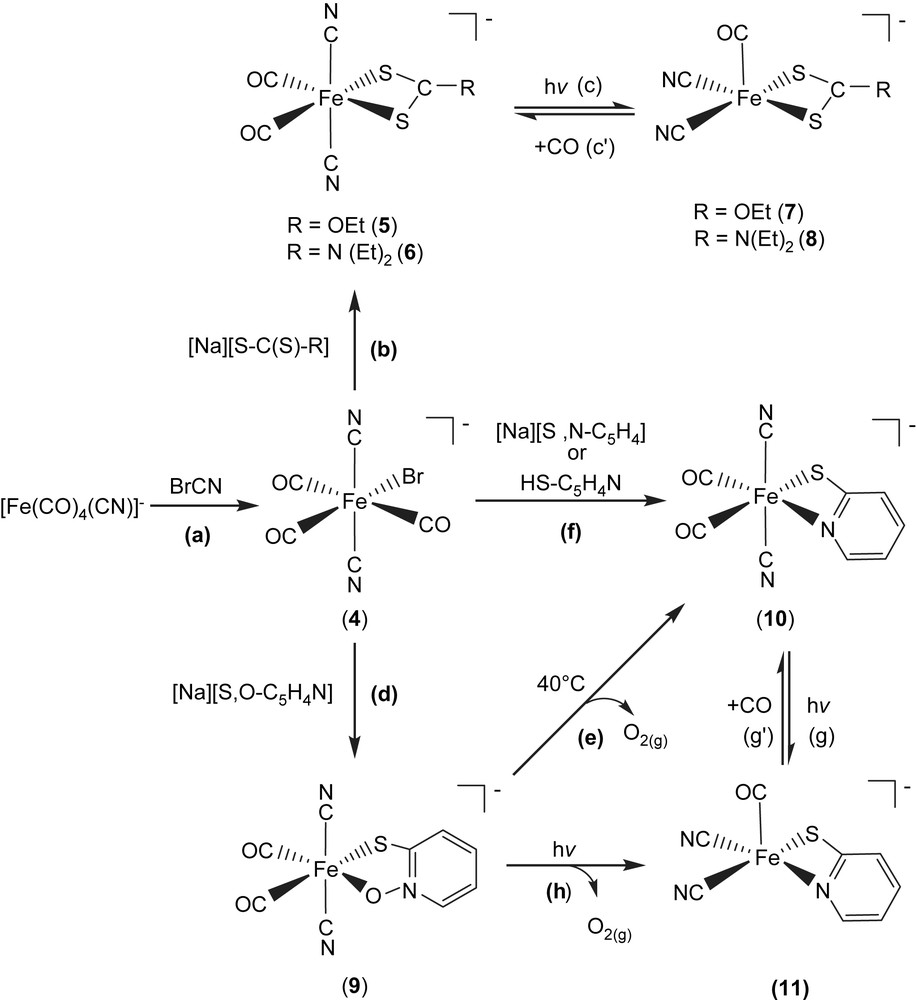
Upon cyanogen bromide reacted with [Fe(CO)4(CN)]− [13] in THF at room temperature, air-stable hexacoordinated Fe(II) complex [PPN][(CN)2(CO)3Fe(Br)] (4) was isolated (Scheme 2a). Subsequent reactions of complex 4 with [Na][S–C(S)–R] (R = OEt, N(Et)2) in THF/CH2Cl2 produced the air and thermally stable, yellow trans,cis-[PPN][(CN)2(CO)2Fe(S,S–C–R)] complexes (R = OEt (5), N(Et)2 (6)) individually (Scheme 2b) [17]. The IR νCO/νCN data of complexes 4–6 are shown in Table 2 [17]. Upon photolysis of THF solution of complex 5 (or 6) at room temperature, the IR νCO and νCN stretching frequencies appeared is consistent with the formation of the coordinatively unsaturated [PPN][(CN)2(CO)Fe(S,S–COEt)] (7) ([(CN)2(CO)Fe(S,S–CN(Et)2)]− (8)) (Table 2) with two cyanide ligands occupying cis positions (Scheme 2c) [17]. On the basis of density functional theory (DFT) calculation [17], it was found that the square pyramidal complexes 7 and 8 having vacant sites trans to the CO ligand are relatively stable [17]. The anionic complexes 7, 8 and Ni–A/Ni–C states in [NiFe] hydrogenase from D. gigas exhibit a similar one-band pattern in the νCO region and two-band pattern in the νCN region individually [17,18], but at different positions, 1996 vs (νCO), 2113 w, 2105 w (νCN) cm−1 (THF) for complex 7, 1985 vs (νCO), 2109 w, 2102 w (νCN) for complex 8 (Table 2), and 1947 vs (νCO), 2093 w, 2083 w (νCN) cm−1 for Ni–A state [NiFe] hydrogenases isolated from D. gigas, which may be accounted for by the distinct electronic effects between [S,S–C–R]− and cysteine ligands [18]. Also, this result is consistent with the conclusion, reported by Darensbourg et al., that the CO vibrational frequency to the electron density changes around Fe(II) center is about 2.6 times more sensitive than that for CN− [18c].
Selected infrared data for complexes 4–11 [17]
| Compounds | νCN, cm−1 (THF) | νCO, cm−1 (THF) |
| 4 | 2139 vw, 2127vw | 2099 m, 2056s, 2035 m |
| 5 | 2122 vw, 2112 w | 2038 vs, 1984vs |
| 6 | 2119 vw, 2112 w | 2027 vs, 1973 vs |
| 7 | 2113 w, 2105 w | 1996 vs |
| 8 | 2109 w, 2102 w | 1985 vs |
| 9 | 2121 vw, 2109 w | 2041 vs, 1982 vs |
| 10 | 2124 vw, 2113 w | 2036 vs, 1983 vs |
| 11 | 2111 w, 2103 w | 1996 vs |
In a similar fashion, when a THF solution of complex 4 was treated with 1 equiv of [Na][S,O–C5H4N], the d6 Fe(II) trans,cis-[PPN][(CN)2(CO)2Fe(S,O–C5H4N)] (9) with a five-membered ring (bidentate, S,O-bound) was isolated (Scheme 2d). Subsequently, the four-membered ring trans,cis-[PPN][(CN)2(CO)2Fe(S,N–C5H4)] (10) (bidentate, S,N-bound) along with the presumed O2 (g) was obtained via thermolytic conversion of complex 9 on heating (40 °C) in THF (Scheme 2e). The S,N-bound complex 10 was alternatively obtained via ligand-substitution reaction, a straightforward reaction of complex 4 with 1 equiv of [Na][S,N–C5H4] (or HS–C5H4N) in THF (Scheme 2f). To further add credibility to the CO ligand reversibly bound to the FeII site in these model compounds, a straightforward photolysis of THF solution of complex 10 was also conducted at room temperature, formation of five-coordinated [(CN)2(CO)Fe(S,N–C5H4)]− (11) complex with two cyanide ligands occupying cis positions was observed (Scheme 2g). Obviously, photolysis of THF solutions of complexes 5, 6, and 10 at room temperature led to the formation of coordinatively unsaturated iron(II)–thiolate dicyanocarbonyl complexes 7, 8, and 11, respectively, with two cyanides occupying cis positions and a vacant site preference trans to the CO ligand [17]. Additionally, density functional theory (DFT) calculations also suggest the architecture of five-coordinated complexes 7, 8, and 11 with a vacant site trans to the CO ligand and two CN− ligands occupying cis positions serves as a conformational preference [17]. Presumably, the strong σ-donor, weak π-acceptor CN− ligands play a major role in creating and stabilizing a five-coordinate iron(II) center with a vacant coordination site [17–19]. The reversibility of CO ligand binding demonstrates that complexes 5/7, 6/8, and 10/11 are photochemically interconvertible. This result may implicate the involvement of iron site in the mechanism of hydrogen activation, and can be useful in exploring the key step in H2 uptake mechanism in [NiFe] hydrogenases. The vibrational spectra of the [Fe(II)(CN)2(CO)] and [Fe(II)(CN)2(CO)2] units (νCO and νCN) found in complexes 5–8 may be regarded as spectroscopic references for a variety of [NiFe] hydrogenase enzymatic states, and the exogenously added CO inhibited state, respectively [8–12].
Scrutiny of the coordination chemistry of iron(II)–thiolate cyanocarbonyls [Fe(II)(CO)x(CN)y(SR)z]n– reveals that certain combinations of thiolates and cyanide ligands (3 ≤ y + z ≤ 4) bound to Fe(II) play an important role in stabilizing the [Fe(II)(CO)x(CN)y(SR)z]n– species [14,17,19], as summarized in Table 3, and this could point the way to understand the reasons for Nature's choice of combinations of these ligands in hydrogenases.
