

1 Introduction
Mimicry of nucleosides is an active research field, because of the many potential applications in chemical biology and drug discovery. Probably the most popular application consists of ATP-competitive kinase inhibitors [1]. Naturally occurring ATP-competitive inhibitors are usually composed of a heterocyclic, nucleobase-like part connected to a sugar ribose via a glycosidic bond. Apart from several specificity and toxicity issues, the glycosidic bond and the carbohydrate moiety often create stability and bioavailability problems in pharmacologically relevant conditions [2]. Many of the known synthetic ATP-competitive kinase inhibitors consist of heterocyclic structures capable of replacing the nucleobase in the conserved ATP-binding site of the enzymes [3]. The affinity and possibly the specificity of such molecules could be improved by sugar-like appendages capable of engaging the ribose-binding portion of the active site, while being devoid of the above-mentioned stability and bioavailability issues connected to glycosidic linkages and sugars. This concept is supported by the synthetic indolocarbazole SRN-003-556 (1) (compare with the naturally occurring, glycosidic bond-containing K252a (2), Fig. 1), which is an in vivo active, potent kinase inhibitor [4].

Structure of SRN-003-556 (1) and K252a (2).
Here, we report on the general synthetic approaches capable of affording two classes of molecular structures, bearing 5- and 6-membered carbocyclic polyols of the general formula A or B shown in Chart 1. These examples contain indolinone (Z CH2) or benzimidazolone (Z NH) rings as the nucleobase decoy. Such heterocycles have been used previously in the synthesis of ATP-competitive kinase inhibitors [5] and represent therefore an interesting model system.
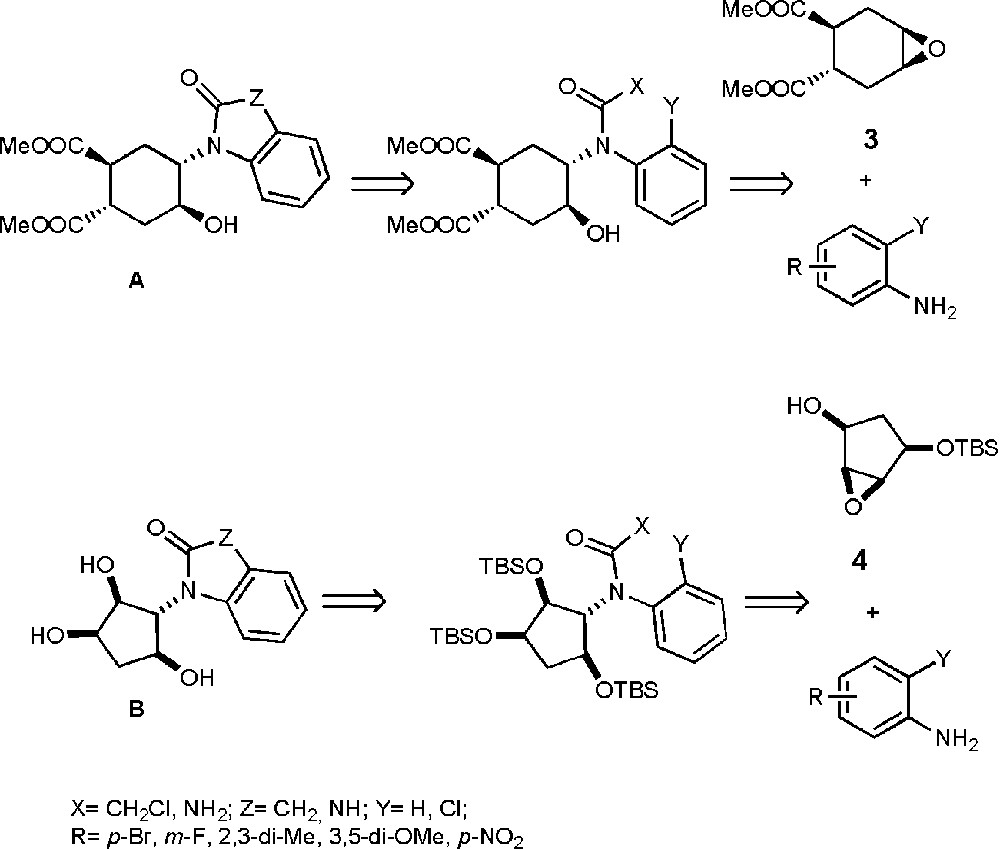
General structures of the targets and retrosynthetic approaches.
2 Results and discussion
2.1 Epoxide opening
The envisioned modular strategy (Chart 1), allows the access to chemical diversity via the opening of two epoxides, enantiomerically pure 3 [6a] and racemic 4 (The epoxide 4 was obtained by epoxidation of trans 4-tert-butyl-dimethyl-silanyloxy)-cyclopent-2-enol [7] (see the experimental section) with different anilines. Further N-functionalization and Pd-catalyzed ring formation yield the target nucleobase-sugar mimics. Known epoxide 3 was chosen because it is easily available in enantiopure form and has been used for the synthesis of sugar mimics [6]. Easily available epoxide 4 was introduced as a closer mimic of naturally occurring sugars.
The opening of epoxides with anilines in the presence of Lewis acid promoters is amply documented [8a–g], and a brief screening was performed to identify the best reaction conditions for substrates 3 and 4, using aniline 5a or 2-chloroaniline 5b as nucleophiles (Scheme 1). Results are detailed in Table 1.
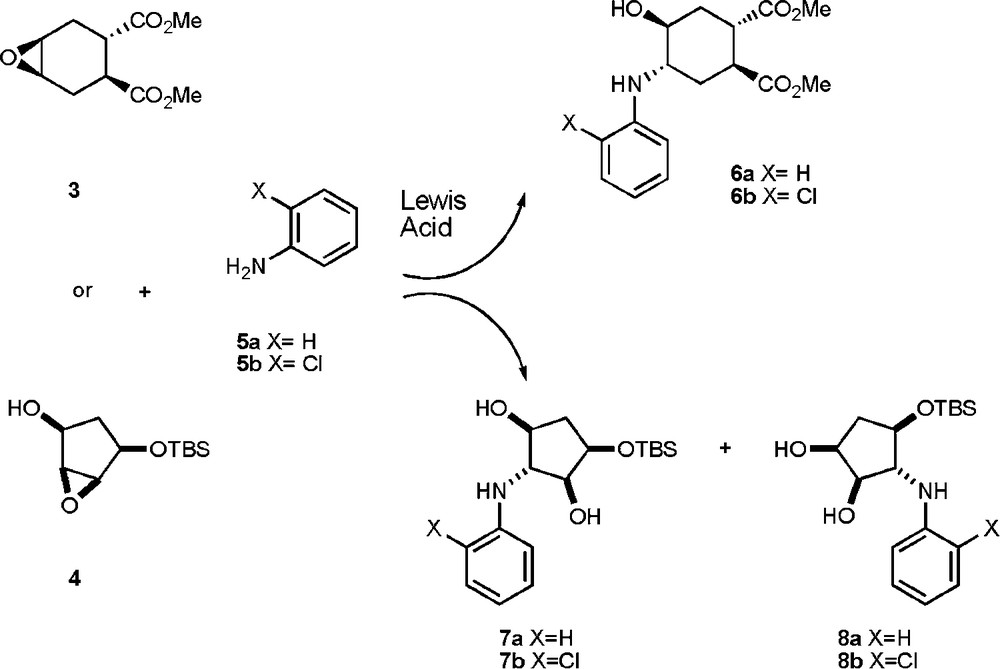
Opening of epoxides 3 and 4 with anilines 5a,b.
Opening of epoxides 3[a] and 4[b] with anilines 5a,b.
| Entry | Epoxide | Aniline | Promoter | Reaction time (h) | Compound/yield (%)[c] |
| 1 | 3 | 5a | Bi(OTf)3 | 18 | 6a/55 |
| 2 | 3 | 5a | Yb(OTf)3 | 18 | 6a/77 |
| 3 | 3 | 5a | InBr3 | 2.5 | 6a/94 |
| 4 | 3 | 5b | BiCl3 | 72 | 6b/47 |
| 5 | 3 | 5b | Bi(OTf)3 | 18 | 6b/65 |
| 6 | 3 | 5b | Yb(OTf)3 | 18 | 6b/83 |
| 7 | 3 | 5b | InBr3 | 2.5 | 6b/85 |
| 8 | 3 | 5b[d] | InBr3 | 2.5 | 6b/91 |
| 9 | 4 | 5a | InBr3 | 8 | 7a/54 8a/22 |
| 10 | 4 | 5a | Bi(OTf)3 | 48 | 7a/n.i.[e] 8a/n.i.[e] |
| 11 | 4 | 5a | BiCl3 | 48 | 7a/27[f] 8a/23[f] |
| 12 | 4 | 5a | Yb(OTf)3 | 8 | 7a/67[f] 8a/15[f] |
| 13 | 4 | 5b | InBr3 | 8 | 7b/50 8b/25 |
Both regioisomers 7 and 8 (structures assigned by nOe experiments) were formed with epoxide 4, whereas the C2’ symmetry axis of compound 3 led only to isomer 6. Somewhat surprisingly, yields for the reaction with epoxide 3 were consistently higher with 2-chloroaniline 5b than with 5a. Both epoxides 3 and 4 (the latter requiring stronger reaction conditions) reacted with good yields in presence of InBr3 [8a] (entries 3, 7, 8, 9, 13, Table 1) or Yb(OTf)3 [8b], (entries 2, 6, 12, Table 1). Bi-based catalysts [8c,8d] were less performing under all the examined reaction conditions (entries 1, 4, 5, 10, 11, Table 1).
Eventually, InBr3 was selected as the catalyst for further experiments. Although its performance was overall similar to Yb(OTf)3, we were often able with InBr3 to significantly reduce reaction times (compare entries 2 and 3, Table 1). Anilines 5c–h were then used with the same experimental protocols to test the versatility of our strategy (Scheme 2).

Opening of epoxides 3 and 4 with anilines 5c-h.
Using epoxide 3, yields were good to excellent with anilines 5c–f (entries 1–4), i.e. comparable to those obtained with 5a,b. Anilines 5 g and 5 h gave moderate yields (entries 5–6), probably due to the poor nucleophilicity of their nitrogen atom. Epoxide 4 confirmed its overall lower activity, even under stronger reaction conditions, and did not react with deactivated anilines 5 g and 5 h (entries 11–12). Yields obtained with anilines 5cf were nevertheless good (entries 7–10). Results are reported in Table 2.
Opening of epoxides 3[a] and 4[b] with anilines 5c-h (Scheme 2).
| Entry | Epoxide | Aniline | Reaction time (h) | Compound/yield (%)[c] |
| 1 | 3 | 5c | 15 | 6c/82 |
| 2 | 3 | 5d | 4 | 6d/98 |
| 3 | 3 | 5e | 15 | 6e/97 |
| 4 | 3 | 5f | 5 | 6f/80 |
| 5 | 3 | 5g | 72 | 6g/43 |
| 6 | 3 | 5h | 48 | 6h/40 |
| 7 | 4 | 5c | 48 | 7c/40 8c/30 |
| 8 | 4 | 5d | 24 | 7d/64 8d/21 |
| 9 | 4 | 5e | 30 | 7e/52 8e/14 |
| 10 | 4 | 5f | 24 | 7f/58 8f/22 |
| 11 | 4 | 5g | X | 7g/n.i.d 8g/n.i.d |
| 12 | 4 | 5h | X | 7h/n.i.d 8h/n.i.d |
2.2 Synthesis of indolinones 14 and 16
We then proceeded to the synthesis of indolinones 14 and 16 starting respectively from 6a and a mixture of compounds 7a and 8a. It must be noted that the formation of both regioisomers from the opening of epoxide 4 (Scheme 1) is of no relevance from now on, as the two regioisomers are converted into a single racemate after the first reaction step (step a, Scheme 3).

Synthesis of indolinones 11 and 12.
Sequential protection of the free hydroxyl groups (step a, Scheme 3) and chloroacetylation of the fully O-protected aniline (step b, Scheme 3) afforded respectively 9 and 10 in good to excellent yields. Both compounds could be used without purification in the subsequent cyclization to the indolinone ring using typical Buchwald's conditions [9] (step c, Scheme 3). O-protected indolinones 11 and 12 were isolated respectively in 47 and 70% unoptimized yield. With indolinone 11, 30% of starting material was recovered and could be recycled.
With protected compounds 11 and 12 in hands, we decided to perform an aldolisation on the free CH2 position of the heterocycle in order to obtain 3-alkylidene products, as similar heterocycles are known to be active on kinases in the literature [10]. Therefore, we performed an aldol condensation (step a, Scheme 4) according to established protocols [10]. 3-(Benzylidenyl)indolin-2-ones 14 and 16 were obtained in an overall 85 and 59% yield respectively, and in a ≈ 60/40 E/Z ratio, after deprotection (step b, Scheme 4). Unfortunately, but not unexpectedly [10], single geometric isomers isolated by chromatography quickly equilibrated to the original E/Z ratio in solution.
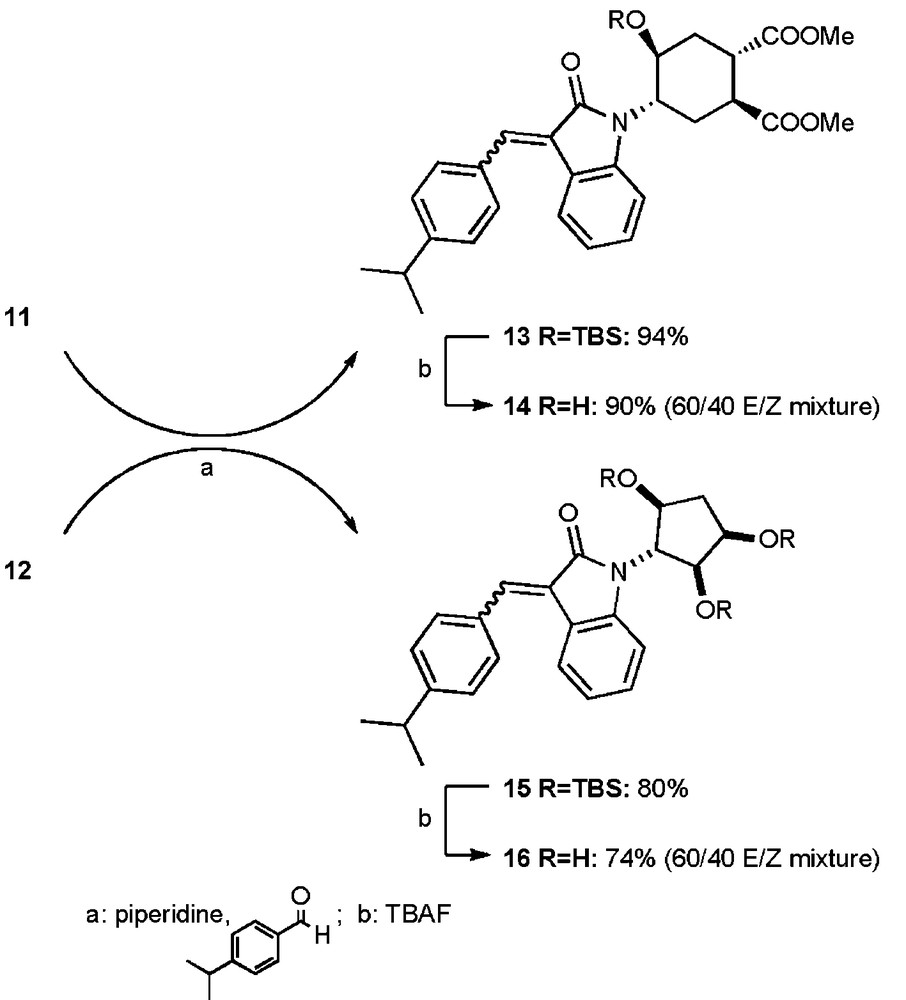
Synthesis of compound 14 and 16.
2.3 Synthesis of benzimidazolones 21 and 23
The synthesis of benzimidazolones 21 and 23 (Scheme 6) entailed a key Pd-catalyzed cyclization of intermediate ureas 19 and 20. Protection of 6b or of a mixture of compounds 7b and 8b (step a, Scheme 5), followed by urea formation with chlorosulfonylisocyanate (step b, Scheme 5) yielded respectively 19 and 20 in good overall yields. It has to be noted that O-deprotection occurred during urea formation from compound 17, while the same Si-protecting group was not removed from compound 18.
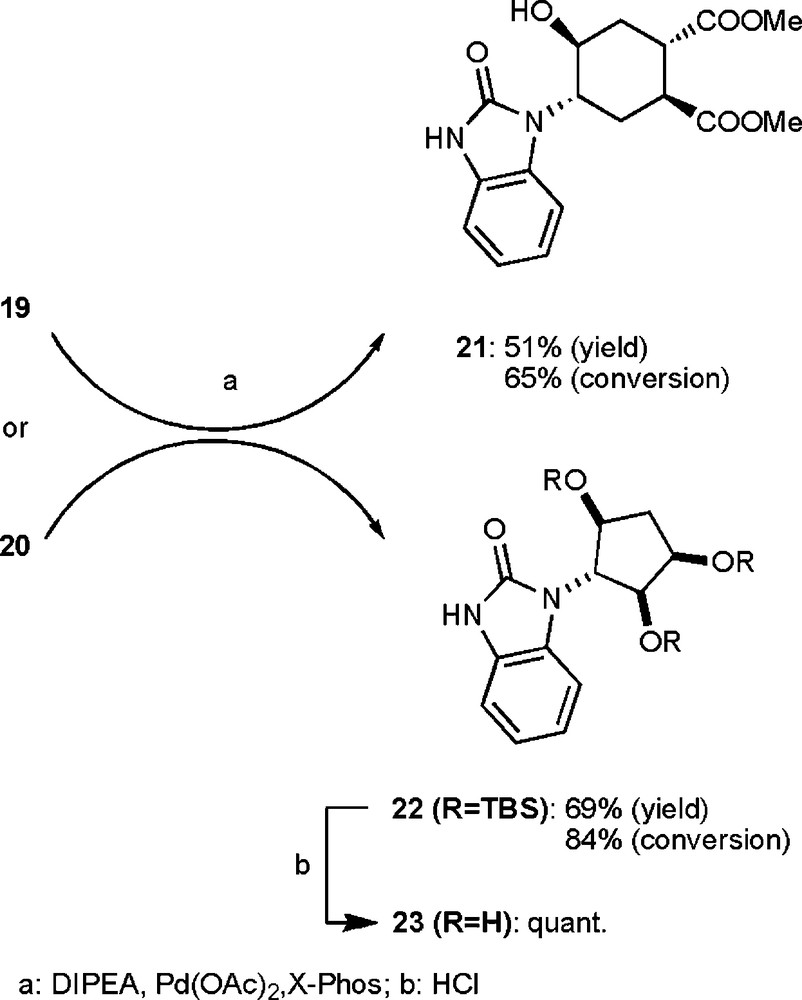
Synthesis of benzimidazolones 21 and 23.

Synthesis of ureas 19 and 20.
Cyclization of 19 and 20 was achieved by a slight modification of a strategy, recently proposed by McLaughlin et al. [11], using Pd(OAc)2 and Buchwald's X-Phos ligand (step a, Scheme 6) [12]. Benzimidazolones 21 and 22 were isolated in good yield and some starting material was recovered to be recycled. Final O-deprotection of compound 22 (step b, Scheme 6) yielded benzimidazolone 23 in quantitative yield.
3 Biology
We have tested compounds 16, 21 and 23 on a panel of 48 kinases (single point determination, 20 μM) Best results on selected 12 kinases are reported on Chart 2. In particular, compound 16 shows promising results on several kinases (TRK-B and VEGF-R2 in particular). Consequently, we measured the IC50 of compound 16 on several kinases, which obtained a 24 μM IC50 value for both TRK-B and VEGF-R2, and a 50 μM IC50 value for IKK-2.

