

1 Introduction
The coordination chemistry of oximes has attracted huge interest, reflected in several reviews covering structural [1–3], synthetic and reactivity [4,5] as well as magnetic [3,6] aspects. A significant recent development in the field of molecular magnetism refers to the discovery of two large families of trinuclear (e.g. [MnIII3(μ3-O)(O2CR′)3(R-pao)3]+ [7] and [MnIII3(μ3-O)(O2CR′)(R-sao)3(solvent)3–5] [8]) and hexanuclear (e.g. [MnIII6(μ3-O)2(O2CR′)2(R-sao)6(solvent)4–6] [9]) oxime-based MnIII single-molecule magnets (SMMs), where H-Hpao stands for pyridine-2-aldoxime and H-H2sao for salicylaldoxime. A salient feature of these complexes is that it is possible to switch the pairwise exchange between two Mn(III) from antiferromagnetic to ferromagnetic by increasing the Mn–N–O–Mn torsion angles via derivatization of the parent oximes [8,9]. In this way, the ground state spin values can be changed from S = 2 to S = 6 for {MnIII3} complexes and from S = 4 to S = 12 for {MnIII6}. Very high effective anisotropy barriers have been determined for the mentioned hexanuclear complexes, the record being 86.4 K for [Mn6(μ3-O)2(Et-sao)6(O2CPh(Me)2)2(EtOH)6] [10]. This record has not been exceeded yet among d-transition metal complexes, except theoretically in an iron(II) complex [11] and at low temperature in a photo-excited cobalt(II)-carbene dimer (effective energy barrier = 96 K) [12]).
Although amidoximes [13] are not merely derivatized aldoximes, we found that salicylamidoxime (H2N-H2sao) forms complexes of the type [Mn6(μ3-O)2(H2N-sao)6(L)2(solvent)4–6] [14] that are similar to those obtained from salicylaldoxime and its alkyl derivatives R-H2sao. However, a distinctive feature of salicylamidoxime-based {Mn6} SMMs relative to salicylaldoxime-based analogues is that quite different Mn–N–O–Mn torsion angles and, consequently, different anisotropy energy barriers can be observed for a given set of ligands, depending on the relative positions of the axial ligands [14]. Data suggest that the minimum value of the Mn–N–O–Mn torsion angles for observing ferromagnetic pairwise exchange is slightly smaller for salicylamidoxime (27°) [14b] than for derivatized salicylaldoximes (31°) [9,15]. On the other hand, the observed intracycle N–H···O hydrogen bond might disfavour large distortion of the Mn–N–O–Mn units [14b], and thus we thought it would be of interest to study the behaviour of dialkylsalicylamidoximes (R2N-H2sao). We report herein four {MnIII6} SMMs based on dimethyl- and diethylsalicylamidoximes:
- • [MnIII6(μ3-O)2(O2CPh)2(Me2N-sao)6(EtOH)4] (1);
- • [MnIII6(μ3-O)2(O2CPh)2(Me2N-sao)6(iPrOH)4] (2);
- • [MnIII6(μ3-O)2(O2CPh)2(Et2N-sao)6(EtOH)4] (3) and;
- • [MnIII6(μ3-O)2(O2CPh)2(Et2N-sao)6(iPrOH)4] (4).
2 Experimental
2.1 Materials
Reagents and solvents were obtained from commercial sources and used as received. Hydroximoyl chloride was prepared according to a literature method. Dialkylsalicylamidoximes have been prepared by reaction of hydroxymoyl chloride [16] with the appropriate dialkylamine (Scheme 1). The {MnIII6} complexes have been obtained by reaction of manganese(II) chloride or bromide with the appropriate derivatized salicylamidoxime in methanol in presence of benzoic acid and triethylamine.
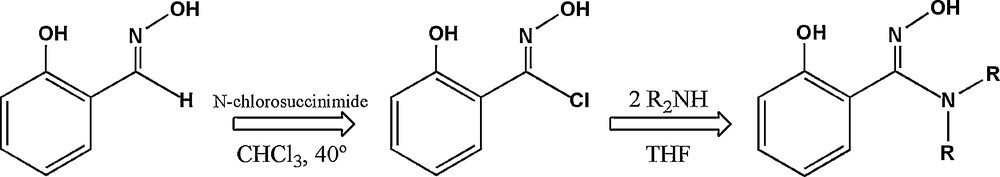
Synthesis of dialkylsalicylamidoximes.