Certain combinations of thiolates and cyanide ligands (3 ≤ y + z ≤ 4) bound to Fe(II) of [FeII(CO)x(CN)y(SR)z]n− complexes
| Compounds | y + Thiolate | Ref. |
| 5 | 2 + Bidentate | [17] |
| 6 | 2 + Bidentate | [17] |
| 7 | 2 + Bidentate | [17] |
| 8 | 2 + Bidentate | [17] |
| [Fe(CO)2(CN)(CS3-S,S)]− | 1 + Bidentate | [19a] |
| [Fe(CO)2(CN)(S(CH2)2S(CH2)2–S-S,S,S)]− | 1 + Tridentate | [19a] |
| [Fe(CO)2(CN)3(SC6H4Br)]2− | 3 + Monodentate | [19a] |
| [Fe(CO)2(CN)2(SPh)2]2− | 2 + 2(Monodentate) | [19b] |
| [Fe(CO)(CN)2(S2C6H4-S,S)]2− | 2 + Bidentate | [19b] |
| [Fe(CO)2(CN)2(S2C6H4-S,S)]2− | 2 + Bidentate | [19b] |
2.2 Dinuclear iron(II)–cyanocarbonyl complexes with bridging ethylthiolates
When complex 4 was reacted directly with [Na][SEt] in THF at room temperature, dinuclear d6 Fe(II) complex [PPN]2[Fe(CN)2(CO)2(μ-SEt)]2 (12) was isolated. Attempts to detect the extremely unstable intermediate, the mononuclear [Fe(CN)2(CO)3(SEt)]−, by FTIR were unsuccessful (Scheme 3a and b). The Fe(II)–Fe(II) distance of 3.505 Å in complex 12 is considerably longer than those reported for the dinuclear iron subcluster (2.6 and 2.62 Å) of the [Fe] hydrogenase active site [3,4]. Here the lack of the third bridging CO ligand and the nearly square planar FeS2Fe core in complex 12, in contrast to butterfly structure of the active-site FeS2Fe core observed in [Fe] hydrogenases, are adopted to rationalize the significantly longer Fe(II)–Fe(II) distance in complex 12. The dinuclear [Fe(CN)2(CO)2(μ-SEt)]22− is electronically dianionic, and therefore both irons should be divalent. Subsequent reactions of complex 12 with [PPN][SEt] in a 1:1 molar ratio in THF produced the thermally stable [PPN][Fe2(CN)2(CO)4(μ-SEt)3] (13), and the known trans-[PPN]2[Fe(CN)4(CO)2] as identified by IR and X-ray diffraction (Scheme 3c) [6c,20,21].
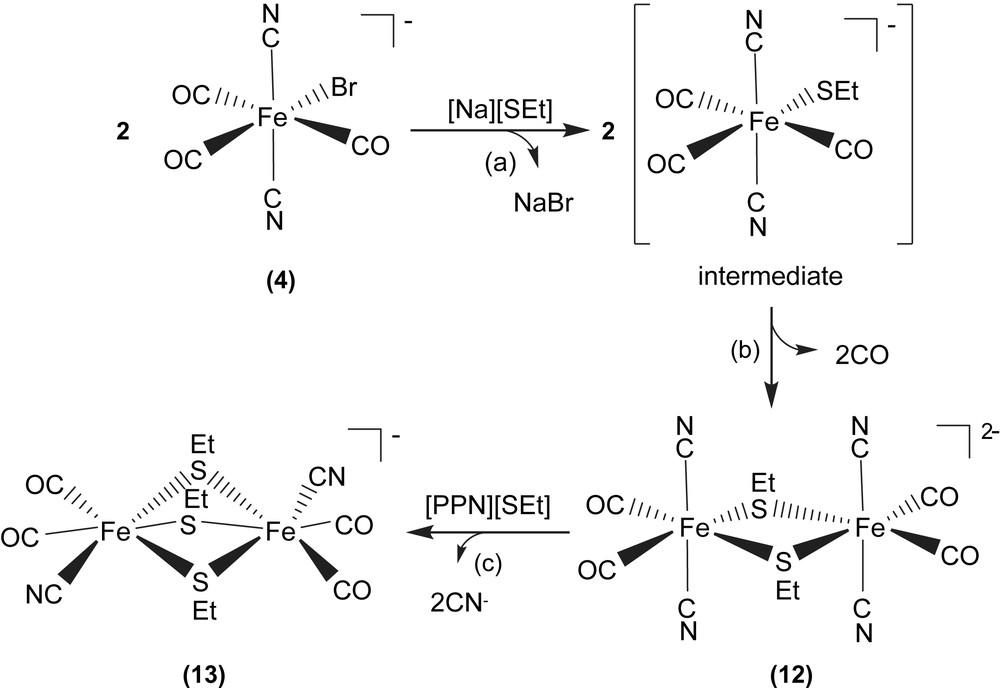
The Fe(II)–Fe(II) distance decreases from 3.505 Å (complex 12) to 3.073 Å (complex 13). The considerably longer Fe(II)–Fe(II) distance of 3.073 Å in complex 13, compared to the reported Fe–Fe distances of 2.6/2.62 Å in [Fe] hydrogenases isolated from Dd and Cp [3,4], can be attributed to the presence of the third bridging ethylthiolate, instead of π-accepting CO-bridging ligand as observed in [Fe] hydrogenases. Also, it is noticed that the Fe(II)–Fe(II) distances of complexes [Fe2(S2C2H4)(μ-CO)(CN)2(CO)2(PPh3)2] containing bridging CO [6c], and [Fe2(S2C2H4)(μ-H)(CO)2(PMe3)4]+ containing bridging hydride [20c] were comparable to 2.6 Å, the reported Fe–Fe distances of [Fe] hydrogenases isolated from Dd and Cp [3,4]. The comparisons of Fe–Fe distances of the reported dinuclear iron–thiolate complexes are collected in Table 4 [3,6c,20].
The selected dinuclear iron–thiolate complexes
| Compounds | Fe–Fe (Å) | νCO; νμ-CO (cm−1) | Ref. |
| D. desulfuricans [Fe] hydrogenase | 2.6 | 2016, 1972, 1963, 1811 | [3] |
| 12 | 3.505 | 2029, 1981 (THF) | [20a] |
| 13 | 3.073 | 2029, 2014, 1968 (THF) | [20a] |
| Fe2(S2C2H4)(μ-CO)(CN)2(CO)2(PPh3)2 | 2.546 | 2004, 1993; 1904 (μ-CO) | [6c] |
| [Fe2(S2C2H4)(μ-H)(CO)2(PMe3)4]+ | 2.610 | 1940, 1928 (CH3CN) | [20b] |
| [Fe2(S2C2H4)(μ-CO)(H)(CO)(PMe3)4]+(μ-CO) | 2.566 | 1940; 1874 (CH3CN) | [20b] |
| [Fe2(MeSCH2C(Me)(CH2S)2)(CN)2(CO)3(μ-CO)]− | 1945, 1978, 2030; 1790 (μ-CO) | [20c,d] | |
| [Fe2(MeSCH2C(Me)(CH2S)2)(CN)2(CO)3(μ-CO)]2− | 1878, 1919, 1957; 1780 (μ-CO) | [20c,d] |
3 Nickel–thiolate complexes
3.1 Heterobimetallic nickel–iron complexes
Interesting challenges are in biomimicry of the [(SCys)2Ni(μ-SCys)2Fe(CN)2(CO)] active site of [NiFe] hydrogenase. Reaction of the [Fe(NO)2(SePh)2]− (14) anion with dimeric [Ni(μ-SCH2CH2SCH2CH2S)]2 in the presence of additional NO2− produced the neutral heterobimetallic [(ON)Ni{(μ-SCH2CH2)2S}Fe(NO)2] (15) (Scheme 4) [22]. According to the Feltham–Enemark notation [23], the distorted tetrahedral iron dinitrosyl group of complex 15 was assigned as {Fe(NO)2}9. The IR νNO stretching frequencies of complex 15 displayed at 1805, 1767 and 1725 cm−1 (CH2Cl2) (1798, 1763 and 1723 cm−1 in THF) are consistent with an assignment for {Ni(NO)}9 Ni0(NO)+ as is known for analogous phosphine derivatives, P3Ni0(NO)+ [24]. The Ni–Fe distance of 2.8 Å in complex 15 is comparable to the natural heterobimetallic Ni–Fe active site found in the [NiFe] hydrogenase enzyme having a Ni–Fe distance of 2.9 Å.