Percentage inhibition of compounds 16, 21 and 23 on kinases.
4 Conclusions
We have devised a practical strategy to tether a 5- and a 6-membered carbocyclic polyol to a heterocyclic structure with the function of a nucleobase decoy. We have shown that this strategy could be extended using different anilines to perform epoxide opening reactions.
The molecules described here, although not yet structurally optimized, are expected to lead to potent and selective ATP-competitive kinase inhibitors, since 3-substituted indolin-2-ones and N-monosubstituted benzimidazolones represent a well-established class of tyrosine kinase inhibitors, which exhibit selectivity toward different receptor tyrosine kinases [5].
Indeed, a preliminary set of tests allowed to determine a medium/micromolar activity on several therapeutically relevant kinases for compound 16, thus providing proof of principle for our mimic concept. The synthesis of a larger set of aryl functionalized indolinones and benzimidazolones bearing variously substituted 5- and 6-membered carbocyclic sugar mimics is ongoing and their test on a panel of different kinases and other nucleotide-binding proteins will be reported in due course.
5 Experimental section
Solvents were dried by standard procedures. Dichloromethane, toluene, methanol, N,N-diisopropylethylamine, piperidine and triethylamine were dried over calcium hydride; iPrOH and 2,6-lutidine were dried over activated molecular sieves. Reactions requiring anhydrous conditions were performed under nitrogen or argon. Thin-layer chromatography (TLC) was carried out with precoated silica gel plates. Purifications were performed by flash chromatography carried out with silica gel (230–400 mesh) or by Biotage SP1™ Purification System. Mass spectra were obtained with an ESI apparatus Bruker Esquire 3000 plus. Optical rotations [α]D were measured in a cell of 1 dm pathlength and 1 mL capacity. 1H and 13C spectra were recorded at 300 K on a 400 MHz spectrometer. Chemical shifts δ for 1H and 13C are expressed in ppm relative to internal Me4Si as standard. Signals were abbreviated as s: singlet; bs: broad singlet; d: doublet; t: triplet; q: quartet; sp: septuplet; m: multiplet.
Each molecule was numbered without following the IUPAC numeration, and the numeration we used is reported on schemes for all the new compounds synthesized.
5.1 Epoxide opening: general procedure for compound 3
To a 0.8 M solution of 3[6a] (1 mol equiv.) in CH2Cl2 (technical grade) aniline 5a–h (1.5 mol equiv.) and InBr3 (0.1 mol equiv.) were added. The reaction mixture was stirred for 2.5 h at room temperature, monitoring the reaction progression by TLC (6/4 petroleum ether/EtOAc). After reaction completion the solution was evaporated and all the compounds were isolated by flash chromatography or by Biotage SP1™.
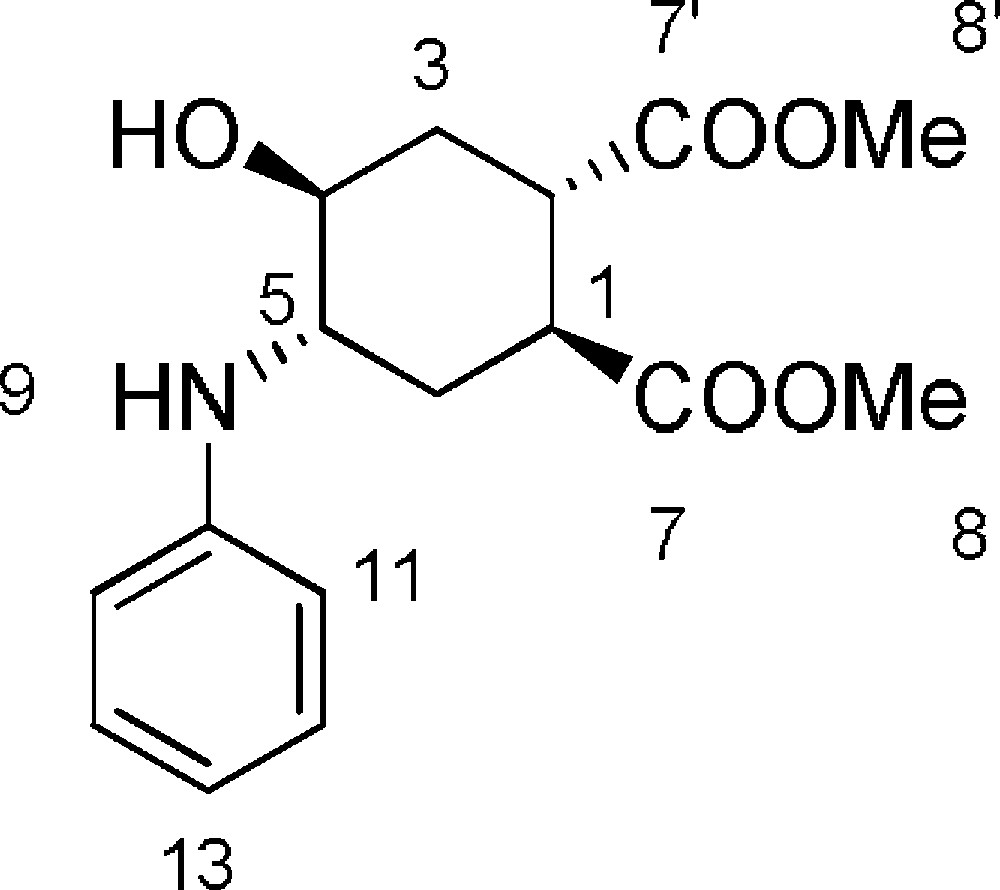
5.2 (1S,2S,4S,5S)-dimethyl-4-hydroxy-5-(phenylamino)cyclohexane-1,2-dicarboxylate 6a (Fig. 2. – S1)
Starting from 240 mg (1.14 mmol) of epoxide 3, we obtained a crude reaction product which was purified by flash chromatography using petroleum ether/EtOAc (60/40) to afford 6a as a colorless oil (329 mg, 94%). Rf = 0.2 (DCM/EtOAc 65/35). 1H-NMR (400 MHz, CDCl3): 1.65–1.72 (m, 1 H, 6ax-H); 1.81–1.89 (m, 1 H, 3ax-H); 2.21–2.30 (m, 1 H, 3eq-H); 2.38–2.43 (m, 1 H, 6eq-H); 3.09–3.16 (m, 1 H, 1-H); 3.25–3.32 (m, 1 H, 2-H); 3.46–3.52 (m, 1 H, 5-H); 3.73 (s, 3 H, 8-H or 8’-H); 3.75 (s, 3 H, 8-H or 8’-H); 3.75–3.81 (m, 1 H, 4-H); 6.73 (d, J11–12 = 7.4 Hz, 2 H, 11-H); 6.78 (t, J12–13 = 7.4 Hz, 1 H, 13-H); 7.21 (t, J11–12 = J12–13 = 7.4 Hz, 2 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): 28.3 (6-C); 31.1 (3-C); 39.9 (2-C); 40.3 (1-C); 52.2 (8-C + 8’-C); 54.4 (5-C); 68.6 (4-C); 114.0 (11-C); 118.6 (13-C); 129.5 (12-C); 146.5 (10-C); 174.1 (7-C or 7’-C); 174.5 (7-C or 7’-C). MS (ESI+): 308.0 [M + H+] [α]D = + 40,0 [c = 0.1, MeOH].
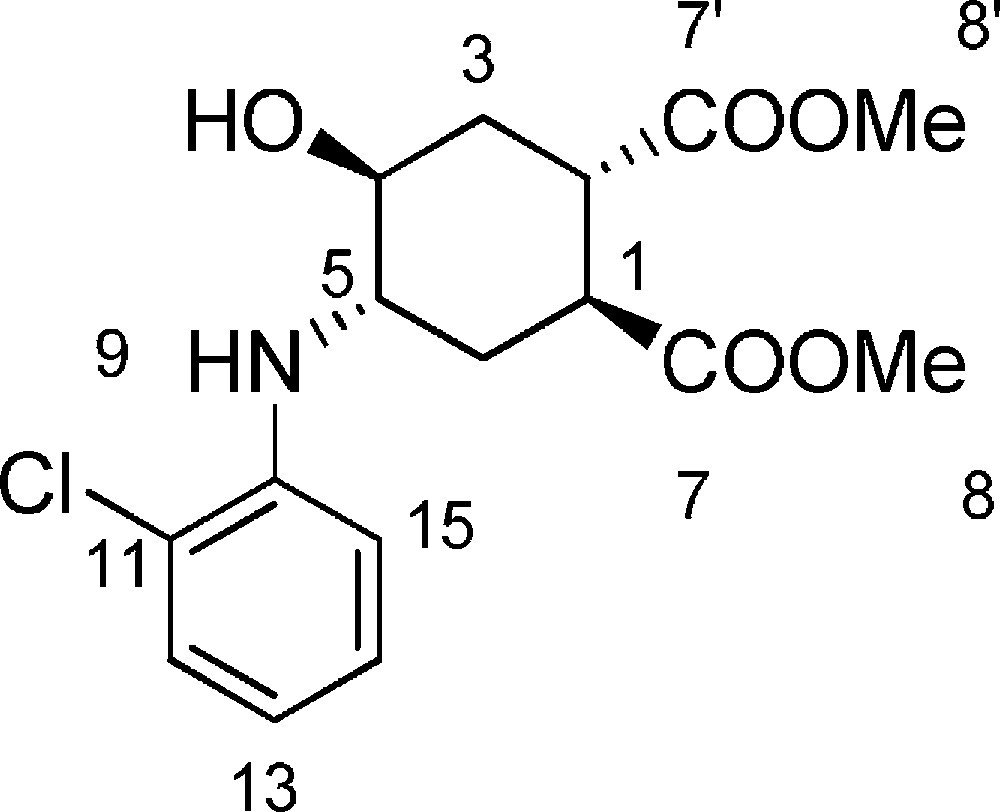
5.3 (1S,2S,4S,5S)-dimethyl-4-(2-chlorophenylamino)-5-hydroxycyclohexane-1,2-dicarboxylate 6b (Fig. 2. – S2)
Starting from 357¦mg (1.67¦mmol) of epoxide 3, we obtained a crude reaction product which was purified by flash chromatography using n-hexane/EtOAc (60/40) to afford 6b as a colorless oil (520¦mg, 91%). Rf¦=¦0.2 (CHCl3/EtOAc 9/1). 1H-NMR (400¦MHz, CDCl3): 1.68-1.77 (m, 1 H, 3ax-H); 1.80-1.88 (m, 1 H, 6ax-H); 2.29-2.42 (m, 2 H, 3eq-H¦+¦6eq-H); 3.16-3.20 (m, 1 H, 2-H); 3.30-3.33 (m, 1 H, 1-H); 3.53-3.58 (m, 1 H, 4-H); 3.70 (s, 3 H, 8-H or 8’-H); 3.75 (s, 3 H, 8-H or 8’-H); 3.77-3.88 (m, 1 H, 5-H); 6.73 (t, J¦=¦7.6¦Hz, 1 H, 13-H); 6.88 (d, J¦=¦8.0¦Hz, 1 H, 15-H); 7.18 (t, J¦=¦7.6¦Hz, 1 H, 14-H); 7.26-7.31 (m, 1 H, 12-H). 13C-NMR (100.61¦MHz, CDCl3): 28.2 (6-C); 31.1 (3-C); 40.2 (2-C); 40.6 (1-C); 52.3 (8-C¦+¦8’-C); 55.5 (5-C); 68.8 (4-C); 112.8 (15-C); 118.8 (13-C); 120.9 (11-C); 127.7 (14-C); 129.8 (12-C); 142.1 (10-C); 173.9 (7-C or 7’-C); 174.1 (7-C or 7’-C). MS (ESI+): 705.2 [2M¦+¦Na]+.
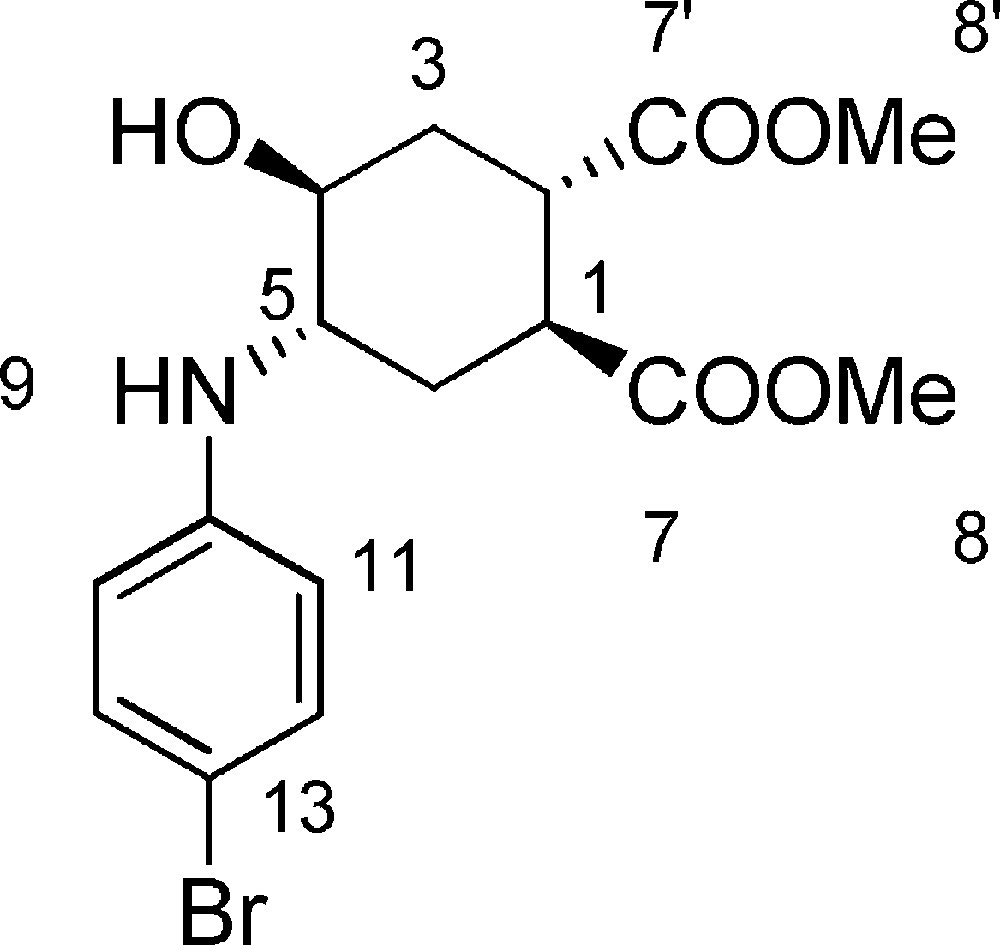
5.4 (1S,2S,4S,5S)-dimethyl-4-(4-bromophenylamino)-5-hydroxycyclohexane-1,2-dicarboxylate 6c (Fig. 2. – S3)
Starting from 103 mg (0.48 mmol) of epoxide 3, we obtained a crude reaction product which was purified by Biotage SP1™ using n-hexane/AcOEt (from 100/0 to 20/80) to afford 6c as a colorless oil (152 mg, 82%). Rf = 0.3 (Hexane/EtOAc 7/3). 1H-NMR (400 MHz, CDCl3): 1.64 (ddd, Jgem = 13.5 Hz, J = 7.2 Hz, J = 4.3 Hz, 1 H, 3ax-H); 1.81 (ddd, Jgem = 13.8 Hz, J = 7.4 Hz, J = 4.6 Hz, 1 H, 6ax-H); 2.14 (ddd, Jgem = 13.8 Hz, J = 7.8 Hz, J = 3.1 Hz, 1 H, 6eq-H); 2.30 (ddd, Jgem = 13.5 Hz, J = 7.8 Hz, J = 3.9 Hz, 1 H, 3eq-H); 2.83 (bs, 1 H, 4-OH); 2.96–3.10 (m, 1 H, 2-H); 3.18–3.26 (m, 1 H, 1-H); 3.32–3.47 (m, 1 H, 4-H); 3.68 (s, 3 H, 8-H or 8’-H); 3.70 (s, 3 H, 8-H or 8’-H); 3.70–3.83 (bs, 1 H, 5-H); 6.53 (d, J = 8.9 Hz, 2 H, 10-H); 7.21 (d, J = 8.9 Hz, 2 H, 11-H). 13C-NMR (100.61 MHz, CDCl3): 28.2 (3-C); 31.2 (6-C); 39.8 (1-C); 40.2 (2-C); 52.4 (8-C + 8’-C); 53.9 (4-C); 68.3 (5-C); 109.6 (10-C); 115.2 (11-C); 132.2 (12-C); 146.1 (13-C); 174.3 (7-C or 7’-C); 174.8 (7-C or 7’-C). MS (ESI+): 386 [M + H]+.
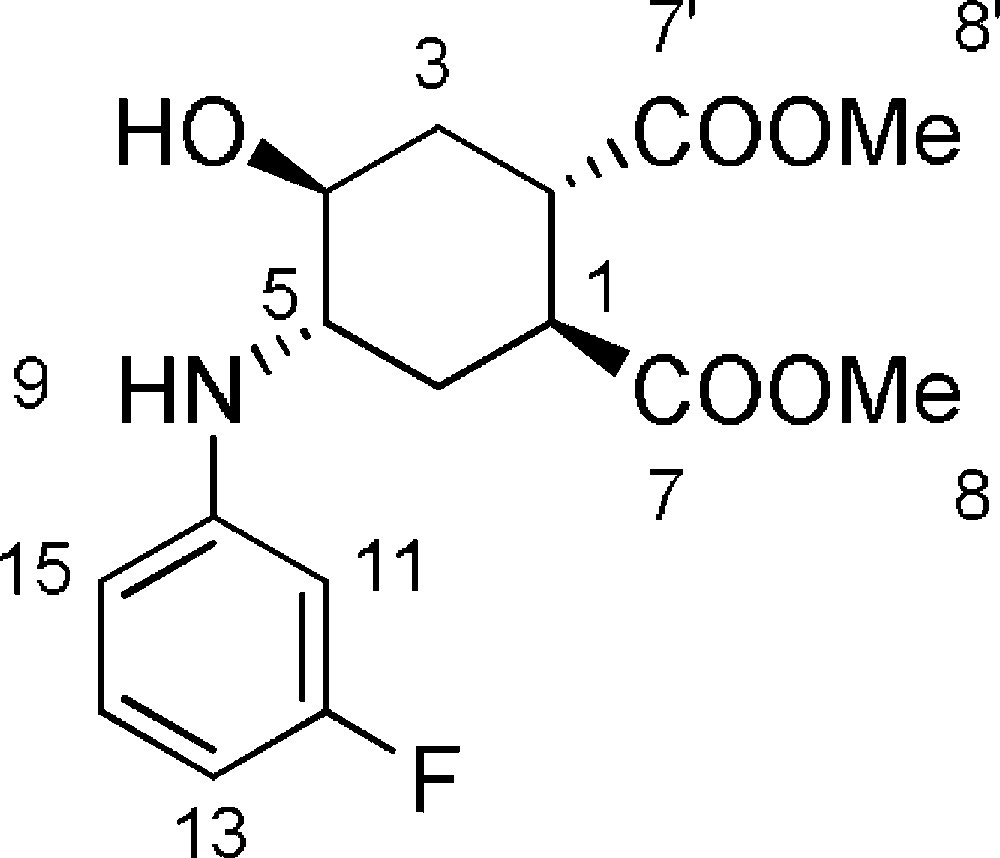
5.5 (1S,2S,4S,5S)-dimethyl-4-(3-fluorophenylamino)-5-hydroxycyclohexane-1,2-dicarboxylate 6d (Fig. 2. – S4)
Starting from 100 mg (0.47 mmol) of epoxide 3, we obtained a crude reaction product which was purified by flash chromatography using n-hexane/AcOEt (from 100/0 to 50/50) to afford 6d as a colorless oil (151 mg, 98%). Rf = 0.3 (Hexane/EtOAc 6/4). 1H-NMR (400 MHz, CDCl3): 1.68 (ddd, Jgem = 13.8 Hz, J = 7.1 Hz, J = 4.3 Hz, 1 H, 3ax-H); 1.86 (ddd, Jgem = 13.7 Hz, J = 7.3 Hz, J = 4.6 Hz, 1 H, 6ax-H); 2.17(ddd, Jgem = 13.7 Hz, J = 8.1 Hz, J = 3.8 Hz, 1 H, 6eq-H); 2.36 (ddd, Jgem = 13.8 Hz, J = 8.1 Hz, J = 3.8 Hz, 1 H, 3eq-H); 2.90 (bs, 1 H, OH); 3.08 (ddd, J = 7.6 Hz, J = 7.3 Hz, J = 4.4 Hz, 1 H, 2-H); 3.26 (ddd, J = 7.4 Hz, J = 7.3 Hz, J = 4.8 Hz, 1 H, 2-H); 3.95 (dd, J = 10.3 Hz, J = 6.6 Hz, 1 H, 4-H); 3.71 (s, 3 H, 8-H or 8’-H); 3.74 (s, 3 H, 8-H or 8’-H); 3.78–3.85 (bs, 1 H, 5-H); 6.36–6.46 (m, 3 H, 11-H + 13-H + 15-H); 7.05–7.12 (m, 1 H, 14-H). 13C-NMR (100.61 MHz, CDCl3): 28.2 (3-C); 31.1 (6-C); 39.7 (1-C); 40.1 (2-C); 52.2 (8-C + 8’-C); 53.8 (4-C); 68.1 (5-C); 100.0 + 104.3 + 109.3 (11-C + 13-C + 15-C); 130.4 (14-C); 148.9 (10-C); 164.2 (d, J = 244 Hz, 12-C); 174.2 (7-C or 7’-C); 174.8 (7-C or 7’-C). MS (ESI+): 326 [M + H]+, 348 [M + Na]+.