2.2 Synthesis of the complexes
2.2.1 [MnIII6(μ3-O)2(O2CPh)2(Me2N-sao)6(EtOH)4] (1)
To a solution of dimethylsalicylamidoxime (0.541 g, 3.0 mmol) and benzoic acid (1.099 g, 9.0 mmol) in MeOH (140 mL) was added a solution of MnCl2·4H2O (0.594 g, 3.0 mmol) MeOH (20 mL). Then Et3N (3 mL) was added dropwise and the resulting brown-green solution was refluxed for 1.5 h upon. The resulting dark green precipitate was collected by filtration (0.5 g) and dissolved in chloroform (15 mL) which was then layered with ethanol. Small polyhedral crystals of 1·2CHCl3 were obtained within 2 weeks. Yield: 40%. Anal. calcd. (found) for C76H94N12O22Mn6 (1): C 49.1 (49.1), H 5.1 (5.1), N 9.0 (9.1), Mn 17.8 (18.2).
2.2.2 [MnIII6(μ3-O)2(O2CPh)2(Me2N-sao)6(iPrOH)4] (2)
2 was prepared analogously to 1 except that the chloroform solution of the crude precipitate was layered with isopropanol. A polycrystalline sample was obtained within 2 weeks. Yield: 50%. Anal. calcd. (found) for C80H102N12O22Mn6 (2): C 50.2 (50.3), H 5.4 (5.2), N 8.8 (8.8), Mn 17.2 (17.5)%.
2.2.3 [MnIII6(μ3-O)2(O2CPh)2(Et2N-sao)6(EtOH)4] (3)
This compound was prepared analogously to 1 and 2, starting from a solution of diethylsalicylamidoxime (0.625 g, 3.0 mmol) and benzoic acid (1.099 g, 9.0 mmol) in MeOH (130 mL). The crude precipitate (0.7 g) was dissolved in chloroform (15 mL) and the solution was layered with ethanol. Dark green plates were obtained within 3 weeks. Yield: 30%. Anal. calcd. (found) for C88H118N12O22Mn6 (3): C 52.2 (51.7), H 5.9 (5.8), N 8.2 (8.5), Mn 16.3 (17.3)%.
2.2.4 [MnIII6(μ3-O)2(O2CPh)2(Et2N-sao)6(iPrOH)4] (4)
This compound was prepared analogously to 3 except that MnBr2·4H2O was used in place of MnCl2·4H2O. The crude precipitate (0.8 g) was dissolved in chloroform (15 mL) and the solution was layered with isopropanol. Dark green polyhedral crystals were obtained within 3 weeks. Yield: 40%. Anal. calcd. (found) for C92H126N12O22Mn6 (4): C 54.0 (53.7), H 6.1 (6.1), N 8.1 (8.0), Mn 16.3 (16.4)%.
2.3 Analytical and physical measurements
Elemental analyses were performed by the Service de microanalyse of UPMC or by the Service central d’analyse of the CNRS (Solaize). IR spectra were obtained from KBr pellets on a Biorad FT 165 spectrometer or a Bruker Tensor 27 instrument equipped with a Harrick ATR. Magnetic studies were performed on Quantum Design PPMS-5S and MPMS-XL SQUID magnetometers equipped with a 5 T or a 7 T dc magnet, respectively. The polycrystalline samples were introduced at 200 K to avoid desolvation when pumping at room temperature. Magnetic susceptibility data were collected in the temperature range 2–200 K under an applied magnetic induction of 100 G. Diamagnetic corrections were applied using Pascal's constants [17]. Variable field and temperature dc magnetization data were collected in the 2–7 T magnetic induction range and 2–10 K temperature range. The M vs μ0 H/T data were fitted assuming that S is an exact quantum number, using the program ANISOFIT 2.0 [18] with the Hamiltonian H = DSz2 + gμBSB where μB is the Bohr magneton, g the Landé factor, D the axial zero-field splitting parameter, S the ground state spin operator and B the magnetic induction vector. Alternating current data were collected in the 2–10 K range using a 3 G ac field oscillating at 1–1000 Hz.
2.4 Crystallographic data collection and structure determination
Crystals of 1, 3 and 4 were taken out from their mother liquor. All crystals were mounted onto glass fibers and immediately transferred in a cold nitrogen gas stream on a Bruker-Nonius Kappa-CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Unit-cell parameters determination, data collection strategy and integration were carried out with the Nonius EVAL-14 suite of program [19]. Data were corrected from absorption by a multiscan method [20]. The structures were solved by direct methods using the SHELXS-97 [21] or Sir92 [22] program and refined by full-matrix least-squares on F2 using the SHELXL-97 software package [21]. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms of hydroxyl groups were located from difference Fourier maps while the others were introduced at calculated positions. All of them were refined using a riding model. Graphics were done using Diamond [23].
