

1 Introduction
In the vast majority of molecules and materials, the specific value provided by the presence of benzene rings owes much to their aromaticity-related properties of either nature, intrinsic (rigidity, insulating/conjugating bridge effects, hexagonal paving of the graphene or carbon nanotube surface,…), or extrinsic (substitution reactivity, complexation to transition metals, π–π-stacking and intercalation in DNA grooves,…). Although the chemical properties of the C6 benzene ring are unique, a topologically equivalent expanded version is the C18 carbo-benzene ring that inevitably deserved consideration [1]. Many theoretical studies have thus supported the relevance of the analogy by showing that the carbo-benzene ring is just slightly less aromatic than its parent ring [2], and experimental observations have confirmed these previsions (synthesis of several stable examples exhibiting a cyclically delocalized structure and NMR deshielding of peripheral protons) [2b,3].
Most of the known carbo-benzene representatives are decorated by phenyl or trimethylsilylethynyl groups, and by, at most, one additional kind of substituent in either octupolar (A, R = H, tBu, 4-tBu–C6H4) [1a,4], dipolar (B, R = H) or quadrupolar (C: R = H, CCSiMe3) arrangement (Scheme 1) [3,5]. They have all been obtained by reductive aromatization of hexaoxy-[6]pericyclynes.[3,5,6] Pericyclynes were exemplified first by Scott et al. in the hydrocarbon series with CMe2 vertices [7], and more recently by Ueda et al. [1a], and by us in the functional series with various CR(OR′) vertices [3,6,8,9], The latter were synthesized through multistep routes, where the ring formation occurs in a one-pot-two-step [(18−n)+n] process from a C18−n ω-bisterminal diyne and a 1,n-dicarbonyl dielectrophile (n = 4, 7, 10) (Scheme 2) [3].
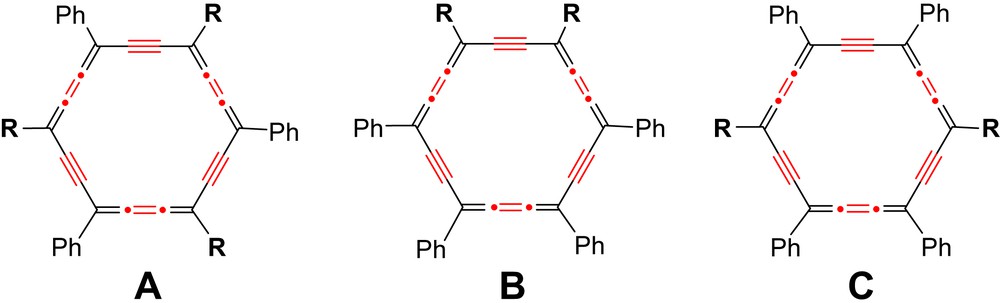
Examples of substitution patterns of carbo-benzenes bearing two different kinds of substituents, Ph and R: A, octupolar; B, dipolar, C, quadrupolar.
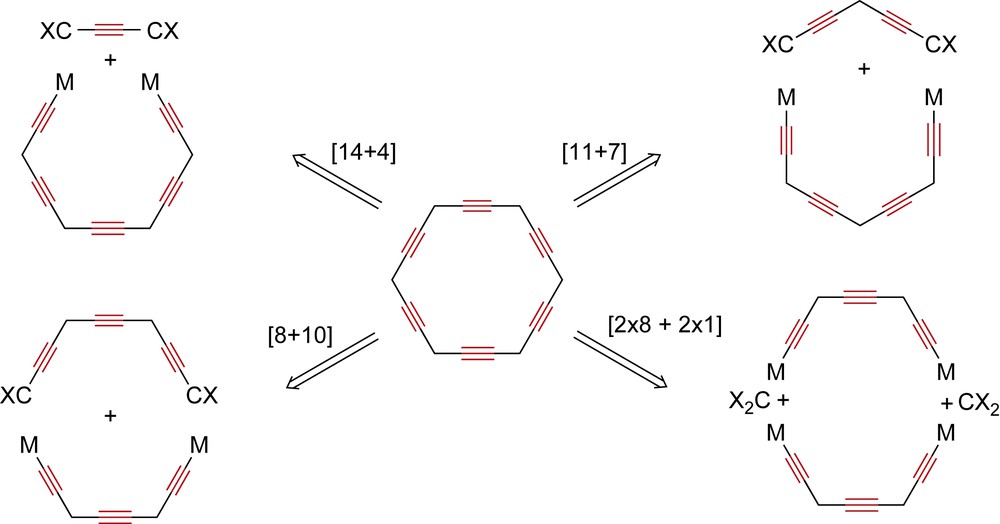
Possible strategies for the C18 ring formation of [6]pericyclynes, through a one-pot process for the formation of two or four endocyclic bonds.
One prevailing criterion of synthetic efficiency is the versatility of the strategy, allowing access to different carbo-benzenes through a maximum number of steps in common. Within this prospect, the m-[6]pericyclynetrione 1 proved to be a valuable last-but-one precursor of octupolar carbo-benzenes of type A [1a,4]. The analogous [6]pericyclynedione 2 can thus be naturally envisioned as a versatile precursor of quadrupolar carbo-benzenes of type C (Scheme 3) [9].
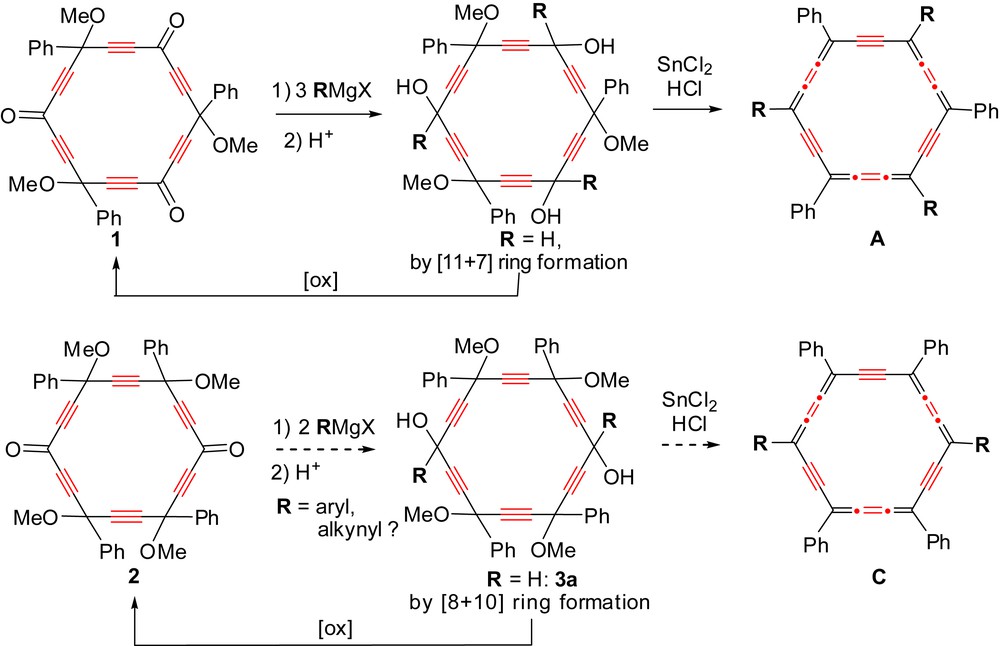
Versatility of [6]pericyclynetrione and [6]pericyclynedione precursors of octupolar and quadrupolar carbo-benzene derivatives, respectively.
The [8 + 10] route (Scheme 2) to 2 via [6]pericyclynediol 3a (Scheme 4) was previously shown to be quite efficient regarding the length (nine steps from commercially available trans-dibenzoylethene) and the divergence/convergence level (triyne 4 is also the precursor of dialdehyde 5) [9]. In spite of this success, improvements were required regarding two additional criteria.
- (i) The yields, and in particular of the ring formation step 4 + 5 → 3a (12% reported) [9].
- (ii) The availability of the reactants and reagents. Efforts in this sense are described in the following report. Three related challenges are also naturally addressed.
- (iii) The generalization of the [8 + 10] ring formation process to substrates of higher and lower oxidation states than dialdehyde 5, namely diester 6a and ditosylate 7, respectively.
- (iv) The possibility of an even shorter route via a direct four-step-one-pot [2 × 8 + 2 × 1] ring formation process (Scheme 2) from two molecules of 4 and two phosgene equivalents.
- (v) The resolution of the five diastereoisomers of pericyclynedione 2.
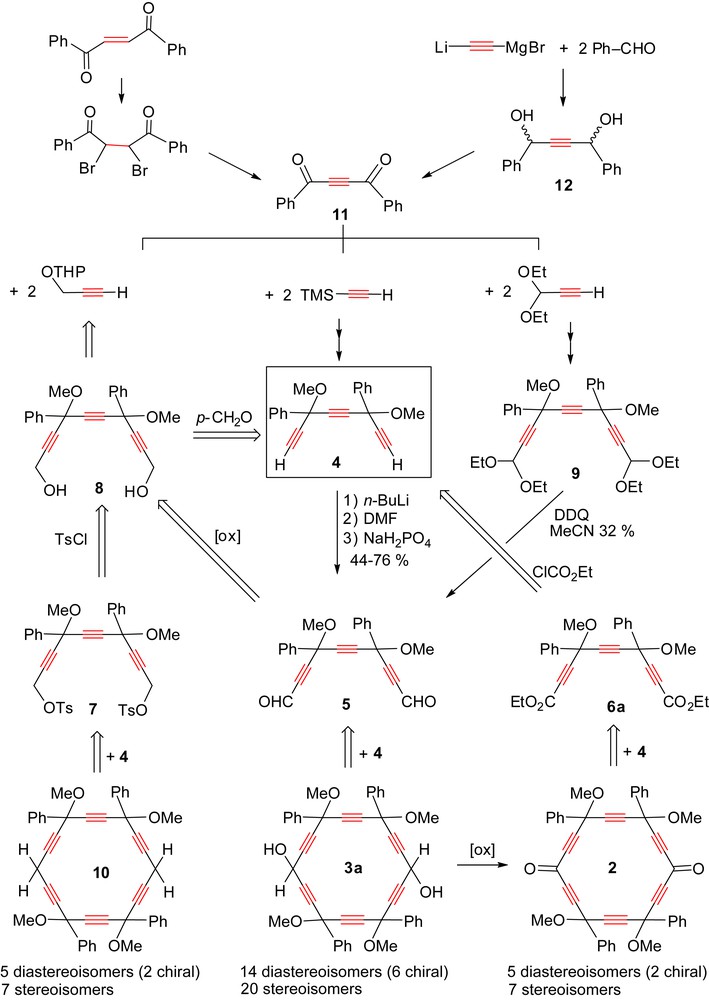
Synoptic view of the state-of-the-art and proposed investigations for the synthesis of tetraphenyl-[6]pericyclyne derivatives by a [8 + 10] route from the pivotal triyne 4 and three types of dioxygenated C10 dielectrophiles 5–7. The number of stereoisomers of the targeted [6]pericyclyne derivatives 2, 3a, 10 depends on the oxidation level of the opposite unsubstituted vertices (CH2, CHOH, CO).