5.6 (1S,2S,4S,5S)-dimethyl-4-(2,3-dimethylphenylamino)-5-hydroxycyclohexane-1,2-dicarboxylate 6e (Fig. 2. – S5)
Starting from 107 mg (0.5 mmol) of epoxide 3, we obtained a crude reaction product which was purified by flash chromatography using n-hexane/AcOEt (from 100/0 to 60/40) to afford 6e as a brown oil (163 mg, 97%). Rf = 0.25 (Hexane/EtOAc 7/3). 1H-NMR (400 MHz, CDCl3): 1.71 (ddd, Jgem = 13.6 Hz, J = 6.8 Hz, J = 4.3 Hz, 1 H, 3ax-H); 1.88 (ddd, Jgem = 13.8 Hz, J = 7.1 Hz, J = 4.6 Hz, 1 H, 6ax-H); 2.05 (s, 3 H, 17-H); 2.13–2.24 (m, 1 H, 6eq-H); 2.28 (s, 3 H, 16-H); 2.38 (ddd, Jgem = 13.6 Hz, J = 8.2 Hz, J = 3.7 Hz, 1 H, 3eq-H); 3.04–3.13 (m, 1 H, 2-H); 3.23–3.31 (m, 1 H, 1-H); 3.48–3.57 (m, 1 H, 4-H); 3.72 (s, 3 H, 8-H or 8’-H); 3.74 (s, 3 H, 8-H or 8’-H); 3.80–3.85 (bs, 1 H, 5-H); 6.60–6.68 (m, 2 H, 13-H + 15-H); 7.02 (t, J = 7.8 Hz, 1 H, 14-H). 13C-NMR (100.61 MHz, CDCl3): 12.7 (17-C); 20.8 (16-C); 28.6 (3-C); 31.1 (6-C); 39.7 (1-C); 40.3 (2-C); 52.1 (8-C + 8’-C); 53.8 (4-C); 68.3 (5-C); 108.9 (15-C); 120.1 (13-C); 121.2 (11-C); 126.3 (14-C); 136.9 (12-C); 144.7 (10-C); 174.3 (7-C or 7’-C); 174.8 (7-C or 7’-C). MS (ESI+): 336 [M + H]+, 358 [M + Na]+.
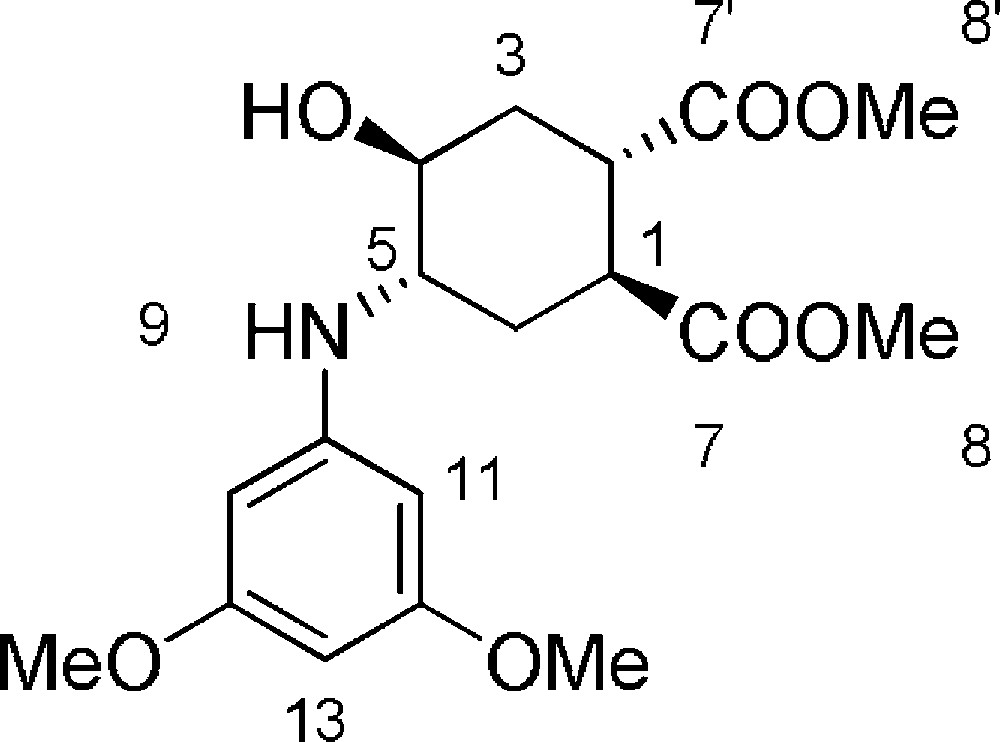
5.7 (1S,2S,4S,5S)-dimethyl-4-(3,5-dimethoxyphenylamino)-5-hydroxycyclohexane-1,2-dicarboxylate 6f (Fig. 2. – S6)
Starting from 120 mg (0.56 mmol) of epoxide 3, we obtained a crude reaction product which was purified by Biotage SP1™ using n-hexane/AcOEt (from 100/0 to 20/80) to afford 6f as a colorless oil (164 mg, 80%). Rf = 0.3 (Hexane/EtOAc 7/3). 1H-NMR (400 MHz, CDCl3): 1.64 (ddd, Jgem = 12.9 Hz, J = 8.1 Hz, J = 4.4 Hz, 1 H, 3ax-H); 1.81 (ddd, Jgem = 13.9 Hz, J = 8.1 Hz, J = 4.8 Hz, 1 H, 6ax-H); 2.16 (bs, 1 H, 4-OH); 2.24 (ddd, Jgem = 13.9 Hz, J = 6.8 Hz, J = 3.6 Hz, 1 H, 6eq-H); 2.41 (ddd, Jgem = 13.5 Hz, J = 6.9 Hz, J = 3.6 Hz, 1 H, 3eq-H); 3.01 (dd, J = 11.0 Hz, J = 6.1 Hz, 1 H, 2-H); 3.28 (dd, J = 11.6 Hz, J = 5.6 Hz, 1 H, 1-H); 3.43 (ddd, J = 7.7 Hz, J = 7.6 Hz, J = 4.0 Hz, 1 H, 4-H); 3.52 (bs, 1 H, 5-H); 3.72 (s, 3 H, 8-H or 8’-H); 3.75 (s, 3 H, 8-H or 8’-H); 3.76 (s, 6 H, 14-H); 5.89 (d, J = 2.1 Hz, 2 H, 11-H); 5.90–5.93 (m, 1 H, 13-H). 13C-NMR (100.61 MHz, CDCl3): 28.3 (6-C); 31.1 (3-C); 39.7 (1-C); 40.2 (2-C); 52.2 (8-C + 8’-C); 53.7 (4-C); 55.2 (11-C); 68.1 (5-C); 90.3 (13-C); 92.3 (11-C); 149.0 (10-C); 161.8 (12-C); 174.3 (7-C or 7’-C); 174.9 (7-C or 7’-C). MS (ESI+): 368 [M + H]+, 390 [M + Na]+.

5.8 (1S,2S,4S,5S)-dimethyl-4-hydroxy-5-(4-nitrophenylamino)cyclohexane-1,2-dicarboxylate 6g (Fig. 2. – S6)
Starting from 98 mg (457 mmol) of epoxide 3, we obtained a crude reaction product which was purified by flash chromatography using n-hexane/AcOEt (from 100/0 to 50/50) to afford 6 g as a colorless oil (69 mg, 43%). Rf = 0.3 (Hexane/EtOAc 6/4). 1H-NMR (400 MHz, CDCl3): 1.65 (ddd, Jgem = 12.6 Hz, J = 7.7 Hz, J = 4.4 Hz, 1 H, 3ax-H); 1.77 (ddd, Jgem = 12.9 Hz, J = 7.6 Hz, J = 4.7 Hz, 1 H, 6ax-H); 2.13 (ddd, Jgem = 12.9 Hz, J = 6.9 Hz, J = 3.0 Hz, 1 H, 6eq-H); 2.31 (ddd, Jgem = 12.6 Hz, J = 7.7 Hz, J = 4.1 Hz, 1 H, 3eq-H); 2.78 (bs, 1 H, OH); 3.05 (dd, J = 11.0 Hz, J = 6.8 Hz, 1 H, 2-H); 3.22 (dd, J = 11.4 Hz, J = 6.5 Hz, 1 H, 2-H); 3.54 (dt, J = 11.1 Hz, J = 7.2 Hz, 1 H, 4-H); 3.63 (s, 3 H, 8-H or 8’-H); 3.68 (s, 3 H, 8-H or 8’-H); 3.80 (ddd, J = 7.3 Hz, J = 7.1 Hz, J = 3.3 Hz, 1 H, 5-H); 4.93 (d, J = 7.5 Hz, 1 H, 9-H); 6.56 (d, J = 9.2 Hz, 2 H, 11-H); 7.94 (d, J = 9.2 Hz, 2 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): 28.1 (3-C); 31.4 (6-C); 39.9 (1-C); 40.1 (2-C); 52.3 (8-C or 8’-C); 52.4 (8-C or 8’-C); 53.7 (4-C); 68.0 (5-C); 111.6 (11-C); 126.5 (12-C); 138.0 (13-C); 153.0 (9-C); 174.0 (7-C or 7’-C); 174.5 (7-C or 7’-C). MS (ESI+): 353 [M + H]+, 375 [M + Na]+.
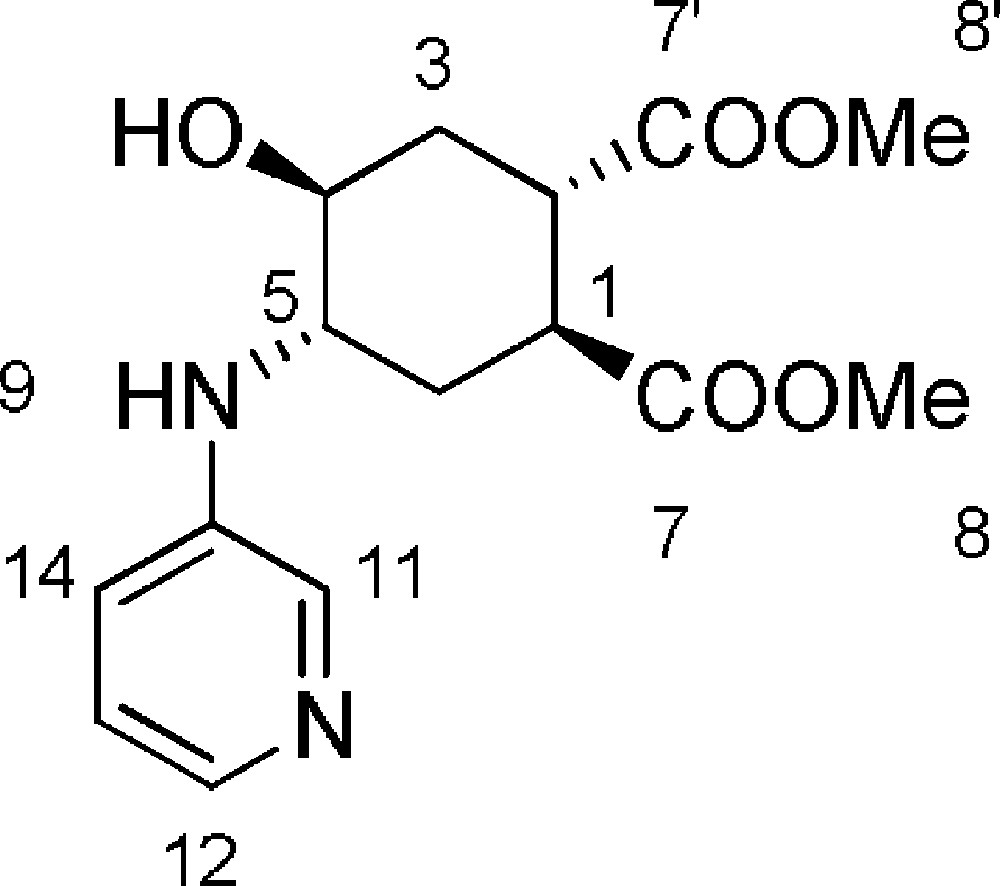
5.9 (1S,2S,4S,5S)-dimethyl-4-hydroxy-5-(pyridin-3-ylamino)cyclohexane-1,2-dicarboxylate 6h (Fig. 2. – S8)
Starting from 120 mg (0.56 mmol) of epoxide 3, we obtained a crude reaction product which was purified with Biotage SP1™ using CH2Cl2/MeOH (from 90/10 to 80/20) to afford 6 h as a white solid (69 mg, 40%). Rf = 0.1 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CD3OD): 1.77 (ddd, Jgem = 13.9 Hz, J = 11.3 Hz, J = 5.2 Hz, 1 H, 3ax-H); 2.34 (dt, Jgem = J = 13.3 Hz, J = 5.1 Hz, 1 H, 6ax-H); 2.52–2.64 (m, 2 H, 3eq-H + 6eq-H); 3.42–3.50 (m, 1 H, 2-H); 3.50–3.56 (m, 1 H, 1-H); 3.84 (s, 3 H, 8-H or 8’-H); 3.85 (s, 3 H, 8-H or 8’-H); 4.05 (dt, J = 10.9 Hz, J = 4.6 Hz, 1 H, 4-H); 4.42 (ddd, J = 12.9 Hz, J = 9.6 Hz, J = 3.5 Hz, 1 H, 5-H); 7.66–7.73 (m, 2 H, 13-H + 14-H); 8.06–8.16 (m, 1 H, 12-H); 8.16–8.26 (m, 1 H, 11-H). 13C-NMR (100.61 MHz, CD3OD): 30.6 (6-C); 33.6 (3-C); 41.9 (1-C); 42.1 (2-C); 52.2 (8-C + 8’-C); 69.5 (4-C); 75.2 (5-C); 128.4 (11-C); 129.0 (13-C or 14-C); 129.4 (13-C or 14-C); 131.5 (12-C); 150.5 (10-C); 174.1 (7-C or 7’-C); 174.4 (7-C or 7’-C). MS (ESI+): 309 [M + H]+.