Crystallographic data for 1, 3 and 4 are summarized in Table 1. Metrical parameters relevant for the discussion of the magnetic properties are listed in Table 2. Bond lengths and bond angles are gathered in Tables S1 for 1, S2 for 3 and S3 for 4.
Crystal data and structure refinement for 1·2CHCl3, 3 and 4.
| Compound | 1·2CHCl3 | 3 | 4 |
| Formula | C78H96Cl6Mn6N12O22 | C88H118Mn6N12O22 | C92H126Mn6N12O22 |
| Formula weight | 2096.01 | 2025.58 | 2081.69 |
| Temperature (K) | 200 | 200 | 200 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group | P 21/n | ||
| Z | 1 | 2 | 1 |
| a (Å) | 13.8646(10) | 14.2679(17) | 13.370(2) |
| b (Å) | 14.0687(12) | 22.150(3) | 14.8788(15) |
| c (Å) | 14.3634(10) | 16.5298(14) | 15.747(2) |
| α(°) | 99.049(6) | 90 | 11.536(10) |
| β(°) | 118.343(6) | 115.124(8) | 106.607(17) |
| γ(°) | 106.634(6) | 90 | 102.280(11) |
| V(Å3) | 2215.2(3) | 4729.8(9) | 2609.0(8) |
| Dc (g.cm−3) | 1.571 | 1.422 | 1.325 |
| F(000) | 1076 | 2112 | 1088 |
| μ (cm−1) | 1.088 | 0.852 | 0.774 |
| Refln. collected/obs. | 10108/5800 | 13721/7986 | 15034/7805 |
| Goodness-of-fit on F2 | 1.036 | 1.010 | 1.000 |
| R1 [I > 2σ(I)]a | 0.0725 | 0.0457 | 0.0480 |
| wR2 b,c | 0.2130 | 0.1387 | 0.1484 |
a R1 = ∑||Fo|–|Fc||/∑|Fo|.
b wR2 = {∑[w(Fo2–Fc2)2]/[(w(Fo2)2]}1/2.
c w = 1/[σ2(Fo2) + (aP)2 + bP] with P = [Fo2 + 2Fc2]/3.
Selected structural parameters for compounds 1·2CHCl3, 3 and 4.
| Compound | α/° a | ϕ/° b | h/Å c | Mn-Oph, Mn-Oox (Å) | Type d |
| 1·2CHCl3 | 52.2(4), 35.2(5), 8.84(5) | 99.2(2) | 0.147(4) | 2.333(4), 2.354(5) | b |
| 3 | 50.2(1), 37.9(2), 30.1(2) | 98.09(7) | 0.103(1) | 2.3416(17), 2.3941(16) | b |
| 4 | 50.3(2), 36.4(2), 32.0(2) | 99.78(9) | 0.109(2) | 2.3673(16), 2.4293(17) | b |
3 Results and discussion
3.1 Crystal structure of 1, 3 and 4
The molecular structure of 1 is shown in Fig. 1 and those of 3 and 4 are given in Supplementary Information (Fig. S1, S2). The numbering scheme directly relevant to the discussion is shown in Scheme 2.
