

1 Introduction
There is currently active interest in the development of molecular electronic devices that can be used as optical and/or magnetic data storage media. Compounds of specific interest are bistable molecular materials having two nearly degenerated states with different optical and/or magnetic properties [1]. These complexes have an appreciable sensitivity to the environment, so an external perturbation, such as temperature or irradiation, may lead to an interconversion between the two degenerated electronic states, settling down the conceptual basis of a molecular device. Examples of electronic labile complexes are mixed valence [2] spin-crossover [3] and valence tautomeric complexes [4]. The comprehension and understanding of the characteristic features for all these complexes have been developed in parallel over the last decades, a large interest in the scientific community as demonstrated by the large amount of examples developed for each case.
Valence tautomeric (VT) metal complexes with at least two redox-active centers, the metal ion and an electroactive ligand, are characterized by the existence of two electronic isomers (valence tautomers) with different charge distributions, and consequently, different optical, electric and magnetic properties [5]. The interconversion between the different electronic isomers is accomplished by a reversible intramolecular electron transfer involving the metal ion and the redox-active ligand. A schematic representation of valence tautomerism together with the corresponding potential energy curve plotted as a function of the nuclear coordinate is shown in Fig. 1.
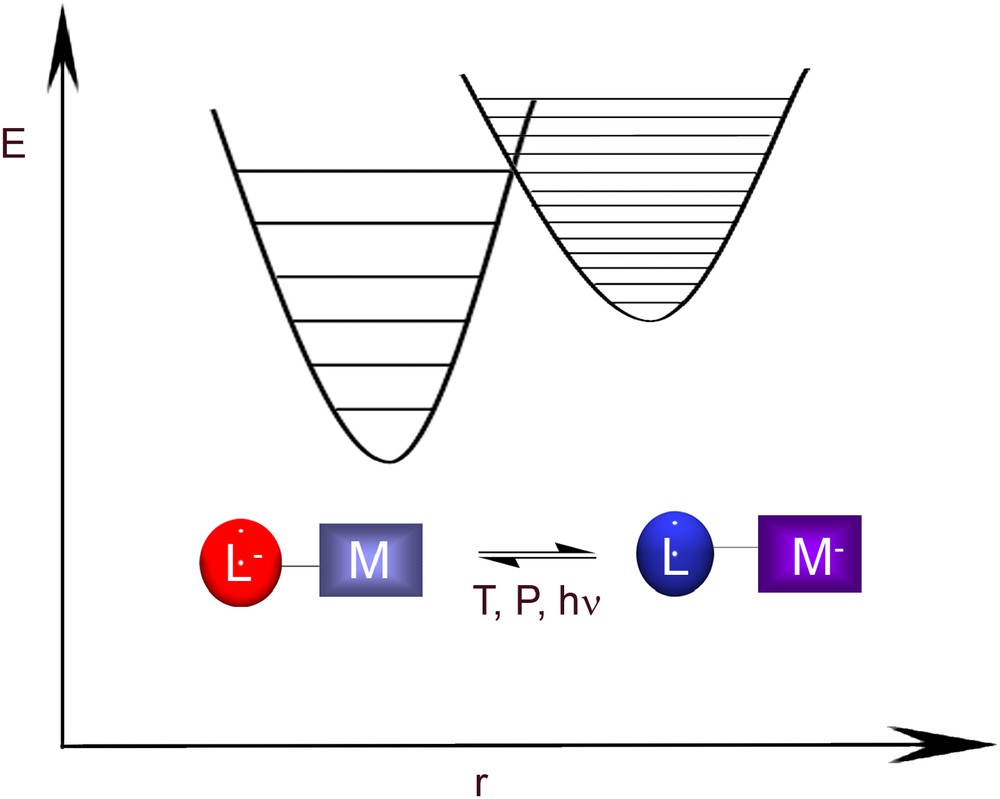
Schematic representation of valence tautomerism involving a redox-active ligand and a transition metal ion together with the corresponding potential energy curve plotted as a function of the nuclear coordinate.
One of the first descriptions of valence tautomerism and an excellent example of the above mentioned charge distribution sensitivity is exhibited by the cobalt bis(quinone) complex [CoIII(3,5-DTBCat)(3,5-DTBSQ)(bpy)] (1), [6] where 3,5-DTBCat2− and 3,5-DTBSQ− refer, respectively, to the catecholate (DTBCat2−) and semiquinonate (DTBSQ−) forms of 3,5-di-tert-butyl-o-quinone, and bpy is 2,2′-bipyridine. In solution, the equilibrium in Eq. (1) can be induced by variations of temperature and monitored by magnetic measurements and spectroscopic techniques such as UV–vis, NMR and/or EPR.
| [CoIII(3,5-DTBCat)(3,5-DTBSQ)(bpy)] ⇌ [CoII(3,5-DTBSQ)2(bpy)] | (1) |
For instance, the UV–vis spectrum of a solution of complex 1 in toluene at low temperatures shows a band at 600 nm characteristic of the [CoIII(3,5-DTBCat)(3,5-DTBSQ)(bpy)] (1, ls-Co(III)) tautomer. An increase of temperature promotes an intramolecular electron transfer so the 1, ls-Co(III) converts into the 1, hs-Co(II) as one of the ligands is reduced by one electron from a DTBSQ− to a DTBCat2− ligand. As a consequence, the intensity of the 600 nm band decreases and a band at 770 nm characteristic of the 1, hs-Co(II) tautomer increases in intensity. Accompanying the optical changes, variations of the magnetic response are also observed.
This interconversion is an entropy-driven process, in analogy to the extensively studied low-spin to high-spin-crossover phenomena. The large entropy gain arises from: (1) a gain in electronic entropy due to the higher spin state degeneracy of the hs-Co(II) form; and (2) the higher density of vibrational states of the hs-Co(II) form due its longer metal–ligand bond lengths [7]. Thus, thermal population of tautomeric states is dictated by the Gibbs free energy expression shown in Eq. (2). At low temperatures, TΔS is negligible compared to ΔH, and consequently if ΔH > kT only the ls-Co(III) state is populated. An increase of the temperature will increase the TΔS contribution, making it non-negligible and favouring the population of the hs-Co(II) state, up to a critical temperature Tc where ΔG = 0 and ΔH = TΔS. A further increase of the temperature will change the sign of ΔG.
| ΔG = ΔGhs-Co(II) − ΔGls-Co(III) = ΔH − TΔS | (2) |
Moreover, since valence tautomeric complexes are electronically labile, they exhibit significant vibronic interactions and therefore an appreciable sensitivity to the environment. As a consequence, intramolecular electron transfer can be induced not only by temperature variations but also by irradiation or pressure. For instance, Hendrickson et al. have reported results of the first picosecond time-resolved optical experiments on valence tautomeric complexes in solution [8] and in the solid state after illumination at low temperatures [9–11]. Light-induced VT has also been observed in polynuclear complexes. For instance, Sato et al. reported not only photoswitching but also hysteresis effects around room temperature (310 and 297 K) for complex [{Co(tpa)}2(DHBQ)](PF6)3 (2), (where DHBQ is the deprotonated 2,5-dihydroxy-1,4benzoquinone), for which intermolecular interactions are enhanced by the presence of significant π–π interactions [12]. Some years before, Dei et al. [13] realized additional femtosecond spectroscopic experiments in a simple cobalt–dioxolene derivative, [Co(Me4Cyclam)DTBSQ]+ Me4Cyclam = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacycletetradecane (3) showing the involvement of at least three states in a two-step VT process in solution.