2 Results and discussion
The state-of-the-art and the addressed challenges for the synthesis of functional pericyclynes, and in particular the pivotal [6]pericyclynedione 2, are summarized in Scheme 4 [3]. The results are presented below in four sections: (i) the synthesis of C10 dielectrophiles (dialdehyde 5, diester 6a, and ditosylate 7), (ii) the investigation of ring formation processes to [6]pericyclynes 3a, 10 or pericyclynedione 2 from triyne 4 and the various C10 dielectrophiles 5–7, (iii) the resolution of the five diastereoisomers of [6]pericyclynedione 2, and (iv) the reactivity of pericyclynedione 2.
2.1 Synthesis of C10 dielectrophilic synthons: dialdehyde 5, diester 6a and ditosylate 7
Dibenzoylacetylene 11 has been used as a key C4 brick for the synthesis of functional [5]- and [6]-pericyclynes by various [(15 + n) − n] and [(18 − n) + n] strategies, respectively [3,8,9]. It was hitherto prepared in two steps (dibromination and dehydrobromination) from commercially available trans-dibenzoylethene [10], but it could be alternatively obtained from cheaper reactants, namely benzaldehyde and the mixed lithium–bromomagnesium diacetylide, thus giving the diol 12 in 60% yield [11]. Amongst efficient mild oxidizing agents of primary and secondary alcohols, a cheap alternative to the Dess-Martin reagent is the so-called IBX reagent, which was prepared from iodobenzoic acid and oxone® in 79–81% yield [12]. Diol 12 was converted to diketone 11 by oxidation with either catalytic IBX and oxone® [13] or with MnO2 in 66% and 63% yield, respectively. It is worth noting here that a η2-Co2(CO)6 complex of diol 12 was previously prepared by reaction of phenylmagnesium bromide with the corresponding butynedial complex [14].
The diketone 11 was then converted to a meso/dl mixture of the C8 triyne 4 in three steps and 67% yield [8,9]. Both diketone 11 and triyne 4 were subsequently used for the preparation of various C10 dioxygenated dielectrophiles.
2.1.1 Synthesis of the C10 triynedial 5
Triynedial 5 has been first prepared by direct formylation, i.e. by treatment of diyne 4 with n-BuLi and DMF. The presence of monoaldehyde among the products required purification by column chromatography, allowing us to isolate the dialdehyde 5 in 32% yield only [8]. An alternative route was thus developed in five steps through the bisacetal 9 obtained by addition of the lithium salt of propiolaldehyde diethylacetal to diketone 11 [8]. Deprotection of the acetal ends proved to be difficult, and could be achieved under neutral conditions only (DDQ/CH3CN/H2O) in poorly reproducible crude yields. The triynedial product 5 could be purified by column chromatography, but partial decomposition on silica gel resulted in a dramatic decrease of the yield [8]. In order to circumvent this sensitivity, a method for the generation of crude-but-clean 5 was required. It could thus be generated by oxidation of diol 8 with MnO2 (pre-activated by heating overnight at 150 °C under vacuum) [9,15]. In THF as solvent, oxidation was not complete (monoaldehyde was always present) and 5 was produced in poor crude yields (<30%). In chlorinated solvents however (CH2Cl2 or ClCH2CH2Cl), trinynedial was generated in a spectroscopically pure state (according to NMR analysis). Nevertheless, after filtration and evaporation, the weighting yields remained erratic (20–60%) depending on the temperature (20–80 °C), the reaction time (2–8 h), and the number of equivalents and addition rate of MnO2 (40–60 equivalents added in one, two or three portions). The weight loss could be attributed to adsorption of over-oxidized products on the MnO2 powder (treatment of the final solid with 2 N NaOH, followed by filtration, HCl re-acidification and extraction in diethylether gave a residue, the 1H NMR spectrum of which was compatible with the dicarboxylic acid of diester 6a).
Other classical oxidizing agents proved inefficient (oxalyl chloride/DMSO [16], DCC/Py/TFA [17], BaMnO4/CuSO4/Al2O3,… [18]). In refluxing ethyl acetate, treatment of triynediol 8 with IBX (3–8 equivalents) leads to mixtures of the corresponding mono- and dialdehydes, even after quite long time (≥7 h). An alternative solvent of similar boiling temperature (83 °C) is 1,2-dichloroethane (DCE): when the reaction was conducted in refluxing DCE in the presence of six equivalents of IBX, simple filtration and evaporation afforded pure triynedial 5 in 83% yield (Scheme 5). Although no obvious explanation is proposed, this result provides a further illustration of the astonishing “solvent effects” of DCE [19].
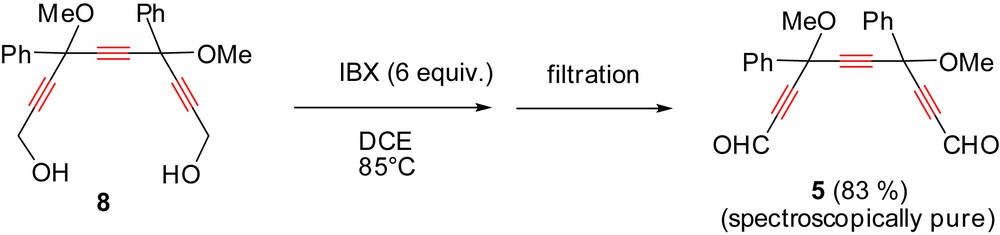
Simple and efficient process for the preparation of spectroscopically pure triynedial 5 without chromatographic purification.
The excess of IBX and its reduced form IBA can both be recovered at the end of the reaction by simple filtration. This mixture can then be easily re-oxidized by treatment with oxone® in water at 70 °C for 3 h. This recycled oxidative agent can be re-used without any problem [12c].
2.1.2 Synthesis of the C10 triynediester 6a
Triyne 4 was deprotonated twice with n-BuLi in THF at −78 °C and then reacted with an excess of ethyl chloroformate (Scheme 6). After treatment, the meso/dl mixture of diester 6a was isolated in 79% yield. The side product 6b resulting from the addition of two acetylides to the same ester group could also be isolated in 8% yield. No pericyclynedione 2 was however observed. Direct double carboxylation of triyne 4 thus occurs in a much more selective way than does direct double formylation of the same substrate (see Section 2.1.1) [8].
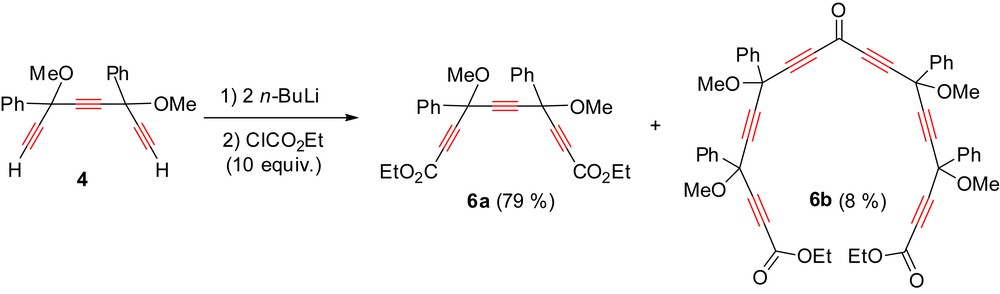
Double carboxylation of triyne 4.
2.1.3 Synthesis of the C10 triynediol 8 and ditosylate 7
Triynediol 8 was previously prepared in excellent yield by bubbling formaldehyde gas into a solution of the dilithium salt of triyne 4 [15]. We found that it can be more conveniently obtained in 95% yield by simply adding solid para-formaldehyde to this solution.
Diol 8 can also be prepared via its THP-protected precursor 13 (Scheme 7). Indeed, reacting two equivalents of the lithium salt of THP-protected propargylic alcohol with diketone 11 gave triynediol 13 in 87% yield. The latter was deprotonated with n-BuLi, then O-silylated to bis(silylether) 14 (71% yield) or O-methylated to triynediether 15 (88% yield). Treatment of the diether 15 with PPTS (pyridinium p-toluenesulfonate) in methanol allowed for a selective cleavage of the THP groups, affording diol-diether 8 in 80% yield.
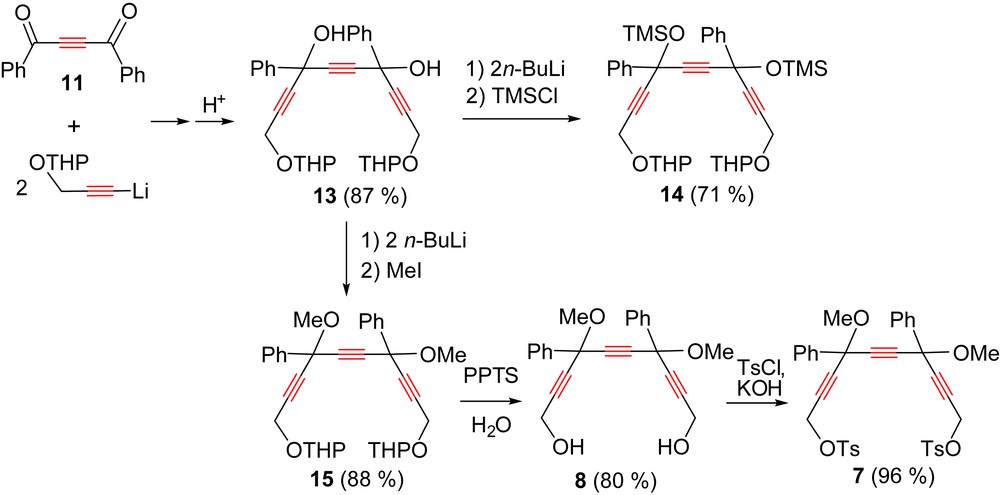
Synthesis of the triynediol 8 and ditosylate thereof via THP-protected propargylic alcohol derivatives.