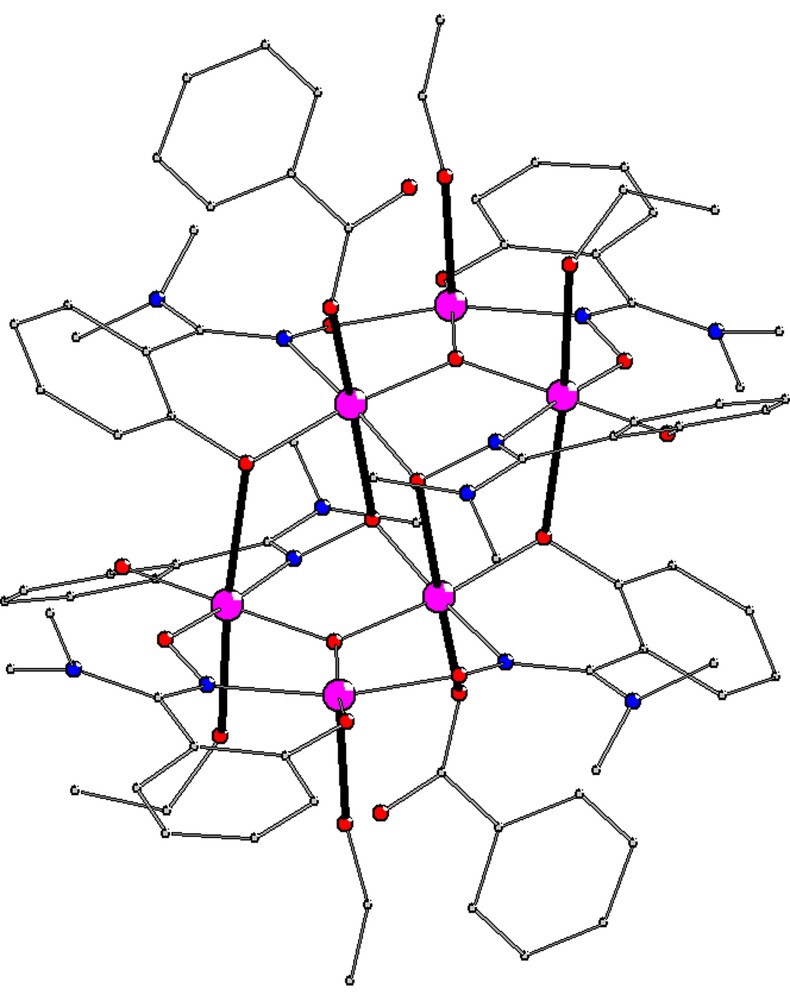
Molecular structure of the {Mn6} cluster in 1·2CHCl3 highlighting the elongated Mn-Oaxial bonds as heavy lines. (For a colour version, the reader is referred to the web version of this article.)
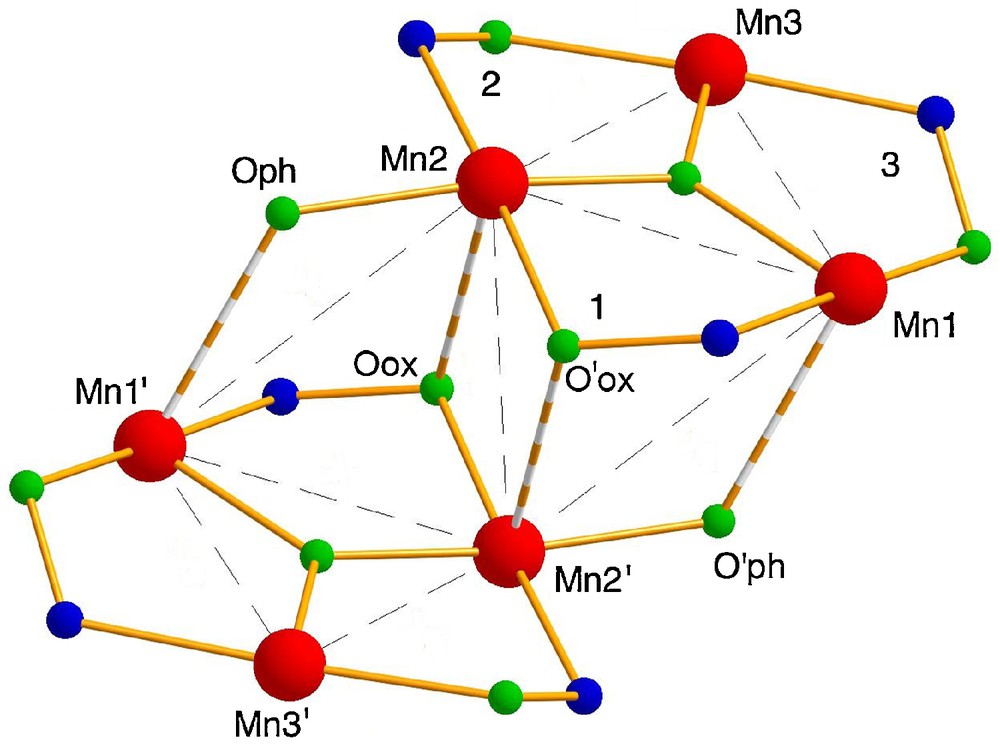
Atom and torsion angle numbering scheme (note that Mn2 and Mn3 are permuted in comparison with Ref. [14a]).