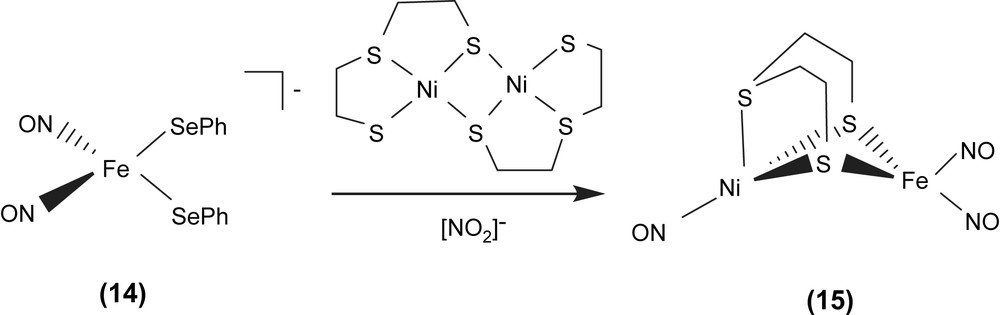
In particular, complex [Fe(CO)2(CN)2(μ-pdt)Ni(S2CNEt2)]− with bridging 1,3-propanedithiolate ligand and iron center coordinated by two CO and two CN−, the precise model of the active site of the reduced-form [NiFe] hydrogenase, was isolated and characterized by IR and single-crystal X-ray structure [22f]. The selected heterodinuclear Ni–Fe–thiolate complexes are collected in Table 5.
The selected heterodinuclear Ni–Fe–thiolate complexes
| Compounds | Ni/Fe valence | Ni–Fe distance (Å) | Ref. |
| D. gigasox | 3+, 2+ | 2.9 | [1a,1b] |
| 15 | 0, 1+ | 2.8 | [22a] |
| [{Fe(‘S3’)(CO)(PMe3)2-S,S′}Ni(‘S2’)]a | 2+, 2+ | 3.32 | [22b] |
| [Ni(L)Fe(CO)4]b | 0, 2+ | 3.76 | [22c] |
| [{Ni(SCH2CH2CH2S)(dppe)-S,S′}Fe(CO)3]c | 0, 2+ | 2.47 | [22d] |
| [{Fe(NS3)(CO)2-S,S′}NiCl(dppe)]d | 2+, 2+ | 3.31 | [22e] |
| [Fe(CO)2(CN)2(μ-pdt)Ni(S2CNEt2)]−e | 2+, 2+ | 2.9 | [22f] |
| [(dppe)Ni(pdt)Fe(Cp)(CO)]+ | 2.78 | [22g] |
a ‘S3’2− = bis(2-mercaptophenyl)sulfide(2−); ‘S2’2− = 1,2-benzenedithiolate.
b L = N,N′-bis(2-mercaptoethyl)-1,5-diaza-cyclooctane.
c dppe = 1,2-bis(diphenyl-phosphino)ethane.
d NS3 N(CH2CH2S)33−.
e pdt = 1,3-propanedithiolate.
3.2 Mononuclear nickel(II) carbonyl complexes
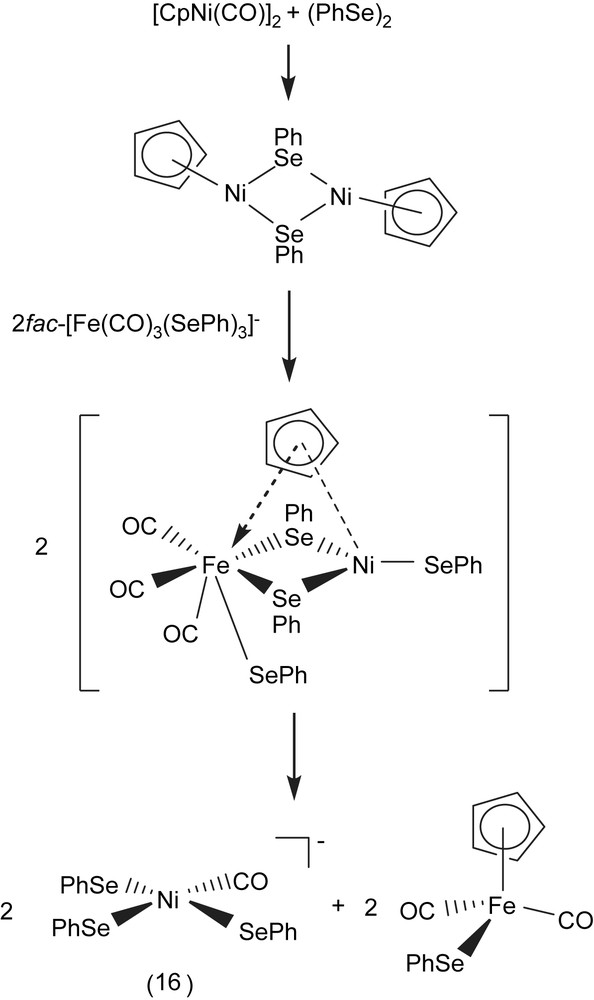
A THF mixture solution of [CpNi(CO)]2 and (PhSe)2 added to the acetonitrile (THF) solution of fac-[PPN][Fe(CO)3(SePh)3] (1:1:2 molar ratio) yielded [PPN][Ni(CO)(SePh)3] (16) and byproduct [CpFe(CO)2(SePh)] [25,26]. The sequences of reactions given in Scheme 5 rationalizes the formation of complex 16 and [CpFe(CO)2(SePh)]. The oxidative addition of diphenyldiselenide across the Ni–Ni bond of [CpNi(CO)]2, and nucleophilic attack of an incoming tridentate motif fac-[Fe(CO)3(SePh)3]− resulting in the bridged-selenolate cleavage was presumed to be accompanied by cyclopentadienyl [C5H5]− ligand-transfer from Ni(II) to Fe(II), the concomitant Fe(II)–SePh bond cleavage, and a labile carbonyl ligand shifting from Fe(II) to Ni(II) led to the formation of [Ni(CO)(SePh)3]− and CpFe(CO)2(SePh) [26]. Compared to the νCO band of pentacoordinated Ni(I)/Ni(II) carbonyl complexes [Ni(NS3tBu)(CO)]+ (2026 cm−1) [27], [Ni(DAPA)(SePh)2(CO)]– (2024 cm−1) [28], and [Ni(PS3∗)(CO)]− (2029 cm−1) [29], complex 16 displays a IR νCO band at 2023 cm−1. When a THF solution of [Ni(II)(CO)(SePh)3]− was purged with 13CO, the IR νCO peak at 2023 cm−1 immediately shifts to a single absorbance at 1976 cm−1. Reaction of complex 16 with 1 equiv of (PhS)2 in THF results in the formation of the extremely air-sensitive [Ni(CO)(SPh)(SePh)2]− (compared to [Ni(CO)(SePh)3]−). In a similar fashion, [Ni(CO)(SePh)3]− was reacted with (PhS)2 in a 1:2 molar ratio to yield [Ni(CO)(SPh)2(SePh)]− [30].
3.3 Mononuclear nickel(II)–thiolate complexes with pendant thiol
Study on the structures and reactivity of Ni(II)–thiol and Ni(III)–thiolate complexes may be useful in interpreting the catalytic cycle of H2 activation in [NiFe] hydrogenase. In model compounds, an electrochemical study provided evidence of formation of a Ni(III)–H species generated by one-electron reduction of a nickel(II) macrocyclic complex accompanied by protonation [31]. Reaction of complex 16 with tris(2-phenylthiol)phosphine (P(o-C6H4SH)3) produced the four-coordinated [PPN][Ni(SePh)(P(o-C6H4S)2(o-C6H4SH))] (17) [32]. Complex 17 containing pendant thiol stabilized by an intramolecular S–H proton directly interacting with both nickel and sulfur atoms was synthesized and characterized by IR, 1H NMR, UV–vis, and X-ray diffraction (Scheme 6). The infrared KBr solid spectrum of complex 17 shows one broad stretching band (2273 cm−1) in the νS–H region. The lower energy νS–H band of complex 17 shifted by ∼215 cm−1 from that of the free ligand P(o-C6H4SH)3 (2488 cm−1) implies the specific [Ni–S⋯H–S]/[Ni⋯H–S] interactions [32,33]. Intramolecular [Ni–S⋯H–S]/[Ni⋯H–S] interactions in complex 17 also result in the 1H NMR chemical shift of the SH group, shifting from δ 4.07 (d) ppm (CDCl3) in free ligand P(o-C6H4SH)3 to δ 8.079 (d) ppm (S–H) (CD2Cl2) in complex 17 [32,33]. The intramolecular interaction is best interpreted in terms of the thiol proton interacting with both sulfur and nickel atoms, where the proton is bound to sulfur and “attracted” by the Ni(II) and the second sulfur atom, that is, a combination of [Ni–S⋯H–S]/[Ni⋯H–S] interactions. The H/D exchange reaction between complex 17 and D2O occurred in CH3CN–THF solution (1:3 volume ratio) at 5 °C as indicated by IR and 2H NMR. The IR νS–H at 2273 br cm−1 shifting to absorbance at 1676 br cm−1 (νS–D, KBr) is roughly consistent with the calculated position, based only on the difference in masses between S–H and S–D. The interactions [Ni–S⋯H–S]/[Ni⋯H–S] may contribute to the stabilization of Ni(II)–thiol complex 17.