Examples of pressure-induced VT have also been reported although in less extent than thermally-driven or light-induced transitions, mainly due to experimental difficulties [14]. In these experiments, the increase of the molecular size on passing from the low- to the high-spin isomer due to the population of antibonding orbitals is used to favour the low-spin isomer after application of pressure as an external stimulus. The first example of a pressure-driven solid state valence tautomeric transformation was reported by Verdaguer and Hendrickson [15] who studied the non-solvated complex [CoII(3,5-DTBSQ)2(phen)] (4) and its related solvated form 4·C6H5CH3 by EXAFS and XANES. Recently, pressure-induced valence tautomerism has also been monitored by changes on the magnetic response [16]. A nice example is complex [Co(cth)(Phendiox)]PF6·H2O (5), where cth is a macrocycle and phendiox corresponds to the dioxolene neutral form (catecholate) of 9,10-dihydroxyphenanthrene. This complex has been shown by X-ray diffraction experiments to shrink its unit cell volume by more than 4% along the VT process. Systems with these characteristics have potential interest as pressure sensors [17]. A schematic representation of the representative complexes 1, 2, 4 and 5 as well as a brief summary of their VT characteristics is shown in Table 1.
Representative examples of valence tautomeric complexes induced by different external stimuli applied and their interconversion characteristics
| Ext. stimuli | Representative compounds | Matrix | Charact. techniques | Tc/Pc or temperature rangea | Reference | ||
| Temperature-induced | Solid | Magnetism, EPR, X-ray diffraction | 310 K | [6] | |||
| Liquid | Electronic spectra, 1H NMR, Magnetism, EPR | 273 K | [17] | ||||
| [71a] | |||||||
| Light-induced | Solid | Irrad. at 532 nm (5 K and 30 min) | 310 Tc↑–297 Tc↓ | [12] | |||
| Magnetic charact. | Thermal hysteresis 13 K | ||||||
| Pressure-induced | Solid | Diamond cell | 4 | 240 K, 0.37 GPa | [15] | ||
| XANES | |||||||
| EXAFS | 4·C6H5CH3 | – K, 1.1 GPa | |||||
| Solid | Magnetic charac. | 150–340 K | [16] | ||||
| Hydrostatic pressure cell | Tc↑ as P↑ |
a Tc (critical temperature) and Pc (critical pressure), in VT are defined as the temperature and pressure at which there are equal amounts of both electronic isomers at atm. pressure or room temperature. When we refer to range temperature is due that the Tc (which is the term more used in these cases) is not calculated in the reference. For strictly obtain this value is required the thermodvnamics parameters ΔH and ΔS.
In summary, the interest for studying this family of complexes is considerable [18]. First, they are unique model systems which provide insight into the basic factors affecting intramolecular electron transfer in coordination complexes. Second, from an applied perspective, the large changes in the optical, structural, and magnetic properties that often accompany the valence tautomeric interconversion have potential applications in bistable molecular materials and devices. For this reason, since first reported by Pierpont et al. in 1980 [6], several new tautomeric complexes especially over the last few years, have been described. For most of these cases, observation of valence tautomerism has been traditionally associated almost exclusively to the matching paper of the redox-active ligand and the transition metal ion electronic orbital. However, several other factors (structural or environmental) have also been detected to play a crucial role. For this reason we have structured this review into two main sections. First, we will give a brief revision of the different families of VT complexes so far reported, with special emphasis on the families of complexes derived from catecholate and phenoxylate ligands that are by far the ligands most often used in VT. In Section 2 we will revise in detail the different structural and environmental factors that clearly influence VT processes. Even though different reviews have already been reported in the literature that compile several examples of VT organized according to the nature of their redox-active ligand and the transition metal ion, this is the first revision that systematically faces the crucial effect of such additional structural and environmental factors on the observation of valence tautomerism.
2 Redox-active ligands and transition metal ions
The term of non-innocent ligand, directly related to their role and the influence on the final electronic distribution of certain transition metal complexes, was first pointed out by Jørgensen [19]. This concept can be used when both the metal-centered and ligand-centered electronic frontier orbitals are similar in energies. Within such a scenario, the final oxidation state of the complex and the internal electronic distribution may be modulated, becoming a great subject of investigation both by theoretical [20] and experimental means [21]. This investigation is of interest not only for the field of valence tautomerism but also for several other fields such as the study of complexes bearing non-innocent ligands that perform physiological functions in biological systems. For instance, transition metal complexes in which one or more of the ligands is present as a phenoxyl radical have attracted much interest due to their broad occurrence in technical processes [22] and enzymatic metalloproteins such as galactose oxidase (GAO) [23] or glyoxal oxidase (GLO) [24]. A schematic representation of the active site of both metalloproteins is shown in Fig. 2.
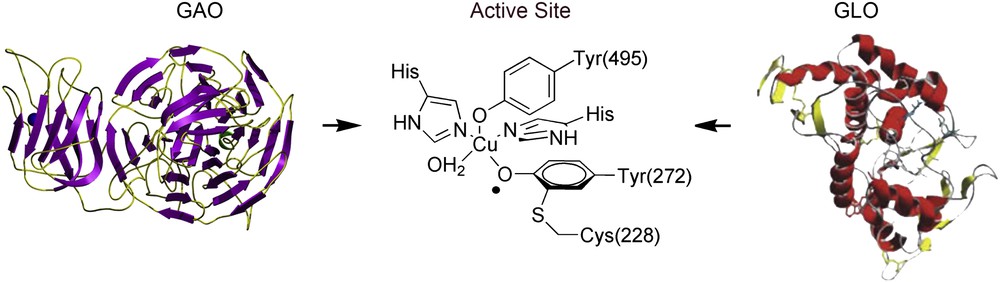
Representation of the phenoxyl radical acting as an active site in enzymatic metalloproteins as galactose oxidase (GAO) and glyoxal oxidase (GLO). Pictures of the proteins were extracted from personal scientific web page of Prof. Mike McPherson: www.astbury.leeds.ac.uk/gallery/leedspix.html (GAO) and Int. Microbiol. 8 (2005) 195 (GLO) rights authorised by © International Microbiology.
So far, many different types of redox-active ligands have been described in the literature [25]; among them are quinone-type, crown-ethers [26], ferrocene [27] and tetrathiafulvalene derivatives [28]. A schematic drawing of the different redox-active ligands previously mentioned is shown in Scheme 1. π-Conjugated polymers and/or oligomers bearing coordinating sites, such as polyanilines and polythiophenes, have also been successfully used with this aim [29]. However, we must emphasize that even though the number of redox-active ligands is considerable, those exhibiting valence tautomerism are rather limited since they must simultaneously satisfy two conditions: (1) the degree of covalency in the interaction between metal ion and electroactive ligand must be low; and (2) the energy of their frontier orbitals must be similar [7,18]. In other words, it is necessary that these complexes exhibit localized electronic structures, low orbital mixing, and a small energy difference between the two electronic tautomers. Most of the valence tautomeric complexes so far reported are based on quinone or phenoxylate ligands with a series of transition metal ions. Nevertheless, in the last few years the number of electroactive ligands inducing valence tautomerism is being expanded by including new electroactive ligands such as polychlorotriphenylmethyl radicals (PTM) [30] and tetraphenylporphyrin (TTP) [31]. Among them, redox-active catecholate and phenoxyl are by far the ligands most often used in VT, the reason why both families are revised next (for a detailed description of the different families of VT complexes so far reported, please see Refs. [4,5]).