5.10 (1S,2S,4S,5S)-dimethyl-4-(tert-butyldimethylsilyloxy)-5-(2-chloro-N-phenylacetamido)cyclohexane-1,2-dicarboxylate 9 (Fig. 2. – S9)
To a solution of compound 6a (273 mg, 0.89 mmol) and dry 2,6-lutidine (190 mg, 1.78 mmol) in dry CH2Cl2 (4.4 mL), at 0 °C and under N2, TBDMSOTf (398 mg, 1.51 mmol) was added. The reaction was stirred at room temperature for 2.5 h and monitored by TLC (9/1 CH2Cl2/EtOAc). After reaction completion, the solvent was evaporated under reduced pressure and the crude was taken up with Et2O, washed with H2O and dried with Na2SO4. The solvent was evaporated under reduced pressure and the resulting crude silylether intermediate (345 mg, 92%) was used for the following reaction without any further purification. Rf = 0.1 (DCM/EtOAc 9/1). MS (ESI+): 422.2 [M + H+].
To a solution of the silylether intermediate (390 mg, 0.93 mmol) and dry 2,6-lutidine (222 μg, 1.87 mmol) in dry toluene (2 mL), chloroacetylchloride (181 mg, 1.6 mmol) was added under nitrogen and at room temperature. The reaction was stirred while monitoring by TLC (8/2 petroleum ether/EtOAc), then the solution was diluted with Et2O and the organic phase was washed with water. The solvent was dried with Na2SO4 and evaporated under reduced pressure, to yield a crude that was purified by flash chromatography (95/5 CH2Cl2/AcOEt), affording compound 9 (452 mg, 98%). Rf = 0.4 (Hexane/EtOAc 8/2). 1H-NMR (400 MHz, CDCl3): 0.16 (s, 3 H, 9-H); 0.18 (s, 3 H, 9-H); 0.95 (s, 9H, 11-H); 1.61 (m, 1 H, 6ax-H); 2.25–2.38 (m, 3 H, 3eq-H + 3ax-H + 6eq-H); 3.05–3.11 (m, 1 H, 2-H); 3.20–3.26 (m, 1 H, 1-H); 3.65 (s 3 H, 8-H or 8′-H); 3.72 (s, 3 H, 8-H or 8′-H); 3.75 (m, 3 H, 4-H + 2 × 17-H.); 4.41–4.51 (m, 1 H, 5-H); 7.27–7.38 (m, 2 H, Ar-H); 7,40–7.50 (m, 3 H, Ar-H). 13C-NMR (100.61 MHz, CDCl3): −4.3 (9-C); −4.2 (9-C); 18.0 (10-C); 25.9 (11-C); 26.4 (3-C); 33.5 (6-C); 40.6 (1-C + 2-C); 43.3 (17-C); 52.2 (8-C or 8′-C); 64.1 (4-C); 128.8 (Ar-C); 129.9 (Ar-C); 141.4 (12-C); 165.8 (16-C); 173.4(7-C or 7′-C); 173.7 (7-C or 7′-C). MS (ESI+): 498.2 [M+H+]. [α]D = −174.0 [MeOH, c = 1.0].
5.11 (1S,2S,4S,5S)-dimethyl-4-(tert-butyldimethylsilyloxy)-5-(2-oxoindolin-1-yl)cyclohexane-1,2-dicarboxylate 11 (Fig. 2. – S10)
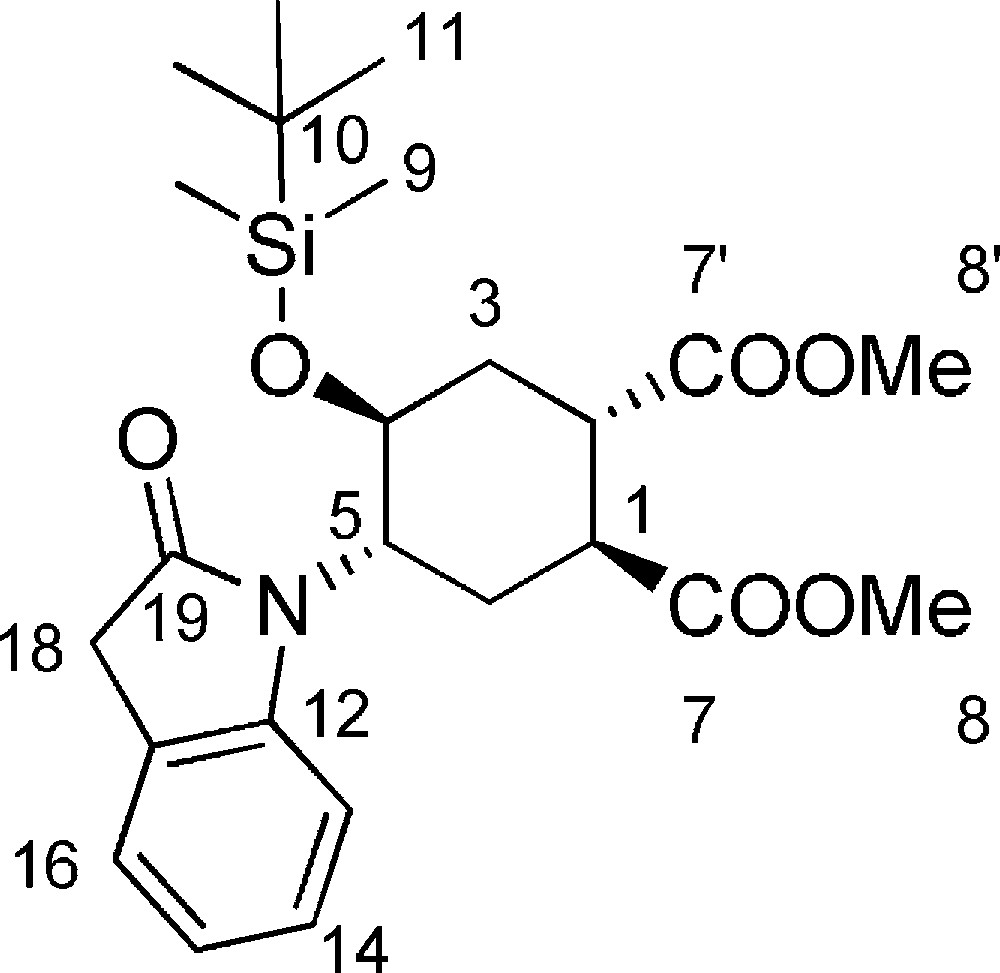
In a Schlenk reactor, under argon atmosphere and at room temperature, 9 (109 mg, 0.22 mmol), Pd(OAc)2 (5 mg, 0.022 mmol) and di-tert-butyl-diphenyl-phosphine (14 mg, 0.48 mmol) were dissolved in dry toluene (240 μL), and dry DIPEA (63 μL, 0.36 mmol) was added. The reaction mixture was stirred at 100 °C for ca. 6 h, monitoring by TLC (95/5 CH2Cl2/AcOH). After reaction completion, the reaction mixture was taken up with EtOAc (2 mL), filtered on a pad of celite, and the organic phase was evaporated under reduced pressure. The crude was purified by flash chromatography (95/5 CH2Cl2/AcOH) affording compound 11 (48 mg, 47%). Starting material 9 (33 mg, 68% conversion) was also recovered, and could be recycled. Rf = 0.4 (Hexane/EtOAc 8/2). 1H-NMR (400 MHz, CDCl3): −0.30 (s, 3 H, 9-H); −0.01 (s, 3 H, 9-H); 0.66 (s, 9H, 11-H); 1.65 (dt, Jgem = J1–6ax = 15.6 Hz, J5–6ax = 4.8 Hz, 1 H, 6ax-H); 2.21 (td, Jgem = 12.0 Hz, J2–3eq = J3eq–4 = 2.0 Hz, 1 H, 3eq-H); 2.44 (td, Jgem = 15.6 Hz, J1–6eq = J5–6eq = 2.0 Hz, 1 H, 6eq-H); 2.72 (dt, Jgem = J3ax–2 = 13.5 Hz, J3ax–4 = 4.6 Hz, 1 H, 3ax-H); 3.40 (bs, 2 H, 1-H + 2-H); 3.46 (s, 2 H, 18-H); 3.82 (s 3 H, 8-H or 8’-H); 3.83 (s, 3 H, 8-H or 8’-H); 3.98–4.07 (m, 1 H, 5-H); 4.54 (dt, J = 11.0 Hz, J3ax–4 = 4.6 Hz, 1 H, 4-H); 6.93 (d, J15–16 = 7.8 Hz, 1 H, 16-H); 7.00 (t, J13–14 = J14–15 = 7.8 Hz, 1 H, 14-H); 7.22 (t, J14–15 = J15–16 = 7.8 Hz, 1 H, 15-H); 7.18–7.28 (m, 1 H, 13-H). 13C-NMR (100.61 MHz, CDCl3): −5.5(9-C); −4.7 (9-C); 17.5 (10-C); 25.3(3-C); 25.4 (11-C); 33.6 (6-C); 36.3 (18-C); 41.0 (2-C); 41.2 (1-C); 52.3 (8-C or 8’-C); 52.4 (8-C or 8’-C); 55.5 (5-C); 66.3 (4-C); 109.2 (16-C); 121.7 (14-C); 124.2 (15-C); 124.4 (17-C); 127.6 (13-C); 145.2 (12-C); 173.4 (19-C); 173.5 (7-C or 7’-C); 175.2 (7-C or 7’-C). MS (ESI+): 462.0 [M + H+]. [α]D = + 175.0 [c = 1.5, MeOH].
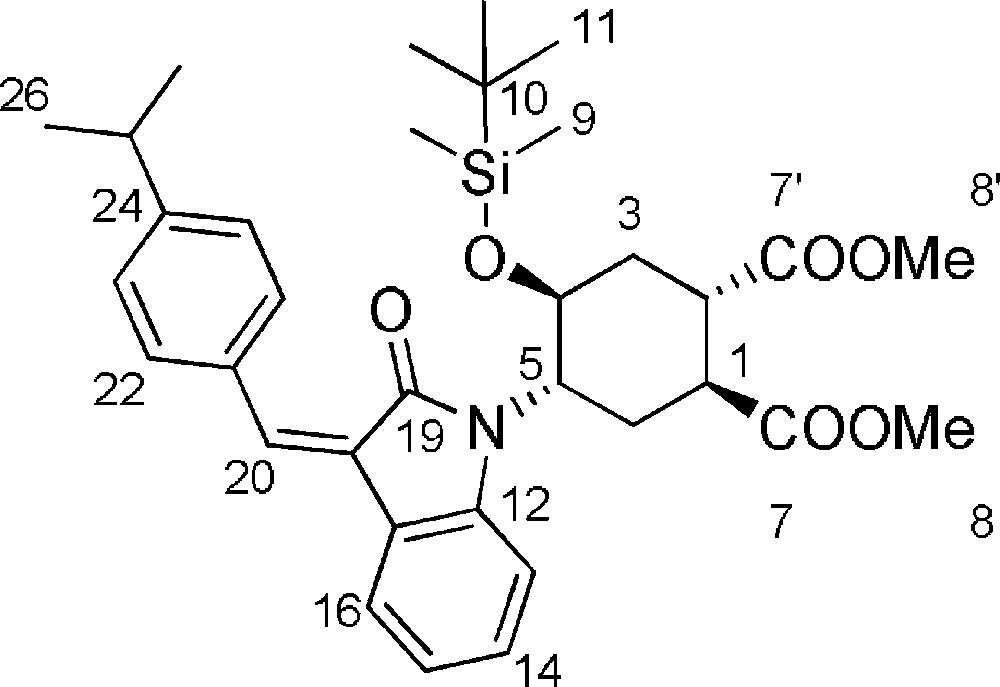
5.12 (1S,2S,4S,5S)-dimethyl-4-(tert-butyldimethylsilyloxy)-5-((E and Z)-3-(4-isopropylbenzylidene)-2-oxoindolin-1-yl)cyclohexane-1,2-dicarboxylate 13
To a solution of 11 (67 mg, 0.164 mmol) in dry methanol (290 μL), under nitrogen atmosphere and at room temperature, 4-isopropyl-benzaldehyde (27 μL, 0.175 mmol) and piperidine (6 μL, 0.06 mmol) were added. The reaction mixture was stirred ad 65 °C monitoring the reaction progression by TLC (85/15 petroleum ether/EtOAc). After reaction completion, the solvent was evaporated under reduced pressure and the silylether 13 was isolated by flash chromatography (85/15 petroleum ether/EtOAc) as a E/Z isomeric mixture (251 mg, 94%).
5.12.1 Z isomer (Fig. 2. – S11)
Rf = 0.34 (Hexane/EtOAc 9/1). 1H-NMR (400 MHz, CDCl3): −0.33 (s, 3 H, 9-H); −0.03 (s, 3 H, 9-H); 0.66 (s, 9 H, 11-H); 1.30 (d, J25–26 = 7.2 Hz, 6 H, 26-H); 1.60–1.72 (m, 1 H, 3ax-H); 2.20–2.30 (m, 1 H, 6eq-H); 2.42–2.51 (m, 1 H, 3eq-H); 2.79–2.90 (m, 1 H, 6ax-H); 2.92–3.01 (sp, J25–26 = 7.2 Hz, 1 H, 25-H); 3.37–3.45 (m, 2 H, 1-H + 2-H); 3.82 (s, 3 H, 8-H or 8’-H); 3.84 (s, 3 H, 8-H or 8’-H); 4.08–4.02 (m, 1 H, 5-H); 4.53–4.60 (m, 1 H, 4-H); 6.92 (d, J15–16 = 8.0 Hz, 1 H, 16-H); 7.01 (dt, J = 7.6 Hz, J = 0.9 Hz, 1 H, 14-H); 7.24 (dt, J = 7.8 Hz, J = 1.2 Hz, 1 H, 15-H); 7.32 (d, J22–23 = 8.0 Hz, 2 H, 23-H); 7.50 (s, 1 H, 20-H); 8.22 (d, J22–23 = 8.0 Hz, 2 H, 22-H). MS (ESI+): 592 [M + H+].
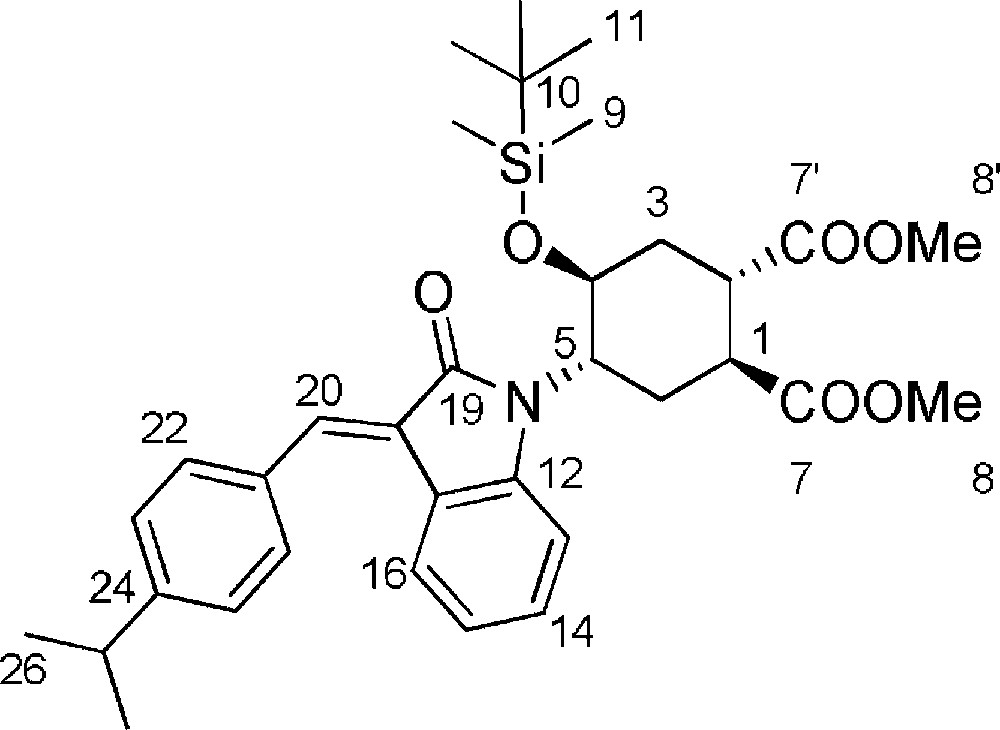
5.12.2 E isomer (Fig. 2. – S12)
Rf = 0.34 (Hexane/EtOAc 9/1). 1H-NMR (400 MHz, CDCl3): −0.28 (s, 3 H, 3-H); 0.06 (s, 3 H, 9-H); 0.66 (s, 9 H, 11-H); 1.32 (d, J25–26 = 6.8 Hz, 6 H, 26-H); 1.58–1.71 (m, 1 H, 3ax-H); 2.21–2.30 (m, 1 H, 6eq-H); 2.44–2.51 (m, 1 H, 3eq-H); 2.79–2.89 (m, 1 H, 6’-H); 2.98 (sp, J25–26 = 6.8 Hz, 1 H, 25-H); 3.40 (bs, 1 H, 2-H); 3.44 (bs, 1 H, 1-H); 3.83 (s, 3 H, 8-H or 8’-H); 3.85 (s, 3 H, 8-H or 8’-H); 3.97–4.06 (m, 1 H, 5-H); 4.51–4.62 (m, 1 H, 4-H); 6.84–6.89 (m, 1 H, 14-H); 6.92–6.97 (m, 1 H, 16-H); 7.21–7.28 (m, 1 H, 15-H); 7.34 (d, J22–23 = 8.0 Hz, 2 H, 23-H); 7.60 (d, J22–23 = 8.0 Hz, 2 H, 22-H); 7.72 (d, J13–14 = 7.2 Hz, 1 H, 13-H); 7.75 (s, 1 H, 20-H). MS (ESI+): 592 [M + H+].