The diol 8 was finally cleanly converted to the corresponding meso/dl mixture of ditosylate 7 in 96% yield by reaction with p-tolylsulfonyl chloride in the presence of potassium hydroxide.
2.2 [8 + 10] Ring formation of [6]pericyclynes from various C10 dielectrophiles
The ideal route to pericyclynedione 2 would rely on a “supramolecular” [2 × 8 + 2 × 1] process (Scheme 2) involving two equivalents of the dianion of triyne 4 and two equivalents of phosgene. However, reaction of the dilithium salt of 4 with diethylcarbonate ((EtO)2CO) or triphosgene (CCl3–O–C(O)–O–CCl3) failed to produce the expected pericyclynedione, giving only undetermined polymeric materials. Similarly, reaction of two equivalents of the dilithium salt of 4 with two equivalents of HC(O)OEt did not allow for the isolation of the expected pericyclynediol 3a: only undetermined polymeric compounds were obtained.
These results, simply explained by the obvious difficulty to control a four-step-one-pot [2 × 8 + 2 × 1] process, prompted us to turn back to the more classical strategy based on two-step-one-pot [8 + 10] processes (Scheme 2).
The diester 6a was first envisioned as a possible C10 synthon in a two-step two-component process. Nevertheless, reaction of the dilithium or dimagnesium salt of triyne 4 with 6a (see Scheme 4) gave polymeric materials only.
The ditosylate 7 has then been reacted with triyne 4 (see Scheme 4) in the presence of the CuI/NaI/K2CO3/DMF system [20,21], which was previously envisioned for the generation of [5]- and [10]-pericyclynes from 1,4-ditosyloxybut-2-yne and a C11 bisterminal tetrayne [22]. None of the products could be, however, unambiguously identified in the present case. This result is to be related to the well-documented versatile reactivity of propargylic halides and pseudo-halides [23,24].
The dialdehyde 5 has been previously used as a C10 dielectrophile in poorly selective [5 + 10] and [8 + 10] ring formation processes [8,9], and its reactivity was thus resumed in more details. Using the dilithium salts of a 1,4-pentadiyne and triyne 4 as dinucleophiles, the corresponding [5]pericyclynediol and [6]pericyclynediol 3a were obtained in 18% and 12% isolated yield, respectively. In the latter case, a considerable amount of the acyclic nonaynediol adduct 3b was also formed, and oxidation of the carbinol vertices was then carried out from a 70:30 3a:3b mixture, to give the corresponding mixture of diketones, from which pericyclynedione 2 could be isolated in 20% yield (Scheme 8) [9]. The conditions required to be optimized. It was found that using the di(bromomagnesium) salt of triyne 4 (instead of the dilithium one) in diluted conditions (3 × 10−3 M), pericyclynediol 3a was formed in a better yield (31%). This improvement can also be partly attributed to the modification of the purification method, with the choice of optimized eluting system for the silica gel chromatography (hexane/CH2Cl2/AcOEt 7/2/1, then 5/3/2). Oxidation could then be carried out from a pure sample of 3a with MnO2 or IBX to give pericyclynedione 2 in 72% or 70% yield, respectively (Scheme 8). It is, however, noteworthy that the use of MnO2 gave more reproducible results than IBX. The pericyclynedione 2 was thus finally prepared in 9 steps from commercially available dibenzoylethene and 7% overall yield.
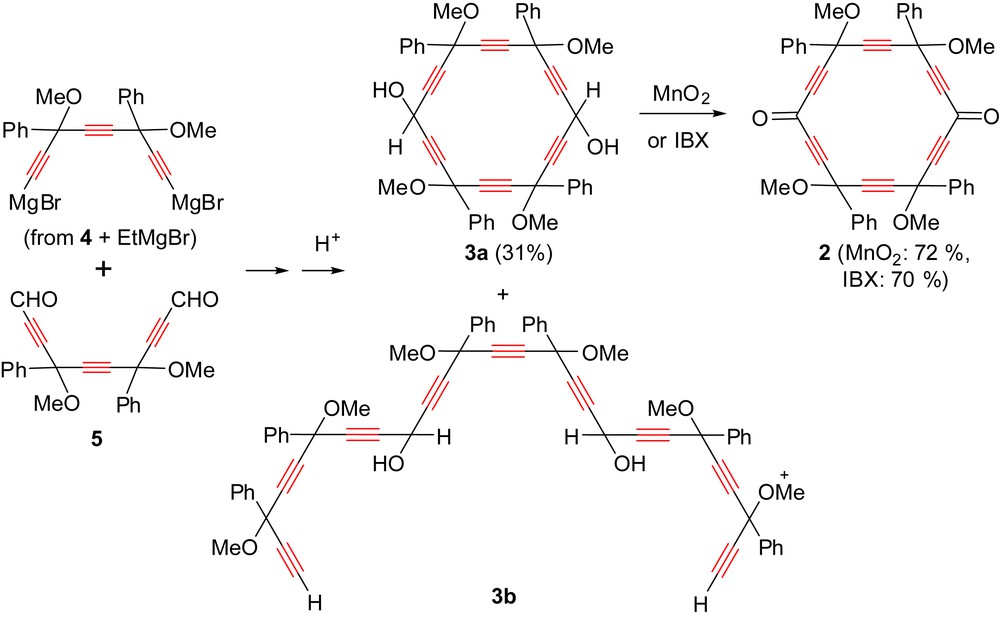
Optimized access to [6]pericyclynedione 2 via a [8 + 10] ring formation process.
2.3 Stereochemical resolution of [6]pericyclynedione 2
Replacement of CH2 vertices of [n]cycloalkanes by potentially stereogenic CRX vertices induces peculiar stereochemical manifolds [25]. For example, removal of one type of potentially stereogenic vertices in pericyclynedione 2 results in a dramatic decrease of the number of diastereoisomers (Scheme 9).
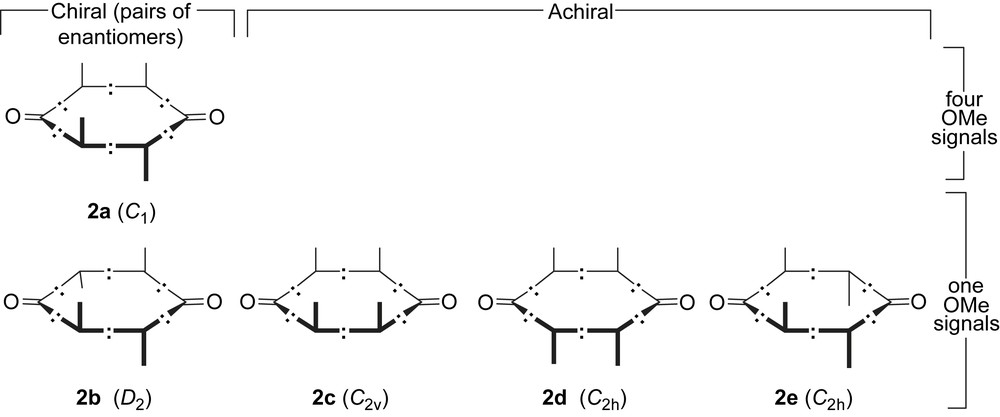
The five diastereoisomers of pericyclynedione 2. Butyn-1,4-diyl edges are featured as –:–. Nacked vertical lines and opposite blanks represent methoxy groups and phenyl groups.
The resolution of 2 was thus a reasonable challenge. Although the five diastereoisomers exhibit a single TLC spot under heptane/ethyl acetate (7/3) eluting conditions, three distinct spots can be observed by eluting with heptane/dichloromethane (8/2). Three stereoisomers could be finally separated by analytical HPLC techniques (petroleum ether/dichloromethane 3/7), the two others coming out at the same retention time with the selected column (Fig. 1).
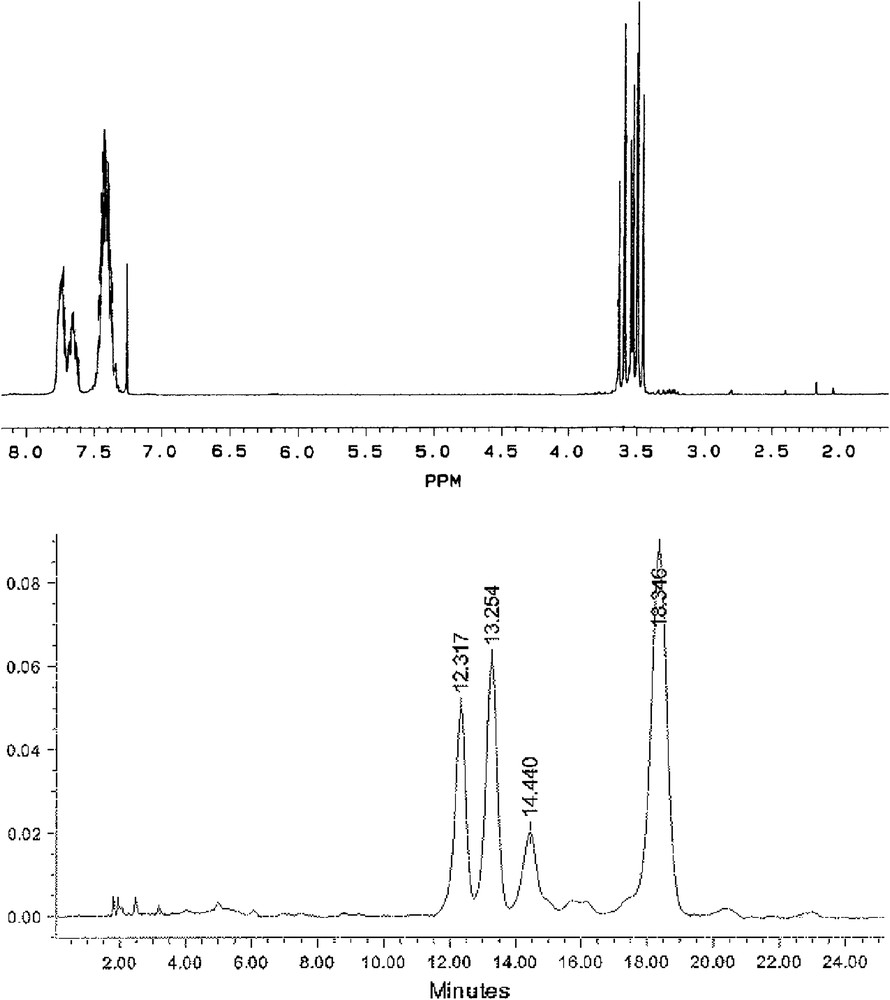
1H NMR spectrum of the mixture of the five stereoisomers of 2 (top) and their analytical HPLC chromatogram (petroleum ether/CH2Cl2 3/7) (bottom).