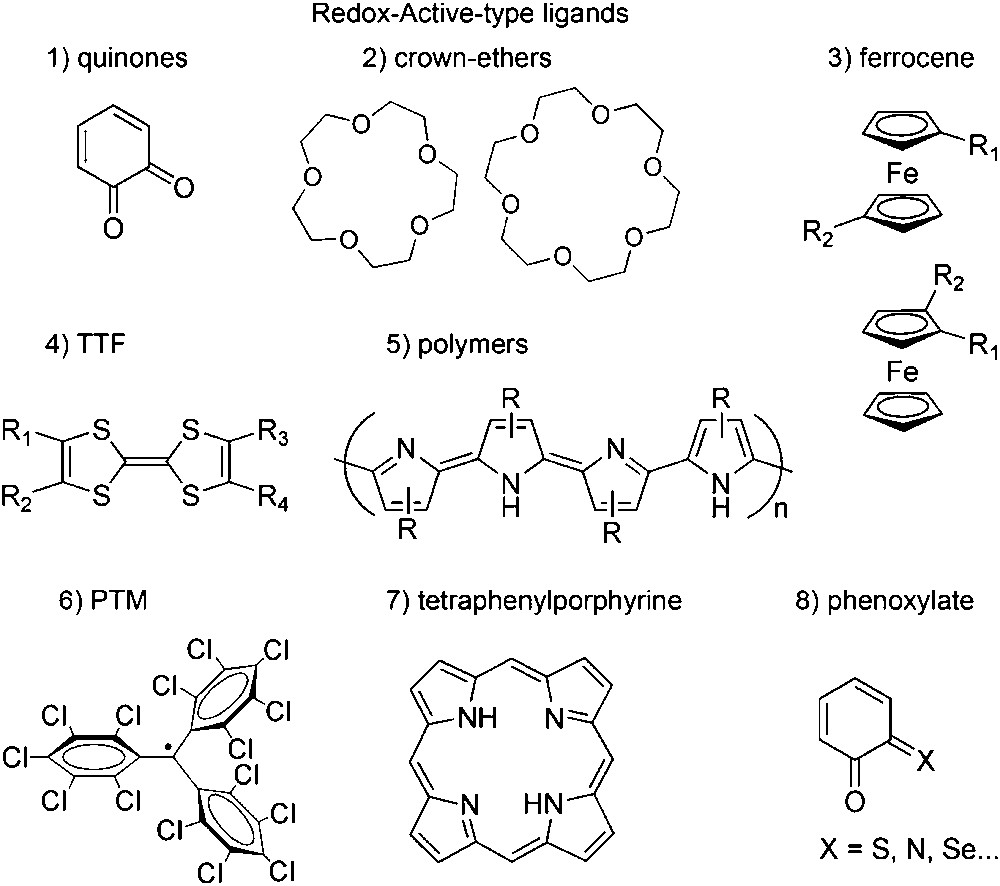
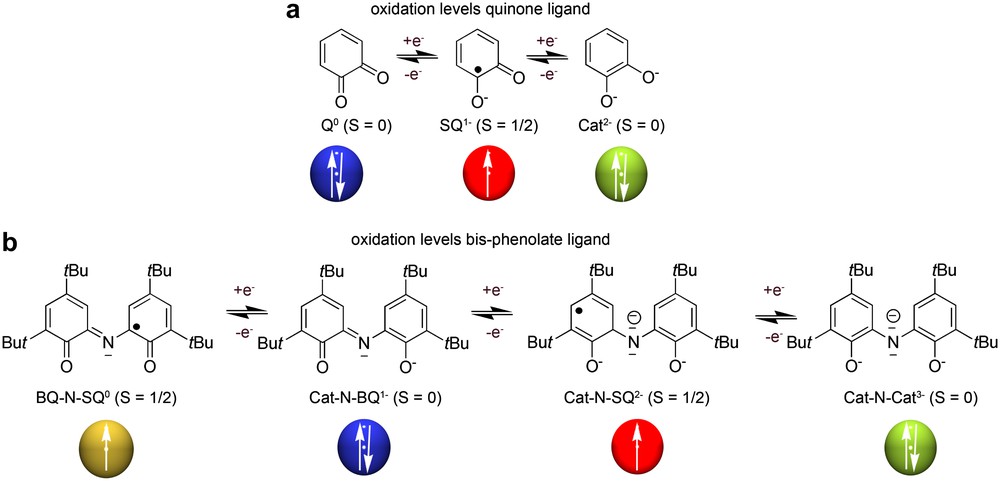
Quinones. Most of the ligands so far shown to exhibit valence tautomerism are quinone or quinone-based ligands with a series of transition metal ions. Thanks to their rich redox activity, o-quinone ligands may exist as neutral quinones (Q), radical semiquinones (SQ−) or dianionic catecholates (Cat2−) (see Scheme 2), although they usually are only found in the SQ− and Cat2− forms due to the limiting binding ability of the quinone ligand [32].
As far as the metal ion is concerned, one of the most effective in showing valence tautomerism is the pair Co(II)/Co(III). In other words, cobalt complexes bearing quinone-based ligands have been shown to exhibit not only localized structures but also a good matching between the energy of its frontier orbitals with those of the redox-active ligand. For this reason, several mononuclear complexes exhibiting VT have been described. The simplest cobalt–quinone complexes undergoing valence tautomerism belong to the family of ionic complexes with the general formula [ML(diox)]Y, where L = tetraazamacrocycle ancillary ligand, diox = 9,10-dioxophenantrene and Y = PF6, BPh4, I [16]. However, most cobalt complexes shown to exhibit valence tautomerism are of the general formula [Co(DTBQ)2 (N-N)], where N-N is a diazine ligand, DTBQ = 3,5- and 3,6-di-tert-butylcatecholato (DTBCat2−) or semiquinone (DTBSQ−) form [6,17,34]. In addition to monomeric complexes, a further step was made in the synthesis of binuclear cobalt–dioxolene complexes. For instance, the use of a modified (diimine)-bipyridine ligand as electronic coupling of two cobalt centers led to the description of the first dinuclear VT complex with the three possible tautomeric forms existing below 500 K [35]. Pierpont et al. also studied modified catecholate ligands to tune the critical temperature (Tc), defined as the temperature at which there are equal amounts of both electronic isomers, in a related family of dinuclear complexes [36]. Similar results where observed for another cobalt bis(dioxolene) complex by near-IR absorption spectroscopy in a thin amorphous film [37]. Simultaneously, Dei et al. reported the binuclear complex [{Co(cth)}2(DHBQ)](PF6)3 (6) that undergoes a gradual thermal tautomeric transition at around 175 K as well as a quantitative VT photoconversion [38].
Finally, in addition to mono- and binuclear, different polynuclear (mostly polymeric) tautomeric complexes have also been reported. Pierpont et al. [17] reported the polymer [Co(pyz)(3,6-DTBQ)2]n (7) that exhibited a temperature-induced tautomeric interconversion in the solid state that was derived in a mechanical process due to variations in the bond lengths. Hysteresis was recently reported by Schultz et al. [39] in the coordination polymer with analytical formula [Co(phen)L]·0.5CH2Cl2 (8) where L = 3,5-bis(3′,4′-dihydroxy-5′-tert-butylphenyl)-1-tert-butylbenzene. This cobalt–dioxolene polymer exhibits the necessary cooperative properties that leads to thermal hysteresis in a valence tautomeric equilibrium, even though the transition could be classified as gradual. Finally, very recently, a new strategy based on coordination polymerization and precipitation in a poor solvent to produce cross-linked valence tautomeric metal–organic nanospheres has been reported [40]. A schematic representation of the nanospheres formed along the polymerization process is shown in Fig. 3.