These {Mn6} clusters all display a centrosymmetrical {MnIII6(μ3-O)2(R2N-sao)6}2+ core (Scheme 2) quite similar to that of other complexes of the type [MnIII6(μ3-O)2(O2CR)2(X-sao)6(solvent)4–6] (X = H, Me, Et, NH2) [9,10,14]. The coordination spheres of the Mn centres are completed by two terminal benzoates and by four alcohol molecules in such a way that Mn1 and Mn2 (and thus Mn1′ and Mn2′) achieve six-coordination while Mn3 and Mn3′ are five-coordinate. Jahn-Teller elongated axes run approximately parallel to each other and perpendicular to the {Mn3} triangles. While four structural types a, b, c and d are in principle possible according to the location of the terminal carboxylates (a, trans to coordinated solvent, b, trans to oximate, c, trans to phenolate, d, bridging between phenolate and oximate) [14], compounds 1, 3 and 4 all belong to type b, i.e. benzoates are coordinated to Mn2 and Mn2′. This contrasts with related complexes of underivatized salicylamidoxime which predominantly adopt type a or type c [14], but is reminiscent of those with derivatized methyl- and ethylsalicylaldoximes which all belong to type b with the exception of [Mn6O2(O2CCPh3)2(Me-sao)6(EtOH)4] [9]. It was expected that going from NH2 to NR2, that is increasing the steric bulk of the salicylamidoximate ligand, would result in a larger structural distortion of the {Mn6} core, that is higher Mn–N–O–Mn torsion angles. This is achieved for the largest Mn1–N–O–Mn2 distortion angle – labelled 1 –which, in 1, 3 and 4, is larger than 50°, higher than the underivatized analogues [14] or the R-sao derivatives [9b]. However, the other Mn–N–O–Mn torsion angles in 3 and 4 are comparable to those of the unique ligand type b example among complexes based on underivatized salicylamidoxime [14] and to the R-sao derivatives [9b], and in 1, one of them is even much lower (Table 2).
A possible explanation of the similarity of the Mn–N–O–Mn values for derivatized and underivatized salicylamidoximes {Mn6} complexes is the presence of close contacts between the oximate oxygen and the hydrogen atoms either of the amine in underivatized compounds or of the methyl and ethyl group in the derivatized samples. The intra-triangle Mn-neighbours distances and bond angles are very similar to the ones of other {MnIII6} complexes (Tables S1–3). The h distance between the central oxygen O(1) and the Mn3 plane (ranging from 0.103 to 0.147 Å, Table 2) is also close to previously observed distances in {MnIII6} clusters. The same observation holds for inter-triangle distances (corresponding to the elongated Jahn-Teller axes) and angles (close to 90°) (Tables S1–3).
3.2 Magnetic properties
Variable-temperature dc magnetic susceptibility data were collected on polycrystalline samples in the temperature range 2–200 K under an applied induction of 100 G. The results are plotted as χMT vs T curves in Fig. 2.
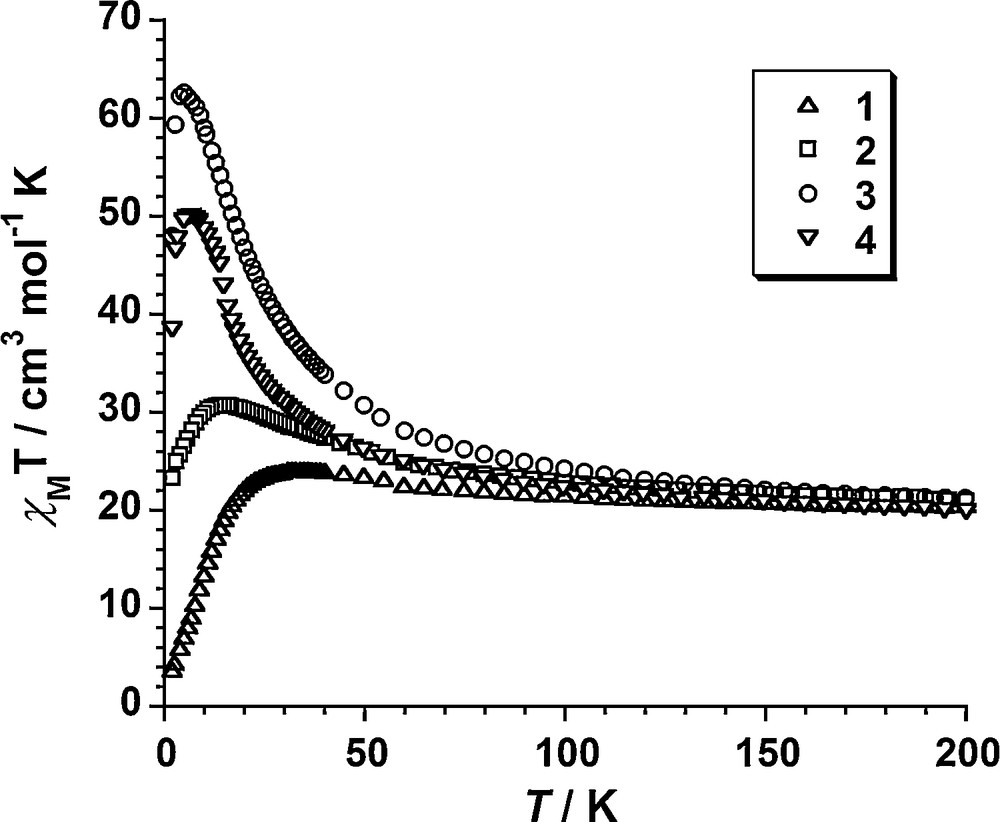
Thermal variation of the molar susceptibility as χMT for polycrystalline samples of compounds 1–4.