5.13 (1S,2S,4S,5S)-dimethyl-4-hydroxy-5-(3-(4-isopropylbenzylidene)-2-oxoindolin-1-yl)cyclohexane-1,2-dicarboxylate 14 (Fig. 2. – S13)
A 1 M solution of TBAF in THF (100 μL, 0.84 mmol) was added to intermediate silylether 13 (14.5 mg, 0.21 mmol). The reaction mixture was stirred at room temperature for 16 h, monitoring the reaction progression by TLC (85/15 CH2Cl2/EtOAc). After reaction completion the mixture was taken up with Et2O and washed with saturated NH4Cl. The organic phase, dried with Na2SO4, was evaporated under reduced pressure and the crude product was purified by flash chromatography (85/15 CH2Cl2/EtOAc) affording compound 14 (10.5 mg, 90%) as a ≈ 40/60 (Z/E) isomeric mixture of Z and E isomers which reequilibrated after any purification attempt. Rf = 0.2 + 0.4 (DCM/EtOAc 85/15). 1H-NMR (400 MHz, CDCl3): 1.33 (d, J22–23 = 6.8 Hz, 6 H, 23-H); 1.62–1.78 (m, 1 H, 3eq-H); 2.15–2.40 (m, 2 H, 6ax-H + 6eq-H); 2.58–2.66 (m, 1 H, 3ax-H); 3.32 (bs, 1 H, 1-H); 3.36 (bs, 1 H, 2-H); 3.82 (s, 3 H, 8-H or 8’-H); 3.84 (s, 3 H, 8-H or 8’-H); 3.95–4.01 (m, 1 H, 5-H); 4.70–4.86 (m, 1 H, 4-H); 6.43–6.49 (m, Ar-H, Z isomer); 6.67–6.74 (m, Ar-H, Z isomer); 6.77–6.90 (m, Ar-H, E and Z isomers); 6.95–7.15 (m, Ar-H, E and Z isomers); 7.19–7.28 (m, Ar-H, E and Z isomers); 7.31 (m, Ar-H, Z isomer); 7.46 (m, Ar-H, Z isomer); 7.52 (m, Ar-H, E isomer); 7.67 (m, Ar-H, E isomer); 7.70 (m, Ar-H, E isomer); 8.10 (m, Ar-H, Z isomer). MS (ESI+): 478.0 [M + H+].
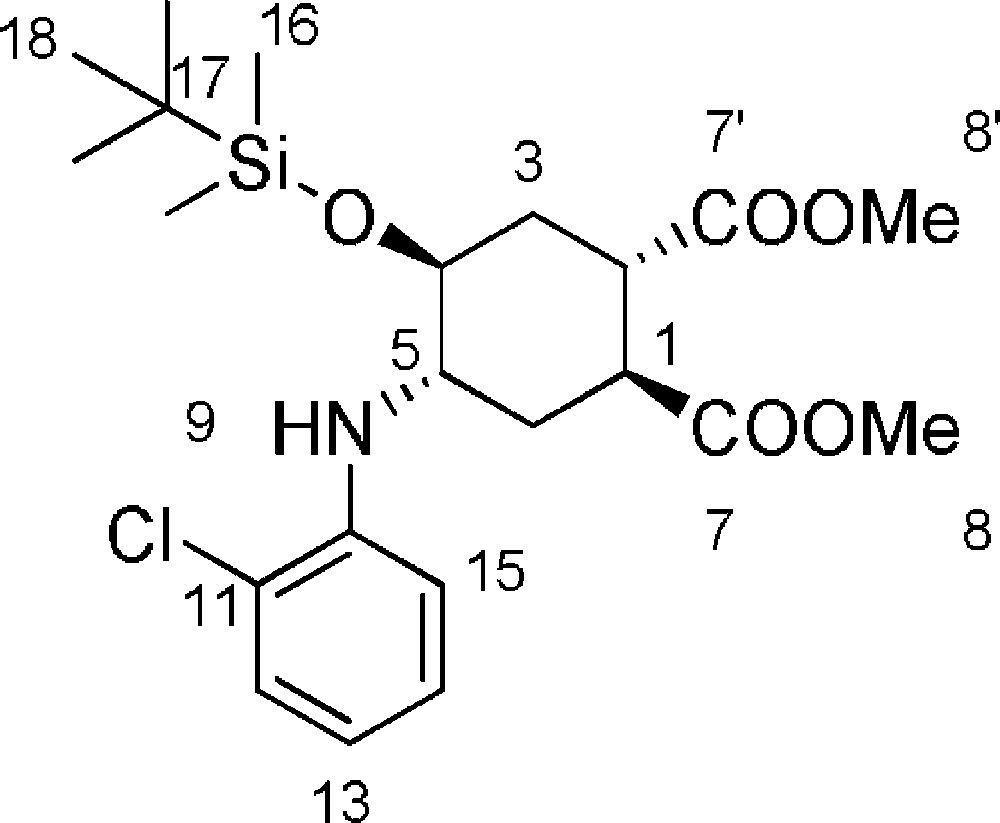
5.14 (1S,2S,4S,5S)-dimethyl-4-(tert-butyldimethylsilyloxy)-5-(2-chlorophenylamino)cyclohexane-1,2-dicarboxylate 17 (Fig. 2. – S14)
To a solution of compound 6b (205 mg, 0.6 mmol) and 2,6-lutidine (140 μL, 1.2 mmol) in dry CH2Cl2 (2.4 mL), at 0 °C and under N2, TBDMSOTf (330 μL, 1.44 mmol) was added. The reaction mixture was stirred at room temperature for 2.5 h, monitoring the reaction progression by TLC (9/1 CH2Cl2/EtOAc). After reaction completion, the solvent was evaporated under reduced pressure, the crude was taken up with Et2O, washed with H2O and dried with Na2SO4. The solvent was evaporated under reduced pressure. The crude was purified by flash chromatography on silica gel (9/1 petroleum ether/EtOAc), affording 17 (232 mg, 85%) that was used as such. A sample of the silylether was further purified by flash chromatography (94/6 petroluem ether/EtOAc) for analytical characterization. Rf = 0.2 (Hexane/EtOAc 6/4). 1H-NMR (400 MHz, CDCl3): 0.00 (s, 6 H, 16-H); 0.80 (s, 9 H, 18-H); 1.70–1.85 (m, 3 H, 3ax-H + 6eq-H + 6ax-H); 2.10 (m, 1 H, 3eq-H); 2.82–2.89 (m, 1 H, 2-H); 3.02–3.09 (m, 1 H, 1-H); 3.36–3.42 (m, 1 H, 4-H); 3.60 (s, 6 H, 8-H and 8’-H); 3.82–3.89 (m, 1 H, 5-H); 6.60 (t, J12–13 = J13–14 = 8.0 Hz, 1 H, 13-H); 6.68 (d, J14–15 = 8.0 Hz, 1 H, 15-H); 7.05 (t, J13–14 = J14–15 = 8.0 Hz, 1 H, 14-H); 7.20 (m, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.7 (16-C); −4.6 (16-C); 17.9 (17-C); 25.8 (18-C); 27.7 (6-C); 31.5 (3-C); 39.0 (2-C); 39.6 (1-C); 52.1 (8-C + 8’-C); 53.0 (5-C); 67.8 (4-C); 112.5 (15-C); 119.0 (13-C); 127.9 (14-C); 129.5 (12-C); 142.1 (10-C); 173.9 (7-C or 7’-C); 174.1 (7-C or 7’-C).
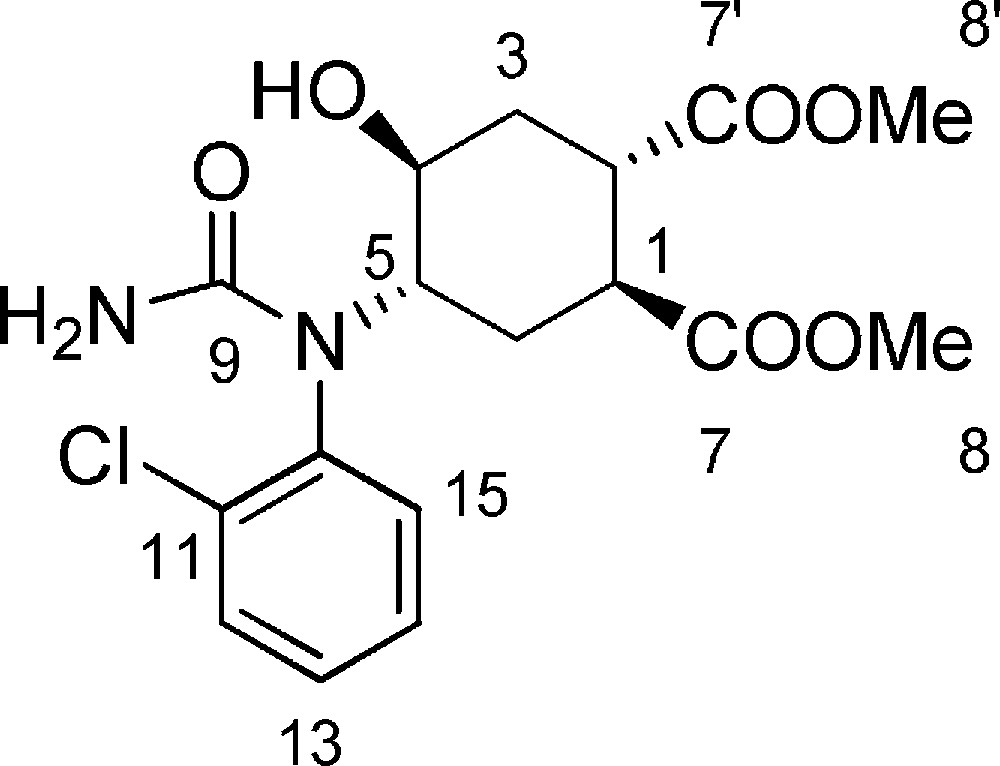
5.15 (1S,2S,4S,5S)-dimethyl-4-(1-(2-chlorophenyl)ureido)-5-hydroxy-cyclohexane-1,2-dicarboxylate 18 (Fig. 2. – S15)
A solution of chlorosulfonyl isocyanate (70 μL, 0.81 mmol) in dry THF (560 μL), under N2, was cooled to −10 °C and added dropwise over 30 minutes (using a syringe pump) to a solution of the 17 (232 mg, 0.51 mmol) in dry THF (1.2 mL). After 20 minutes a second portion of chlorosulfonyl isocyanate (30 μL, 0.34 mmol) was added dropwise. The reaction progression was monitored by TLC (95/5 CH2Cl2/MeOH). After reaction completion, water (120 μL) was added, and the mixture stirred for 30 minutes at room temperature. The solution was treated with 3 M NaOH until pH 8–9, and the organic phase washed twice with brine and dried with Na2SO4. The solvent was evaporated under reduced pressure and the crude was purified by flash chromatography on silica gel (93/7 CH2Cl2/MeOH), affording 18 (158 mg, 81%). Rf = 0.3 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CDCl3): 1.23 (td, Jgem = J5–6ax = 13.0 Hz, J1–6ax = 4.0 Hz, 1 H, 6ax-H); 1.77 (td, Jgem = J3ax–4 = 13.6 Hz, J2–3ax = 5.6 Hz, 1 H, 3ax-H); 2.38–2.52 (m, 2 H, 3eq-H + 6eq-H); 3.12 (bs, 1 H, 1-H); 3.27 (bs, 1 H, 2-H); 3.70–3.55 (m, 4H, 4-H + 8-H or 8’-H); 3.77 (s, 3 H, 8-H or 8’-H); 4.43–4.52 (m, 1 H, 5-H); 7.38 (m, 2 H, 13-H + 15-H); 7.46 (m, 1 H, 14-H); 7.55 (m, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): 26.5 (6-C); 33.20 (3-C); 40.5 (2-C); 40.7 (1-C); 52.1 (8-C or 8’-C); 52.5 (8-C or 8’-C); 58.4 (5-C); 70.0 (4-C); 128.6 (15-C); 130.4 (13-C); 131.1 (12-C); 132.3 (14-C); 135.3 (11-C); 135.7 (10-C); 159.3 (9-C); 173.3 (7-C or 7’-C); 173.6 (7-C or 7’-C). MS (ESI+): 791.2 [2M + Na+]. [α]D = −26.7 [c = 1.0, CHCl3]. HRMS: C17H21N2O6ClNa: calcd. 407.09804; found 407.09781.
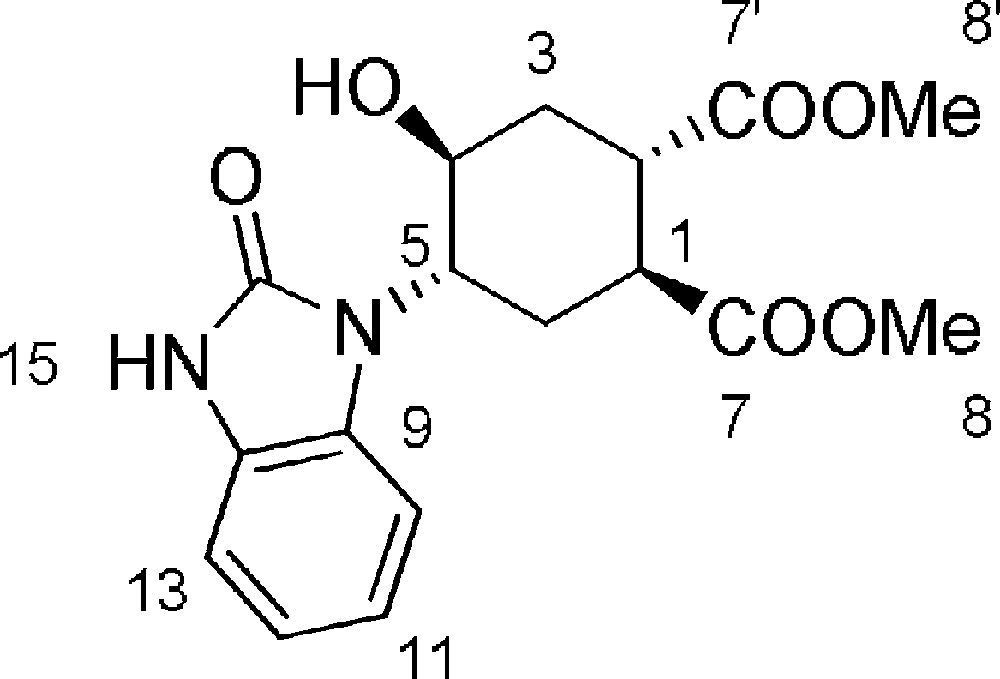
5.16 (1S,2S,4S,5S)-dimethyl-4-hydroxy-5-(2-oxo-2,3-dihydro-1H-benzo[d]imidazol-1-yl)cyclohexane-1,2-dicarboxylate 21 (Fig. 2. – S16)
A solution of 18 (157.9 mg, 0.41 mmol) and iPr2EtN (215 μL, 1.23 mmol) in dry iPrOH (1.8 mL) (dried on molecular sieves and purged with N2) was stirred under N2 atmosphere. The reaction mixture was purged with N2 for 15 minutes and then a first aliquot of X-Phos [9] (2-dicyclohexylphosphino-2’,4’,6’-triisopropylbiphenyl) (24.4 mg, 0.06 mmol) and Pd(OAc)2 (4.6 mg, 0.02 mmol) were added. The reaction mixture was purged with N2 for further 15 minutes and the solution was heated to 83 °C. After 4 h a second aliquot of X-Phos (24.4 mg, 0.06 mmol) and Pd(OAc)2 (4.6 mg, 0.02 mmol) in iPrOH (300 μL) was added. The reaction was stirred at 83 °C for 24 h, monitoring its progression by TLC (EtOAc). The solvent was evaporated under reduced pressure, the residue taken up with CH2Cl2, washed once with H2O and dried with Na2SO4. The crude was purified by flash chromatography on silica gel (EtOAc), affording 21 (74 mg, 51%) and recovered starting material 18 (34 mg, 65% conversion). Rf = 0.2 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CDCl3): 1.78 (td, Jgem = J3ax–4 = 13.2 Hz, J2–6ax = 5.0 Hz, 1 H, 3ax-H); 2.41 (m, 2 H, 6eq-H + 6ax-H); 2.62 (dt, Jgem = 13.7 Hz, J3eq–4 = J2–3eq = 2.0 Hz, 1 H, 3eq-H); 3.40 (bs, 1 H, 2-H); 3.47 (bs, 1 H, 1-H); 3.83 (s, 3 H, 8-H or 8’-H); 3.84 (s, 3 H, 8-H or 8’-H); 4.18 (m, 1 H, 4-H); 4.65 (dt, J4–5 = J3eq-5 = 11.2 Hz, J3ax-5 = 4.3 Hz, 1 H, 5-H); 7.01–7.12 (m, 3 H, 10-H + 11-H + 13-H); 7.23 (d, J = 7.4 Hz, 1 H, 10-H). 9.10 (bs, 1 H, 15-H). 13C-NMR (Hetcor, CDCl3, 400 MHz): 26.8 (6-C); 32.4 (3-C); 40.7 (2-C); 41.1 (1-C); 52.4(8-C and 8’-C); 56.9 (5-C); 65.9 (4-C); 108.9 (15-C); 109.5 (13-C); 121.5 (12-C). MS (ESI+): 719.8 [2M + Na+]. [α]D = + 21.0 [c = 1.5, CHCl3]. HRMS: C17H20N2O6Na: calcd. 371.12136; found 371.12189.
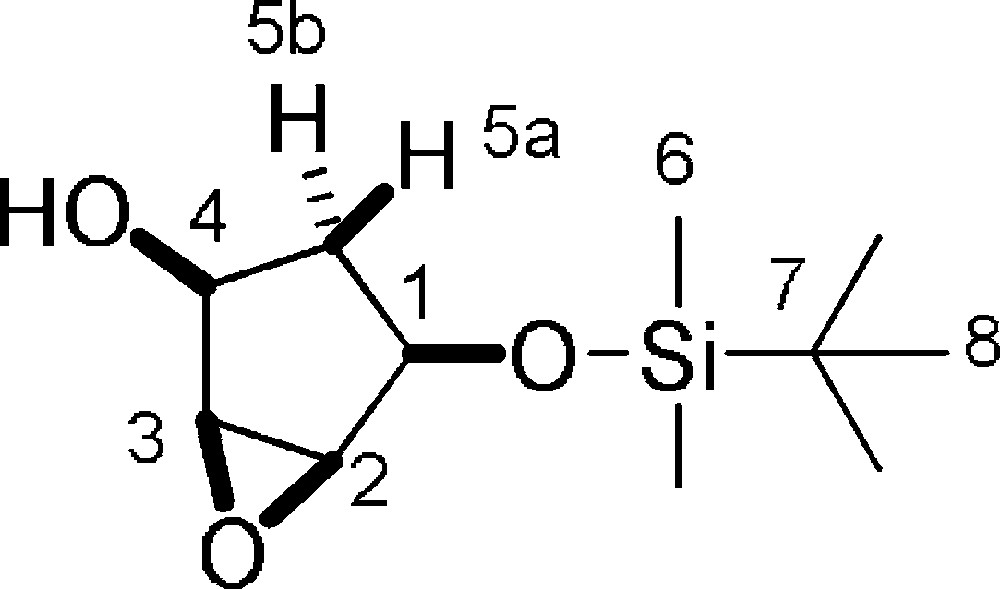
5.17 (±)-(1S,2S,4R,5S)-4-(tert-butyldimethylsilyloxy)-6-oxabicyclo[3.1.0]hexan-2-ol 4 (Fig. 2. – S17)
To a stirred solution of commercially available (±)-(1S,4R)-4-(tert-butyldimethylsilyloxy)cyclopent-2-enol (3.5 g, 16.33 mmol) in CH2Cl2 (40 mL) 3-chloroperbenzoic acid (5.63 g, 32.7 mmol) was added and the reaction was stirred for 3 h at room temperature. The reaction mixture was diluted with CH2Cl2 (40 mL) and quenched by saturated Na2CO3. The organic layer was washed (2 × 60 mL) with saturated Na2CO3, then with brine (2 × 60 mL), was dried on Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified on silica gel (n-hexane/EtOAc from 20/80 to 50/50) to afford epoxide 4 (3.21 g, 85%) as a white solid. Rf = 0.5 (Hexane/EtOAc 6/4). 1H-NMR (400 MHz, CDCl3): 0.08 (s, 3 H, 3 × 6-H); 0.10 (s, 3 H, 3 × 6-H); 0.91 (s, 9 H, 9 × 8-H); 1.30 (dt, Jgem = 12.6 Hz, J1–5a = J4–5a = 8.4 Hz, 1 H, 5a-H); 2.19 (dt, Jgem = 12.6 Hz, J1–5b = J4–5b = 7.6 Hz, 1 H, 5b-H); 3.44 (dd, J1–2 = 1.4 Hz, J2–3 = 2.9 Hz, 1 H, 2-H); 3.48 (dd, J3–4 = 1.4 Hz, J2–3 = 2.9 Hz, 1 H, 3-H); 4.02–4.08 (m, 1 H, 4-H); 4.10–4.16 (m, 1 H, 1-H). 13C-NMR (100.61 MHz, CDCl3): −4.8 (6-C); −4.7 (6-C); 18.0 (7-C); 25.7 (8-C); 34.4 (5-C); 57.3 (3-C); 58.3 (2-C); 69.9 (4-C); 70.6 (1-C). MS (ESI+): 253 [M + Na]+; 231 [M + H]+.
5.18 Epoxide opening: general procedure for compound 4
To a 0.061 M solution of 4 [14] (1 mol equiv.) in CHCl3 anilines 5a–h (1.5 mol equiv.) and InBr3 (0.5 mol equiv.) were added. The reaction mixture was stirred between 10 h to 48 h at reflux, monitoring the reaction progression by TLC (6/4 petroleum ether/EtOAc). After reaction completion the solution was evaporated and all the compounds were isolated by flash chromatography or with Biotage SP1™.
5.19 (±)-(1S,2R,3S,4R)-4-(tert-butyldimethylsilyloxy)-2-(phenylamino)cyclopentane-1,3-diol 7a and (1S,2R,3R,4R)-4-(tert-butyldimethylsilyloxy)-3-(phenylamino)cyclopentane-1,2-diol 8a
Starting from 500 mg (2.17 mmol) of epoxide 4, we obtained a crude reaction product which was purified by Biotage SP1™ (n-hexane/EtOAc from 90/10 to 0/100; SNAP 100 g column) to give 7a (377 mg, 54%) and 8a (155 mg, 22%) as brown oils.
5.19.1 7a (Fig. 2. – S18)
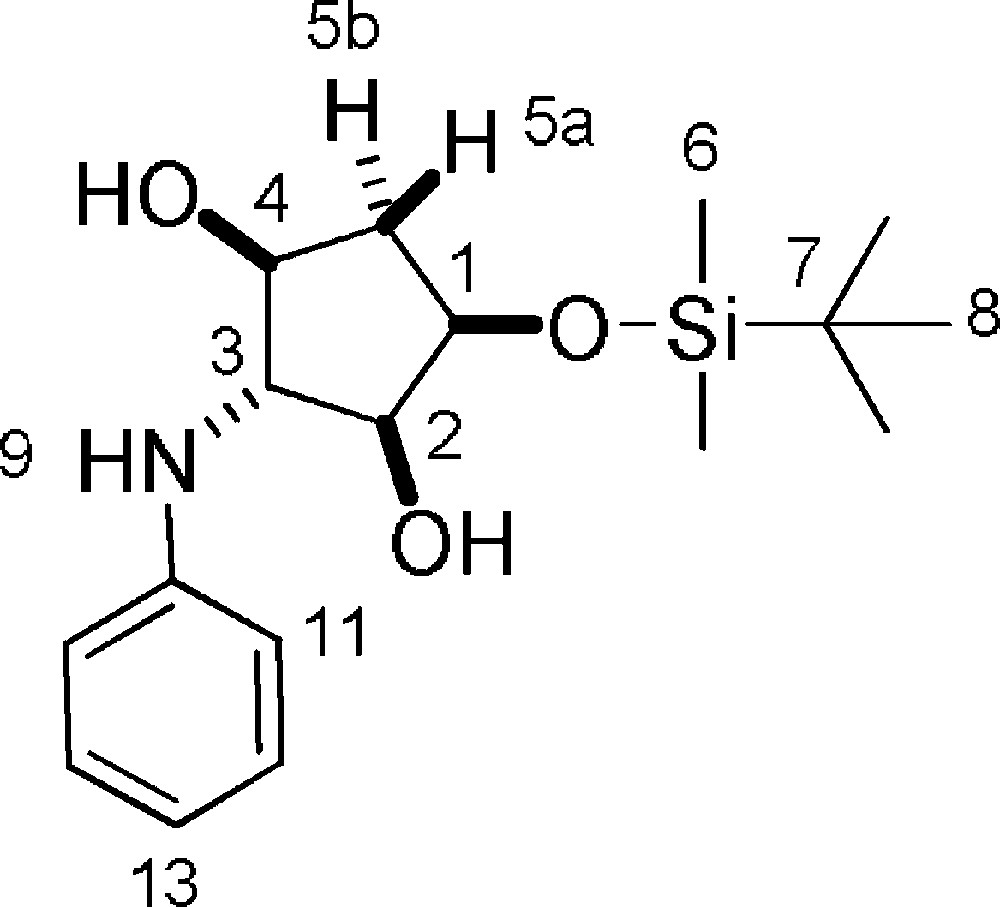
Rf = 0.5 (Hexane/EtOAc 5/5). 1H-NMR (400 MHz, CDCl3): 0.14 (s, 3 H, 3 × 6-H); 0.15 (s, 3 H, 3 × 6-H); 0.94 (s, 9 H, 9 × 8-H); 1.87 (ddt, Jgem = 14.3 Hz, J = 4.3 Hz, J = 1.1 Hz, 1 H, 5a-H); 2.26 (ddd, Jgem = 14.3 Hz, J = 6.5 Hz, J = 5.7 Hz, 1 H, 5b-H); 2.74–2.75 (2 × bs, 2 H, 2-OH + 4-OH); 3.61 (t, J = 3.5 Hz, 1 H, 3-H); 3.71–3.80 (dd + bs, J = 10.3 Hz, J = 4.8 Hz, 2 H, 9-NH + 2-H); 3.90 (bs, 1 H, 4-H); 4.24 (dd, J = 9.8 Hz, J = 4.8 Hz, 1 H, 1-H); 6.75 (d, J12–13 = 7.3 Hz, 2 H, 13-H); 6.79 (d, J11–12 = 8.7 Hz, 2 H, 11-H); 7.20 (dd, J11–12 = 8.7 Hz, J12–13 = 7.3 Hz, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.4 (7-C); 26.1 (8-C); 40.2 (5-C); 69.2 (3-C); 73.5 (1-C); 76.8 (4-C); 79.1 (2-C); 114.1 (11-C); 118.5 (13-C); 129.6 (12-C); 147.6 (10-C). MS (ESI+): 324 [M + H]+; 346 [M + Na]+.
5.19.2 8a (Fig. 2. – S19)
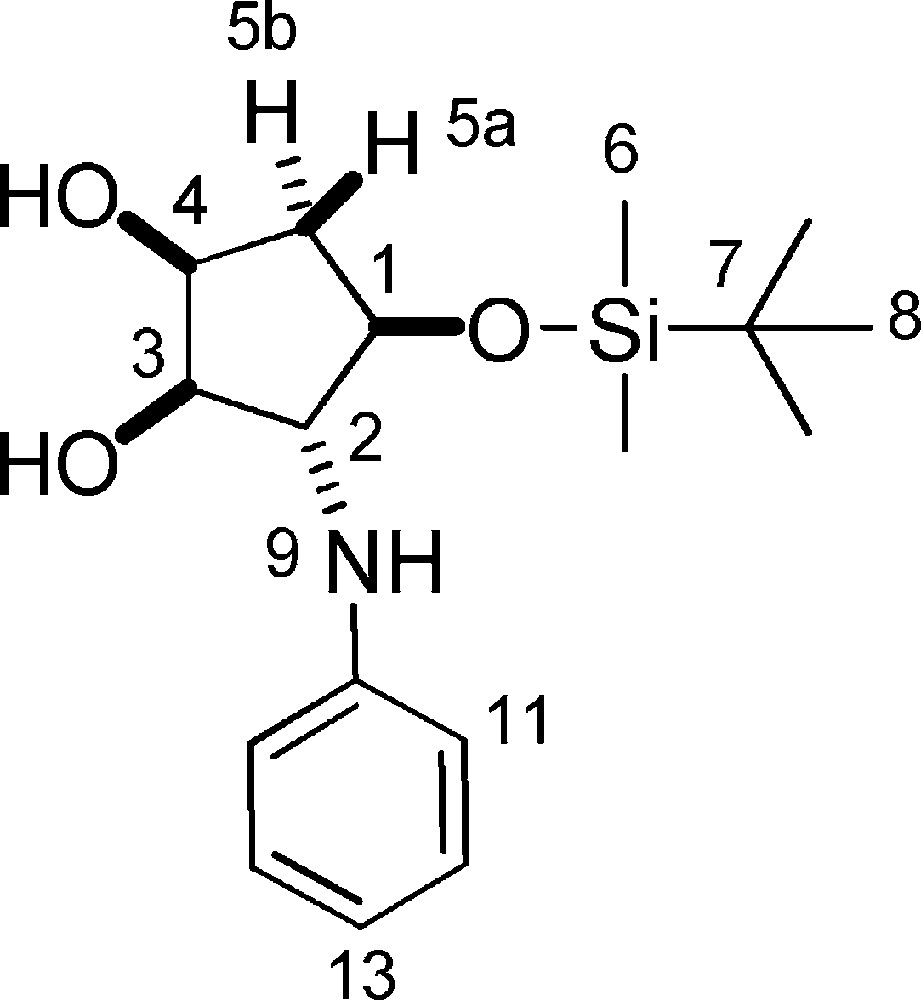
Rf = 0.2 (Hexane/EtOAc 5/5). 1H-NMR (400 MHz, CDCl3): 0.08 (s, 3 H, 3 × 6-H); 0.09 (s, 3 H, 3 × 6-H); 0.91 (s, 9 H, 9 × 8-H); 1.91 (ddd, Jgem = 14.6 Hz, J4–5a = 4.3 Hz, J1–5a = 2.8 Hz, 1 H, 5a-H); 2.17 (ddd, Jgem = 14.3 Hz, J1–5b = J4–5b = 5.6 Hz, 1 H, 5b-H); 3.10 (bs, 1 H, OH); 3.20 (bs, 1 H, OH); 3.65–3.70 (m, 1 H, 2-H); 3.77 (t, J2–3 = J3–4 = 4.9 Hz, 1 H, 3-H); 4.00 (ddd, J1–5b = 4.6 Hz, J1–5a = J1–2 = 2.8 Hz, 1 H, 1-H); 4.13–4.20 (m, 1 H, 4-H); 6.72 (d, J11–12 = 8.6 Hz, 1 H, 11-H); 6.76 (d, J12–13 = 7.3 Hz, 1 H, 13-H); 7.18 (dd, J11–12 = 8.6 Hz, J12–13 = 7.3 Hz, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.2 (7-C); 26.1 (8-C); 39.8 (5-C); 68.7 (2-C); 72.8 (4-C); 78.0 (1-C); 79.6 (3-C); 114.1 (11-C); 118.5 (13-C); 129.6 (12-C); 148.0 (10-C). MS (ESI+): 324 [M + H]+; 346 [M + Na]+.
5.20 (±)-(1S,2S,4R,5S)-4-(tert-butyldimethylsilyloxy)-2-(2-chlorophenyamino)cyclopentane-1,3-diol 7b and (±)-(1S,2R,3R,4R)-4-(tert-butyldimethylsilyloxy)-3-(2-chlorophenylamino)cyclopentane-1,2-diol 8b
Starting from 500 mg (2.17 mmol) of epoxide 4, we obtained a crude reaction product which was purified by Biotage SP1™ (CH2Cl2/MeOH from 98/2 to 80/20; SNAP 50 g column) to give 7b (388 mg, 50%) and 8b (195 mg, 25%) as colorless oils.
5.20.1 7b (Fig. 2. – S20)
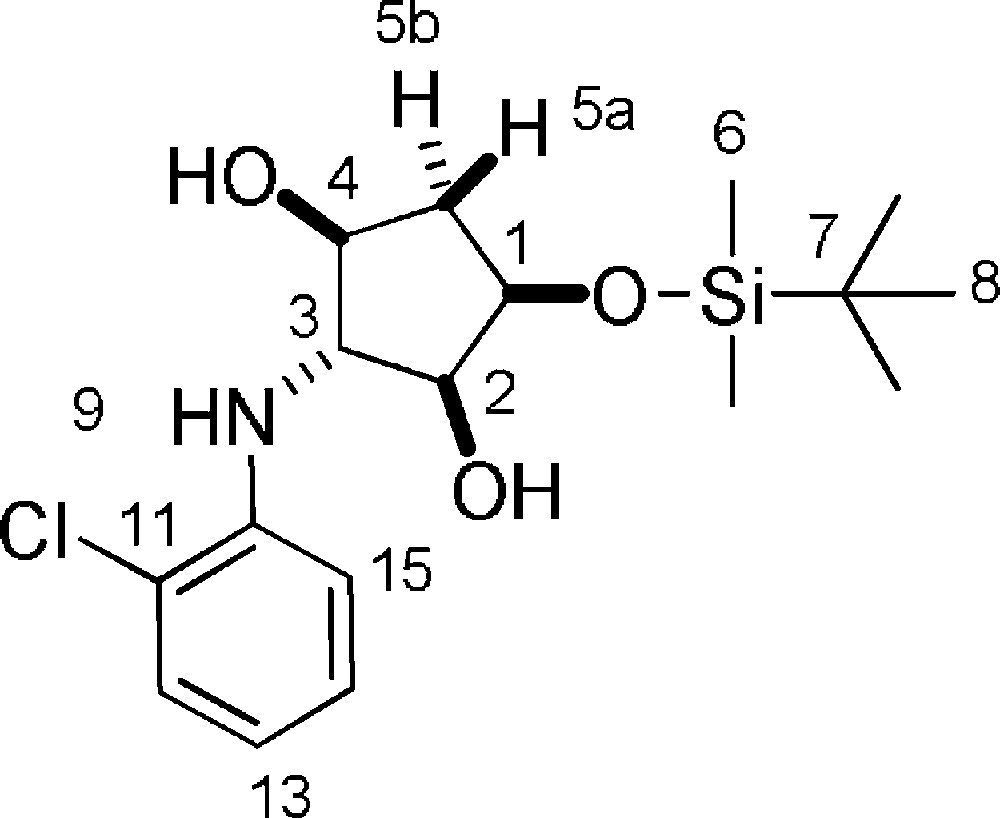
Rf = 0.9 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CDCl3): 0.15 (s, 3 H, 3 × 6-H); 0.16 (s, 3 H, 3 × 6-H); 0.94 (s, 9 H, 9 × 8-H); 1.90 (ddd, Jgem = 14.3 Hz, J = 4.2 Hz, J = 1,3 Hz, 1 H, 5a-H); 2.28 (ddd, Jgem = 14.3 Hz, J 1–5b = 6.5 Hz, J4–5b = 5.5 Hz, 1 H, 5b-H); 2.66 (d, J = 9.2 Hz, 1 H, 4-OH); 2.74 (d, J = 6.8 Hz, 1 H, 2-OH); 3.59–3.68 (m, 1 H, 3-H); 3.83 (dt, 1 H, 2-H, J1–2 = J2–3 = 4.7 Hz, J2-OH = 6.8 Hz); 3.95 (ddd, J = 9.8 Hz, J = 7.1 Hz, J = 3.7 Hz, 1 H, 4-H); 4.27 (dd, J = 9.6 Hz, J = 4.7 Hz, 1 H, 1-H); 4.32 (d, J = 4.3 Hz, 1 H, 9-NH); 6.68 (ddd, J = 7.8 Hz, J = 7.4 Hz, J = 1.5 Hz, 1 H, 13-H); 7.01 (dd, J = 8.2 Hz, J = 1.4 Hz, 1 H, 15-H); 7.15–7.21 (m, 1 H, 14-H); 7.26 (dd, J = 7.8 Hz, J = 1.4 Hz, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.4 (7-C); 26.1 (8-C); 40.1 (5-C); 69.4 (3-C); 73.5 (1-C); 76.6 (4-C); 79.1 (2-C); 113.5 (15-C); 118.7 (13-C); 120.0 (11-C); 128.4 (14-C); 129.5 (12-C); 147.0 (10-C). MS (ESI+): 358 [M + H]+.
5.20.2 8b (Fig. 2. – S21)
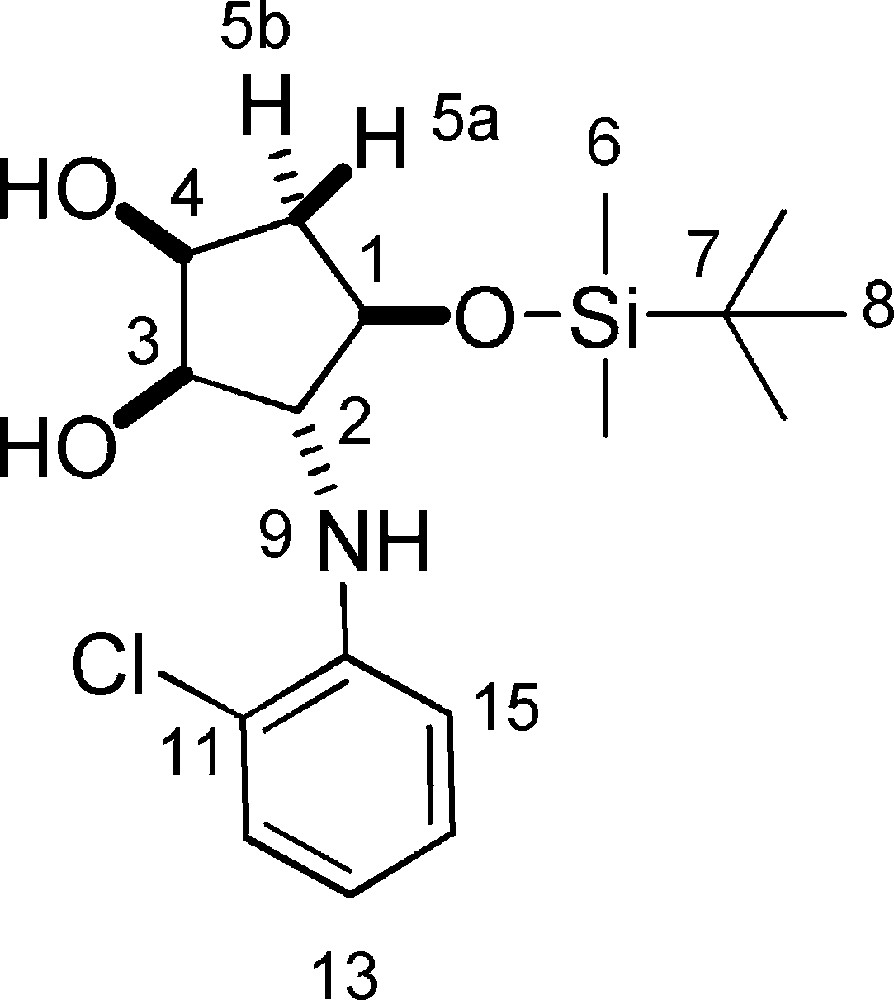
Rf = 0.5 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CDCl3): 0.14 (s, 3 H, 3 × 6-H); 0.15 (s, 3 H, 3 × 6-H); 0.90 (s, 9 H, 9 × 8-H); 1.90–1.98 (m, 1 H, 5a-H); 2.22 (dt, Jgem = 14.6 Hz, J = 5.7 Hz, 1 H, 5b-H); 2.74 (d, J = 6.8 Hz, 1 H, 2-OH); 3.06 (d, J = 8.0 Hz, 1 H, 4-OH); 3.68–3.76 (m, 1 H, 3-H); 3.79–3.88 (m, 1 H, 2-H); 4.00–4.05 (m, 1 H, 4-H); 4.11–4.34 (m, 1 H, 1-H); 6.62–6.72 (m, 1 H, Ar-H); 6.91–6.95 (m, 1 H, Ar-H); 7.09–7.17 (m, 1 H); 7.24 (dd, J = 7.9 Hz, J = 1.5 Hz, 1 H, Ar-H). 13C-NMR (100.61 MHz, CDCl3): −4.8 (6-C); −4.5 (6-C); 18.1 (7-C); 25.9 (8-C); 39.7 (5-C); 68.3 (3-C); 72.4 (1-C); 77.8 (4-C); 79.2 (2-C); 113.1 (15-C); 118.2 (13-C); 119.6 (11-C); 128.0 (14-C); 129.3 (12-C); 143.5 (10-C). MS (ESI+): 358 [M + H]+.
5.21 (±)-(1S,2R,4S,5R)-2-(4-Bromophenylamino)-4-(tert-butyldimethylsilyloxy)cyclopentane-1,3-diol 7c and (±)-(1S,2R,4S,5R)-3-(4-bromophenylamino)-4-(tert-Butyldimethylsilyloxy) cyclopentane-1,2-diol 8c
Starting from 70 mg (0.304 mmol) of epoxide 4, we obtained a crude reaction product which was purified by Biotage SP1™ (CH2Cl2/EtOAc from 100/0 to 20/80; SNAP 50 g column) to give a mixture of 7c (49 mg, 40%) and 8c (36 mg, 30%) as brown oils.
5.21.1 7c (Fig. 2. – S22)
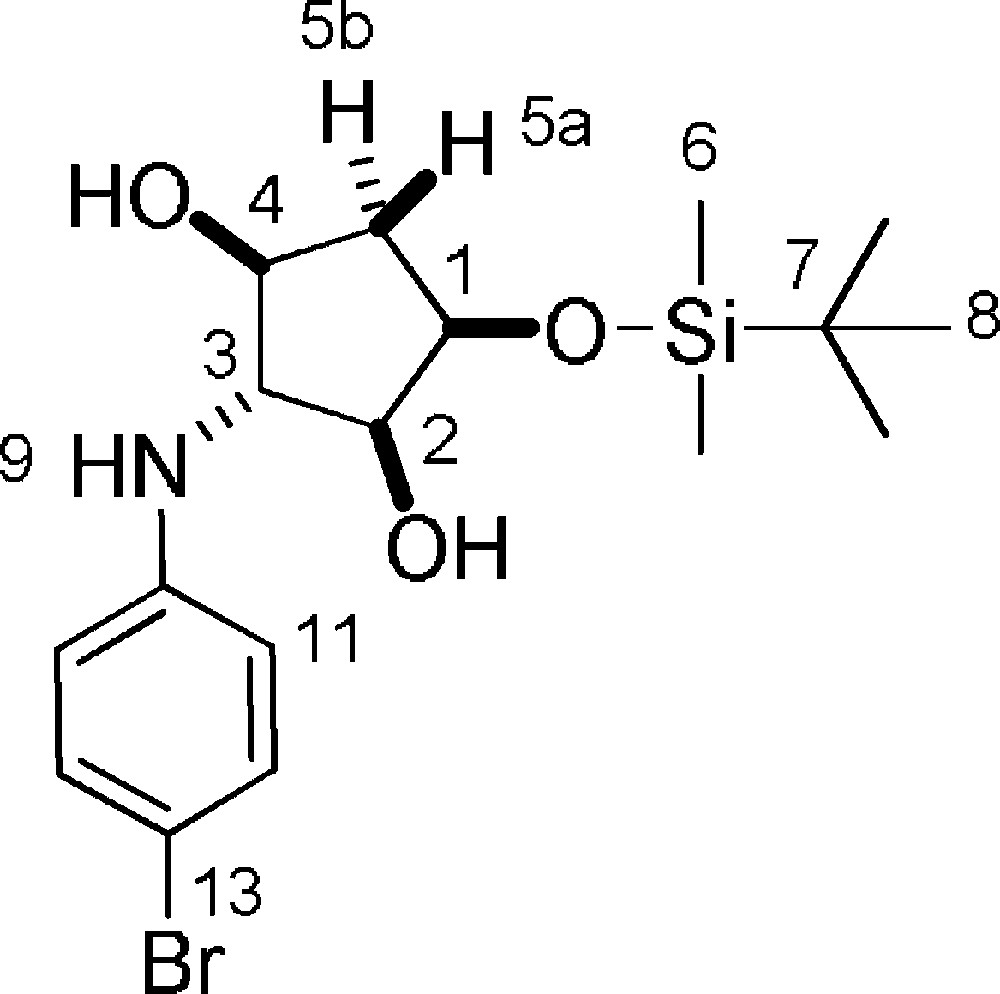
Rf = 0.9 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CDCl3): 0.14 (s, 3 H, 3 × 6-H); 0.15 (s, 3 H, 3 × 6-H); 0.93 (s, 9 H, 9 × 8-H); 1.86 (ddd, Jgem = 14.3 Hz, J = 4.3 Hz, J = 1,3 Hz, 1 H, 5a-H,); 2.23 (ddd, Jgem = 14.3 Hz, J = 6.6 Hz, J4–5b = 5.5 Hz, 1 H, 5b-H); 3.53 (dd, J2–3 = J3–4 = 3,7 Hz, 1 H, 3-H); 3.73 (t, J1–2 = J2–3 = 4.8 Hz, 1 H, 2-H); 3.82–3.90 (m, 1 H, 4-H); 4.21 (dd, J = 4.8 Hz, J = 9.5 Hz, 1 H, 1-H); 6.67 (d, J11–12 = 8.9 Hz, 2 H, 11-H); 7.26 (d, J11–12 = 8.9 Hz, 2 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.4 (7-C); 26.1 (8-C); 40.1 (5-C); 69.4 (3-C); 73.4 (1-C); 76.5 (4-C); 79.1 (2-C); 110.2 (13-C); 115.7 (11-C); 132.3 (12-C); 147.0 (10-C). MS (ESI+): 402 [M + H]+; 424 [M + Na]+.
5.22 8c (Fig. 2. – S23)