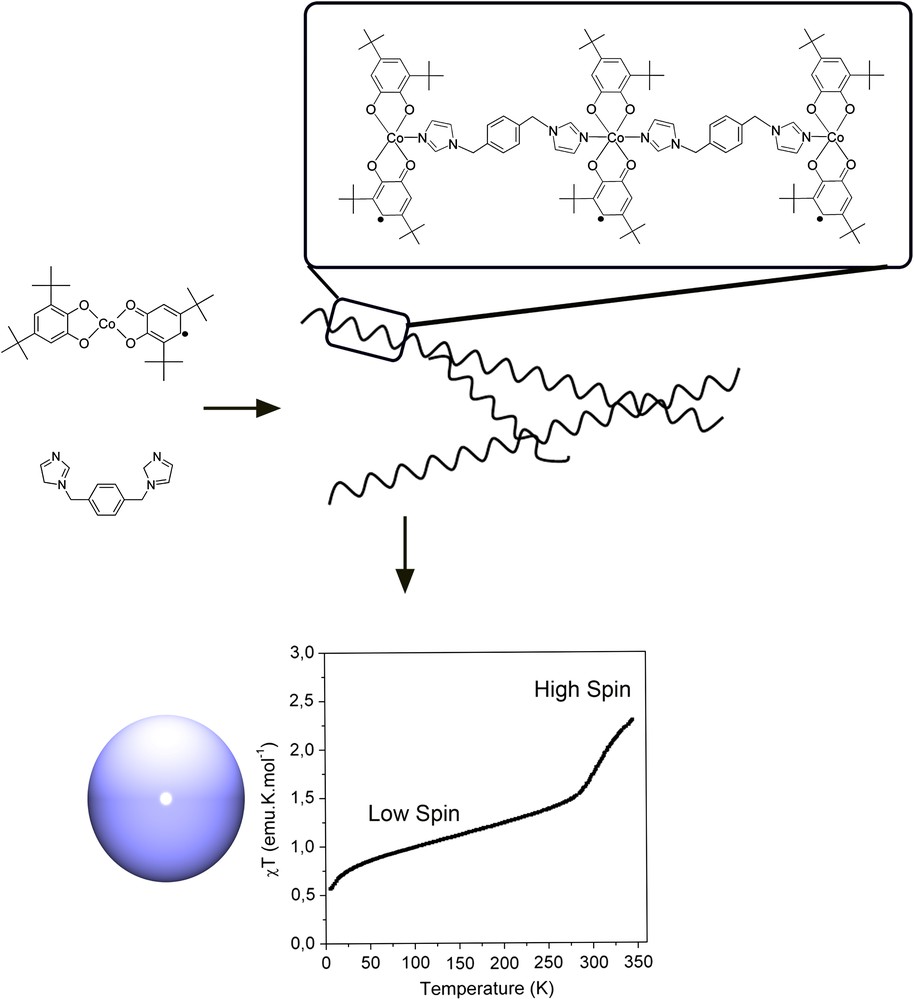
Coordination polymerization technique used to obtain valence tautomeric nano-scale metal–organic particles with general formula [Co(3,5-DBSQ)(DBCat)(N-N)] where N-N stands for the 1,4-bis(imidazol-1-ylmethyl)benzene ligand. Representation of the χT values as a function of temperature for the amorphous nanosphere, showing the transition between both electronic isomers, Ref. [40].
In addition to the cobalt ion, valence tautomerism has also been observed for different transition metal complexes such as manganese, nickel and copper. A few years after the first report of VT on a cobalt complex, a similar temperature-dependent equilibrium was observed for a related manganese complex [41]. Complex [Mn(py)2(3,5-DTBCat)2] (9) experienced a reversible colour change from the intense purple of the Mn(IV) form to the pale green-brown characteristic of the Mn(II) form. The interconversion takes place through a Mn(III) form, which usually exhibits similar colour than the Mn(IV) form, as confirmed later on in solid state [42] and in solution [43,44]. Valence tautomerism has also been reported in a tretraazamacrocycle complex of manganese with a single o-benzoquinone ligand [45].
In addition to manganese, copper complexes containing dioxolene ligands may also yield under the right conditions the valence tautomeric interconversion shown in Eq. (3) [46].
| [CuII(DTBCat)(L)] ⇌ [CuI(DTBSQ)(L)] | (3) |
One of the first examples of valence tautomerism involving the Cu(I)/Cu(II) pair was reported in a copper containing enzyme that catalyzes the oxidation of amines to aldehydes [47], an important process in relevant biological functions such as growth regulation and tissue maturation. Similar studies have also been made by Speiers et al. who described the existence of valence tautomerism for the species [Cu(py)2(PhenQ)2] [48] (10) and Kaim et al. who reported the use of a weak π-acceptor thioether in a Cu(I)–semiquinone complex to induce valence tautomerism as a paramagnetic compound associated to amine oxidase enzymes [49,79].
Finally, very recently, two new examples of nickel and iron–quinone complexes have been reported to exhibit valence tautomerism. Tanaka et al. [50] reported complexes [Ni(PyBz2)(DTBSQ)]PF6 (11) and [Ni(MePyBz2)(DTBCat)]PF6 (12) as the first examples for successful control of valence tautomerism between the Ni(II)–SQ and Ni(III)–Cat frameworks. On the other hand, Banerjee et al. [51] have reported a semiquinone/catecholate based mixed valence complex [Fe(bispicen)(Cl4Cat)(Cl4SQ)]·DMF (13), where valence tautomerism has been followed by electronic absorption spectroscopy. This is the first example where a mixed valence semiquinone/catecholate iron(III) complex undergoes intramolecular electron transfer.
Phenoxyl ligands. Phenoxyl radicals are monovalent oxygen radical species that exhibit delocalization of the unpaired electron over the aromatic ring with ortho and para substituents that give steric protection [52]. For instance, it has been shown that tert-butyl substituents at the ortho and para positions of the phenolates facilitate one-electron oxidation to the corresponding phenoxyl radicals, because these substituents decrease the oxidation potential of the phenolates and provide enough steric bulkiness to suppress bimolecular decay reactions of the generated phenoxyl radicals [53]. Several groups have prepared metal–phenoxyl complexes giving new insights into the chemical factors that govern the generation and stability of this type of radicals [54] among them worth to mention is Wieghardt et al. For instance, they have established that bidentate O,N-coordinated o-aminophenolato ligands can be found in one of the following different protonation and oxidation levels bounded to a transition metal ion: o-imidophenolate(2−) anions, o-iminobenzosemiquinonate (1−) π radical monoanions or even o-iminobenzoquinone. All these forms can exist in coordination compounds as confirmed by high quality low temperature X-ray crystallography [55]. In addition, o-iminobenzosemiquinonate (1−) anions are paramagnetic (S = 1/2) ligands that couple either ferro- or antiferromagnetically when coordinated to a paramagnetic transition metal ion, depending on the reciprocal symmetry of the SOMO ligand orbital of the 3d magnetic orbital of the metal ion. Following this approximation, a large number of new complexes bearing non-innocent phenoxyl ligands have been reported, not only in the aminophenolate case (O,N-) [56] or (N,N-) [57] but also with other donor atoms such as S (S,N-, S,S- or S,O-) [58] or with different metal ions (Fe, Cu, Co, Mn, Ni, Rh, Ru, Pt, Pd as representative examples) [59,57c]. However, and in spite of the large number of complexes so far reported, only a few of such examples have been shown to exhibit valence tauto-merism. For instance, Wieghardt et al. [60] described an octahedral {Fe-NO}7, [LPrFe(NO)·2.5CH2Cl2] (14) (LPr = 1-isopropyl-4,7-(4-tert-butyl-2-mercaptobenzyl)-1.4,7-triazacyclononane), with a pentadentate macrocyclic pendent arm ligand exhibiting the first temperature-dependent S = 1/2 to S = 3/2 spin equilibrium in the solid state (see Fig. 4). The first example of valence tautomeric equilibria for a N,S-coordinated o-aminothiophenolate cobalt and S,S-coordinated o-dithiolate iron-complexes in solution was also recently reported [61]. Almost simultaneously, Shimazaki et al. reported the one-electron oxidized form of a new mononuclear nickel(II)-bis-(salicylidene)diamine complex (15), derived from H2tert-Bu-salcn ligand, that exhibits valence tautomerism [62], and a few years later a similar complex (16) but now with the H2tert-Bu-salen was shown to exhibit a structurally-induced transition on the translation from a square planar to a octahedral geometry [63]. Finally, Yamauchi et al. [64] have recently shown additional studies based on VT for mononuclear nickel(II) complex with same ligand-type and complexes where the metal is replaced by platinum and palladium(II).
