

1 Introduction
For some time, we have been attempting to make polymetallic clusters of transition metal ions with the purpose of synthesizing Single-Molecule Magnets [1]. Our approach, like many others [2], was to employ flexible organic bridging ligands in self-assembly processes and was particularly focused on using manganese [3]. Recently we instigated an alternative approach: a project involving the use of derivatised salicylaldoximes in which we use not only self-assembly, but the deliberate targeted structural distortion of molecules as a means of enhancing SMM properties [4–8]. Phenolic oximes, with the generic structure shown in Scheme 1, have existed for decades with uses not only academically in the formation of polynuclear transition metal complexes, but as industrial metal extractants and anti-corrosive agents in protective coatings [9,10]. In all of the oxime-based Mn clusters we have isolated, the exchange between the metal centres is very weak and typically only a few wavenumbers (<1–2 cm−1) in magnitude. This has allowed us to switch antiferromagnetic pairwise exchange interactions to ferromagnetic pairwise exchange interactions via minor structural modifications of the magnetic core. Specifically, changing the R-group (Scheme 1) allows us to distort the planarity of the Mn–N–O–Mn moiety and switch the exchange [4–8]. Here we demonstrate this phenomenon in the family of hexametallic compounds of general formula [Mn6O2(R-sao)6(O2C-th)2L4–6] (where R = H (1), Et (2); HO2C-th = 3-thiophene carboxylic acid; and L = EtOH, H2O) in which the structures differ only in the identity of the oxime present.
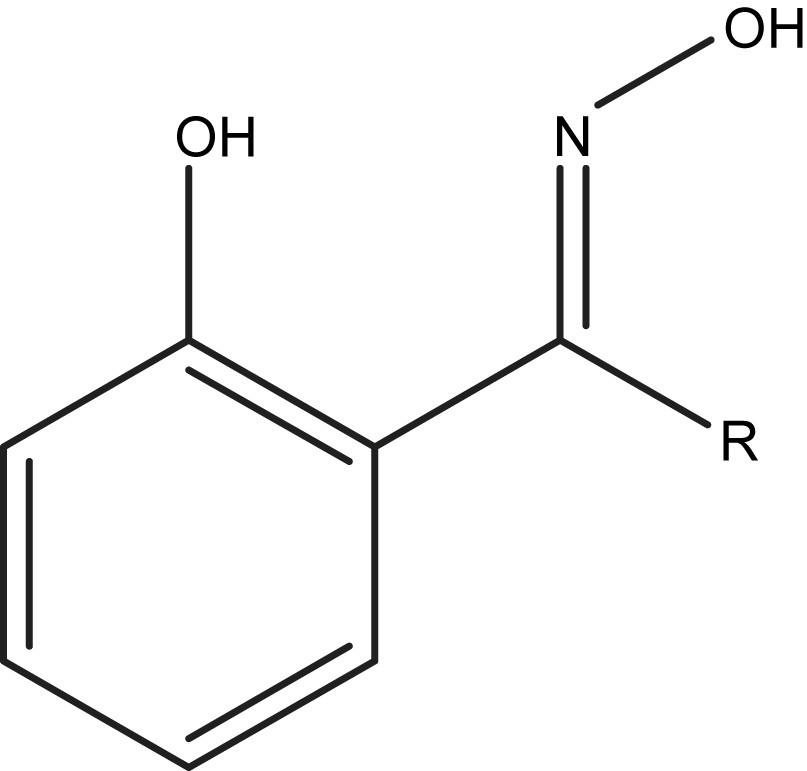
The structure of the phenolic oxime R–saoH2; R = H (saoH2); R = Me (Me–saoH2); R = Et (Et–saoH2).
2 Experimental section
2.1 Materials and physical measurements
All manipulations were performed under aerobic conditions using materials as received (reagent grade). Caution! Although we encountered no problems care should be taken when using the potentially explosive perchlorate anion. The derivatised oximes (Scheme 1) were synthesized as described elsewhere [11]. Variable temperature, solid-state direct current (dc) and alternating current (ac) magnetic susceptibility data down to 1.8 K were collected on a Quantum Design MPMS-XL SQUID magnetometer. Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants.