The χMT values at 200 K (20.5 for 1, 21.0 for 2, 20.6 for 3, 19.9 for 4, in cm3 mol−1 K) are much higher than those expected for six independent Mn(III) (χMT = 18, g = 2). They gradually increase with decreasing temperature, reaching maxima of 24.1, 30.8, 62.6 and 50.5 cm3 mol−1 K at 30.5, 14.9, 4.9 and 6.8 K for 1, 2, 3 and 4, respectively. The initial values and the gradual increase upon cooling is indicative of ferromagnetic interactions within the {MnIII6} clusters while the decrease at the lowest temperatures is likely to be due to zero-field splitting and/or (weak) intermolecular antiferromagnetic exchange interactions. The maximum χMT values lie between the values 10 cm3 mol−1 K and 78 cm3 mol−1 K expected for S = 4 and S = 12 ground spin states, which suggests, at least for compounds 1 and 2, competing anti- and ferromagnetic exchange interactions and intermediate ground spin states. Indeed, M/NμB vs μ0H/T data indicate an S = 12 ground state for both 3 and 4, a S = 9 ground state for 2 and an S = 6 ground state for 1 (Fig. 3 and Supplementary content).
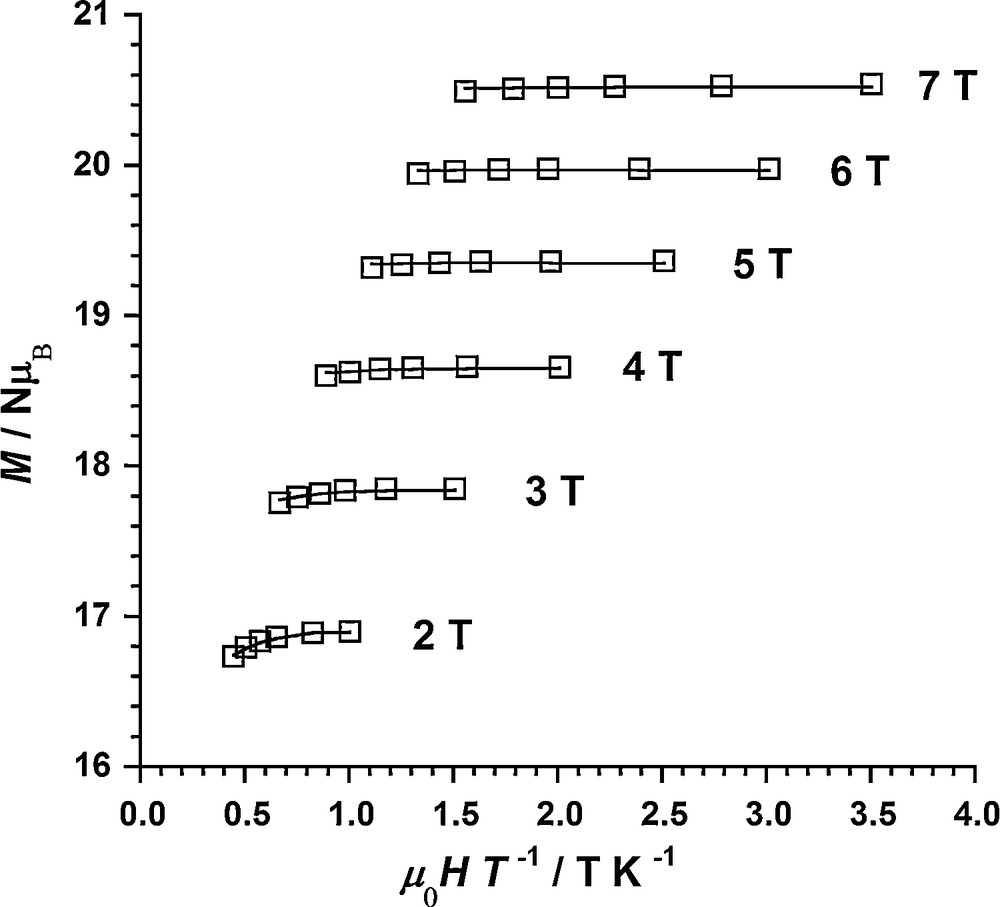
Isofield magnetization vs μ0H/T plot for compound 3.