Semi-preparative HPLC was thus performed under the same eluting conditions but using a different column (semi-preparative Sunfire column 5 μm 150 mm × 19 mm, Waters), through which the retention times are about twice those of the analytical column used. It allowed for the separation of three good quality fractions A, B, C, the corresponding 1H NMR spectra and HPLC chromatograms being depicted in Fig. 2.
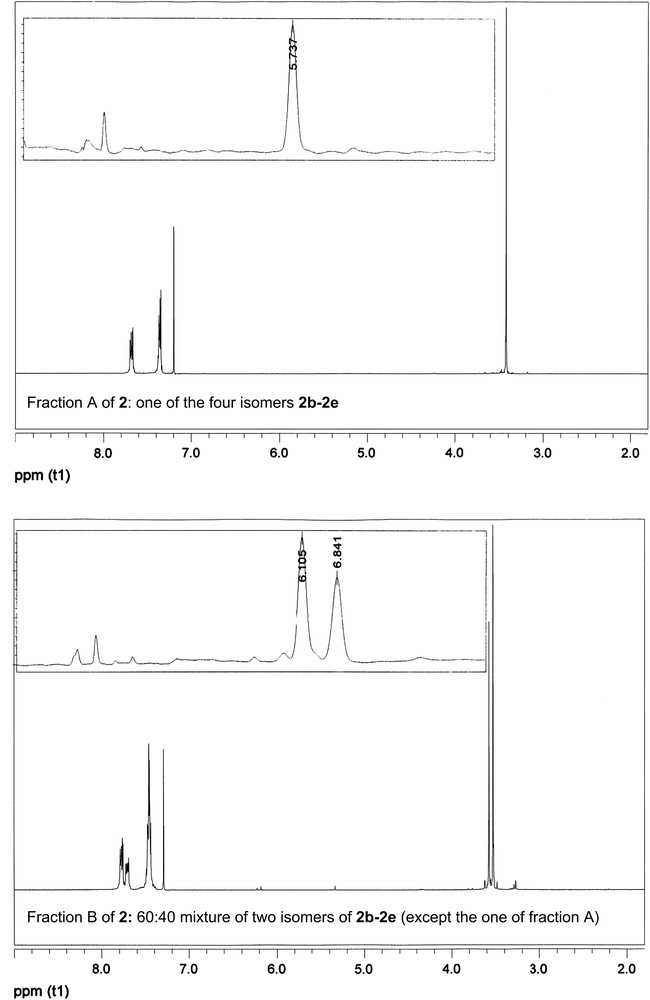
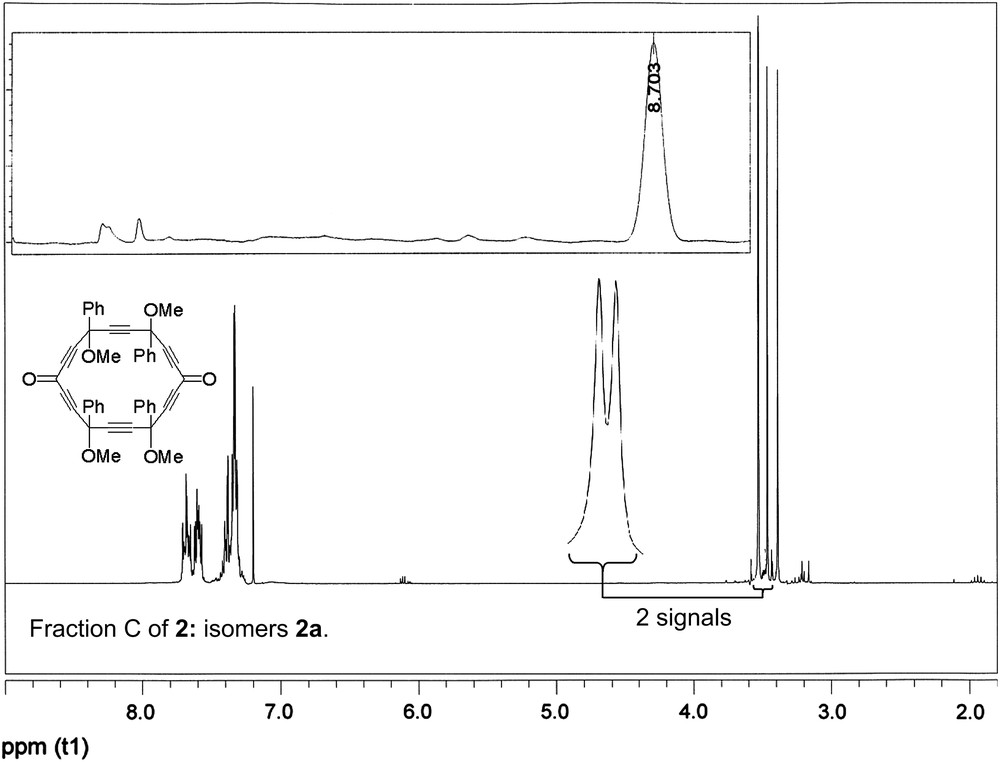
1H NMR spectra and corresponding HPLC traces of the three good quality fractions A (top), B (middle), C (bottom) of the semi-preparative HPLC separation of 2 (mixture of isomers 2a–2e: Scheme 9).
On the basis of its single 1H NMR OCH3 resonance, the less polar fraction A with a single HPLC peak (Rt = 5.74 min) corresponds to one of the four achiral stereoisomers 2b–e (Scheme 9). The second fraction B with two HPLC peaks (Rt = 6.11, 6.84 min) exhibits only two 1H NMR OCH3 resonances and can thus be assigned to a 60:40 mixture of two isomers amongst 2b–e, except the isomer of fraction A. The third fraction C consists in a single peak in the semi-preparative HPLC chromatogram (Rt = 8.70 min) and thus results from an unexpected separation of one of the two isomers coming out at roughly identical retention times on the analytical column (Fig. 1). Fraction C exhibits four 1H NMR OCH3 resonances of identical integrals, which can be unambiguously attributed to the four non-equivalent methoxy groups of the chiral isomer 2a.
The chair conformation of a [6]pericyclyne ring was established by X-ray diffraction analysis on two instances: the C18Me12 hydrocarbon representative with CMe2 vertices [26], and the all trans-isomer of the functional representative with C(OMe) (CCH) vertices [6]. The potential energy surface of [6]pericyclyne itself in the gas phase was calculated (at either MM or DFT level) to be very flat, with several quasi-isoenergetic conformational minimas (chair, boat, twisted) [7b,27]. The flat D6h conformation was predicted to be not a minimum, but the presence of two sp2 vertices might suffice to “flatten” the [6]pericyclynedione ring. Several attempts at X-ray diffraction analysis of single crystals of 2 proved however unsuccessful. Enhancement of the diffracting power was attempted by coordination to an Ag+ cation. Indeed, Scott et al. reported that dodecamethyl-[6]pericyclyne may act as a ligand of silver ions [7b]. However, no [2/Ag]+ complex could be characterized after treatment of one of the isomers of pericyclynedione 2 (fraction A) with a quasi-stoichiometric amount (0.9 equiv.) of AgOTf in THF at room temperature. This failure can be attributed to the weak coordinating power of the four triple bonds adjacent to the keto vertices of 2: the hexagonal-coordination picture proposed by Scott would thus require sufficiently donating triple bonds, as they are made by the six CMe2 vertices of the [Ag(C18Me12)]+ complex [28].
2.4 Reactivity of pericyclynedione 2
Reaction of 2 with two equivalents of trimethylsilylacetylenemagnesium bromide afforded the mono-adduct (16) and bis-adduct (17) in 43% and 22% isolated yields, respectively (Scheme 10). The bis-adduct is actually a regioisomer of [6]pericyclynediol 18, previously obtained by a [14 + 4] ring formation route (Scheme 2), and converted to the quadrupolar dialkynyl carbo-benzene 19 [9]. Whereas 18 possesses 20 diastereoisomers (and 36 stereoisomers), 17, just as 3a, possesses 14 diastereoisomers (and 20 stereoisomers) only.