2.2 General synthetic methodology for complexes 1 and 2
Method 1. To pale pink solutions of Mn(ClO4)2·6H2O in EtOH were added equivalent amounts of the derivatised oximes (R-saoH2), the carboxylic acid (HO2C-th) and CH3ONa (or NEt4OH). The solutions were left stirring for ∼30 min, filtered and then left to slowly evaporate.
Method 2. The sodium salt of the carboxylic acid (NaO2C-th) was treated with equivalent amounts of Mn(ClO4)2·6H2O, the derivatised oxime and CH3ONa (or NEt4OH) in EtOH. Single crystals were grown upon slow evaporation.
Method 3. Method 1 and/or method 2 was repeated using a solvent mixture of 1:1 EtOH/CH2Cl2. After stirring for ∼30 min, the solution was filtered and layered with diethyl ether (Et2O).
In each case suitable crystals grew after a period of 3–5 days. For both compounds the yields vary from minimum of 30% to a maximum of 50%. CHN analysis, Found (Calculated). 1 (Mn6C52H44N6S2O22): C, 43.02 (42.81); H, 3.17 (3.02); N, 5.81 (5.76) %. 2 (Mn6C85H107N7S2O26): C, 50.45 (50.27); H, 5.37 (5.27); N, 4.60 (4.83) %.
2.3 X-ray crystallography and structure solution
Diffraction data were collected at 150 K on a Bruker Smart Apex CCDC diffractometer, equipped with an Oxford Cryosystems LT device, using Mo radiation. See Table 1 and CIF files for full details.
Crystallographic data for complexes 1 and 2
| 1 | 2 | |
| Formula | C63H70Cl6N6O24S2Mn6 | C85H109N7O26S2Mn6 |
| Mw | 1901.73 | 2038.53 |
| Crystal size, mm | 0.86 × 0.62 × 0.41 | 0.49 × 0.45 × 0.23 |
| Crystal system | Triclinic | Triclinic |
| Space group | P-1 | P-1 |
| a, Å | 11.5076(8) | 12.8647(5) |
| b, Å | 13.8717(9) | 13.1373(5) |
| c, Å | 14.1965(9) | 16.1689(6) |
| α, deg | 117.336(3) | 92.116(2) |
| β, deg | 90.196(3) | 101.617(2) |
| γ, deg | 109.517(3) | 114.076(2) |
| V, Å3 | 1863.9(2) | 2422.07(17) |
| Z | 1 | 1 |
| Dc, g cm−3 | 1.694 | 1.398 |
| μ, (Mo Kα), mm−1 | 1.337 | 0.876 |
| T, K | 150(2) | 150(2) |
| λ, Å | 0.71073 | 0.71073 |
| Meas/indep (Rint) reflns. | 41,028, 10,196(0.0489) | 29,271, 11,556(0.00) |
| Observed data [I > 4σ(I)] | 8614, 0.0394 | 6357, 0.0847 |
| R1 [I > 2σ(I)], wR2 [all data] | 0.0462, 0.0557 | 0.0785, 0.000168 |
| GOF on F2 | 1.0960 | 1.0236 |
| Δρ (max/min), e Å−3 | −1.19, 1.34 | 1.05, −1.34 |
3 Results and discussion
Both complexes, [Mn6O2(sao)6(O2C-th)2(H2O)4]·2CH2Cl2·Et2O (1·2CH2Cl2·Et2O), and [Mn6O2(Et-sao)6(O2C-th)2(EtOH)6]·Et-saoH2 (2·Et-saoH2), display very similar molecular structures, crystallizing in the triclinic space group P-1, each possessing an inversion centre. They can be described (Fig. 1) as consisting of two parallel off-set, stacked [MnIII3(μ3-O)]7+ triangular sub-units linked via two ‘central’ oximate O-atoms and two ‘peripheral’ phenoxide O-atoms (O′ph), leading to a [MnIII6(μ3-O)2(μ3-ONR)2(μ-ONR)4]8+ core. The bridging between neighbouring Mn ions within each triangle occurs through an NO oximate group, such that each Mn2 pair forms a –Mn–N–O–Mn– moiety, and thus the Mn3 triangle a (–Mn–O–N–)3 ring. In both complexes the coordination spheres of the Mn ions are completed by two terminal carboxylates (one on each triangle; except for complex 1 where the carboxylate is bridging in a η1:η1:μ fashion), a phenoxide O-atom, and by terminal alcohol solvate molecules or H2O molecules. All Mn ions are in the 3+ oxidation state, as confirmed by a combination of bond-length considerations, BVS calculations and charge-balance. All are six-coordinate adopting distorted octahedral geometry, except for two MnIII atoms in 1 (Mn3 and Mn3′) which are five-coordinate adopting square-based pyramidal geometry. All Jahn–Teller (JT) axes are approximately co-parallel, perpendicular to the [Mn3O]7+ planes.