Rf = 0.5 (DCM/MeOH 9/1). 1H-NMR (400 MHz, CDCl3): 0.06 (s, 3 H, 3 × 6-H); 0.08 (s, 3 H, 3 × 6-H); 0.90 (s, 9 H, 9 × 8-H); 1.87–1.96 (m, 1 H, 5a-H); 2.17 (dt, Jgem = 14.6 Hz, J1–5b = J4–5b = 5.5 Hz, 1 H, 5b-H); 3.58–3.64 (m, 1 H, 2-H); 3.75 (t, J1–2 = J2–3 = 5.03 Hz, 1 H, 3-H); 3.94–3.99 (m, 1 H, 1-H); 4.13–4.19 (m, 1 H, 4-H); 6.61 (d, J11–12 = 8.8 Hz, 2 H, 11-H); 7.25 (d, J11–12 = 8.8 Hz, 2 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.2 (7-C); 26.1 (8-C); 39.8 (5-C); 68.9 (2-C); 72.7 (1-C); 77.9 (4-C); 80.0 (3-C); 110.1 (13-C); 115.7 (11-C); 132.2 (12-C); 146.0 (10-C). MS (ESI+): 402 [M + H]+; 424 [M + Na]+.
(±)-(1S,2R,3S,4R)-4-(tert-butyldimethylsilyloxy)-2-(3-fluorophenylamino)cyclopentane-1,3-diol 7d and (±)-(1S,2R,3R,4R)-4-(tert-butyldimethylsilyloxy)-3-(3-fluorophenylamino)cyclopentane-1,2-diol 8d: Starting from 70 mg (0.304 mmol) of epoxide 4, we obtained a crude reaction product which was purified by flash chromatography (first CH2Cl2/n-hexane 1/1, then CH2Cl2/AcOEt from 95/5 to 70/30) to give 7d (66 mg, 64%) and 8d (22 mg, 21%) as a brown oil.
5.22.1 7d (Fig. 2. – S24)
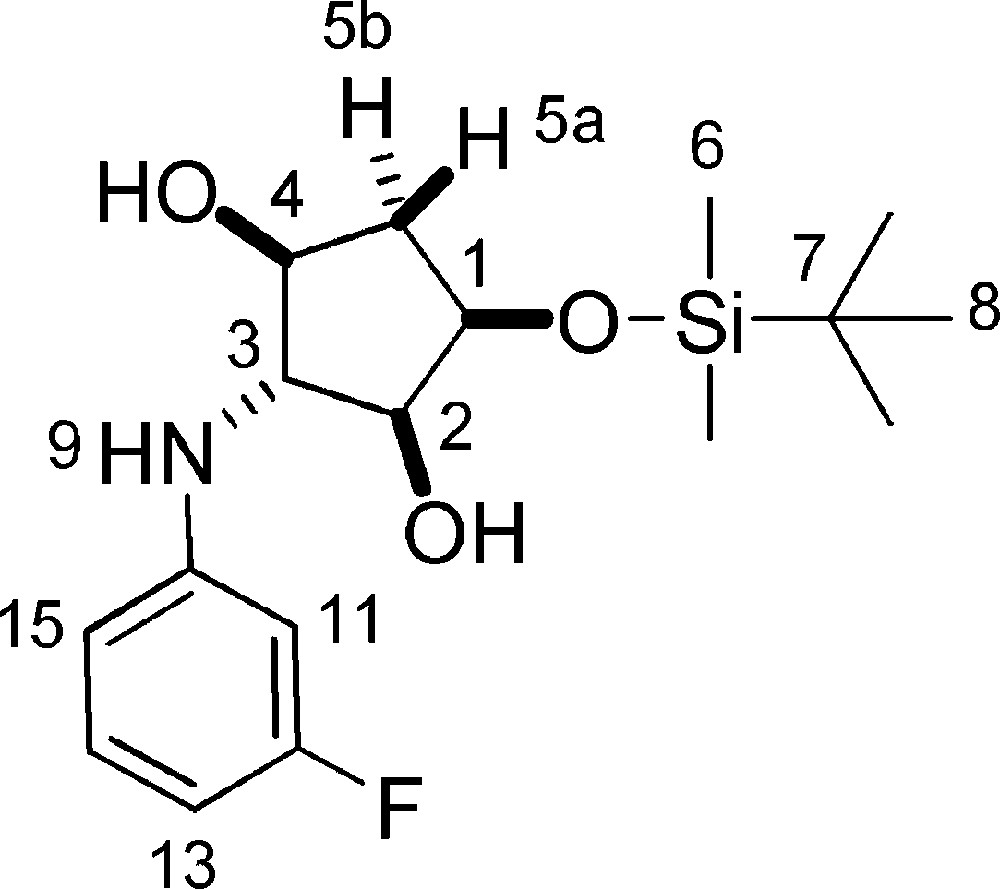
Rf = 0.5 (DCM/Hex 5/5). 1H-NMR (400 MHz, CDCl3): 0.14 (s, 3 H, 3 × 6-H); 0.15 (s, 3 H, 3 × 6-H); 0.93 (s, 9 H, 9 × 8-H); 1.87 (ddd, Jgem = 14.3 Hz, J = 4.3 Hz, J = 1,2 Hz, 1 H, 5a-H); 2.27 (ddd, Jgem = 14.3 Hz, J = 6.9 Hz, J = 5.5 Hz, 1 H, 5b-H); 2.74 (d, J = 7.1 Hz, 1 H, 2-OH); 3.58 (t, J2–3 = J3–4 = 4.2 Hz, 1 H, 3-H); 3.79–3.86 (m, 1 H, 2-H); 3.93–4.02 (m, 1 H, 4-H); 4.23 (dd, J = 4.8 Hz, J = 9.5 Hz, 1 H, 1-H); 6.54 (dt, J = 1.5 Hz, J = 8.3 Hz, 1 H, Ar-H); 6.61–6.70 (m, 2 H, Ar-H + 11-H); 7.11–7.22 (m, 1 H, Ar-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.4 (7-C); 26.1 (8-C); 40.1 (5-C); 69.3 (3-C); 73.2 (1-C); 76.4 (4-C); 78.8 (2-C); 101.0 (d, J = 25.8 Hz, Ar-C); 104.8 (d, J = 21.8 Hz, Ar-C); 109.9 (d, J = 2.4 Hz, Ar-C); 130.7 (d, J = 0.1 Hz, 15-C); 149.8 (d, J = 10.9 Hz, 10-C); 164.3 (d, J = 245.4 Hz, 12-C). MS (ESI+): 342 [M + H]+; 364 [M + Na]+.
5.22.2 8d (Fig. 2. – S25)