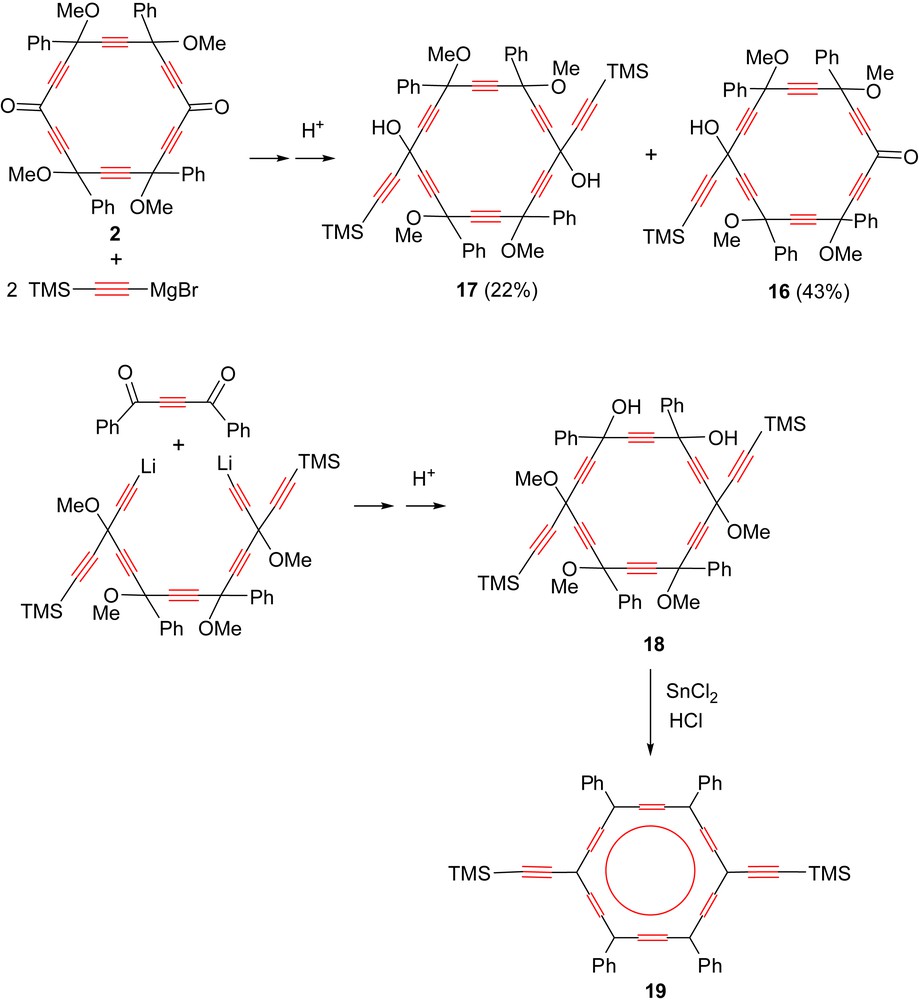
Syntheses of regioisomeric dialkynyl-[6]pericyclynediols 17 (this work) and 18 [9]. The latter was shown to undergo reductive aromatization to the quadrupolar carbo-benzene 19 [9].
As indicated by the multiplicity of the NMR signals of the crude material, the dialkynyl-[6]pericyclynediol 17 was a priori obtained as a mixture of the 14 diastereoisomers (Scheme 10). In the course of the chromatographic purification however, a fraction exhibited remarkably simple and sharp 1H and 13C{1H}-J-Mod NMR spectra, with just the number of signals corresponding to a maximum number of geometrically non-equivalent nuclei (Fig. 3). In particular, four OCH3 signals, two OH signals, and two Si(CH3)3 signals with identical integrals for each kind, allowed us to assign the sample to a unique diatereoisomer amongst those of C1 maximal symmetry, i.e. 17c–f (Scheme 11: the chiral isomers 17a–b would exhibit twice less signals). Although the exact stereochemistry of the sample remains to be determined, partial assignment of the NMR resonances can be attempted. The OCH3 signals indeed occur at 3.37, 3.50, 3.56 and 3.59 ppm: the latter chemical shifts are particularly close to each other. In each diastereoisomer 17c–f, one may also graphically identify two OCH3 environments being more similar to each other (up to seven bonds away, and denoted as a and a′ in Scheme 11) than any other pair of OCH3 environments (up to four bonds away only). The quasi-degenerate resonances at 3.56 and 3.59 ppm may thus be assigned to methoxy groups at position types a and a′ (Fig. 3, Scheme 11). In the 13C-{1H}-J-Mod spectrum, only the sp3 carbon vertices of either kind, benzylic (one signal instead of four) or trispropargylic (one signal instead of two), are accidentally degenerate (Fig. 3). It is also noteworthy that the 12 non-equivalent endocyclic sp-carbon atoms resonate at 12 different chemical shifts.
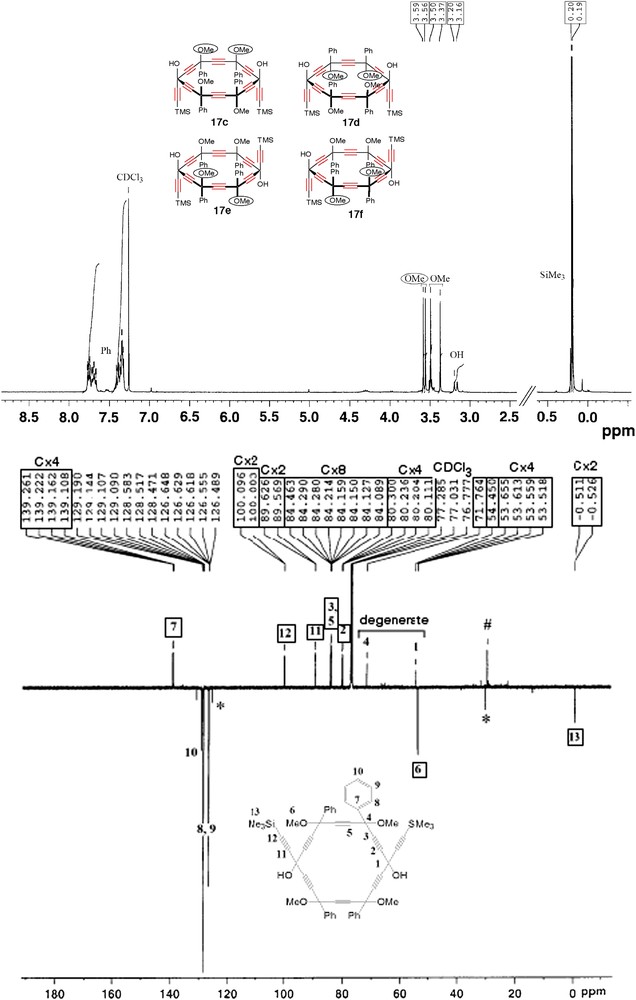
1H NMR spectrum (Top, 300 MHz,) and 13C{1H} J-Mod spectrum of a single diasteroisomer of pericyclynediol 17 in CDCl3 solution.
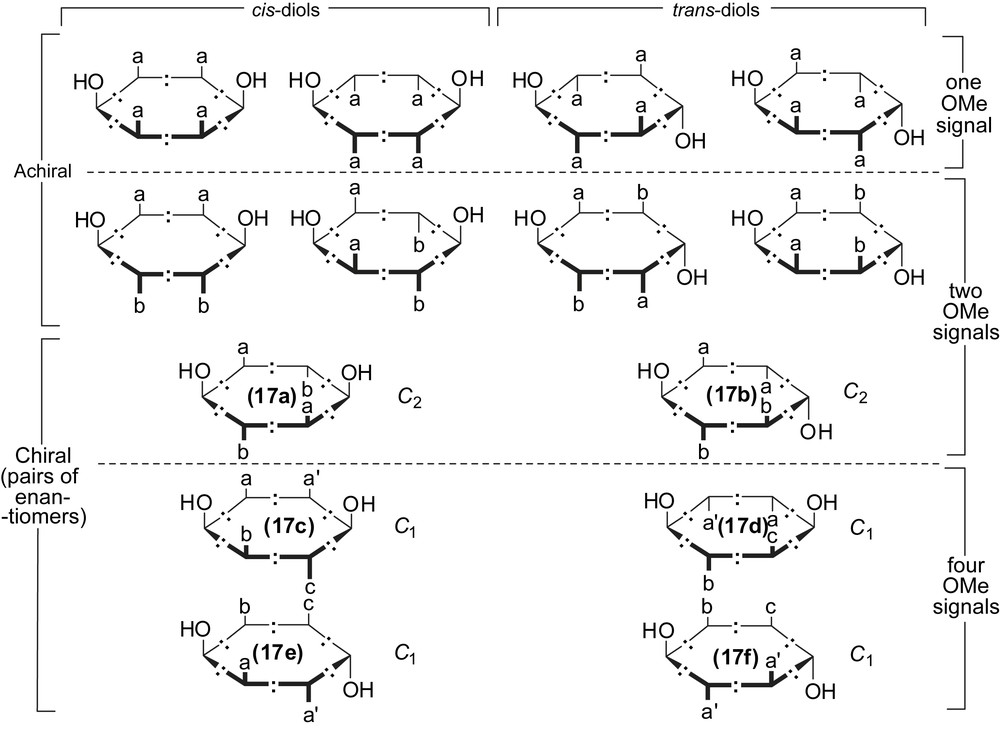
The 14 diastereoisomers of pericyclynediols 3a or 17 with two types of potentially stereogenic heterodisubstituted vertices: eight are achiral, six are chiral and occur as pairs of enantiomers (20 stereoisomers in a whole). Butyn-1,4-diyl edges are featured as –:–. Vertical lines and opposite blanks represent methoxy groups and phenyl groups respectively. Opposite blanks to OH-terminated vertical lines represent H (3a) or trimethysilylalkynyl (17) substituents. In each chiral isomer, the topographically equivalent substituents are denoted as a, a′, b, c depending of their chemical (geometrical) equivalence class: the types a and a′ are distinct, but are in similar stereochemical environments up to seven bonds away.
3 Conclusion
An efficient and versatile synthesis of functional [6]pericyclynes has thus been achieved via the tetraphenyl-[6]pericyclynedione 2, the synthesis of which, based on a [8 + 10] ring formation process, has been optimized. Although the [6]pericyclyne derivatives 2, 3a, 17 were obtained as complex mixtures of diastereoisomers, their partial resolution has been achieved in two instances (2 and 17). Despite these synthetic improvements, the reductive aromatization of the ultimate [6]pericyclynediols bis-adducts remains the limiting step towards carbo-benzenes, and alternative strategies based on simultaneous C18 ring formation and aromatization processes are being currently investigated.