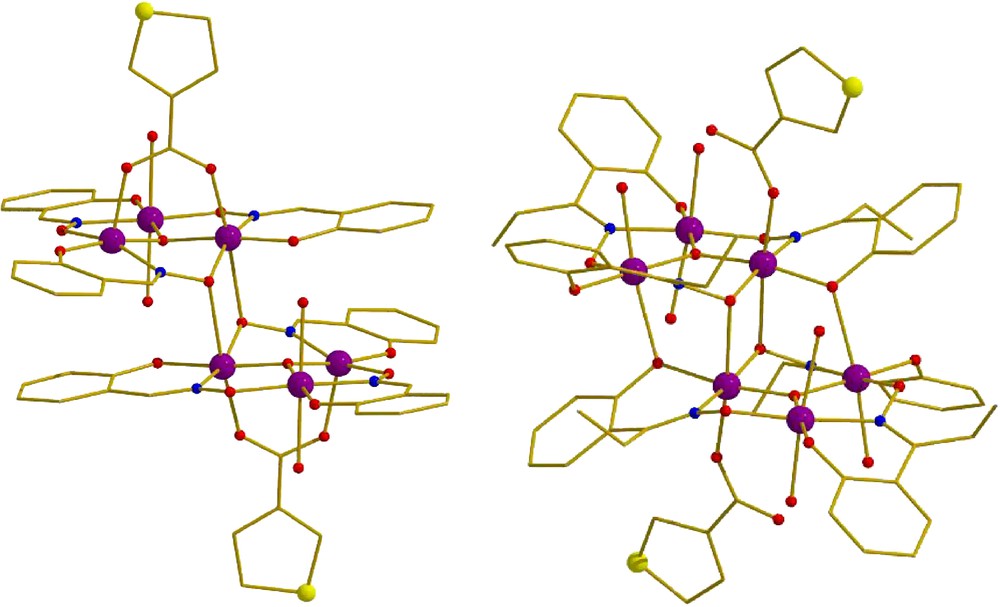
The molecular structures of complexes 1 (left) and 2 (right); colour code: Mn = purple, O = red, N = blue, S = yellow. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Besides the carboxylate coordination mode, there are two other characteristic and important structural differences between the two reported complexes: (a) the distance of Mn3–O′ph (and its symmetry equivalent, as shown in Figs. 1 and 2) ranges from a minimum of 2.432 Å in 2 to a maximum of 3.630 Å in 1. This results in the formation of an extra bridge between the two triangular rings, which is present in 2, and absent in 1; (b) the Mn–O–N–Mn torsion angles (Fig. 2) within each triangular sub-unit range from a minimum value of 2.87° in 1 to a maximum value of 42.31° in 2.
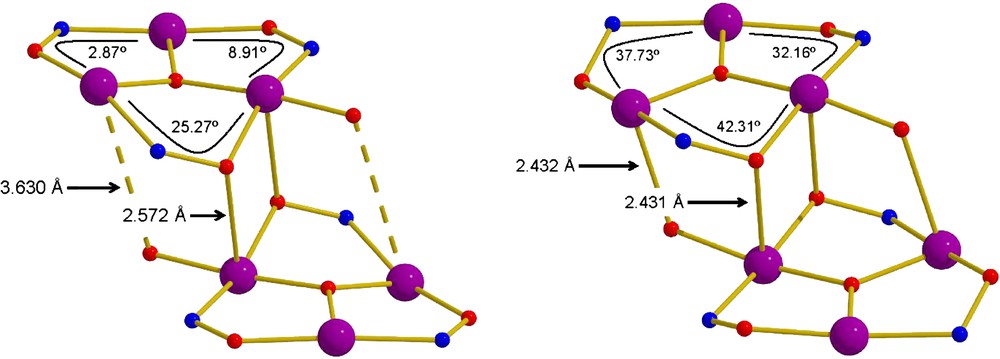
The cores of complexes 1 (left) and 2 (right) highlighting the differences in the bond lengths and torsion angles. The dashed line represents the Mn3⋯O′ph distance. See text for details.