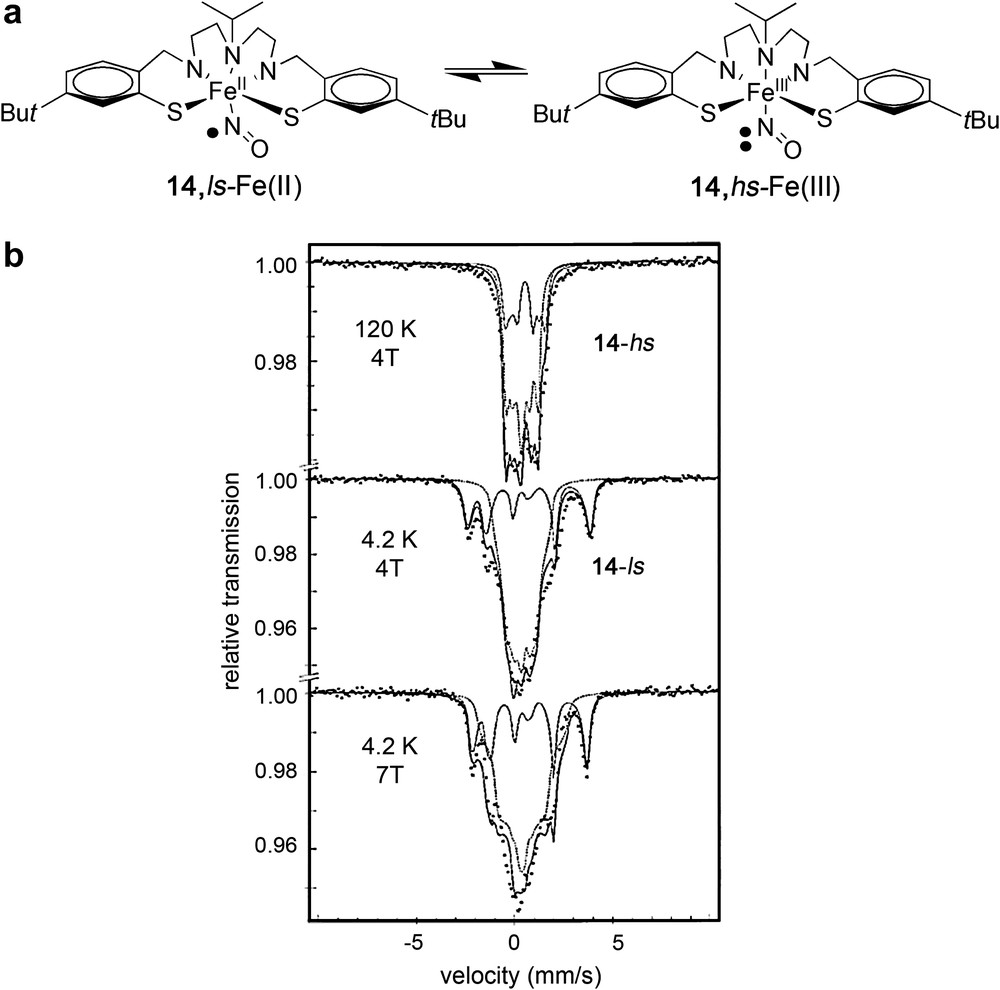
(a) Schematic representation of the valence tautomerism transition in the octahedral {FeNO}7 nitrosyl complex 14 (from S = 1/2 to S = 3/2). (b) Mössbauer spectra at different temperatures and fields (Ref. [60]).
A further step has been the study and characterization of O,N,O-coordinated type ligands containing two phenolate donor groups such as ligand BQ-N-SQ (also termed 2-[(3,5-bis(1,1-dimethylethyl)-6-hydroxyl-2,4-cyclohexadien-1-ylidene)amino]-4,6-bis(1,1-dimethylethyl)-phenol) [65]. These ligands, in addition to producing phenoxyl radicals in the presence of air, exhibit better chelating capabilities and good π-donor atoms that stabilize higher oxidation states [66]. The synthesis and mechanical features for the obtaining of the ligand BQ-N-SQ was first described in 1975 by Girgis and Balch [65c] treating the 3,5-di-tert-butyl-catechol with aqueous ammonia in the presence of a divalent metal ion under aerobic conditions. Since then, a considerable interest has increased in the scientific community for this family of complexes. Thanks to their rich redox activity, these ligands may exist at least in four different oxidation states (see Scheme 2) although structural and magnetic characterization of the complexes bearing this ligand suggest that they can be mostly found as MIV(Cat-N-SQ)2, MIII(Cat-N-SQ)(Cat-N-BQ) and MII(Cat-N-BQ)2. Other unusual interesting trends of the electronic distribution along different members of this family of complexes have also been found [65c,d,67]. Recently, Wieghardt et al. [68] showed the stabilization of the trianionic (Cat-N-Cat)3− form in a Cu(II) metal complex. Moreover, in the quest for dinuclear MII–phenoxyl radical complexes playing an active role in the aerobic catalysis of alcohols, two new zinc-complexes [69] [Zn(Cat-N-BQ)2] (17) and its isomeric red and paramagnetic form [Zn(Cat-N-SQ)(BQ-N-SQ)] (18) have been published. The last complex represents the first example where the oxidation levels of the two coordinated ligands differ formally by two electrons although the stability of this product is relatively low.
As far as valence tautomerism is concerned, the first complex of this family to be shown to exhibit VT was complex [CoIII(Cat-N-BQ)(Cat-N-SQ)] (19). Although first reported in 1988 by Pierpont et al. no evidence for tautomeric interconversion was shown to take place at that time [67a] Ten years later, the temperature-dependence of the spectral and magnetic properties of solutions of this compound in non-polar solvents suggested the existence of the valence tautomeric equilibrium shown in Eq. (4) [67d]. A few years later the interconversion was also shown to take place in the solid state [82] though at much higher temperatures.
| [CoIII(Cat-N-BQ)(Cat-N-SQ)] ⇌ [CoII(Cat-N-BQ)2] | (4) |
Afterwards, valence tautomerism has also been reported for the related manganese complex [MnIV(Cat-N-SQ)2] (20) [70] whereas its solid-state structural features are in agreement with the electronic distribution that involves the manganese atom in its oxidation state (IV), its solution temperature-dependence shows that this complex exhibits valence tautomerism involving three different tautomeric isomers associated to the three different oxidation states of the manganese ion: Mn(IV), Mn(III) and Mn(II) [67a,b].
The advantages of valence tautomeric Schiff base complexes over transition metal complexes with o-quinone ligands are considerable. First, valence tautomeric Schiff base complexes display higher stabilities relative to atmospheric oxygen atmosphere in solution and solid state. Second, the differences between the optical properties of isomers involved in the valence tautomerism of the cobalt Schiff base complex are enhanced when compared to those observed for cobalt complexes with o-quinone ligands. Third, the Schiff base ligand exhibits a richer electrochemical behaviour since it can exist in different oxidation forms, ranging from +1 to −3 (see Scheme 2b), which may lead to stable coordination complexes with several metal ions in a variety of oxidation states [69].
3 Valence tautomerism: beyond electroactive ligands and metal ions
As shown in the previous section, the presence of VT in a given complex has been traditionally associated almost exclusively to the role played by the redox-active ligand and the transition metal ion. More specifically to the degree of covalency in the interaction between them, which must be low, and the matching between the energy of their frontier orbitals, which must be similar [18]. However, the existence of several other factors that can modulate the observation of VT has also been detected along all these years of research in the field. In other words, the same complex may exhibit or not, or the VT process can be shifted to much higher temperatures, depending on other structural and environmental parameters such as those schematized in Fig. 5. Such factors can be grouped into three main areas of influence that are counterligand, charge-induced and environmental effects. It is the aim of the following section to revise more in detail such factors, which must be taken seriously into consideration for a proper evaluation of potential VT candidates.
Schematic representation of the different factors that have been shown to modulate valence tautomerism represented as casual ordered/disordered puzzles. Such representation is intended to emphasize the need for a good understanding of the effect of such parameters to fully understand and predict VT (i.e., the need for a proper connection of all the puzzles pieces).