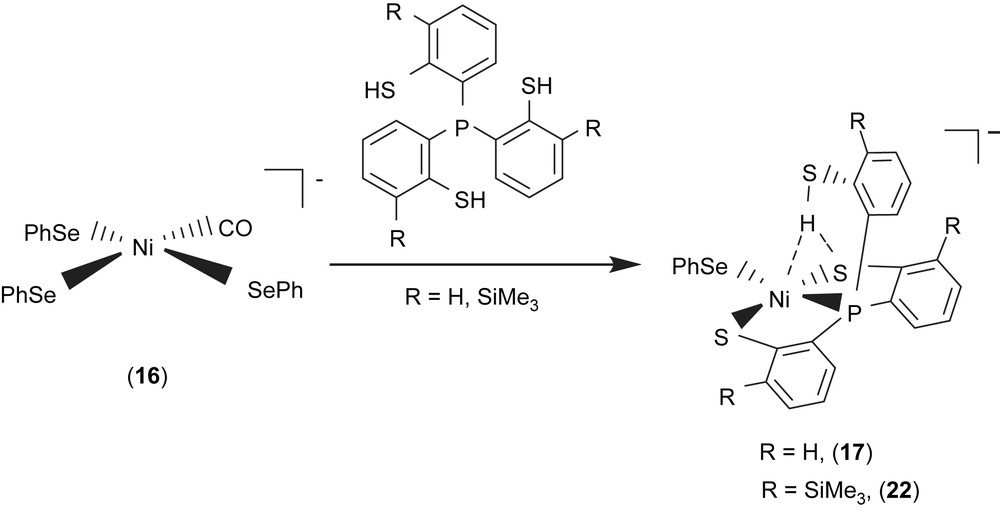
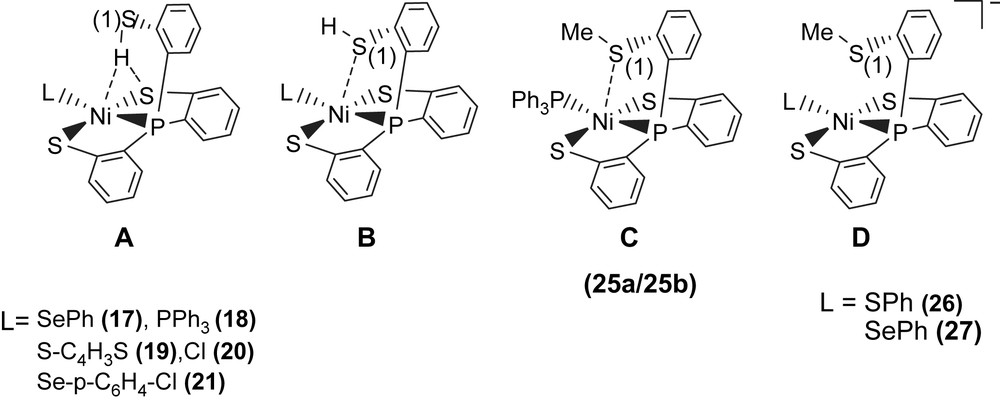
A comparison of IR νS–H and 1H NMR data of analogues [PPN][Ni(X)(P(o-C6H4S)2(o-C6H4SH))] (X = PPh3 (18), S–C4H3S (19), Cl (20), Se–p-C6H4–Cl (21)) and SiMe3-substituted complexes [PPN][Ni(X)(P(o-C6H3–3-SiMe3–2-S)2(o-C6H3–3-SiMe3–2-SH))] (X = SePh (22), Cl (23)) with intramolecular [Ni–S⋯H–S]/[Ni⋯H–S] interactions is provided in Table 6 (Scheme 6) [32,34–36]. These results show that electronic modulations of the monodentate ligand of complexes 17–23 are capable of inducing different degree of interacting strength of the intramolecular [Ni–S⋯H–S] and [Ni⋯H–S] interactions.
Selected infrared and 1H NMR data for complexes 17–23 and 35
| Compounds | νS–H, cm−1 (KBr) | 1H NMR, δ | Ref. |
| P(o-C6H4SH)3 | 2488 | 4.07 (d) (CDCl3) | [33] |
| P(C6H3–3-SiMe3–2-SH)3 | 2466 | 4.58 (br) (CDCl3) | [33] |
| 17 | 2273 | 8.079 (d) (CD2Cl2) | [32] |
| 18a | 2393 | 5.908 (s) (CDCl3) | [35] |
| 18b | 2240 | 5.908 (s) (CDCl3) | [35] |
| 19 | 2283 | 8.39 (d) (C4D8O) | [32] |
| 20 | 2300 | 5.808 (s) (CDCl3) | [35] |
| 21 | 2288 | 8.39 (d) (C4D8O) | [35] |
| 22a | 2250 | 8.59 (br) (C4D8O) | [39] |
| 22b | 2137 | 8.59 (br) (C4D8O) | [39] |
| 23a | 2287 | 8.54 (br) (C4D8O) | [39] |
| 23b | 2235 | 8.54 (br) (C4D8O) | [39] |
| 35 | 2385 | 6.24 (d) (CDCl3) | [39] |
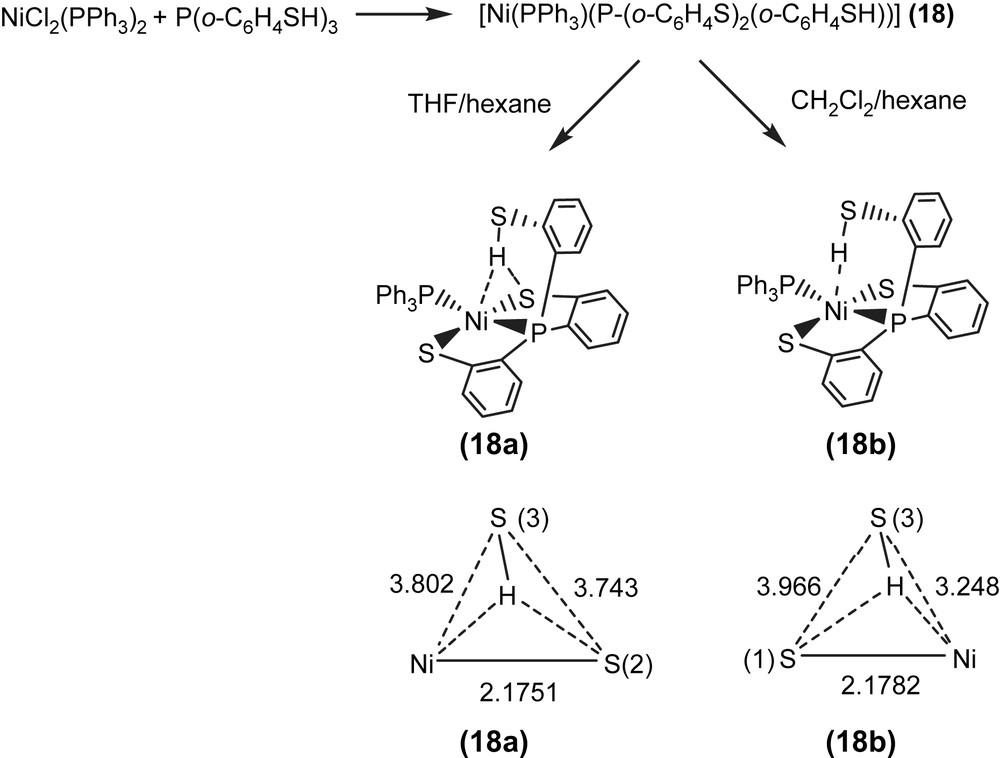
Alternatively, reaction of NiCl2(PPh3)2 and tris(2-phenylthiol)phosphine (P(o-C6H4SH)3) in THF also led to the isolation of the red-brown four-coordinated Ni(II) [Ni(PPh3)(P(o-C6H4S)2(o-C6H4SH))] (18) (Scheme 7) [35]. When a THF solution of complex 18 was recrystallized with THF–hexane, complex 18a displaying a broad νS–H stretching band at 2393 cm−1 (KBr) was isolated and characterized by X-ray crystallography (Scheme 7). In contrast, the diffusion of hexane into a CH2Cl2 solution of complex 18 yielded complex 18b with a broad νS–H stretching band at 2240 cm−1 (KBr), characterized by single-crystal diffraction (Scheme 7). The IR spectra of complexes 18a and 18b display broad νS–H stretching bands at 2393 and 2240 cm−1 (KBr), which are ∼90 and ∼250 cm−1 lower than the free ligand 2488 cm−1 (KBr), respectively. This lower νS–H stretching frequency may imply the existence of intramolecular [Ni–S⋯H–S]/[Ni⋯H–S] interactions. In complex 18a, the acute (C–S–H) bond angle of 95.04° and the similar distances of Ni⋯S(3) (3.802 Å) and S(2)⋯S(3) (3.743 Å) suggested that the proton of S(3)–H points toward the middle of the Ni and S(2) atoms, and interacts with both sulfur (S(2)) and nickel atoms (i.e., a combination of the intramolecular [Ni–S⋯H–S]/[Ni⋯H–S] interactions, Scheme 7). In contrast, the Ni⋯S(3) distance of 3.248 Å is significantly shorter than the S(1)⋯S(3) distance of 3.966 Å in complex 18b. Compared to complex 18a, the acute (C–S–H) bond angle of 99.64° and a lower νS–H stretching band (2240 cm−1 (KBr)) in complex 18b may indicate that the thiol proton tends to interact directly with the nickel atom (i.e., a [Ni⋯H–S] interaction) (Scheme 7) [35]. Similarly, two forms of crystalline products 22a/23a (plate-shaped, red-brown crystals) and 22b/23b (block-shaped, dark-red crystals) were also obtained when 22/23 were recrystallized from THF–diethyl ether and THF–hexane, respectively, at room temperature [36].