In 1 there are extensive inter-molecular interactions propagated primarily through the terminally bound water molecules (Fig. 3). These H-bond to both the phenolic (O⋯O, 2.958 Å) and oximic (O⋯O, 2.988 Å) O atoms on neighbouring Mn6 molecules and to Et2O molecules of crystallization (O⋯O, 2.782 Å). The result is a 1-D H-bonded chain of Mn6 molecules running along the a-axis of the crystal with the solvent molecules in the channels between the chains. For 2 there are no significant inter-molecular interactions between the individual Mn6 molecules due to the presence of a molecule of Et-saoH2 (disordered over an inversion centre) in the lattice that sits in between adjacent cluster molecules, essentially acting as a protective sheath (Fig. 3). The only H-bonds are of the intra-molecular variety, between the terminally bound solvent molecules (EtOH) and the O-atom of the carboxylate (O⋯O, 2.675–2.744 Å).

The packing of 1 (top) and 2 (bottom) in the crystal; 1 viewed down the b-axis and 2 viewed down the a-axis.
4 Magnetochemistry
4.1 Dc magnetic susceptibility studies
Direct current magnetic susceptibility studies were performed on polycrystalline samples of complexes 1 and 2 in the 2–300 K range in an applied field of 0.1 T. The results are plotted as the χMT product versus T in Fig. 4. The χMT values at 300 K are 17.74 and 17.31 cm3 K mol−1 for 1 and 2, respectively, close to the spin-only (g = 2) value of 18 cm3 K mol−1 expected for a [Mn6] unit comprising six high-spin MnIII ions. For complex 1 the room temperature χMT value of 17.74 cm3 K mol−1 decreases gradually and continually as the temperature decreases reaching a minimum value of 7.01 cm3 K mol−1 at 2 K. This behaviour is consistent with the presence of dominant antiferromagnetic interactions between the metal centres with the low temperature value indicating a relatively small (S = 4) spin ground state. For 2, the room temperature χMT value of 17.31 cm3 K mol−1 increases gradually and continually as the temperature decreases reaching a maximum value of 57.47 cm3 K mol−1 at 3 K. This behaviour is consistent with the presence of dominant ferromagnetic interactions between the metal centres with the low temperature value indicating a relatively large (S = 12) spin ground state.
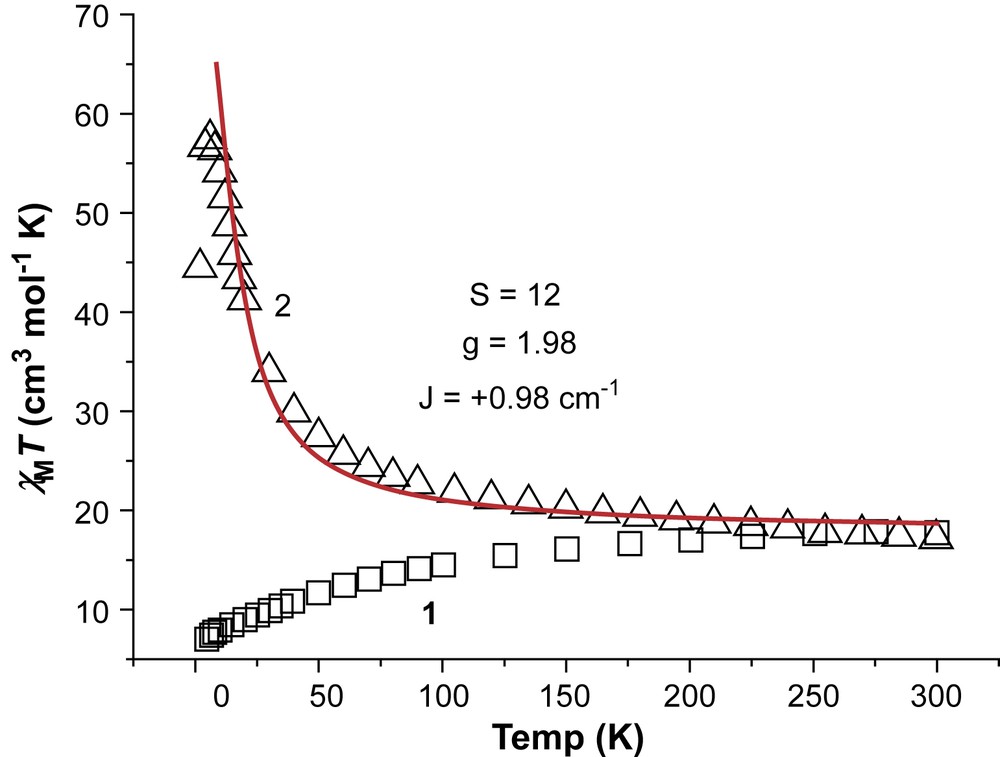
Plot of χMT versus T for complexes 1 and 2. The solid line represents a fit of the data for 2. See text for details.