3.1 Counterligand effect
Most of the work devoted to elucidate the role played by the counterligand has been made on the family of complexes with the general formula [CoIII(3,5-DTBCat)(3,5-DTBSQ)(N-N)]. Even though the N-N diazine ligand is not directly involved in the electron transfer process, Hendrickson et al. [71] Pierpont et al. [17] and Dei et al. [73] have concluded that it can be used to systematically shift the Tc for the equilibrium shown in Eq. (5) by changing its donor abilities.
| [CoIII(3,5-DTBCat)(3,5-DTBSQ)(N-N)] ⇌ [CoII(3,5-DTBSQ)2(N-N)] | (5) |
For instance, the systematic variation on the series of complexes with the general formula [Co(py2X)(3,5-DTBSQ)2] (X = O, S, Se and Te) has been shown to clearly influence Tc, inducing in some of the cases even a large hysteresis effect [72a]. Sato et al. [74] reported complex [CoIII(3,5-DTBSQ)(3,5-DTBCat)(dpa)] (dpa = N,N-bis(2-pyridylmethyl)amine) (21) with one of the highest Tc values (around 380 K) so far reported for a VT complex, a fact that was attributed to the strong coordination field of the counterligand used (dpa) (see Fig. 6). Another interesting example of the counterligand effect on the valence tautomeric processes has been described by Yamaguchi and Awaga [75] who reported valence tautomerism in the spin-labeled complex [Co(nnbpy)(3,5-DTBSQ)2] (22), where nnbpy is a bipyridine substituted nitronyl nitroxide radical. Even though the temperature-dependence of the magnetic susceptibility indicates a tautomeric interconversion above 250 K as is indicated in Fig. 6b, the ligand radical behaves as a Curie spin over the whole temperature region. Finally, interesting results were obtained for the series of complexes [Co(Cnbpy)(3,5-DTBQ)2] (23) (Fig. 6c). In these systems VT behaviour is tuned by modification of the bpy coligand with the alkyl groups (ranging from 0 to 13 alkyl chan) which favour crystal phase transition (cooperativeness) [76]. All these studies have demonstrated that the charge distribution in metal–quinone complexes is sensitive to the balance of metal and quinone orbital energies, tuned by the appropriate choice of the counterligand. Of special relevance is the possibility to use such modulation for the development of VT complexes that show hysteresis if a future molecular switching device based on these complexes is to be realized.
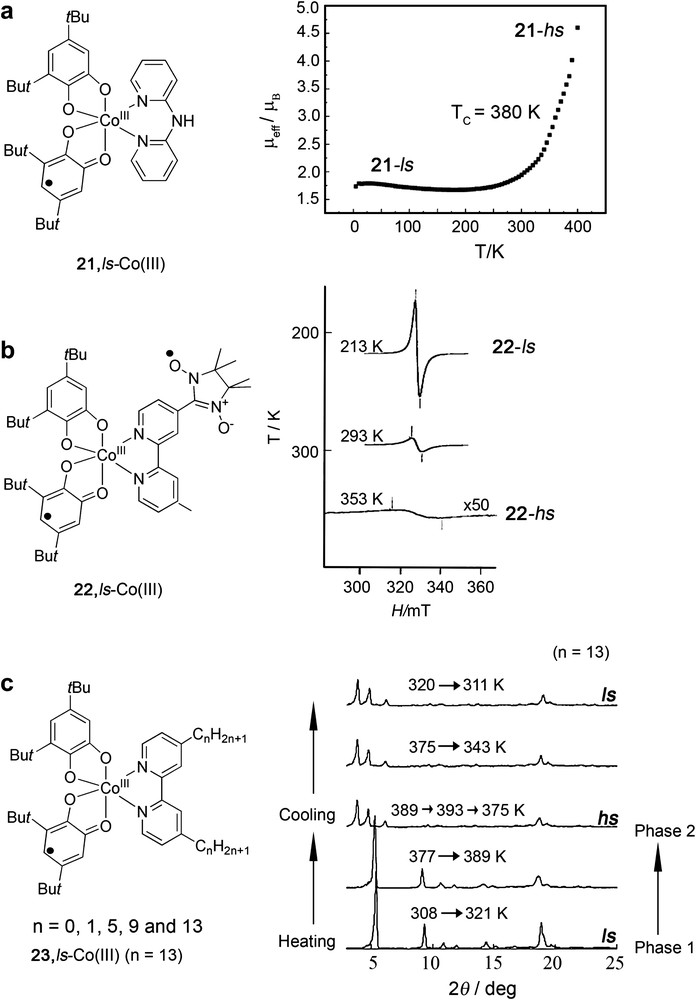
Representative examples of the counterligand effect on the VT for complexes with the general formula [Co(3,5-DTBCat)(3,5-DTBSQ)(N-N)]. A variation of such counterligand may induce the obtaining of a high interconversion temperature (Fig. 6a, Ref. [74]), a considerable shift (Fig. 6b, Ref. [75]) or even modifications of the fraction of both electronic isomers induced by a crystal phase transition (Fig. 6c, Ref. [76]).
The acceptor characteristics of the counterligand in a series of copper–quinone synthetic model with the general formula [(Qn−)Cun+L] have been studied due to their relevance not only for VT purposes but also on other biological or commercial processes. Strong π-acceptor ligands such as L = CO, CNR, PR3 or AsR3 stabilize the Cu(I)–semiquinone form whereas the use of non-π-acceptor ligands, such as amine ligands, favour the Cu(II)–semiquinone form [77]. For instance, Buchanan et al. [78] have demonstrated the possibility to modulate the charge distribution between the two electronic isomers [L2CuII(3,5-DTBCat)] and [L2CuI(3,5-DTBSQ)] by changing the nature of the counterligand from a strong nitrogen donor to a soft phosphine donor. On the other hand, Kaim et al. [79] have also reported the use of a weak π-acceptor thioether to induce sensitive valence tautomerism in paramagnetic copper complexes related to amine oxidase enzymes. In a more recent study, the comparison between two thioether ligands that differ in their π-acceptor capacity and structural rigidity has been used to have a comparative study of both effects [80].
Finally, another example of the relevance of VT systems beyond its potential use for molecular based devices is their influence on the catalytic center of citochrome c oxidase (CcO). The substitution of the tyrosinate cross-linked for a semiquinone group have generated a long-lived imidazolyl-phenoxyl radical [81]. The resulting complexes with the general formula (Cu–imidazolyl-phenoxyl) are created to mimic the CuBII–histidine–tyrosinate center in an attempt to study its catalytic potential on biological processes. A nice example of the formation of such mimic models is complex [(tpa)Cu(Im-hq∗H2)](OTs)2 complex (24). The study of its double deprotonation shows the evidence of the free radical anion (Im-sq*−) and the presence of a valence tautomerism on the coordinative labile copper(I) intermediate.
3.2 Charge-induced effects
3.2.1 Counter-ion effect
One of the few examples where the nature of the counterion has been shown to modify valence tautomerism is that of the ionic (charged) cobalt–quinone complexes with the general formula [ML(diox)]Y, where L is a tetraazamacrocycle ancillary ligand, diox = 9,10-dioxophenantrene and Y = PF6, BPh4, I [16,33]. Such complexes undergo a temperature and pressure-induced VT equilibrium with a transition temperature that varies with the volume and coulombic interaction of the counterion Y. Another example of the counterion effect was reported by Cador et al. [82] in the series of complexes [M(tpy)L]Y, where M = Ni or Co, tpy = 2,2′:6′,2″-terpyridine, L = Cat-N-SQ or Cat-N-BQ and Y = PF6, BPh4. It is found that the critical temperature for the interconversion in the cobalt case is strongly affected by the counterion. Whereas, no VT transition was detected for the tetraphenylborate compound, the related hexafluorophosphate complex exhibits a VT interconversion in solid state around 500 K, as confirmed by magnetic measurements. Contrary, in solution it takes place at lower temperatures, as shown by spectrophotometric measurements.
3.2.2 Redox activity
The redox activity of the ligands not only allows for the existence of valence tautomerism but also for additional switching capacities resulting from an electrochemical process. Such a strategy has been shown to be very useful for the systematic tuning of the critical temperature at which there are equal amounts of both tautomers, as well as to establish arrays of four or six members depending on the redox possibilities of the metal ion involved in the array (Figs. 7 and 8).