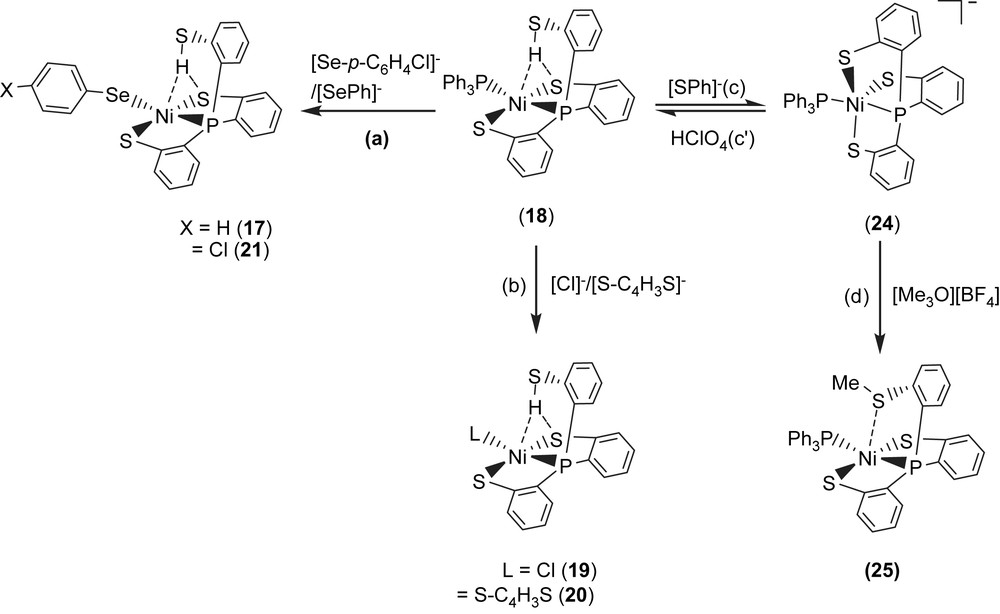
Complex 18 serves as a precursor for the syntheses of the anionic mononuclear Ni(II)–thiol complexes 19–21 via a ligand-displacement reaction. Addition of [SePh]− (or [S–C4H3S]−, [Cl]− and [Se–p-C6H4–Cl]−) to the CH3CN–THF solution (1:3 volume ratio) of complex 18 at 0 °C for 4 h afforded the ligand-displacement complex 17 (or 19–21) identified by IR, NMR, UV–vis and single-crystal X-ray diffraction, respectively, (Scheme 8a and b) [35]. In contrast to the ligand displacemeant occurring in the reaction of complex 18 and the anionic [SePh]− (or [Cl]−/[Se–p-C6H4–Cl]−), addition of [SR]− (R = Ph, Et) to complex 18 yielded mononuclear [PPN][NiII(PPh3)(P(o-C6H4S)3)] (24) by the liberation of thiophenol (or ethanethiol) identified by 1H NMR spectra (Scheme 8c). Protonation of complex 24 in THF at 0 °C by HClO4 produced complex 18 (Scheme 8c′). Treatment of [Me3O][BF4] and complex 24 in a CH3CN–THF solution (1:3 volume ratio) yielded the [NiII(PPh3)(P(o-C6H4S)2(o-C6H4–SCH3))] (25) identified by single-crystal X-ray diffraction (Scheme 8d) [35]. On the basis of the single-crystal X-ray diffraction, two different conformational isomers of complex 25 were isolated. The green crystals of complex 25a were obtained by diffusion of hexane into the THF solution of complex 25 at 0 °C. Interestingly, diffusion of hexane into the THF solution of complex 25 at 30 °C produced green crystals (complex 25a) and red crystals which were identified by single-crystal X-ray diffraction as complex 25b (Scheme 9). Each crystal form shows identical 1H NMR spectroscopic properties (methyl (CH3) proton resonance at 2.021 ppm) (CDCl3)) when complexes 25a and 25b were redissolved, respectively, in CDCl3 at room temperature [35]. Obviously, these two conformational isomers (25a and 25b) are in equilibrium via intramolecular rearrangement controlled by the temperature of solution. In contrast to the temperature-dependent interconversion of the conformational isomers 25a and 25b, the temperature-independent four-coordinated Ni(II)–thioether complexes [Ni(II)(L)(P(o-C6H4S)2(o-C6H4–SCH3))]− (L = SPh (26), SePh (27)) were isolated when complex 25 was reacted with [SPh]− and [SePh]−, respectively, in THF at room temperature overnight (Scheme 10) [35].
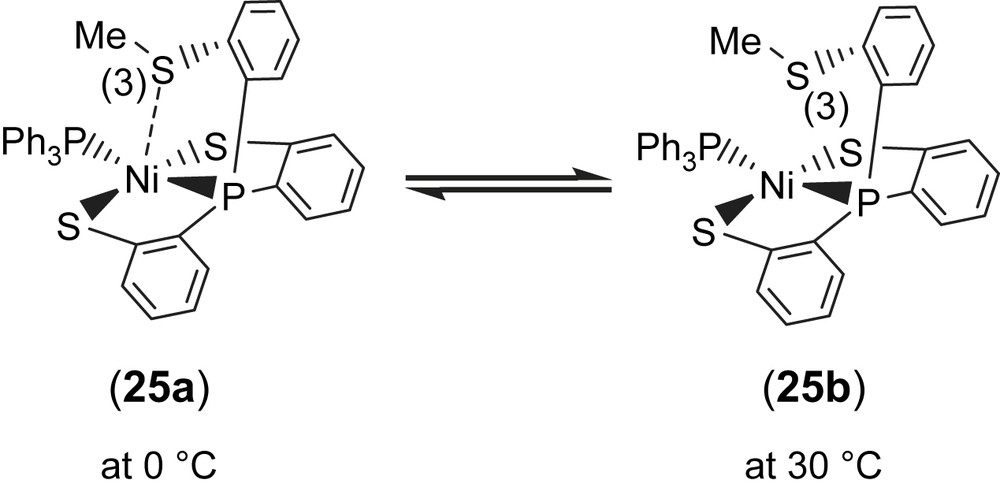
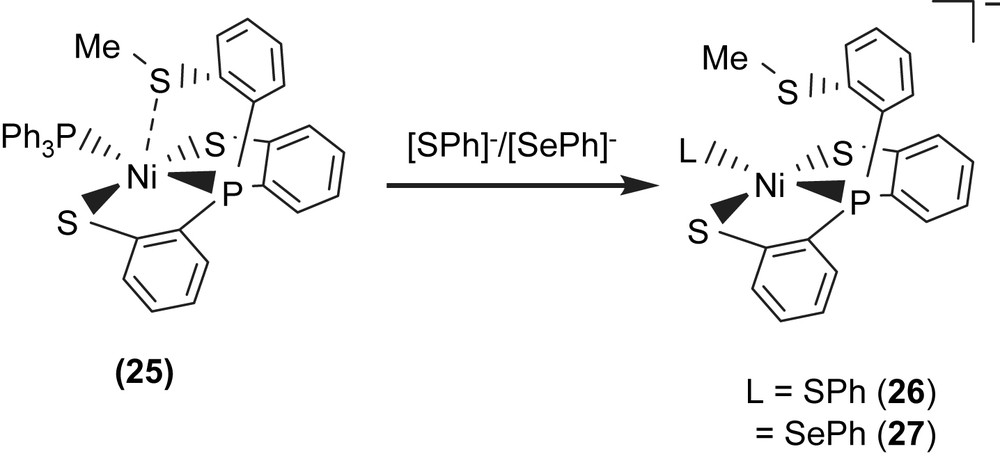
Compared to the Ni⋯S(3) distances of 3.802 and 3.248 Å observed in complexes 18a and 18b (Scheme 7), the single-crystal X-ray structure revealed that thioether sulfur semibonding to Ni(II) (Ni⋯S(3) distance of 2.5397 Å) may exist in complex 25a and that there is no interaction between Ni and S(3) (Ni⋯S(3) distance of 2.9135 Å) in complex 25b (Scheme 9). Obviously, on the basis of the 1H NMR spectra and single-crystal X-ray diffractions, the intramolecular interaction between NiII and the thioether sulfur atom became weaker when the temperature was increased or the more electron-donating monodentate ligand was ligated to the Ni(II) center, as observed in the mononuclear Ni(II)–thioether complexes 25a, 25b, 26 (Ni⋯S(Me) distance of 3.258 Å), and 27 (Ni⋯S(Me) distance of 3.229 Å; Scheme 10). Presumably, increasing the electron density surrounding Ni should have the opposite effect on the interaction with RSH and RSR′. Increasing the electron density on Ni favors the interaction with S–H (Ni acts as a hydrogen-bond acceptor) and disfavors the interaction with S in SRS′ (RSR′ is an electron pair donor) [35].