Inspection of the molecular structure reveals the presence of many exchange pathways between the metal centres in both complexes. In each case we attempted to fit the data with the simplest model possible (i.e. a one-J fits-all model) before introducing more parameters, and only did so if the simple model could in no way reproduce the experimental data. However, our attempts to simulate the data were successful only for complex 2, affording g = 1.98 and J = +0.98 cm−1, employing Hamiltonian (1) and the interaction scheme depicted in Scheme 2. This gives a ground state of S = 12 with S = 11 and S = 10 excited states, 4.65 and 9.39 cm−1 higher in energy, respectively.
| (1) |
| (2) |
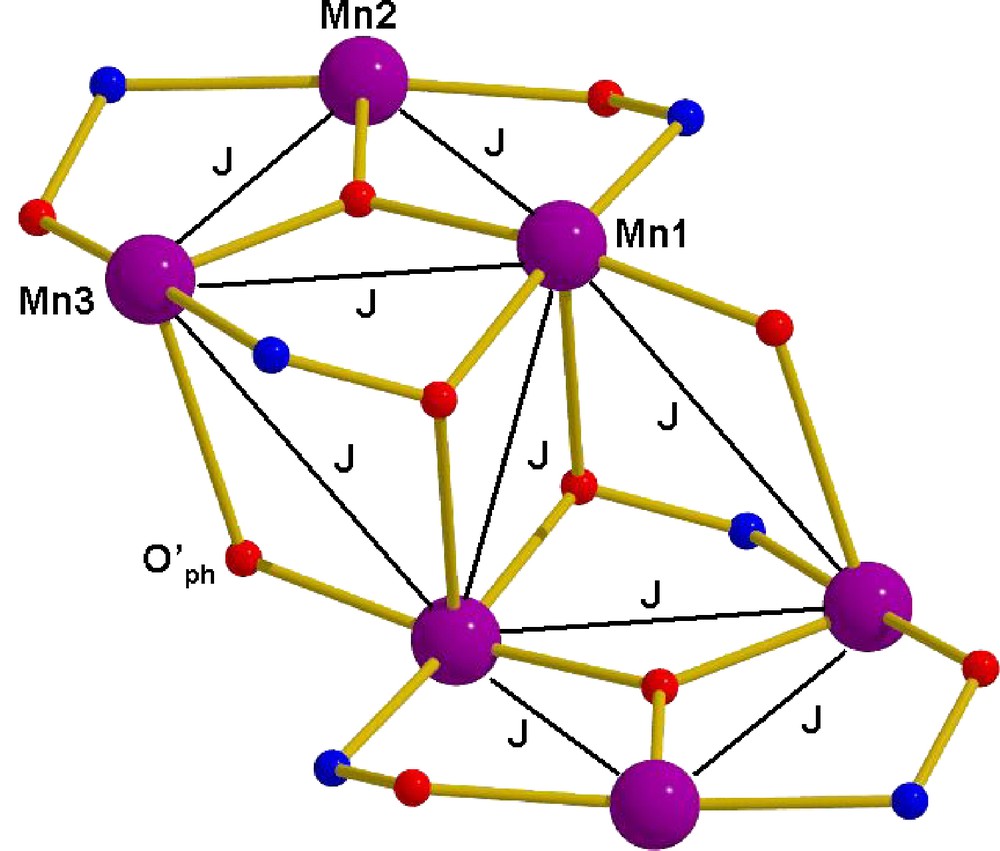
The exchange interaction model used for complex 2, see text for details.