4 Experimental
4.1 General
All reagents were used as their commercially available form (from Acros Organics, Avocado, Aldrich, Lancaster, Strem) without further purification. THF and diethylether were dried and distilled on sodium/benzophenone, pentane on P2O5 and dichloromethane on CaH2. Commercial solutions of EtMgBr are 3 M in diethylether. Commercial solutions of n-BuLi are 1.6 or 2.5 M in hexane. All reactions were conducted under either a nitrogen or argon atmosphere, using Schlenk and vacuum line techniques. Thin Layer Chromatography plates were purchased from SDS (60F254, 0.25 mm) and revealed by treatment with an ethanolic solution of phosphomolybdic acid (20%). Column chromatographies were carried out with SDS silica gel (60 Å, 70–200 μm). Special chromatographies were performed with a Combiflash Graduate ISCO System on Normal Phase Silice packs. The following analytical instruments were used: IR: 0.1 mm CaF2 cell, Perkin–Elmer GX FT-IR; 1H and 13C NMR: Brucker AC 200, AM 250, ARX 250, AV 300, DPX 300 or AMX 400; Mass spectrometry: Quadrupolar Nermag R10-10H. IR absorption frequencies ν are in cm−1. Analytical Alliance HPLC chains, coupled with UV detectors and driven with an Empower (Waters) software. HPLC analytical Sunfire Si column 5 μm 150 mm × 4.6 mm, Waters. Semi-preparative Waters Deltaprep chains, coupled with a UV 486 Waters detector and a Gilson 201 fraction collector. Semi-preparative HPLC columns: Sunfire column 5 μm 150 mm × 19 mm (Waters) and Lichrosphere 10 μm 250 mm × 21 mm (Merck). NMR chemical shifts δ are in ppm, with positive values to high frequency relative to the tetramethylsilane reference; coupling constants J are in Hz. As most compounds were isolated as oily mixtures of diastereoisomers, characteristic assignments are given in order to trace the analytical consistency within the quite homogeneous series of compounds.
4.2 Syntheses
4.2.1 1,4-Diphenylbut-2-yne-1,4-diol (12)
To a commercial solution of ethynyl magnesium bromide (0.5 M in ether, 50 mL, 25 mmol) was added dropwise methyllithium (1.5 M in THF complexed with lithium bromide, 20 mL, 30 mmol) at −78 °C. After stirring for 30 min at −78 °C and 4 h at room temperature, the resulting mixture was added to benzaldehyde (7.96 g, 75 mmol) and stirring was continued overnight. After treatment with saturated aqueous NH4Cl and extraction with Et2O, the organic layer was washed with brine, dried over MgSO4 and concentrated under reduced pressure to give a red oil. Flash chromatography on silica gel (heptane/EtOAc 9/1, then 5/5) followed by recrystallization in toluene, gave 12 as a yellow powder (3.6 g, 15.1 mmol, 60%).
Rf ≈ 0.28 (heptane/AcOEt 5/5). MS (DCI/NH3): m/z [M]+ 238, [M-H2O]+ 220, [PhCO]+ 105, [C6H5]+ 77. 1H NMR (CDCl3): δ 2.22 (d, 3JHH = 5.7 Hz, 2H, OH), 5.59 (d, 3JHH = 5.7 Hz, 2H, CH–OH), 7.36–7.43 (m, 6H, m-, p-C6H5), 7.56–7.7.58 (m, 4H, o-C6H5). 13C{1H} NMR (CDCl3): δ 68.7 (CPhOH), 85.9 (CC), 126.4 (o-C6H5), 127.8 (m-C6H5), 128.1 (p-C6H5), 140.2 (ipso-C6H5).
4.2.2 1,4-Diphenylbut-2-yne-1,4-dione (11)
Procedure A. To a solution of diol 12 (0.370 g, 1.55 mmol) in dichloromethane (35 mL) was added MnO2 (2.7 g, 31.6 mmol) at room temperature. After stirring for 4 h, the suspension was filtered through celite and concentrated under reduced pressure. Purification by recrystallization in ethanol gave 11 as a yellow powder (0.220 g, 0.94 mmol, 63%).
Procedure B. To a solution of diol 12 (2.46 g, 10.33 mmol) in acetonitrile/water (80/40 mL) was added iodozobenzoic acid (IBA: 1.09 g, 4.13 mmol) and oxone® (15.87 g, 25.81 mmol) at room temperature. After stirring for 7 h at 70 °C, the temperature was cooled down to 0 °C and the precipitate was removed by filtration. The solution was then extracted with dichloromethane, and the combined organic layers were washed with saturated aqueous NaHCO3, dried over MgSO4 and concentrated under reduced pressure to give an orange solid. Recrystallization from toluene followed by flash chromatography on silica gel (heptane/EtOAc 9/1) gave 11 as a yellow powder (1.6 g, 6.83 mmol, 66%).
Rf ≈ 0.53 (heptane/AcOEt 5/5). MS (DCI/NH3): m/z [M]+ 234, [M-(PhCO)]+ 129. 1H NMR (CDCl3): δ 7.55–7.61 (m, 4H, m-C6H5), 7.69–7.75 (m, 2H, p-C6H5), 8.21–8.24 (m, 4H, o-C6H5). 13C{1H} NMR (CDCl3): δ 85.8(CC), 128.9 (o-C6H5), 129.7 (m-C6H5), 135.6 (p-C6H5), 165.7 (ipso-C6H5), 175.5 (CO).
4.2.3 4,7-Dimethoxy-4,7-diphenyldeca-2,5,8-triynedial (5)
To a solution of 8 (1.05 g, 2.80 mmol) in 1,2-dichloroethane (150 mL) was added IBX (6.28 g, 22.43 mmol) at room temperature. After refluxing for 8 h, the temperature was cooled down to room temperature and the solid was removed by filtration. The solution was then concentrated under reduced pressure to give 5 as a spectroscopically pure orange oil which crystallized at 4 °C (0.86 g, 2.32 mmol, 83%).
Rf ≈ 0.18 (heptane/EtOAc 8/2). 1H NMR (CDCl3): δ 3.59 (d, 3JHH = 1.2 Hz, 6H, OCH3), 7.43–7.46 (m, 6H, m-, p-C6H5), 7.71–7.75 (m, 4H, o-C6H5), 9.35 (s, 2H, –CHO). 13C NMR (CDCl3): δ 53.9 (OCH3), 71.8 (CPh(OMe)), 83.9, 84.3, 90.6 (CC), 126.2–129.5 (o-, m-, p-C6H5), 137.8 (ipso-C6H5).
4.2.4 Diethyl 4,7-dimethoxy-4,7-diphenyldeca-2,5,8-triynedioate (6a)
To a solution of triyne 4 (0.200 g, 0.636 mmol) in THF (40 mL) was added n-butyllithium (2.3 M, 0.61 mL, 1.34 mmol) at −78 °C. After stirring for 30 min at −78 °C, the resulting mixture was added to a solution of ethyl chloroformate (0.690 g, 6.362 mmol) in THF (20 mL) at −78 °C. Stirring was continued during 2.5 h at −78 °C and 1 h at room temperature. After treatment with saturated aqueous NH4Cl and extraction with Et2O, the organic layer was washed with brine, dried over MgSO4, filtered, and evaporated under reduced pressure. Purification by flash chromatography over silica gel (heptane/AcOEt 9/1) gave 6a as a pale brown solid (0.230 g, 0.502 mmol, 79%), and 6b as a slightly orange oil (21 mg, 0.026 mmol, 8%).
Rf ≈ 0.34 (heptane/EtOAc 7/3). MS (DCI/CH4): m/z [M + C2H5]+ 487, [M]+ 458, [M-OCH3]+ 427. 1H NMR (CDCl3): δ 1.35 (t, 3JHH = 7.2 Hz, 6H, –CH2CH3), 3.60 (s, 6H, OCH3), 4.29 (q, 3JHH = 7.2 Hz, 8H, –CH2CH3), 7.41–7.45 (m, 12H, m-, p-C6H5), 7.73–7.75 (m, 8H, o-C6H5). 13C NMR (CDCl3): δ 14.0 (CH2CH3), 53.9 (OCH3), 62.5 (CH2CH3), 71.8 (CPh(OCH3)), 78.1, 82.5, 82.5, 84.1, 84.2 (CC), 126.4, 126.4, 128.6, 128.7, 129.5 (o-, m-, p-C6H5), 138.3, 138.4 (2s, ipso-C6H5), 152.9 (CO).
4.2.5 Diethyl 4,7,13,16-tetramethoxy-10-oxo-4,7,13,16-tetraphenylnonadeca-2,5,8,11,14,17-hexaynedioate (6b)
By-product of the preparation of diester 6a. Rf ≈ 0.27 (heptane/EtOAc 7/3). MS (DCI/NH3): m/z [M + NH4]+ 816. 1H NMR (CDCl3): δ 1.34 (t, 3JHH = 6.9 Hz, 6H, –CH2CH3), 3.57 (s, 6H, OCH3), 4.28 (q, 3JHH = 6.9 Hz, 8H, –CH2CH3), 7.40–7.41 (m, 12H, m-, p-C6H5), 7.71–7.73 (m, 8H, o-C6H5). 13C{1H} NMR (CDCl3): δ 14.0 (CH2CH3), 53.9, 54.0 (OCH3), 62.5 (CH2CH3), 71.8, 71.9 (CPh(OCH3)), 78.2, 82.3, 83.4, 83.5, 84.7, 84.7, 84.9, 88.8 (CC), 126.4, 128.7, 128.8 (o-, m-C6H5), 129.5, 129.6 (p-C6H5), 138.0, 138.2 (ipso-C6H5), 152.9 (CO).
4.2.6 4,7-Dimethoxy-4,7-diphenyldeca-2,5,8-triyn-1,10-diol (8)
Procedure A. To a solution of 4 (1.22 g, 3.88 mmol) in THF (10 mL) was added dropwise n-butyllithium (3.4 mL, 8.54 mmol) at −78 °C. After stirring for 30 min at −78 °C, the resulting orange mixture was added to solid para-formaldehyde (350 mg, 11.64 mmol) at −78 °C. Stirring was continued during 1 h at −78 °C and 1 h at room temperature. After treatment with saturated aqueous NH4Cl and extraction with Et2O, the organic layer was washed with brine, dried over MgSO4 and concentrated under reduced pressure to give a yellow liquid. Flash chromatography on silica gel (pentane/EtOAc 6/4) gave 8 as a yellow oil (1.38 g, 3.68 mmol, 95%).