In complexes 17–21, the presence of the intramolecular [Ni–S⋯H–S] interaction (or a combination of intramolecular [Ni–S⋯H–S] and [Ni⋯H–S] interactions) was verified in the solid state by the observation of a lower IR νS–H stretching frequency compared to that of the free ligand P(o-C6H4SH)3 (Table 6), and was subsequently confirmed by single-crystal X-ray diffraction. As shown in Scheme 11, single-crystal X-ray diffraction studies reveal that the Ni⋯S(1) distance falls into the range of 3.609–3.802 Å in Ni(II)–thiol complexes 17, 18a and 19–21 (Scheme 11, type A), compared to those of 2.540 and 2.914 Å in the Ni(II)–thioether complexes 25a and 25b (Scheme 11, type C). In contrast, the long Ni⋯S(1) distances (3.258 and 3.229 Å) found in complexes 26 and 27 indicated the absence of interaction between the Ni and S(1) atoms (Scheme 11, type D). These results rule out the existence of an intramolecular Ni⋯S(1) interaction (Ni⋯S(1) distance of 3.609–3.802 Å) in complexes 17, 18a and 19–21 (Scheme 11, type B) and may support the idea that the pendant thiol proton of complexes 17–21 were attracted by both nickel and sulfur of thiolate, resulting in combinations of intramolecular [Ni–S⋯H–S] and [Ni⋯H–S] interactions (Scheme 11, type A) [35].
3.4 Mononuclear nickel(III)–thiolate complexes
The mononuclear Ni(III) complex [PPN][Ni(SePh)(P(o-C6H4S)3)] (28) identified by 1H NMR, EPR, UV–vis, SQUID and X-ray diffraction was obtained upon addition of dry O2 to complex 17 (Scheme 12a). The 4.2 K EPR spectrum of complex 28 exhibits high rhombicities with three principal g values of 2.304, 2.091, and 2.0. There is no 31P hyperfine coupling observed under various conditions, even in ENDOR detection. The average g value (gav = 2.132) that is much higher than the spin-only g value 2.0023 indicates large orbital contribution to the paramagnetism, suggesting that the unpaired electron is associated primarily with the nickel ion [32,36]. The 1H NMR spectrum of complex 28 at 298 K shows the expected paramagnetic properties. The effective magnetic moment in the solid state by SQUID magnetometer was 1.91 μB for complex 28, which is consistent with the NiIII having a low-spin d7 electronic configuration in a distorted trigonal bipyramidal ligand field [32,36]. The 4.2 K EPR signals of complex 28 with three principal g values of 2.304, 2.091, and 2.0 are comparable with the g values near 2.31, 2.24, and 2.01 (for Ni–A) and 2.33, 2.16, and 2.01 (for Ni–B) ascribed to Ni(III) observed in the oxidized Ni–A as well as Ni–B states of [NiFe] hydrogenases, a formal Ni(III) in a electronic configuration. The most noteworthy characteristic of complex 28 is the existence of a reversible, metal-based reduction with the Ni(III/II) redox potential E1/2 = −0.67 V vs Ag/AgCl (ΔEp = 0.07 V, scan rate = 0.1 V/s) in MeCN, which is comparable to the Ni(III/II) redox potentials, ca. −390 to −640 mV vs SCE of [NiFe] hydrogenases [37]. The stable mononuclear Ni(III)–thiolate complex 28 is soluble in CH2Cl2/THF–CH3CN, and form a thermally sensitive, air-sensitive solution in organic solvents. On the basis of this investigation and related observations, it is presumed that the polarizable anionic thiolate/selenolate donor ligand and a relatively rigid tetradentate ligand in complex 28 place Ni(III) in an optimum electronic condition to minimize the possibility of autoreduction of Ni(III) and to prevent the formation of polynuclear nickel–thiolate species.
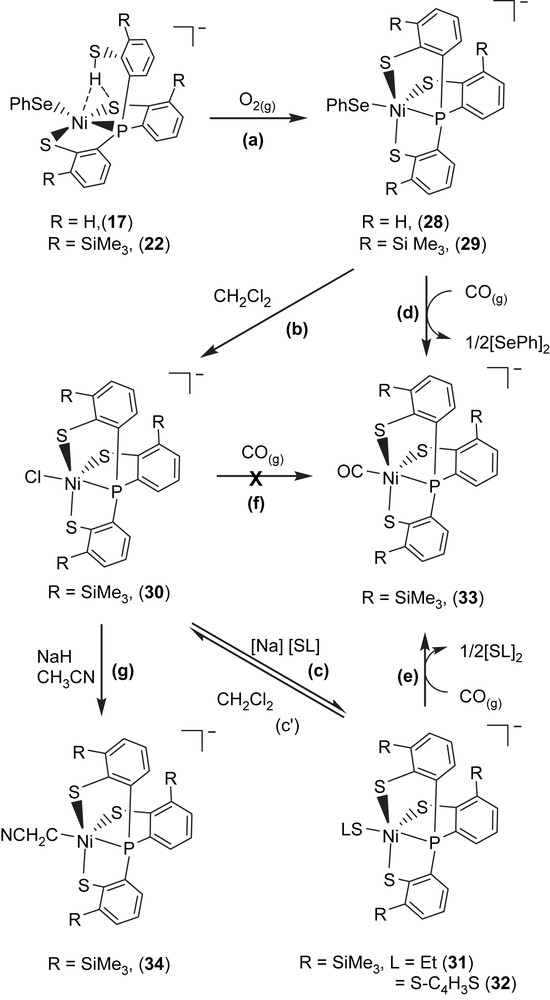
In a similar fashion, the mononuclear Ni(III) complex [PPN][Ni(SePh)(P(o-C6H3–3-SiMe3–2-S)3)] (29) was obtained upon addition of dry O2 to complex 22 [36]. As shown in Scheme 12b, addition of CH2Cl2 to complex 29 in the absence of dioxygen produced [PPN][Ni(III)(Cl)(P(C6H3–3-SiMe3–2-S)3)] (30). In displacement reaction, complexes [PPN][Ni(SR)(P(C6H3–3-SiMe3–2-S)3)] (R = ethyl (31), 2-thienyl (32)) containing coordinated ethylthiolate (or 2-thienylthiolate) were synthesized by adopting complex 30 serving as a precursor (Scheme 12c). The single-crystal X-ray structure shows a Ni(III)–SEt bond length of 2.273(1) Å in complex 31 is in the range of 2.12–2.28 Å for the Ni(III)–S bond lengths of the oxidized D. gigas hydrogenases [8–10]. The EPR spectra of complexes 29–31 are identical at 77 K and exhibit rhombicity with principal g values of 2.31, 2.09, and 2.0 [36].