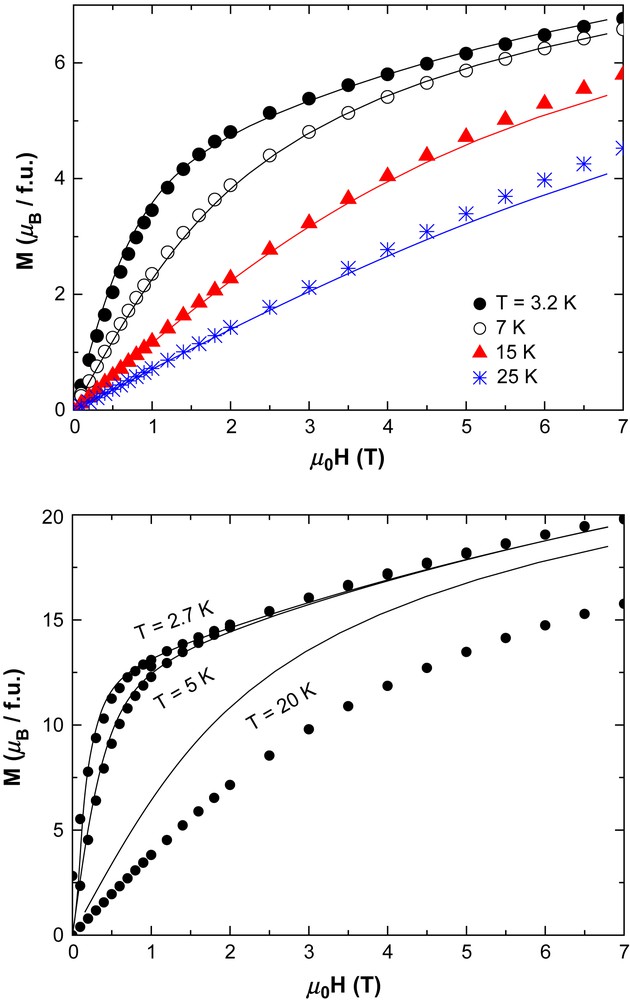
Plots of M versus H for complexes 1 (top) and 2 (bottom). The solid lines are fits to the experimental data with the parameters S = 4, g = 2.0 and D = −1.76 K for 1; S = 12, g = 2.0 and D = −0.62 K for 2.
4.2 Ac magnetic susceptibility studies
Ac susceptibility measurements were performed on complexes 1 and 2 in the 1.8–15 K temperature range in zero applied dc field and a 3.5 G ac field oscillating at 50–1500 Hz. The out-of-phase (χM″) signals for all three complexes are shown in Fig. 6. For 2 we clearly see the appearance of two peaks which are indicative of the presence of Jahn–Teller isomers and slower (high temperature) and faster (low temperature) relaxing species, as seen in the prototype Mn12 SMM [12,13]. In each case the data were then used to construct Arrhenius plots (Fig. 7); the fits of data affording: τ0 = 2.04 × 10−8 s and Ueff = 28 K for 1; τ0 = 1.17 × 10−10 s and Ueff = 55 K for 2FR; and τ0 = 1.46 × 10−10 s and Ueff = 67 K for 2SR (FR = faster relaxing; SR = slower relaxing).

Plots of the out-of-phase χM″ ac susceptibility measurements for 1 (top) and 2 (bottom) at the indicated temperature and frequency ranges.