Procedure B. To a solution of 15 (1.90 g, 3.5 mmol) in MeOH (150 mL) was added PPTS (0.177 g, 0.71 mmol). The resulting mixture was stirred for 2 h at 55 °C, then the solution was evaporated under vacuum and diethylether (60 mL) and H2O (60 mL) were added. The organic layer was washed with saturated NH4Cl (3 × 60 mL). The aqueous layer was then extracted with diethylether (2 × 30 mL), and the combined organic layers were dried with MgSO4, filtered and evaporated to dryness. Purification of the residue by silica gel chromatography (heptane/AcOEt 7/3) allowed for the isolation of pure 8 as a yellow oil (1.05 g, 2.8 mmol, 80%).
Rf ≈ 0.22 (heptane/AcOEt 4/6). MS (DCI/NH3): m/z [M + NH4]+ 392, [M + NH4-MeOH]+ 360, [M + H-MeOH]+ 343. 1H NMR (CDCl3): δ 2.81 (s, 2H, O–H), 3.46, 3.49 (s, 6H, OCH3), 4.27 (s, 4H, CH2–OH), 7.32–7.41 (m, 6H, m-, p-C6H5), 7.69–7.75 (m, 4H, o-C6H5). 13C{1H} NMR (CDCl3): δ 50.7 (CH2OH), 53.2 (OCH3), 71.8 (PhC(OMe)), 82.3 (Ph–C–CC–C–Ph), 84.4 (CC–CH2OH), 85.7 (CC–CH2OH), 125.1–130.4 (m, o-, m-, p-C6H5), 139.6 (ipso-C6H5–COMe). IR (CDCl3): ν 3423 (O–H), 2935 (Csp3–H), 2826 (OCsp3–H), 1599, 1490 and 1453 (CC), 1068 (C–O).
4.2.7 4,7-Diphenyl-1,10-bis(tetrahydro-2H-pyran-2-yloxy)deca-2,5,8-triyn-4,7-diol (13)
To a solution of 2-(prop-2-ynyloxy)-tetrahydro-2H-pyran (2.8 g, 20.0 mmol) in THF (100 mL) under stirring at −78 °C was added n-butyllithium (8.0 mL, 20.0 mmol). The solution was stirred for 25 min at −78 °C, then 25 min at room temperature. After cooling again to −78 °C, a solution of 11 (1.87 g, 8.0 mmol) in THF (60 mL) was added dropwise and the resulting mixture was allowed to warm slowly to room temperature over 3 h, and then stirred overnight. After treatment with saturated NH4Cl, the aqueous layer was extracted with diethylether. The combined organic layers were washed with brine, dried with MgSO4 and evaporated under vacuum. Purification of the residue by silica gel chromatography (heptane/AcOEt 6/4) allowed for the isolation of pure 13 as a yellow oil (3.56 g, 6.92 mmol, 87%).
Rf ≈ 0.33 (heptane/AcOEt 6/4). MS (DCI/NH3): m/z [M + NH4]+ 532. 1H NMR (CDCl3): δ 1.61 (m, 12H, H2, H3 and H4), 3.47 (m, 2H, H5), 3.78 (m, 2H, H5), 4.12 (br s, 2H, OH), 4.31 (s, 4H, C–CH2), 4.80 (br s, 2H, H1), 7.34 (m, 6H, m-, p-C6H5), 7.75 (m, 4H, o-C6H5). 13C{1H} NMR (CDCl3): δ 18.5, 25.0 and 29.8 (C2, C3 and C4), 54.2 (C–CH2), 61.6 (C5), 64.5 (C–C(OH)Ph–C), 81.1, 84.7 and 85.8 (C), 96.6 (C1), 125.8, 128.2 and 128.4 (m, o-, m-, p-C6H5), 141.4 (ipso-C6H5–C–OH). IR (CDCl3): ν 3572 (OH), 3354 (OH), 3088, 3065 and 3032 (Csp2–H), 2947 (Csp3–H), 2871 and 2854 (Csp3–H), 1450, 1119, 1024.
4.2.8 4,7-Diphenyl-1,10-bis(tetrahydro-2H-pyran-2-yloxy)-4,7-bis(trimethylsilyloxy)deca-2,5,8-triyne (14)
To a solution of 13 (0.268 g, 0.52 mmol) in THF (30 mL) at −78 °C was added dropwise n-butyllithium (0.72 mL, 1.15 mmol). After stirring for 20 min at −78 °C, chlorotrimethylsilane (0.15 mL, 1.15 mmol) was added dropwise, then the temperature was allowed to warm to room temperature over 1.5 h. After stirring for an additional 20 min at room temperature, diethylether (20 mL) and saturated NH4Cl (50 mL) were added. The organic layer was washed with saturated NH4Cl (2 × 40 mL). The aqueous layer was extracted with diethylether (2 × 30 mL). The combined organic layers were then dried with MgSO4 and evaporated under vacuum. Purification of the residue by silica gel chromatography (heptane/AcOEt 7/3) gave 14 as an orange oil (0.245 g, 0.37 mmol, 71%).
Rf ≈ 0.34 (heptane/AcOEt 7/3). MS (DCI/NH3): m/z [M + NH4]+ 676. 1H NMR (CDCl3): δ 0.22 (m, 18H, Si(CH3)3), 1.65 (m, 12H, H2, H3 and H4), 3.48 (m, 2H, H5), 3.81 (m, 2H, H5), 4.34 (s, 4H, C–CH2), 4.80 (br s, 2H, H1), 7.33 (m, 6H, m-, p-C6H5), 7.68 (m, 4H, o-C6H5). 13C{1H} NMR (CDCl3): δ 1.4 (Si(CH3)3), 19.1, 25.4 and 30.2 (C2, C3 and C4), 54.1 (C–CH2), 62.0 (C5), 65.7 (C–C(O)Ph–C), 81.7, 85.5 and 86.3 (C), 96.7 (C1), 125.7, 128.0 and 128.1 (m, o-, m-, p-C6H5), 143.3 (ipso-C6H5–C–OH). IR (CDCl3): ν 3088, 3065 and 3032 (Csp2–H), 2949 (Csp3–H), 2854 (Csp3–H), 1252, 1119, 1025.
4.2.9 4,7-Dimethoxy-4,7-diphenyl-1,10-bis(tetrahydro-2H-pyran-2-yloxy)deca-2,5,8-triyne (15)
To a solution of 13 (3.56 g, 6.92 mmol) in THF (120 mL) under stirring at −78 °C was added n-butyllithium (6.1 mL, 15.2 mmol). After 30 min, iodomethane (4.3 mL, 69.2 mmol) and DMSO (4.9 mL, 69.2 mmol) were added at −78 °C, and the resulting mixture was allowed to warm slowly to room temperature. After stirring overnight at room temperature, diethylether (20 mL) and saturated NH4Cl (60 mL) were added. The organic layer was washed with saturated NH4Cl (3 × 60 mL). The aqueous layer was then extracted with diethylether (2 × 30 mL), and the combined organic layers were dried with MgSO4, filtered and concentrated under vacuum. The residue was purified by silica gel chromatography (heptane/AcOEt 7/3), thus giving pure 15 as a yellow oil (3.29 g, 6.07 mmol, 88%).
Rf ≈ 0.36 (heptane/AcOEt 7/3). MS (DCI/NH3): m/z [M + NH4]+ 560. 1H NMR (CDCl3): δ 1.64 (m, 12H, H2, H3 and H4), 3.49 (m, 2H, H5), 3.51 (s, 6H, OCH3), 3.83 (m, 2H, H5), 4.38 (s, 4H, C–CH2), 4.82 (m, 2H, H1), 7.36 (m, 6H, m-, p-C6H5) 7.74 (m, 4H, o-C6H5). 13C{1H} NMR (CDCl3): δ 19.0, 25.3 and 30.2 (C2, C3 and C4), 53.3 (OCH3), 54.2 (C–CH2), 62.0 (C5), 71.8 (C–C(OMe)Ph–C), 82.9, 83.4 and 84.3 (C), 96.8 (C1), 126.5, 128.3 and 128.8 (m, o-, m-, p-C6H5), 140.0 (ipso-C6H5–C–OH).
4.2.10 1,10-(4-Methylbenzenesulfonate)-4,7-dimethoxy-4,7-diphenyldeca-2,5,8-triyne (7)
To a solution of triynediol 8 (0.240 g, 0.64 mmol) in THF (12 mL) was added p-toluenesulfonyl chloride (0.269 g, 1.41 mmol) at room temperature, and then potassium hydroxide (0.575 g, 10.26 mmol) at −30 °C. After stirring for 45 min at −30 °C, the resulting mixture was treated with water (8 mL) and stirred for 10 min at room temperature. After extraction of the aqueous layer with diethylether, the combined organic layers were washed with brine and dried with MgSO4. Concentration under reduced pressure gave 7 as a spectroscopically pure slightly yellow oil (0.420 g, 0.615 mmol, 96%).
MS (DCI/CH4): m/z [M-OMe]+ 651.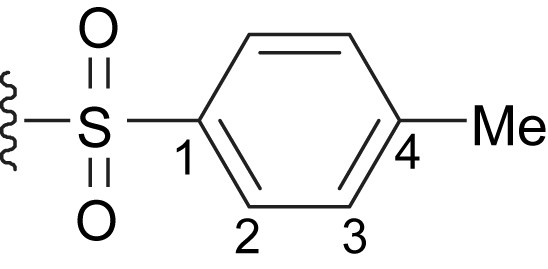
1H NMR (CDCl3): δ 2.33 (s, 6H, PhCH3), 3.43 (s, 6H, OCH3), 4.86 (s, 4H, CH2), 7.21–7.24 (m, 4H; H3), 7.37–7.39 (m, 6H, m-, p-C6H5), 7.60–7.65 (m, 4H, o-C6H5), 7.76–7.79 (m, 4H, H2). 13C{1H} NMR (CDCl3): δ 21.6 (PhCH3), 53.4 (CH2), 57.6 (OCH3), 71.7 (>CPhOMe), 79.1 (C–CC–C), 84.2 (2s, CC–CH2–OTs), 85.8 (2s, C–CH2OTs) 126.4, 128.0, 128.5, 129.17, 130.0 (o-, m-, p-C6H5, C2, C3), 132.8 (ipso-C6H5), 139.1 (C4), 145.3 (C1).