These results are consistent with the Mn–N–O–Mn torsion angle values which are all above the minimum value for observing ferromagnetic interactions in 3 and 4 while one is far below in 1. No conclusion is possible in 2 for which no crystal structure is available. However, it should be noted that the axial anisotropy value for 2 is close to those of 3 and 4 and other previously studied {Mn6} complexes with an S = 12 ground state while it is noticeably higher for 1 (Table 3), as expected for lower spin complexes though not as much as observed for complexes with S = 4 ground state [9,14].
Structural and magnetic data for complexes 1–4.
| α/deg | S a | D/cm−1 a | g a | τ0/s | Ueff/K b | |
| 1 | 52.2(4), 35.2(5), 8.84(5) | 6 | –0.97 | 1.98 | 1.4 × 10−10 | 56 |
| 2 | non available | 9 | –0.43 | 1.98 | 2.5 × 10−11 | 52 |
| 3 | 50.2(1), 37.9(2), 30.1(2) | 12 | –0.44 | 2.00 | 1.0 × 10−11 | 71 |
| 4 | 50.3(2), 36.4(2), 32.0(2) | 12 | –0.39 | 1.99 | 3.8 × 10−11 | 59 |
a From fit of magnetization measurements.
b From ac susceptibility measurements.
Magnetization vs applied dc field data were collected for compounds 1–4 at 2 K. Significant hysteresis was observed for 3 and 4, and spin quantum tunnelling at zero applied field is clearly visible for compound 3 at 2 K (Fig. 4).
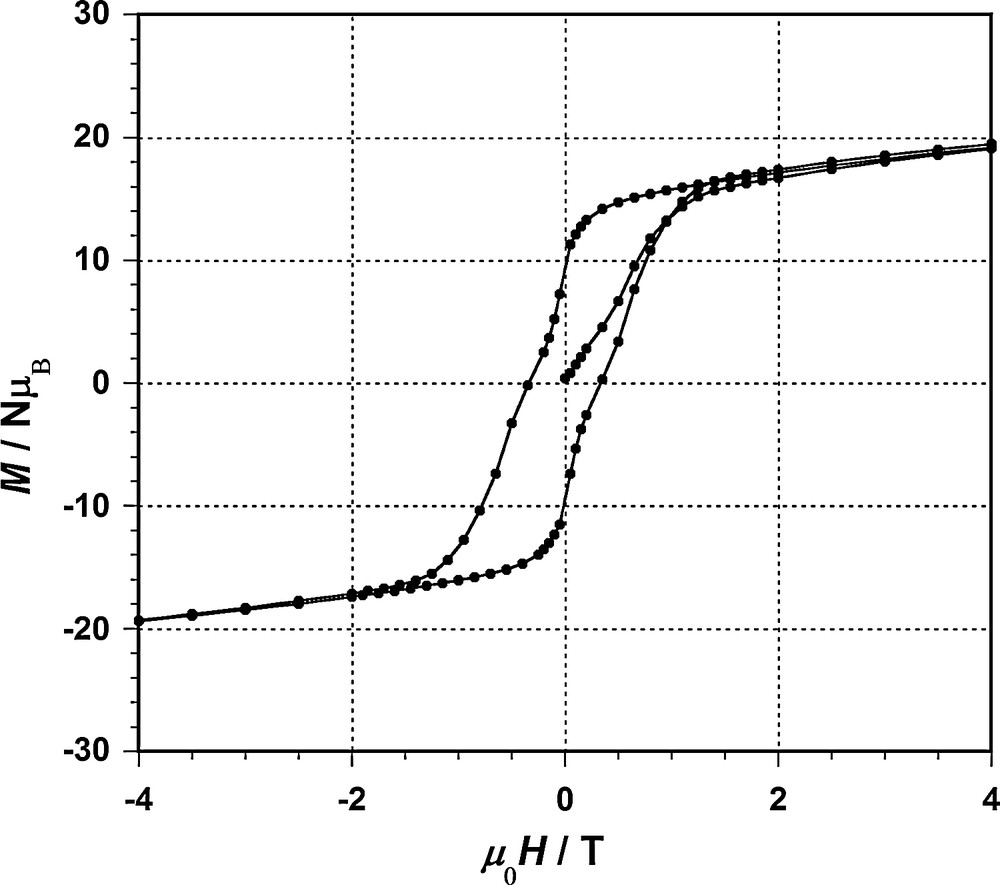
Magnetization vs magnetic induction and hysteresis loop for 3 at 2.0 K.