When a THF–CH3CN (3:1 v/v) solution of complex 29 (or complexes 31 and 32) was treated with CO (1 atm) at ambient temperature, complex 29 (or complexes 31 and 32) completely transformed into complex [PPN][Ni(II)(CO)(P(C6H3–3-SiMe3–2-S)3)] (33) accompanied by the formation of diphenyldiselenide (diethyl disulfide, di(2-thienyl) disulfide) identified by 1H NMR spectrum (Scheme 12d and e). Obviously, CO molecule triggers the reductive elimination of the coordinated phenylselenolate of complex 29 (or ethylthiolate of complex 31, 2-thienylthiolate of complex 32) to yield Ni(II)–CO complex 33 and byproduct (PhSe)2 (or (SEt)2/(2-S–C4H3S)2), relevant to the observation that Ni–C state was converted into the Ni–L form under illumination, which subsequently transformed into Ni–CO state under exogenous CO condition in [NiFe] hydrogenase isolated from D. vulgaris Miyazaki F. [9c,d]. It is presumed that the σ-/π-electron-donating nature of the coordinated thiolates/selenolate converts Ni(III), a weaker π-donor, into a strong π-donor and favors Ni–CO bonding by reductive elimination [26,36,30]. This result implies that the Ni(III)–Cl bond of complex 30 is resistant to undergo reductive elimination, in contrast to the Ni(III)–SePh and Ni(III)–SEt bonds of complexes 29 and 31, respectively (Scheme 12f). The Ni(II)–CO of complex 33 is not photolabile. THF solution of complex 33 is stable under photolysis (λ = 365 nm) for 7 h at ambient temperature. The unprecedented nickel(III)–thiolate complex containing C-bonded cyanomethanide [PPN][Ni(CH2CN)(P(C6H3–3-SiMe3–2-S)3)] (34) was also isolated from reaction of complex 30 and CH3CN in the presence of NaH at ambient temperature (Scheme 12g) [36]. Carbon monoxide did not trigger the reductive elimination of Ni(III)–CH2CN bond, even exposing the CH3CN solution of complex 34 under 1 atm of CO at ambient temperature for 4 days [38]. In contrast to the inertness of complexes 30 and 34 under a CO atmosphere, carbon monoxide did trigger the reductive elimination of the monodentate chalcogenolate ligand of complexes 29, 31, and 32 to yield NiII–CO complex 33 accompanied by the formation of diphenyl/dialkyl dichalcogenides [36]. This result may provide some clues to the transformation mechanism between the Ni–L form and Ni–CO form of [NiFe] hydrogenase isolated from D. vulgaris Miyazaki F. and CO acting as an inhibitor for catalytic activity of [NiFe] hydrogenases [36,37]. It is anticipated that the Ni(III)–alkylthiolate complexes containing an optimum electronic condition around the Ni center may trigger heterolytic H2 cleavage via the cooperation of Ni(III) and the coordinated [SR]− ligand.
4 Dinuclear nickel–thiolate complexes
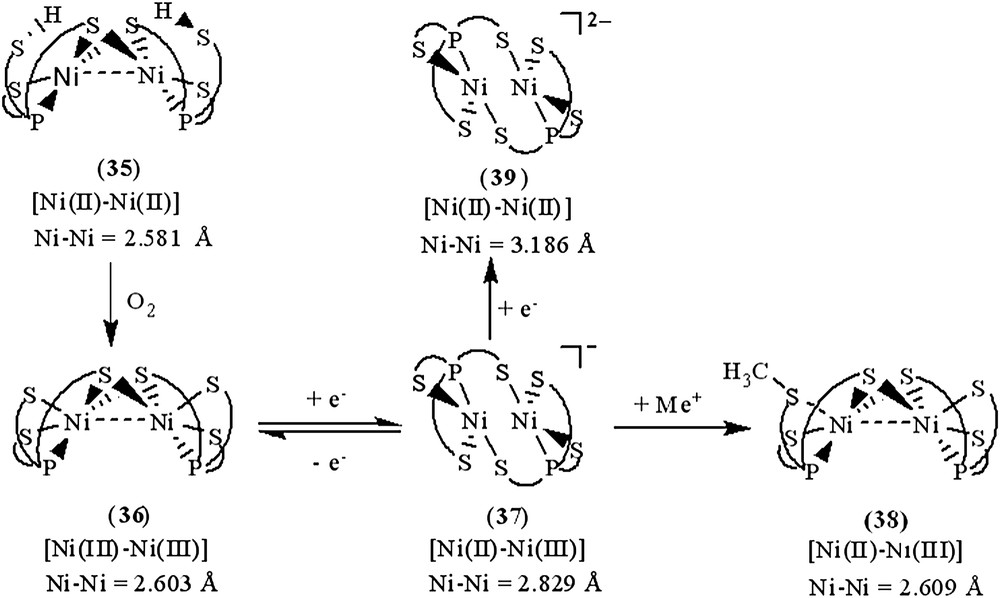
Investigation of the relationship between the core geometry of the bimetallic [Ni–Fe]/[Ni–Ni] center and the oxidation levels of nickel is limited. The neutral dinuclear Ni(II) complex [Ni(P(o-C6H3–3-SiMe3–2-S)2(o-C6H3–3-SiMe3–2-SH))]2 (35) was prepared by ethylation of [PPN][Ni(SePh)(P(o-C6H3–3-SiMe3–2-S)2(o-C6H3–3-SiMe3–2-SH))] (22) (Scheme 13a). Complex 35 exhibits extreme air sensitivity in solution (THF, diethyl ether) [39]. Compared to complexes 22a/22b (IR νS–H 2250 and 2137 cm−1 (KBr)), the higher νS–H stretching frequency (2385 cm−1 (KBr)) of complex 35 implies the absence of intramolecular [Ni–S⋯H–S] interactions (Table 6). The elongation of two Ni⋯S–H distances (3.947(1) and 3.868(1) Å, respectively) in 35, compared to the Ni⋯S–H distance of 3.690(1) and 3.305(1) Å in 22a and 22b, respectively, also implicates the absence of the intramolecular [Ni–S⋯H–S] interactions. Formation of complex 35 via the coordinative association of two [Ni(II)P(o-C6H3–3-SiMe3–2-S)2(o-C6H3–3-SiMe3–2-SH)] units can lend support to the previous proposal that the distinct electron-donating ability of the coordinated ligands may serve to regulate the intramolecular [Ni–S⋯H–S]/[Ni⋯H–S] interactions [35]. The geometry about both of the central Ni(II) can be regarded as slightly distorted square planar and the Ni⋯Ni distance of 2.5808(8) Å is short enough to suggest a certain degree of interaction between two Ni(II) centers of complex 35 [39]. Oxygen oxidation of complex 35 yielded the thermally stable dinuclear Ni(III) [Ni(III)(P(o-C6H3–3-SiMe3–2-S)2(o-C6H3–3-SiMe3–2-μ-S))]2 (36) accompanied by the byproduct H2O identified by 1H NMR (Scheme 13b). Complex 36 in frozen CH2Cl2 shows EPR silent at 77 K. On the basis of 1H NMR, EPR, and magnetic measurements, complex 36 was characterized as a diamagnetic species. Thus, the electronic structure of complex 36 can be best described as two paramagnetic d7 Ni(III) cores with antiferromagnetic coupling (J = −3.13 cm−1), that is, both Ni(III) centers have S = 1/2 spins magnetically coupled to each other possibly via the direct d–d orbital overlap (Ni⋯Ni = 2.6026(7) Å) and the bridging sulfurs. Both complexes 35 and 36 show the butterfly structure of the Ni(μ-S)2Ni unit on going from Ni(II) complex 35 to Ni(III) complex 36 with little Ni–S–Ni bond angles and a Ni⋯Ni distance change (Ni–S–Ni bond angles of 70.59 (4)/70.33 (4)° and a Ni⋯Ni distance of 2.5808 (8) Å for 35, and 70.05 (3)/70.18 (3)° and 2.6026 (7) Å for 36) [39].