4.2.11 4,7,13,16-Tetramethoxy-4,7,13,16-tetraphenylcyclooctadeca-2,5,8,11,14,17-hexayne-1,10-diol (3a)
To a solution of triyne 4 (1.02 g, 3.24 mmol) in THF (200 mL) was added ethylmagnesium bromide (2.16 mL, 6.48 mmol) at 0 °C. The mixture was then stirred for 15 min at 0 °C and 2 h at room temperature (solution A). During that time, dialdehyde 5 (1.20 g, 3.24 mmol) was dissolved in THF (200 mL, solution B). The solutions A and B were simultaneously added into a 2 L flask containing 600 mL THF at 0 °C. The resulting solution was allowed to warm up slowly to room temperature and stirring was continued for 15 h. After treatment with saturated aqueous NH4Cl and extraction with Et2O, the organic layer was washed with brine, dried over MgSO4 and concentrated under reduced pressure. Chromatography on silica gel (heptane/DCM/EtOAc 7/2/1, then 5/3/2) gave 3a as a yellow powder (0.680 g, 0.99 mmol, 31%).
Rf ≈ 0.28 (heptane/AcOEt 5/5). MS (DCI/NH3): m/z [M + NH4]+ = 702, [M + H-MeOH]+ 653. 1H NMR (CDCl3): δ 2.57–2.75 (m, 2H, OH), 3.34–3.57 (m, 12H, OCH3), 5.28–5.34 (m, 2H, CH–OH), 7.32–7.37 (m, 12H, m-, p-C6H5), 7.66–7.74 (m, 8H, o-C6H5). 13C{1H} NMR (CDCl3): δ 52.1 (CHOH), 53.2 (PhC–OCH3), 71.6 (PhC–OCH3), 81.7, 83.3, 83.5, 84.1 (CC), 126.3-128.9 (o-, m-, p-C6H5), 139.0 (ipso-C6H5). IR (CDCl3): ν 3584 (O–H), 2956–2935 (C–H), 2826 (OC–H), 1600, 1490, 1450 (aromatic), 1064 (C–O).
4.2.12 4,7,13,16-Tetramethoxy-4,7,13,16-tetraphenylcyclooctadeca-2,5,8,11,14,17-hexayne-1,10-dione (2)
Procedure A. To a solution of [6]pericyclynediol 3a (0.210 g, 0.307 mmol) in dichloromethane (40 mL) was added MnO2 (0.800 g, 9.20 mmol) at 0 °C. After stirring for 1 h at 0 °C and 2.5 h at room temperature, the solution was filtered through celite and concentrated under reduce pressure. Purification by flash chromatography over silica gel (pentane/Et2O 8/2, then 6/4) gave 2 as a slightly orange solid (0.150 g, 0.220 mmol, 72%).
Procedure B. To a solution of [6]pericyclynediol 3a (0.100 g, 0.146 mmol) in 1,2-dichloroethane (10 mL) was added IBX (0.245 g, 0.876 mmol) at room temperature. After refluxing for 5 h, the medium was cooled down to room temperature and stirring was continued for 5 h. The solid by-product IBA was filtered off and the solution was concentrated under reduced pressure. Purification by flash chromatography over silica gel (heptane/AcOEt 8/2) gave 2 as a beige solid (70 mg, 0.103 mmol, 70%).
Rf ≈ 0.44 (heptane/EtOAc 6/4). MS (DCI/NH3): m/z [M + NH4]+ 698, [M + H-MeOH]+ 649. 1H NMR (CDCl3): δ 3.44–3.62 (m, 12H, OCH3), 7.25–7.44 (m, 12H, m-, p-C6H5), 7.63–7.73 (m, 8H, o-C6H5). 13C{1H} NMR (CDCl3): δ 53.7 (OCH3), 71.8 (CPh(OCH3)), 83.8, 84.0, 84.9, 88.5 (CC), 126.3–128.7 (o-, m-, p-C6H5), 137.4 (ipso-C6H5), 158.9 (CO). IR (CDCl3): ν 2956–2931 (C–H), 2827 (OC–H), 1638 (CO), 1451 (aromatic), 1068 (C–O).
4.2.13 1,10-Bis[(trimethylsilyl)ethynyl]-4,7,13,16-tetramethoxy-4,7,13,16-tetraphenyl-cyclooctadeca-2,5,8,11,14,17-hexayne-1,10-diol (17)
To a solution of trimethylsilylacetylene (0.026 g, 0.264 mmol) in THF (10 mL) at 0 °C was added EtMgBr (0.88 mL, 0.264 mmol). After stirring for 30 min at 0 °C and then for 2 h at room temperature, a solution of 2 (0.060 g, 0.088 mmol) in THF (15 mL) was added at 0 °C. The resulting mixture was stirred for 30 min at 0 °C and overnight at room temperature. After treatment with saturated NH4Cl and dilution with diethylether (20 mL), the organic layer was washed with saturated NH4Cl (2 × 30 mL). The aqueous layer was extracted with diethylether (2 × 20 mL), and the combined organic layers were dried with MgSO4 and concentrated under vacuum. Several purifications by silica gel chromatography (pentane/ether 7/3) allowed for the isolation of 16 as a pale yellow solid (0.033 g, 0.038 mmol, 43%) and 17 (0.015 g, 0.019 mmol, 22%).
Rf ≈ 0.35 (heptane/AcOEt 4/6). MS (DCI/NH3): m/z [M + NH4]+ 894. 1H NMR (CDCl3): δ 0.22 (m, 18H, Si(CH3)3), 3.25–3.65 (m, 14H, OH and OCH3), 7.35–7.41 (m, 12H, m-, p-C6H5), 7.69–7.79 (m, 8H, o-C6H5). 13C{1H} NMR (CDCl3): δ −0.5 (m, Si(CH3)3), 53.6 (m, OCH3), 54.4 (m, C–OH), 71.8 (m, Ph–C–OMe), 80.2 (m, Me3SiCC–C(OH)–C), 84.3 (m, Ph–C(OMe)–C), 89.6 (m, CCSi), 100.1 (m, CC–Si), 126.6 (m, m-C6H5), 128.5 (m, o-C6H5), 129.1 (m, p-C6H5), 139.2 (m, ipso-C6H5).
4.2.14 10-(Trimethylsilyl)ethynyl-10-hydroxy-4,7,13,16-tetramethoxy-4,7,13,16-tetraphenyl-cyclooctadeca-2,5,8,11,14,17-hexayne-1-one (16)
By-product of the preparation of 17.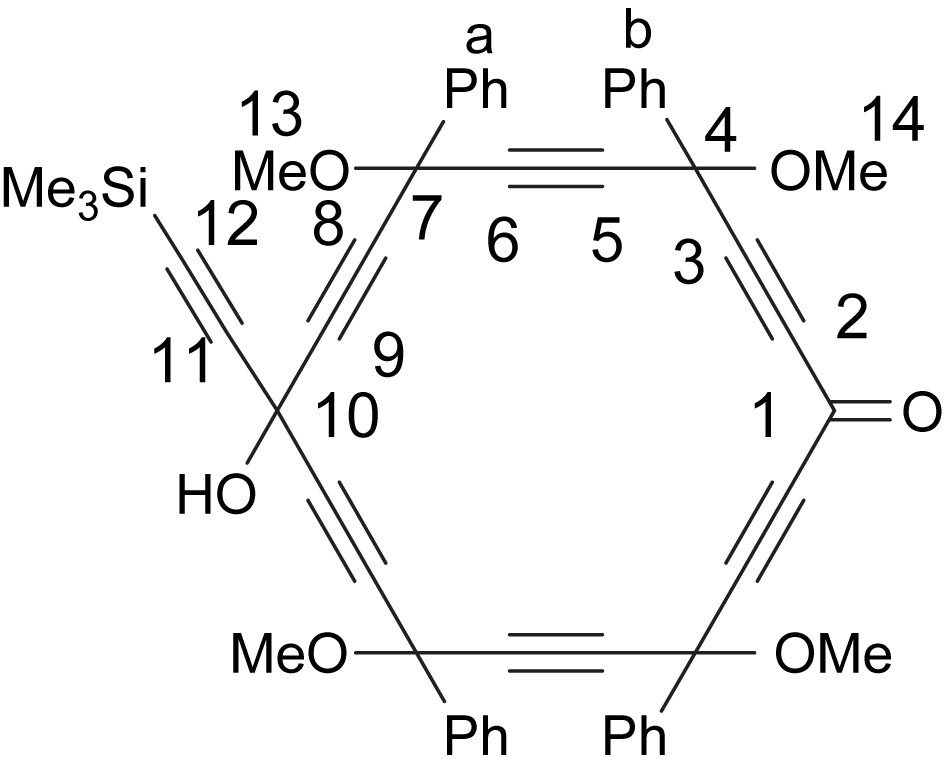
MS (DCI/NH3): m/z [M + NH4]+ 796.3. 1H NMR (CDCl3): δ 0.23 (m, 9H, Si(CH3)3), 3.41–3.64 (m, 12H, OCH3), 7.31–7.45 (m, 12H, m-, p-C6H5), 7.59–7.78 (m, 8H, o-C6H5). 13C{1H} NMR (CDCl3): δ −0.5 (Si(CH3)3), 53.6 (2s, OMe14), 53.8, 53.9 (4s, OMe13), 54.4 (C10), 71.7 (C7), 71.9 (C4), 80.1, 80.2 (3s, C9), 82.5, 82.6, 82.7 and 82.9 (C2), 84.3, 84.4 (4s, C3), 84.8 and 84.9 (C5 or C6), 85.1 (2s, C5 or C6), 85.7, 85.8 (4s, C8), 89.2, 89.4 and 89.7 (C11), 100.3 (C12), 126.55 (5s, Co or Cm of Pha and Phb), 128.5–128.8 (Co or Cm of Pha and Phb), 129.2, 129.3 (4s, Cp of Pha or Phb), 129.5, 129.6 and 129.7 (4s, Cp of Pha or Phb), 137.7, 137.8, 137.9 and 138.0 (Ci of Pha or Phb), 138.7, 138.8, 138.9 (4s, Cp of Pha or Phb), 159.1 (C1).
Acknowledgements
The authors wish to thank the Centre National de la Recherche Scientifique and the Ministère de l'Education Nationale, de la Recherche et de la Technologie for financial support, in particular through ACI fundings.