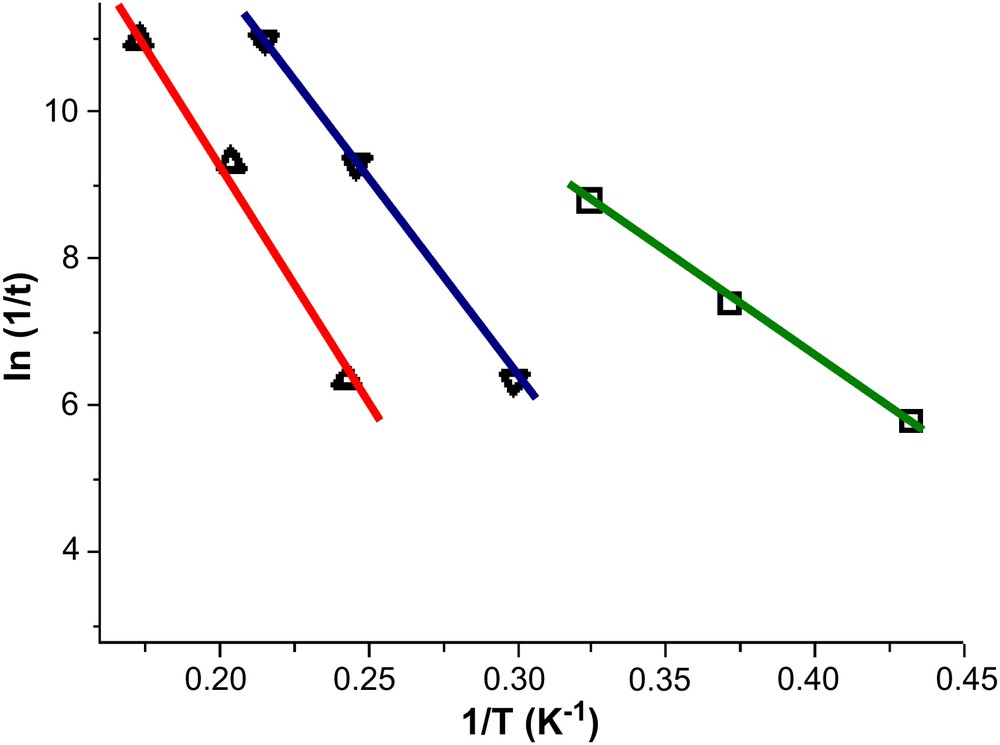
Arrhenius plots for 1 (green) and 2 (blue = FR; red = SR) using powder ac data (bottom right); the solid lines are fits of the data. See text for details. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
5 Concluding remarks
The two major conclusions from the above data are: (1) the dominant magnetic exchange switches from antiferromagnetic to ferromagnetic from 1 to 2, and (2) the barrier to magnetization relaxation has been greatly enhanced from 28 K in 1 to 67 K in 2. The S = 4 ground state in 1 results from the ferromagnetic interaction between two antiferromagnetically coupled (S = 2) MnIII3 triangles [4–8]. The two different pairwise exchange interactions between the metals within each individual triangle are antiferromagnetic and weak; the exchange between triangles is ferromagnetic and weak. The oxide is ∼0.3 Å above the Mn3 plane and the Mn–N–O–Mn torsion angles are 2.87°, 8.91° and 25.27°. In each complex the exchange between the two triangles remains weak and ferromagnetic. On moving to complex 2 the bridging within each triangle changes in two distinct ways: (1) the distance between the phenolic O-atom and the Mn (and its symmetry equivalent, as shown in Fig. 2) ranges from a minimum of 2.432 Å in 2 to a maximum of 3.630 Å in 1. This results in the formation of an extra bridge between the two triangular rings, which is present in 2 and absent in 1; (2) the torsion angles of the oximate groups increase significantly to a maximum of 37.73°, 32.16° and 42.31°. The role of the central oxide in such triangular Mn topologies has always been the promotion of antiferromagnetic exchange between the metal centres. However, recently the triangular oxime-based complex [Mn3O(mpko)3(O2CMe)3](ClO4) (Hmpko = methyl 2-pyridyl ketone oxime) was shown to display ferromagnetic exchange [14]. Initial speculation as to why centred around the possibility of a decreasing magnitude of the AF contribution when the oxide became more non-planar. However, the results here (where the oxide is ∼0.3 Å out of the plane in each case) and elsewhere [4–8,15] suggest the origin (in this family of complexes) to be the non-planarity of the oxime linkage. Thus we can conclude that the deliberate structural distortion induced on a hexanuclear SMM by the introduction of bulkier, “non-planar” oximes has switched the dominant magnetic exchange from antiferromagnetic to ferromagnetic, greatly enhancing the observed SMM properties. The origin of the ferromagnetic ‘switch’ is based upon the following: (a) the exchange in the primary complex 1 is weak and this means that even small structural perturbations will be important in the relative magnitude and sign of J; (b) the dominant ferromagnetic contribution in the individual triangles can be induced by an increase in the Mn–N–O–Mn torsion angles.