Rf = 0.2 (DCM/Hex 5/5). 1H-NMR (400 MHz, CDCl3): 0.07 (s, 3 H, 3 × 6-H); 0.08 (s, 3 H, 3 × 6-H); 0.90 (s, 9 H, 9 × 8-H); 1.92 (ddd, Jgem = 14.7 Hz, J = 4.34 Hz, J = 2.81 Hz, 1 H, 5a-H); 2.17 (dt, Jgem = 14.7 Hz, J1–5b = J4–5b = 5.6 Hz, 1 H, 5b-H); 2.58–3.49 (bs, 2 H, 4-OH + 3-OH); 3.61–3.67 (m, 1 H, 2-H); 3.78 (t, J1–2 = J2–3 = 5.11 Hz, 1 H, 3-H); 3.88–3.95 (m, 1 H, 1-H); 4.11–4.19 (m, 1 H, 4-H); 6.37–6.52 (m, 3 H, Ar-H); 7.10 (dt, J = 8.2 Hz, J = 6.7 Hz, 1 H, 14-H). 13C-NMR (100.61 MHz, CDCl3): −4.7 (6-C); −4.3 (6-C); 18.2 (7-C); 26.1 (8-C); 39.8 (5-C); 68.9 (2-C); 72.7 (4-C); 77.6 (1-C); 79.3 (3-C); 101.1 (d, J = 25.5 Hz, Ar-C); 105.1 (d, J = 21.5 Hz, Ar-C); 110,1 (d, J = 2.3 Hz, Ar-C); 130.6 (d, J = 0.1 Hz, 15-C); 149.1 (d, J = 10.4 Hz, 10-C); 164.3 (d, J = 243.3 Hz, 12-C). MS (ESI+): 342 [M + H]+; 364 [M + Na]+.
5.23 (±)-(1S,2R,3S,4R)-4-(tert-butyldimethylsilyloxy)-2-(2,3-dimethylphenylamino)cyclopentane-1,3-diol 7e and (±)-(1S,2R,3R,4R)-4-(tert-butyldimethylsilyloxy)-3-(2,3-dimethylphenylamino)cyclopentane-1,2-diol 8e
Starting from 50 mg (0.217 mmol) of epoxide 4, we obtained a crude reaction product, which was purified with Biotage SP1™ (n-hexane/AcOEt from 90/10 to 0/100; SNAP 10 g column) to give 7e (40 mg, 52%) and 8e (11 mg, 14%) as a brown oils.
5.23.1 7e (Fig. 2. – S26)

Rf = 0.6 (DCM/Hex 5/5). 1H-NMR (400 MHz, CDCl3): 0.14 (s, 3 H, 3 × 6-H); 0.15 (s, 3 H, 3 × 6-H); 0.94 (s, 9 H, 9 × 8-H); 1.89 (dt, Jgem = 14.3 Hz, J = 3.96 Hz, 1 H, 5a-H); 2.05 (s, 3 H, 3 × 16-H); 2.28 (s + m, 4H, 5b-H + 3 × 17-H); 2.66 (d, J = 9.4 Hz, 1 H, 4-OH); 2.71 (d, J = 6.7 Hz, 1 H, 2-OH); 3.42–3.59 (m, 1 H, 3-H); 3.59–3.67 (m, 1 H, 2-H); 3.83 (dt, J = 4.5 Hz, J = 6.6 Hz, 1 H, 4-H); 3.88–3.98 (m, 1 H, 1-H); 6.65 (d, J11–12 = 7.5 Hz, 1 H, 11-H); 6.84 (d, J12–13 = 8.1 Hz, 1 H, 13-H); 7.07 (dd, J11–12 = 7.5 Hz, J12–13 = 8.1 Hz, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 12.9 (16-C); 18.4 (7-C); 21.0 (17-C); 26.1 (8-C); 40.1 (5-C); 69.4 (3-C); 73.6 (1-C); 77.0 (4-C); 79.3 (2-C); 110.1 (13-C); 120.5 (11-C); 121.1 (14-C); 126.7 (12-C); 136.8 (15-C); 145.9 (10-C). MS (ESI+): 352 [M + H]+; 374 [M + Na]+.
5.23.2 8e (Fig. 2. – S27)
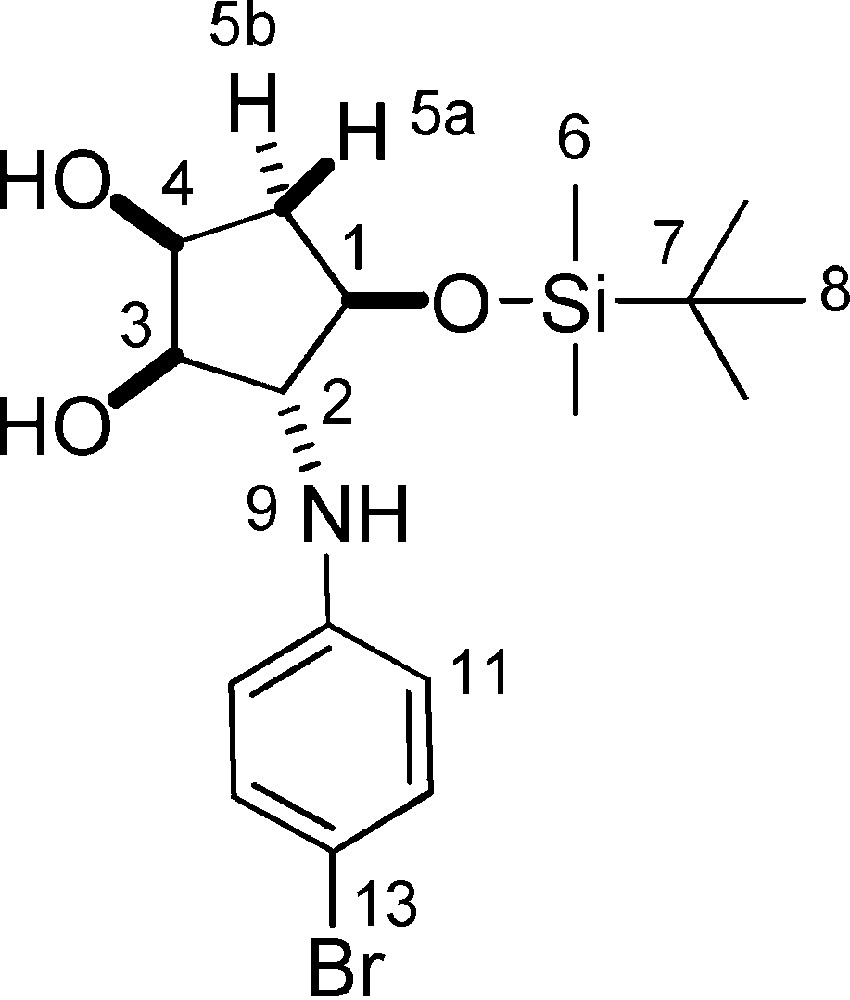
Rf = 0.3 (DCM/Hex 5/5). 1H-NMR (400 MHz, CDCl3): 0.10 (s, 3 H, 3 × 6-H); 0.11 (s, 3 H, 3 × 6-H); 0.92 (s, 9 H, 9 × 8-H); 1.95 (m, 1 H, 5a-H); 2.03 (s, 3 H, 3 × 16-H); 2.17 (dt, Jgem = 14,5 Hz, J1–5b = J4–5b = 5.3 Hz, 1 H, 5b-H); 2.28 (s, 3 H, 3 × 17-H); 3.14 (bs, 3 H, 2-OH + 4-OH + 9-NH); 3.65–3.71 (m, 1 H, 2-H); 3.84 (t, 1 H, 3-H, J2–3 = J3–4 = 4.5 Hz); 4.02–4.08 (m, 1 H, 1-H); 4.17–4.24 (m, 1 H, 4-H); 6.64 (d, J11–12 = 7.5 Hz, 1 H, 11-H); 6.70 (d, J12–13 = 8.0 Hz, 1 H, 13-H); 7.02 (dd, J11–12 = 7.5 Hz, J12–13 = 8.0 Hz, 1 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 12.9 (16-C); 18.2 (7-C); 21.0 (17-C); 26.1 (8-C); 39.7 (5-C); 69.3 (2-C); 73.4 (4-C); 78.3 (1-C); 80.3 (3-C); 110.1 (13-C); 120.5 (11-C); 121.1 (14-C); 126.5 (12-C); 137.0 (15-C); 145.4 (10-C). MS (ESI+): 352 [M + H]+; 374 [M + Na]+.
5.24 (±)-(1S,2R,3S,4R)-4-(tert-butyldimethylsilyloxy)-2-(3,5-dimethoxyphenylamino)cyclopentane-1,3-diol 7f and (±)-(1S,2R,3R,4R)-4-(tert-butyldimethylsilyloxy)-3-(3,5-dimethoxyphenylamino)cyclopentane-1,2-diol 8f
Starting from 100 mg (0.434 mmol) of epoxide 2, we obtained a crude reaction product which was purified with Biotage SP1™ (n-hexane/AcOEt from 90/10 to 0/100; SNAP 50 g column) to give a mixture of 7f (96 mg, 58%) and 8f (36 mg, 22%) as brown oils.
5.24.1 7f (Fig. 2. – S28)
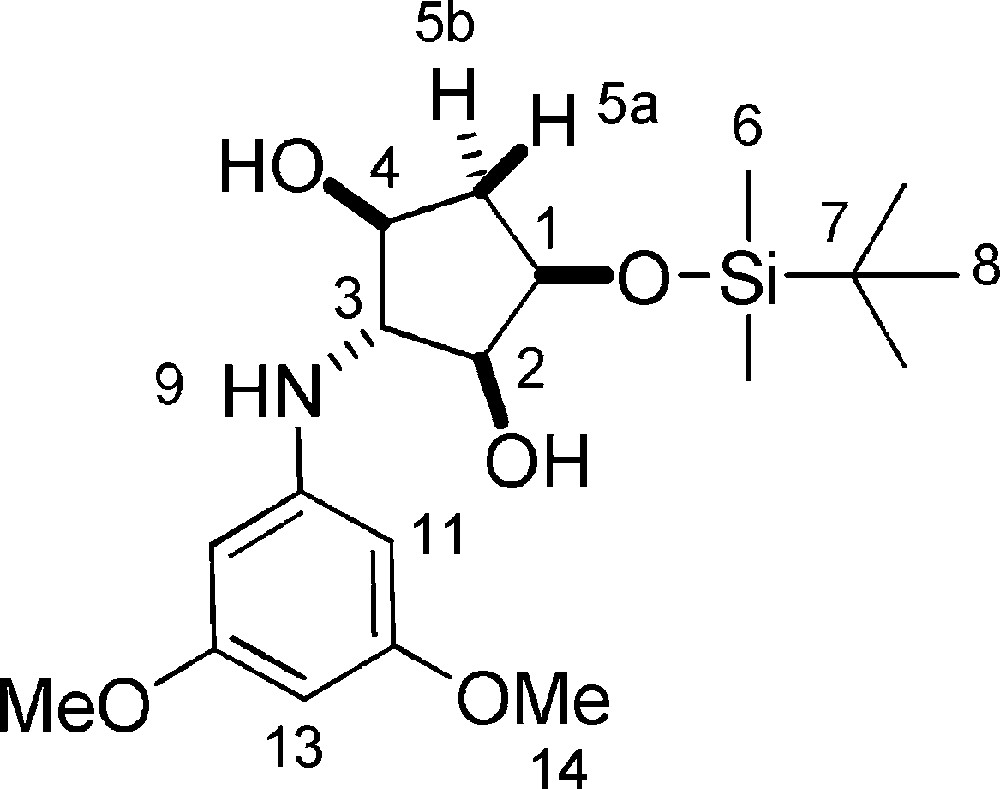
Rf = 0.6 (Hexane/EtOAc 5/5). 1H-NMR (400 MHz, CDCl3): 0.13 (s, 3 H, 3 × 6-H); 0.14 (s, 3 H, 3 × 6-H); 0.93 (s, 9 H, 9 × 8-H); 1.86 (ddt, Jgem = 14.2 Hz, J = 4.4 Hz, J = 1.1 Hz, 1 H, 5a-H); 2.26 (ddd, Jgem = 14.2 Hz, J = 6.6 Hz, J = 5.5 Hz, 1 H, 5b-H); 3.55 (t, J = 3.8 Hz, 1 H, 3-H); 3.74 (s, 3 H, 14-H); 3.75 (s, 3 H, 14-H); 3.76–3.80 (m, 1 H, 2-H); 3.88–3.93 (m, 1 H, 4-H); 4.21 (dd, J = 9.8 Hz, J = 4.8 Hz, 1 H, 1-H); 5.91–5.95 (t, J11–13 = 2.1 Hz, 1 H, 13-H); 6.03 (d, J11–13 = 2.1 Hz, 2 H, 11-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 (6-C); −4.3 (6-C); 18.4 (7-C); 26.1 (8-C); 40.1 (5-C); 55.5 (14-C); 69.6 (3-C); 73.3 (1-C); 76.5 (4-C); 78.9 (2-C); 91.4 (13-C); 94.2 (11-C); 148.5 (10-C); 162.1 (12-C). MS (ESI+): 384 [M + H]+.
5.24.2 8f (Fig. 2. – S29)