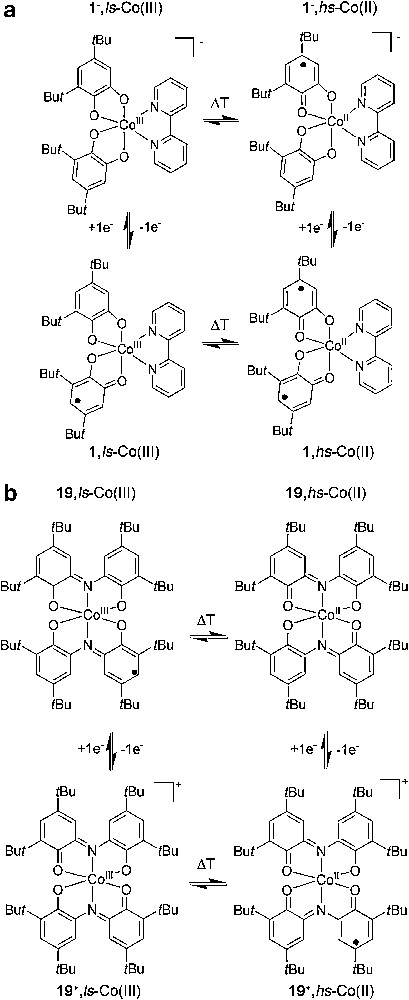
Array of four states showing different optical and magnetic ground states controlled by two temperature-controlled valence tautomeric equilibria and two reversible redox processes for complex [Co(3,5-DTBSQ)(3,5-DTBCat)(bpy) (1) (top) and complex [Co(Cat-N-SQ)(Cat-N-BQ)] (19) (bottom).
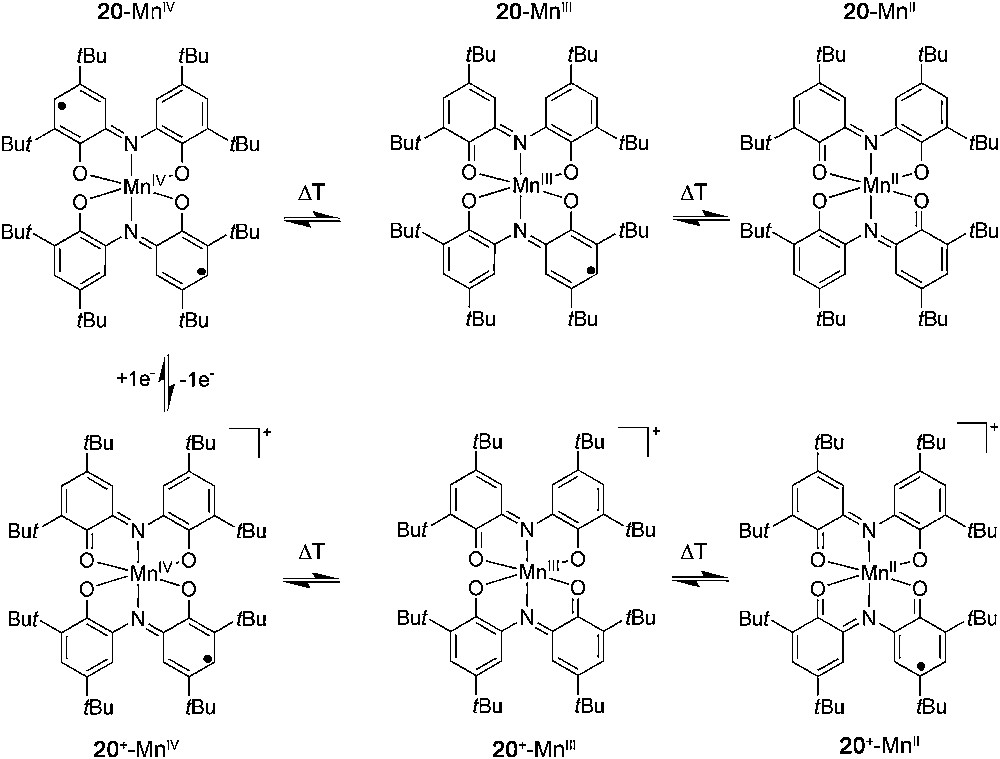
Array of six states showing different optical and magnetic ground states controlled by two temperatures and a reversible redox process for complex [Mn(Cat-N-SQ)(Cat-N-BQ)] (20).
The first example of redox-tuned valence tautomerism was based on the reduction of the complex [CoIII(3,5-DTBCat)(3,5-DTBSQ)(bpy)] (1) [83]. Interestingly, the resulting reduced complex also exhibited a temperature-dependence consistent with the existence of a valence tautomeric equilibrium. This fact allowed for the first time to establish an array of four states (Fig. 7) showing different optical and magnetic ground states controlled by two temperature-controlled valence tautomeric equilibria and two reversible redox processes. The possibility of entering the cycle at each state and advancing through the square array in a clockwise and counter-clockwise direction was also established.
Following this approach, Rovira et al. [84] also established an array of four states (see Fig. 7) based on the oxidation of the tautomeric complex [CoIII(Cat-N-BQ)(Cat-N-SQ)] (19). The charged species [CoIII(Cat-N-BQ)2]+ formed by partial oxidation was generated chemically. Interestingly, its variable-temperature absorption spectra showed the existence of an equilibrium consistent with the existence of a temperature-induced valence tautomerism shown in Eq. (6). This fact allowed the establishment of an array of four states showing different optical and magnetic ground states by using a reversible oxidation process as additional stimulus:
| [CoIII(Cat-N-BQ)2]+ ⇌ [CoII(Cat-N-BQ)(SQ-N-BQ)]+ | (6) |
Later on, three different oxidation states where the manganese metal ion can be most frequently found on a given complex, Mn(IV)–Mn(III)–Mn(II) allowed us to expand the array from four to six members using complex 20 (see Fig. 8). The principal parameters of the VT processes involved in the four and six-member arrays shown in Figs. 7 and 8 are summarized in Table 2.
Redox-tuned valence tautomeric systems in solution at different temperatures for complexes 1, 19 and 20
| Compound | Neutral | Ionic | ||
| VT isomer | Working temp. | VT isomer | Working temp. | |
| 1 | 1, ls-Co(III) | <273 K | 1−, ls-Co(III) | <200 K |
| 1, hs-Co(II) | <273 K | 1−, hs-Co(II) | <305 K | |
| 19 | 19, ls-Co(III) | <300 K | 19+, ls-Co(III) | <328 K |
| 19, hs-Co(II) | >300 K | 19+, hs-Co(II) | >328 K | |
| 20 | 20, Mn(IV) | <5 K | 20+, Mn(IV) | <5 K |
| 20, Mn(III) | 5 K < T > 270 K | 20+, Mn(III) | 5 K < T > 130 K | |
| 20, Mn(II) | >360 K | 20+, Mn(II) | >360 K |
3.3 Environmental effects
In this section, matrix factors (i.e., depending on whether the VT interconversion takes place in solution, solid state or embedded in a polymeric matrix) will be analyzed. As far as the study of VT phenomena in solution is concerned, the influence of the nature of the solvent on the VT equilibrium for the family of complexes [CoIII(Cat)(SQ)(N-N)] has already been shown [71–73]. By choosing the appropriate solvent and controlling the temperature of the solution, the tautomeric equilibrium of this family of complexes can be modulated and one of the two isomers can be made to be the dominant species in solution. Moreover, there is no direct correlation between the Tc values obtained experimentally for each solvent and their corresponding dielectric constant values. This fact evidences that the displacement of the tautomeric equilibrium most probably is not only controlled by ɛ but also by other solute–solvent interactions. As shown, tautomeric interconversion brings changes in the Co–L bond lengths, consequently the endoergic term (Ω) probably has also a reasonable influence. This term, characteristic of each solvent, measures the required work for separating the solvent molecules in order to accommodate them and provide a suitable sized and shaped enclosure.