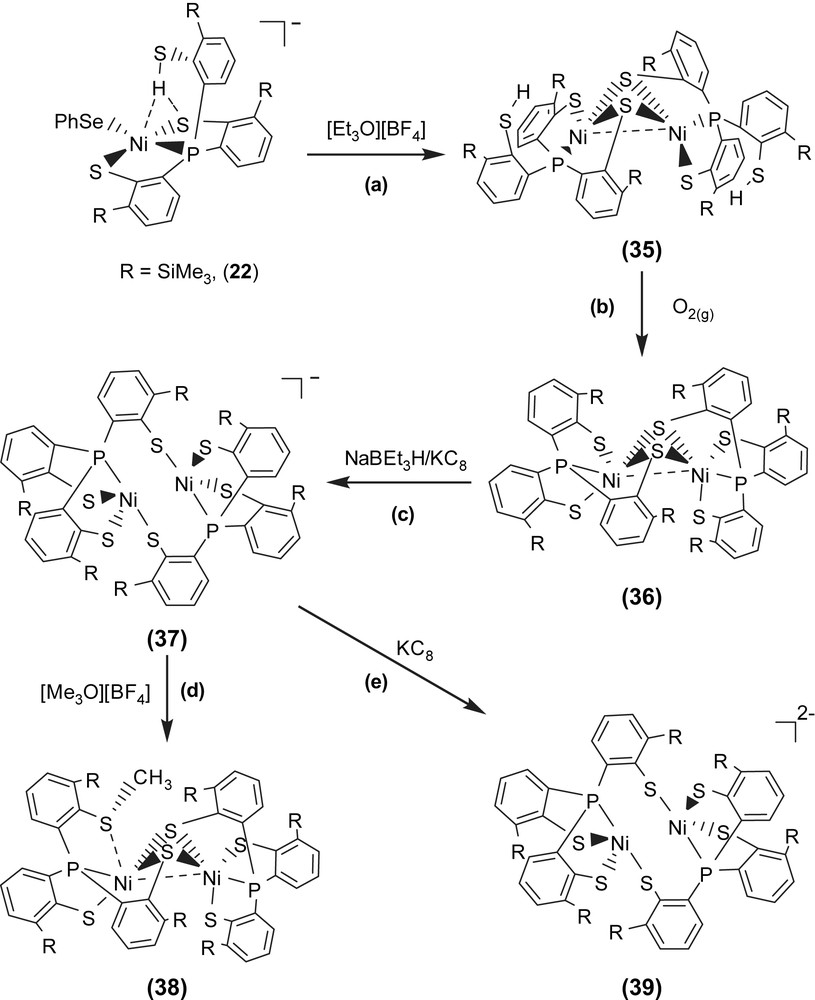
The fully delocalized mixed-valence [Ni(II)Ni(III)] complexes [Ni2(P(o-C6H3–3-SiMe3–2-S)3)2]− (37) and [Ni2(P(o-C6H3–3-SiMe3–2-S)3)(P(o-C6H3–3-SiMe3–2-S)2(o-C6H3–3-SiMe3–2-SCH3))] (38) were isolated upon the reduction of complex 36 and the methylation of complex 37, respectively (Scheme 13c and d). Compared to the rhombic signal with g values of 2.31, 2.09, and 2.0 (77 K) observed in complex 29, the 77 K EPR spectrum of complex 37 exhibits high rhombicities with three principal g values of 2.113, 2.073, and 2.033 (an isotropic signal with a g value of 2.077 at 298 K) [39]. The UV–vis spectrum of complex 37 exhibits an intense absorption around 1124 nm with an extinction coefficient ɛ > 2000 L mol−1 cm−1, which may be ascribed to the intervalence transition of the fully delocalized mixed-valence complexes. These results support that complex 37 adopts a fully delocalized mixed-valence [Ni(III)–Ni(II)] electronic configuration in a distorted square planar ligand field. The geometrical rearrangement from the butterfly structure of [Ni(μ-S)2Ni] unit of complex 36 to the diamond shape of [Ni(S)2Ni] core of complex 37 reflects the effects caused by the addition of one electron. Upon one-electron reduction of complex 36 yielding complex 37, the Ni⋯Ni distance is lengthened by as much as 0.22 Å. The apparently shorter Ni⋯Ni distance (2.6088(11) Å) of complex 36, compared to the Ni⋯Ni distance of 2.829(1) Å in complex 37, indicates a greater extent of Ni⋯Ni interaction in complex 36. The electronic perturbation caused by the sulfur methylation of complex 37 triggers the stronger Ni⋯Ni interaction accompanied by the geometrical rearrangement from the diamond shape of the [Ni(S)2Ni] core of complex 37 to the butterfly structure of the [Ni(μ-S)2Ni] unit of complex 38 to reimburse the deficiency of electron density surrounding [Ni(II)Ni(III)] centers. Interestingly, the frozen-solution EPR spectrum of complex 38 (77 K) in diethyl ether, essentially indistinguishable from those of the 77 K EPR spectrum of the fully delocalized mixed-valence complex 37, exhibits high rhombicities with three principal g values of 2.17, 2.05, and 2.02, which are different from those of the mononuclear d7 Ni(III) complex 29 [37]. These results implicate that complex 38 may exist as the delocalized mixed-valence [Ni(II)–Ni(III)] complex. The lengthening of the Ni⋯Ni distance was expected to be driven by reduction of complex 37; the reduction of complex 37 by 1 equiv of KC8 yielded a dianionic [Ni2(P(o-C6H3–3-SiMe3–2-S)3)2]2− (39) with a Ni⋯Ni distance of 3.186 Å (Scheme 13e) [39].
As shown in Scheme 14, the geometrical (coordination environment) change from distorted square pyramidal to distorted square planar (i.e., from butterfly structure of the [Ni(μ-S)2Ni] core to the diamond shape of the [NiS2Ni] core) occurs upon going from a [d7 Ni(III)–d7 Ni(III)] electronic structure of complex 36 to a fully delocalized mixed-valence [d8 Ni(II)–d7 Ni(III)] electronic structure of complex 37. In particular, the different oxidation levels of the [PS3Ni–NiPS3] units between complexes 36 and 37 reflect the distinctly different Ni⋯Ni distances, 2.603(1) Å for complex 36 and 2.829(1) Å for complex 37, to reach the optimum electron density of the [PS3Ni–NiPS3] core of complexes 36 and 37. The electronic perturbation from the sulfur methylation of complex 37 triggers the stronger Ni⋯Ni interaction and the geometrical rearrangement from the diamond shape of the [NiS2Ni] core to the butterfly structure of [Ni(μ-S)2Ni], resulting in the formation of complex 38 (Scheme 14). Presumably, the shortening of the Ni⋯Ni distance of complex 38 (vs complex 37) was employed to reimburse the electron deficiency induced by methylation, neutralizing the thiolate negative charge. Also, the geometrical change of the nickel centers from distorted tetrahedral to square planar and the lengthening of the Ni⋯Ni distance (from 2.581 to 3.186 Å) occur upon going from a neutral [Ni(II)Ni(II)] electronic structure of complex 35 to a dianionic [Ni(II)Ni(II)] electronic structure of complex 39. Such geometrical differences as determined by the nickel oxidation state in complex 36 versus that in complex 37 and determined by the coordinated ligands in complex 37 versus those in complex 38 (or in complex 35 versus those in complex 39) demand ligands capable of matching the geometrical requirement of the [Ni(SR)2Ni] unit as well as maintaining the preferred coordination number of the nickels (Scheme 14). Obviously, the presence/absence of Ni⋯Ni interaction in cooperation with the geometrical rearrangement were employed by dinuclear complexes 35–39 as an efficient tool to reach an optimum electronic condition to stabilize the dinuclear nickel complexes [39]. These results unambiguously illustrate the aspects of how a coordinated ligand and the electronic states (oxidation states) of two nickel centers function to trigger the geometrical rearrangement, to promote the stability of the dinuclear nickel complexes via the Ni⋯Ni interaction. This result may provide some clues to rationalize the oxidized-form active-site structure of [NiFe] hydrogenases, a heterobimetallic (Scys)2Ni(μ-Scys)2(μ-X)Fe(CO)(CN)2 (X = O2−, HO2−, OH−) with the butterfly structure of the [Ni(μ-Scys)2–Fe] core and a Ni⋯Fe distance of 2.6 Å.
5 Conclusion
Recently, the major progress has been made in solving the active-site structures of [NiFe]/[Fe] hydrogenases by high-resolution single-crystal X-ray diffraction and in elucidating the ways that hydrogenases catalyze reversible hydrogen oxidation [1–12]. Extensive spectroscopic experiments have been performed, as a background for the subsequent modeling studies [1–12]. For the past few years, our studies on the iron(II)–thiolate cyanocarbonyls, the mononuclear/dinuclear nickel(III)–thiolate complexes and the mononuclear nickel(II)–thiolate complexes with pendant thiol have led to the interesting results related to the structure, reactivity and spectroscopic properties of the iron and nickel centers of the active site of [NiFe]/[Fe] hydrogenases.
Acknowledgements
We gratefully acknowledge financial support from the National Science Council of Taiwan.