Alternating current (ac) studies were performed in the 2–10 K range using a 3 G ac field oscillating at 1–1000 Hz. All compounds show out-of-phase susceptibility signals (χM″) revealing a single slow relaxation process. The corresponding anisotropy barriers are 56, 52, 71 and 59 K for 1, 2, 3 and 4 respectively. They are fairly high, but – against our expectations – remain below the effective barriers determined for related {MnIII6) complexes based on underivatized salicylamidoxime [14].
4 Conclusions
Four hexanuclear manganese(III) complexes:
- • [MnIII6(μ3-O)2(O2CPh)2(Me2N-sao)6(EtOH)4] (1);
- • [MnIII6(μ3-O)2(O2CPh)2(Me2N-sao)6(iPrOH)4] (2);
- • [MnIII6(μ3-O)2(O2CPh)2(Et2N-sao)6(EtOH)4] (3) and;
- • [MnIII6(μ3-O)2(O2CPh)2(Et2N-sao)6(iPrOH)4] (4)
have been obtained by the reactions of MnCl2·4H2O with benzoic acid and dimethyl- or diethylsalicylamidoxime in methanol, followed by subsequent workup. The crystal and molecular structures of 1·2CHCl3, 3 and 4 have been determined by single-crystal X-ray diffraction. Manganese (III) ions present different Jahn-Teller distorted elongated octahedral or square pyramidal coordinations with spin S = 2 and different local anisotropies Di. Compounds 1, 3 and 4 display similar centrosymmetrical structures with the benzoate ligands located trans to the oximate oxygen atom that links the {MnIII3} triangles. The adoption of this structural type (type b) undoubtedly reflects the increase in steric bulk of R2N-sao2− (R = Me, Et) compared to H2N-sao2−. Nevertheless, only the Mn–N–O–Mn torsion angle corresponding to the oximate group which connects the two {MnIII3} triangles is significantly increased compared to other {MnIII6} derivatives. The inter-triangle geometry (Mn-Oox-Mn′ angle ≈ 99°) insures ferromagnetic coupling between the {MnIII3} triangles. Within the triangles, the Mn–N–O–Mn torsion angles in 3 and 4 are all above the minimum value for observing ferromagnetic interactions and achieving a S = 12 ground state. Given such torsion angles, the ferromagnetic J coupling constants between Mn(III) ions are expected to be weak as in preceding {MnIII6} complexes and the ground state close to the first excited states. In 1, one of the Mn–N–O–Mn torsion angles is weak, the corresponding coupling constant is expected to be negative as reflected by the intermediate magnetic behaviour. All compounds 1–4 exhibit out-of-phase susceptibility signals (χM″) revealing a single slow relaxation process and single-molecule magnet behaviour with negative D values in line for intermediate spin or S = 12. The corresponding anisotropy barriers (Ueff) are 56, 52, 71 and 59 K for 1, 2, 3 and 4, respectively. These effective values are non negligible but remain below the record value (86.4 K) probably due to the population of excited states close to the ground state and spin quantum tunnelling in the ground state, as beautifully demonstrated in compound 3.
Acknowledgements
This work was supported by the CNRS and the University Pierre et Marie Curie. JML is indebted to spanish Ministerio de Ciencia e Innovación for a postdoctoral grant. The authors are grateful to E. Ruiz (Barcelona) for fruitful discussions and Y. Li for helping magnetic measurements.