These complexes can interconvert between the VT isomers also in the solid state though with significant differences. Indeed, whereas in solution the interconversion takes place over a large temperature range, in a crystalline matrix they can interconvert cooperatively in a narrow temperature range. For instance, it was shown that for the complex 4·C6H5CH3, [8,71] the tautomeric interconversion between the 4, ls-Co(III) and 4, hs-Co(II) isomers can be reversibly driven with temperature and monitored by large changes in the magnetic susceptibilities, with an interconversion that occurs abruptly within a narrow temperature range of ∼30 °C. Similar observations for the differential behaviour of VT complexes in solution and in the solid state were found for complex [CoIII(Cat-N-BQ)(Cat-N-SQ)] (19). In a preliminary work, the UV–vis temperature-dependence of a toluene solution of this complex in solution suggested the existence of a valence tautomeric equilibrium close to 300 K, [67d] as deduced by the ratio between the enthalpy and the entropy changes in solution. However, no tautomerism was observed in the solid state, as evidenced by the lack of changes in the variable-temperature magnetic susceptibility data up to 300 K. However, in a subsequent work [82], the same authors showed that indeed this complex exhibits a tautomeric interconversion in the solid state but at higher temperatures, with a transition that starts at approximately 370–380 K, i.e., at least 150 K higher than in solution. The variations found for the behaviour of these complexes in solution and in the solid state are not atypical. In fact, it is important to emphasize that whereas most of the valence tautomeric complexes thus far reported exhibit a temperature-dependent interconversion in solution, the number of examples exhibiting a valence tautomeric interconversion in the solid state is rather limited. Such variations between the behaviour in solution and solid state have been attributed by Dei et al. to variations of the entropy factor, as deduced by comparison of the thermodynamic quantities obtained for the same VT process in a toluene solution and in solid state. This is not surprising at all. The valence tautomeric interconversion from the ls-Co(III) to the hs-Co(II) leads to a gain in electronic entropy (ΔSelec) due to the higher spin state degeneracy of the hs-Co(II) form and the longer metal–ligand bond lengths of the hs-Co(II) tautomer, which results in lower energy vibrations and a higher density of vibrational states. The spin state degeneracy is not expected to vary in the different solvents under study. However, the variations in the metal–ligand bond lengths, i.e. the density of vibrational states, are expected to be strongly dependent on the surrounding matrix.
Finally, a remarked influence of the solvent nature has been found not only for measurements in solution but also when acting as guest molecules within a given crystalline network (in the solid state). This fact was initially attributed to the effect of solvate molecules that are assumed to impart softness to the lattice, and therefore, to influence any possible phonon relaxation mechanism. For instance, whereas the complex [CoIII(3,5-DTBSQ)(3,5-DTBCAT)(phen)]·C6H5CH3 (4·C6H5CH3) interconverts abruptly within a narrow temperature range of ∼30 °C, a polycrystalline sample recrystallized from ethanol has a μeff value essentially independent of the temperature and close to the value of 1.7 μB expected for the low-spin isomer [85]. In between, for samples recrystallized from acetonitrile and methylene chloride, incomplete transitions were observed. The two paradigms or extremes are the toluene and ethanol solvates whereas in the first of the cases the solvate molecules are favouring a strong cooperative effect that induces an abrupt transition over a short range of temperatures, the ethanol guest molecules are either suppressing or shifting to much higher temperatures the interconversion. The obtaining of a systematic correlation between Tc and the nature of the solvent remains elusive. However, the correlation of Tc found in the different solvate crystallographic forms differs from that observed for the solutions of the same solvent. This is once again not surprising at all, in solution the target molecules are mostly surrounded by solvent molecules, whereas in the crystalline network they are surrounded by other neighbour molecules. All the attempts to obtain different crystallographic phases of complex 19 by recrystallization from different solvents to give more light on this topic were unsuccessful, obtaining the same crystallographic phase in all the cases as confirmed by X-ray powder diffraction. However, the obtaining of an amorphous phase showed that the disorder introduced within the network in this case did not suppress or shift the VT to much higher temperatures, as previously observed for complex 4. For comparison purposes, the X-ray diffraction data obtained for the different crystalline phases of complexes 4 and 19 and the corresponding VT characteristics are shown in Fig. 9.
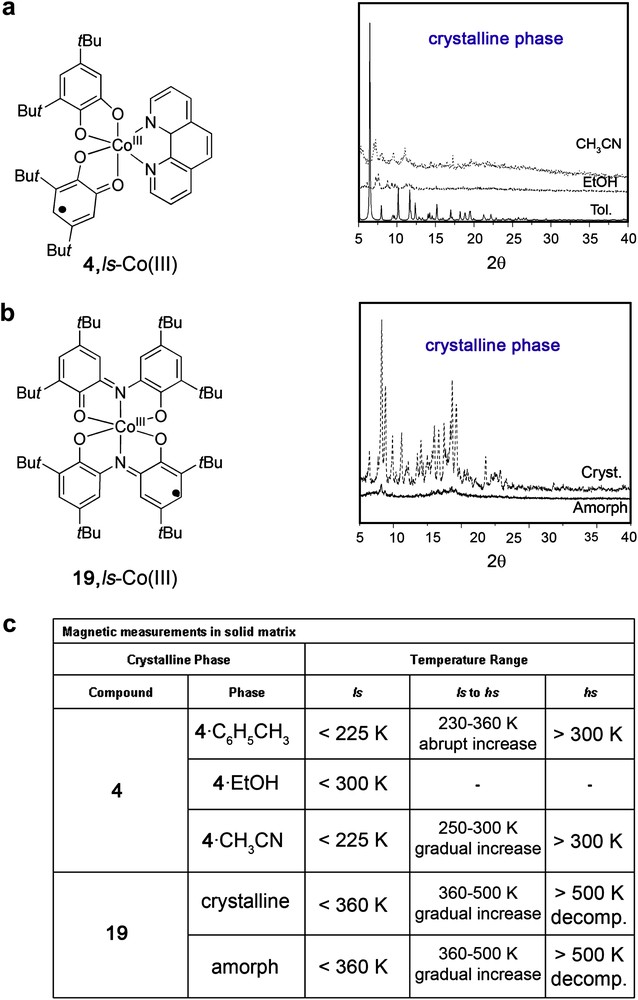
Influence of the crystalline phase on the VT interconversion found for complexes 4(a) and 19(b). The different VT parameters found for each of the cases are summarized in Table (c), Ref. [85].
Other examples of the solvent guest molecule effects on the VT interconversion have been reported by Dei et al. on the family of complexes of general formula [Co(CTH)(Phendiox)]Y·solv, where CTH stands for tetraazamacrocycle and Phendiox for the catecholato or semiquinonato form of 9,10-dioxophenantrene. The critical interconversion temperature was found to depend not only on the nature of the counterion Y but also on the nature of the solvent trapped [86] in the lattice. Moreover, significant hysteresis effects were detected by substituting the solvent guest molecules found within the lattice by its deuterated analogue. Within this scenario, the differential “softness” that the solvent molecules are giving to the crystalline network is clearly affecting the vibrational relaxation of the molecules.
4 Conclusions
The observation of valence tautomerism has been traditionally associated almost exclusively to the role played by two main actors, the redox-active ligand and the transition metal ion. In other words, requirements of potential candidates to exhibit VT were always seek to fulfil the following requirements: (1) the degree of covalency in the interaction between metal ion and electroactive ligand must be low; and (2) the energy of their frontier orbitals must be similar. However, the occurrence of several other factors that can modulate the observation of VT have also been detected during these years of research in the field. In other words, the same complex may exhibit or not, or the VT process be shifted to much higher temperatures, depending on other structural and environmental parameters. Such factors have been divided into three main categories (counterligand, charge and environmental effects) and thoroughly discussed in this review. These results summarized are very important for future investigations in the field. Among others, they reveal the necessity to use a very systematic approach when identifying new VT candidates since the use of an inappropriate solvent may mask the VT interconversion.
Acknowledgments
Authors would like to thank Prof. A. Dei and Prof. D.N. Hendrickson for fruitful discussions and share of knowledge. We also acknowledge financial support of the Ministerio de Educación y Ciencia under grant (MAT2006-13765-C02) and the network of Excellence Magmanet. E.E. thanks the Ministerio de Educación y Ciencia for a predoctoral grant.