f = 0.3 (Hexane/EtOAc 5/5). 1H-NMR (400 MHz, CDCl3): 0.06 (s, 3 H, 3 × 6-H); 0.07 (s, 3 H, 3 × 6-H); 0.88 (s, 9 H, 9 × 8-H); 1.90 (ddd, Jgem = 14.6 Hz, J4–5a = 4.3 Hz, J1–5a = 2.9 Hz, 1 H, 5a-H); 2.17 (dt, Jgem = 14.7 Hz, J1–5b = J4–5b = 5.6 Hz, 1 H, 5b-H); 3.63–3.68 (m, 1 H, 2-H); 3.75 (s, 3 H, 14-H); 3.83 (t, J2–3 = J3–4 = 4.9 Hz, 1 H, 3-H); 4.04 (dt, J1–5b = 5.6 Hz, J1–5a = J1–2 = 2.9 Hz, 1 H, 1-H); 4.13–4.18 (m, 1 H, 4-H); 5.96 (t, J11–13 = 2.1 Hz, 1 H, 13-H); 5.99 (d, J11–13 = 2.1 Hz, 2 H, 11-H). 13C-NMR (100.61 MHz, CDCl3): −5.0 (6-C); −4.7 (6-C); 17.8 (7-C); 25.7 (8-C); 39.5 (5-C); 55.2 (14-C); 69.0 (2-C); 72.4 (4-C); 77.2 (1-C); 78.8 (3-C); 91.2 (13-C); 93.4 (11-C); 148.1 (10-C); 161.8 (12-C). MS (ESI+): 384 [M + H]+.
5.25 (±)-2-chloro-N-phenyl-N-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl) acetamide 10
2,6-Lutidine (0.224 mL, 1.920 mmol) and TBDMSOTf (372 mg, 1.408 mmol) were added at 0 °C to a stirred solution of 7a (207 mg, 0.640 mmol) and 8a (207 mg, 0.640 mmol) in CH2Cl2 (4 mL). The mixture was stirred for 1 h at room temperature, and then the reaction was quenched with a saturated NH4Cl solution. The organic layer was washed once with a saturated NH4Cl solution and twice with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude product was purified by flash chromatography (n-hexane/ CH2Cl2 from 100/0 to 88/12) to give pure silylether intermediate (577 mg, 82%) as a colorless oil.
5.26 Silylether intermediate (Fig. 2. – S30)
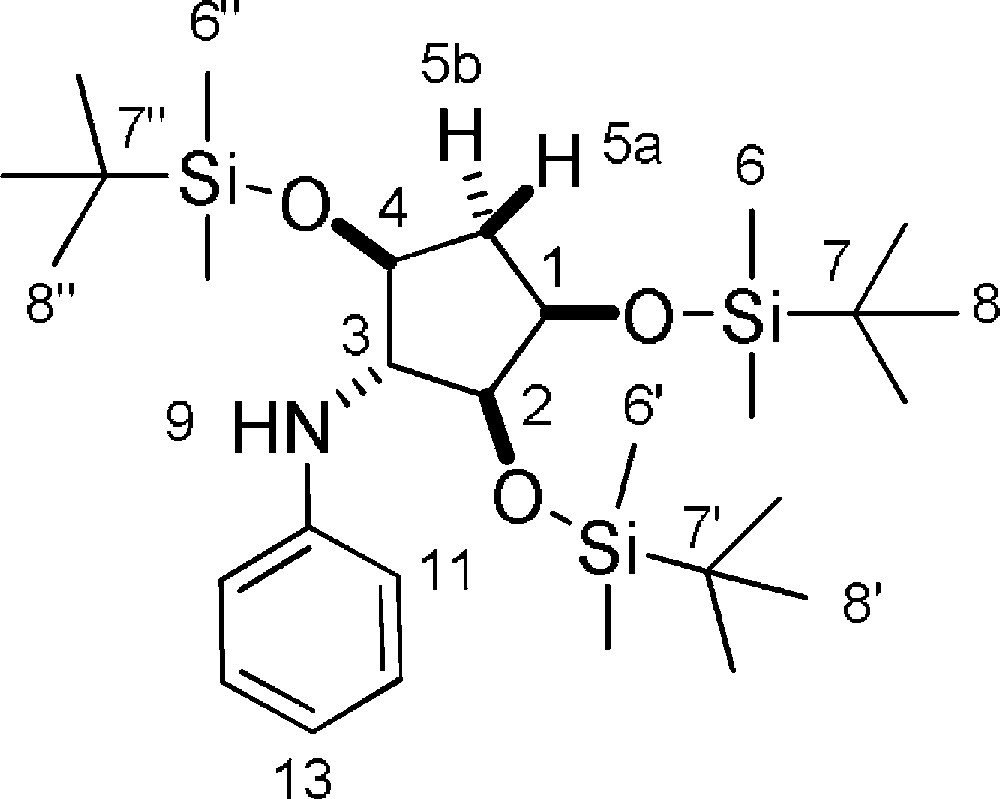
Rf = 0.9 (Hexane/EtOAc 9/1). 1H-NMR (400 MHz, CDCl3): −0.05 + 0.00 + 0.04 + 0.06 + 0.07 + 0.08 (6 s, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.87 + 0.88 + 0.92 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.81 (ddd, Jgem = 13.4 Hz, J1-5a = J4–5a = 5.7 Hz, 1 H, 5a-H); 2.20 (ddd, Jgem = 13.4 Hz, J = 8.2 Hz, J = 5.6 Hz, 1 H, 5b-H); 3.64–3.70 (m, 1 H, 2-H); 3.73 (dd, J2–3 = J3–4 = 5.0 Hz, 1 H, 3-H); 3.81–3.89 (m, 1 H, 4-H); 3.93–4.02 (m, 1 H, 1-H); 6.66–6.82 (m, 3 H, 2 × 11-H + 13-H); 7.15 (dd, J = 8.7 Hz, J = 7.3 Hz, 2 H, 12-H). 13C-NMR (100.61 MHz, CDCl3): −4.6 + −4.4 + −4.2 + −4.1 + −3.9 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 18.2 + 18.48 + 18.49 (7-C + 7’-C + 7”-C); 26.1 + 26.2 + 26.3 (8-C + 8’-C + 8”-C); 40.8 (5-C); 67.6 (3-C, only on HSQC); 73.0 (1-C); 76.6 (4-C, only on HSQC); 78.5 (2-C, only on HSQC); 114.4 (11-C, only on HSQC); 117.9 (13-C, only on HSQC); 129.5 (12-C, only on HSQC); (10-C, not visible on HSQC). MS (ESI+): 552 [M + H]+.
Chloroacetyl chloride (145 μL, 1.812 mmol) and 2,6-lutidine (264 μL, 2.264 mmol) were added to a stirred solution of silylether intermediate (500 mg, 0.906 mmol) in dry toluene (2 mL) under N2 atmosphere. The reaction was stirred for 45 min at room temperature, then was diluted with CH2Cl2 and washed three times with water. The organic layer was dried over Na2SO4, filtered and concentrated under reduced pressure. The crude residue was purified with Biotage SP1™ (n-hexane/EtOAc from 98/2 to 80/20; SNAP 50 g column) to give pure 10 (545 mg, 96%) as a white solid.

Rf = 0.64 (Hexane/EtOAc 9/1). 1H-NMR (400 MHz, CDCl3): 0.03 + 0.04 + 0.05 + 0.07 (6 s, 6 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.84 + 0.90 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.52–1.67 (m, 1 H, 5a-H); 2.24 (ddd, 1 H, 5b-H, Jgem = 13.5 Hz, J = 8.4 Hz, J = 4.7 Hz); 3.81 (d, 1 H, 17a-H, Jgem = 13.2 Hz); 3.88 (d, 1 H, 17b-H, Jgem = 13.2 Hz); 3.93 (dd, 1 H, 3-H, J2-3 = 6.7 Hz, J3–4 = 8.9 Hz); 4.03 (dd, 1 H, 1-H, J = 7.3 Hz, J3–4 = 4.3 Hz); 4.73 (dd, 1 H, 2-H, J = 8.9 Hz, J3–4 = 4.1 Hz); 4.81–4.89 (m, 1 H, 4-H); 7.28–7.43 (m, 5H, Ar-H). 13C-NMR (100.61 MHz, CDCl3): −4.5 + −4.3 + −4.2 + −4.0 + −3.9 + −3.8 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 18.1 + 18.3 + 18.4 (7-C + 7’-C + 7”-C); 26.1 + 26.3 + 26.4 (8-C + 8’-C + 8”-C); 40.9 (5-C); 43.3 (14-C); 69.2 (4-C); 73.0 (1-C); 76.6 (2-C); 78.5 (3-C); 127.9 (Ar-C); 128.2 (Ar-C); 129.7 (Ar-C); 144.6 (10-C); 166.5 (13-C). MS (ESI+): 650 [M + Na]+.

5.27 (±)-1-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl) indolin-2-one 12 (Fig. 2. – S32)
Palladium(II) acetate (18.86 mg, 0.084 mmol) was added under N2 atmosphere to a stirred solution of 10 (264 mg, 0.420 mmol), di-tert-butyl(naphthalen-1-yl)phosphine (252 mg, 0.924 mmol) in toluene (1 mL). The mixture was stirred for 6 h at 100 °C, then filtered through a celite pad and evaporated under reduced pressure. The residue was purified by flash chromatography (n-hexane/CH2Cl2 from 95/5 to 40/60) to give 12 (174 mg, 70%) as a white solid. Rf = 0.3 (Hexane/EtOAc 93/7). 1H-NMR (400 MHz, CDCl3): −0.27 + −0.25 + −0.12 + −0.07 + 0.09 + 0.10 (6 s, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.69 + 0.74 + 0.97 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.66–1.75 (m, 1 H, 5a-H); 2.29 (ddd, 1 H, 5b-H, Jgem = 14.3 Hz, J = 8.6 Hz, J = 4.4 Hz); 3.42 (d, 1 H, 17a-H, Jgem = 22.4 Hz); 3.48 (d, 1 H, 17b-H, Jgem = 22.4 Hz); 4.01–4.08 (m, 1 H, 1-H); 4.39 (dd, 1 H, 3-H, J2–3 = 9.3 Hz, J3–4 = 6.3 Hz); 4.57 (dd, 1 H, 2-H, J2–3 = 9.3 Hz, J1–2 = 4.1 Hz); 4.79–4.86 (m, 1 H, 4-H); 6.89 (d, 1 H, 13-H, J = 8.0 Hz); 6.96 (t, 1 H, 12-H, J = 7.5 Hz); 7.18 (d, 1 H, 10-H, J = 7.3 Hz); 7.20–7.26 (m, 1 H, 11-H). 13C-NMR (100.61 MHz, CDCl3): −5.0 + −4.9 + −4.7 + −4.2 + −4.3 + −4.0 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 18.1 + 18.5 (7-C + 7’-C + 7”-C); 25.9 + 26.0 + 26.3 (8-C + 8’-C + 8”-C); 36.7 (15-C); 40.9 (5-C); 67.7 (3-C); 69.1 (4-C); 72.6 (1-C); 73.5 (2-C); 109.3 (13-C); 122.1 (12-C); 124.3 (14-C); 124.4 (10-C); 128.1 (11-C); 146.5 (9-C); 176.1 (16-C). MS (ESI+): 614 [M + Na]+. HRMS: C31H57NO4Si3Na: calcd. 614.34876; found 614.34797.
5.28 (±)-(Z)-3-(4-isopropylbenzylidene)-1-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl)indolin-2-one 15Z and (±)-(E)-3-(4-isopropylbenzylidene)-1-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl)indolin-2-one 15E
4-Isopropylbenzaldehyde (14 μL, 0.093 mmol) and piperidine (0.8 μL, 8.45 μmol) were added to a stirred solution of 12 (50 mg, 0.084 mmol) in MeOH (5 mL). The reaction was stirred at room temperature for 2 h to give a yellow solution. After reaction completion, the solvent was evaporated under reduced pressure and the residue was purified with Biotage SP1™ (n-hexane/CH2Cl2 from 90/10 to 0/100; SNAP 10 g column) to give 15Z (10 mg, 16%) and 15E (39 mg, 64%), as yellow solids.
5.28.1 15Z (Fig. 2. – S33)

Rf = 0.7 (Hexane/DCM 6/4). 1H-NMR (400 MHz, CDCl3): −0.28 + −0.26 + −0.13 + −0.07 + 0.10 + 0.11 (6 s, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.68 + 0.74 + 0.98 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.28 (d, 6 H, 23-H, J = 6.9 Hz); 1.71 (ddd, 1 H, 5a-H, Jgem = 14.3 Hz, J4–5a = 2.6 Hz, J1–5a = 2.0 Hz); 2.33 (ddd, 1 H, 5b-H, Jgem = 14.3 Hz, J4–5b = 8.5 Hz, J1–5b = 4.3 Hz); 2.96 (sp, 1 H, 22-H, J = 6.9 Hz); 4.08 (ddd, 1 H, 1-H, J1–5b = 4.1 Hz, J1–2 = 4.0 Hz, J1–5a = 2.0 Hz); 4.38–4.52 (m, 1 H, 3-H); 4.69 (dd, 1 H, 2-H, J2–3 = 9.4 Hz, J1–2 = 4.0 Hz); 4.88 (ddd, 1 H, 4-H, J4–5b = 8.5 Hz, J3–4 = 6.3 Hz, J4–5a = 2.6 Hz); 6.88 (d, 1 H, 13-H, J = 7.9 Hz); 6.98 (dt, 1 H, 11-H, J = 7.6 Hz, J = 0.9 Hz); 7.22 (dt, 1 H, 12-H, J = 7.8 Hz, J = 1.2 Hz); 7.32 (d, 2 H, 20-H, J = 8.3 Hz); 7.47 (dd, 1 H, 10-H, J = 7.6 Hz, J = 0.7 Hz); 7.50 (s, 1 H, 17-H) 8.20 (d, 1 H, 19-H, J = 8.3 Hz). 13C-NMR (100.61 MHz, CDCl3): −4.9 + −4.8 + −4.7 + −4.3 + −4.2 + −4.0 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 18.1 + 18.5 (7-C + 7’-C + 7”-C); 24.0 + 24.1 + 24.2 (8-C + 8’-C + 8”-C); 40.9 (5-C); 67.8 (3-C); 69.3 (4-C); 72.7 (1-C); 73.4 (2-C); 109.0 (13-C); 118.9 (10-C); 121.8 (12-C); 127.0 (20-C); 129.9 (11-C); 132.1 (9-C); 132.3 (19-C); 137.0 (17-C); 152.0 (15-C); 169.4(16-C). MS (ESI+): 744 [M + Na]+.
5.28.2 15E (Fig. 2. – S34)

Rf = 0.4 (Hexane/EtOAc 6/4). 1H-NMR (400 MHz, CDCl3): −0.26 + −0.25 + −0.11 + −0.06 + 0.10 + 0.12 (6 s, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.69 + 0.76 + 0.98 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.30 (d, 6 H, 23-H, J = 6.9 Hz); 1.73 (ddd, 1 H, 5a-H, Jgem = 14.1 Hz, J4–5a = 2.9 Hz, J1–5a = 2.6 Hz); 2.32 (ddd, 1 H, 5b-H, Jgem = 14.1 Hz, J4–5b = 8.6 Hz, J1–5b = 4.4 Hz); 2.98 (sp, 1 H, 22-H, J = 6.9 Hz); 4.07–4.11 (m, 1 H, 1-H); 4.42 (dd, 1 H, 3-H, J2–3 = 9.0 Hz, J3–4 = 6.3 Hz); 4.62 (dd, 1 H, 2-H, J2–3 = 9.0 Hz, J1–2 = 3.9 Hz); 4.88 (ddd, 1 H, 4-H, J4–5b = 8.6 Hz, J3–4 = 6.3 Hz, J4–5a = 2.9 Hz); 6.84 (dt, 1 H, 11-H, J = 7.6 Hz, J = 1.0 Hz); 6.91 (d, 1 H, 13-H, J = 8.1 Hz); 7.22 (dt, 1 H, 12-H, J = 7.8 Hz; J = 1.2 Hz); 7.32 (d, 2 H, 20-H, J = 8.4 Hz); 7.63 (d, 1 H, 19-H, J = 7.9 Hz); 7.71 (d, 1 H, 10-H, J = 6.6 Hz); 7.74 (s, 1 H, 17-H). 13C-NMR (100.61 MHz, CDCl3): −4.9 + −4.8 + −4.7 + −4.3 + −4.2 + −4.0 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 18.1 + 18.5 (7-C + 7’-C + 7”-C); 25.9 + 26.0 + 26.3 (8-C + 8’-C + 8”-C); 41.0 (5-C); 67.9 (3-C); 69.7 (4-C); 72.6 (1-C); 73.9 (2-C); 109.4 (13-C); 121.5 (12-C); 122.9 (10-C); 127.0 (20-C); 129.9 (11-C); 130.0 (19-C); 132.9 (9-C); 136.9 (17-C); 151.2 (15-C); 169.4(16-C). MS (ESI+): 744 [M + Na]+.

5.29 (±)-(Z and E)-3-(4-isopropylbenzylidene)-1-((1R,2S,3R,5S)-2,3,5-trihydroxycyclopentyl)indolin-2-one 16
TBAF (0.127 mL, 0.127 mmol, 1 M solution in THF) was added to a stirred solution of 15Z and 15E mixture (30 mg, 0.041 mmol). The solvent was evaporated under reduced pressure and taken up with CH2Cl2. The organic layer was washed four times with water, the combined organic layers were then evaporated under reduced pressure. The residue was purified with Biotage SP1™ (n-hexane/acetone from 88/12 to 0/100; SNAP 10 g column) to give 16 in a Z/E 40/60 mixture (11.6 mg, 74%) as a yellow solid. Rf = 0.3 + 0.2 (Hexane/Acetone 5/5). 1H-NMR (400 MHz, CD3OD): 1.46 (d, 6 H, 23-H Z-isomer, J = 6.9 Hz); 1.48 (d, 6 H, 23-H, Z-isomer, J = 6.9 Hz); 1.94–2.02 (m, 1 H, 5a-H); 2.69 (ddd, 1 H, 5b-H, Jgem = 14.2 Hz, J = 8.4 Hz, J = 5.6 Hz); 3.15 (sp, 1 H, 19-H, J = 6.9 Hz); 4.31–4.38 (m, 1 H, 1-H); 4.64 (dd, 1 H, 3-H, J = 15.5 Hz, J = 7.0 Hz); 4.81 (dd, 1 H, 2-H, J = 8.8 Hz, J = 4.0 Hz); 4.85–4.93 (m, 1 H, 4-H); 7.03–7.12 (m, 1 H, 9-H Z-isomer); 7.18–7.26 (m, 2 H, 9-H Z-isomer + 10-H E-isomer); 7.25–7.31 (d, 1 H, 10-H E-isomer, J = 8.0 Hz); 7.40–7.51 (m, 2 H, Ar-H); 7.52–7.60 (m, 1 H, Ar-H); 7.76–7.92 (m, 3 H, Ar-H); 8.44 (d, 1 H, 14-H Z-isomer, J = 8.3 Hz). 13C-NMR (100.61 MHz, CD3OD): 24.1 + 24.2 (20-C); 35.4 + 35.5 (19-C); 40.2 (5-C); 67.8 + 67.8 (3-C); 69.8 (4-C); 71.0 + 71.1 (1-C); 73.1 (2-C); 109.9 + 110.6 (10-C); 118.9 (10-C); 129.7 (Ar-CH); 130.7 + 130.8 (Ar-CH); 133.3 (CIV); 133.5 (Ar-CH); 133.6 (CIV); 138.3 (Ar-CH); 138.5 (Ar-CH); 143.6 + 145.6 (CIV); 152.6 + 153.2 (12-C); 168.3 + 170.7 (16-C). MS (ESI+): 744 [M + Na]+. HRMS: C23H25NO4Na: calcd. 402.16758; found 402.16725.
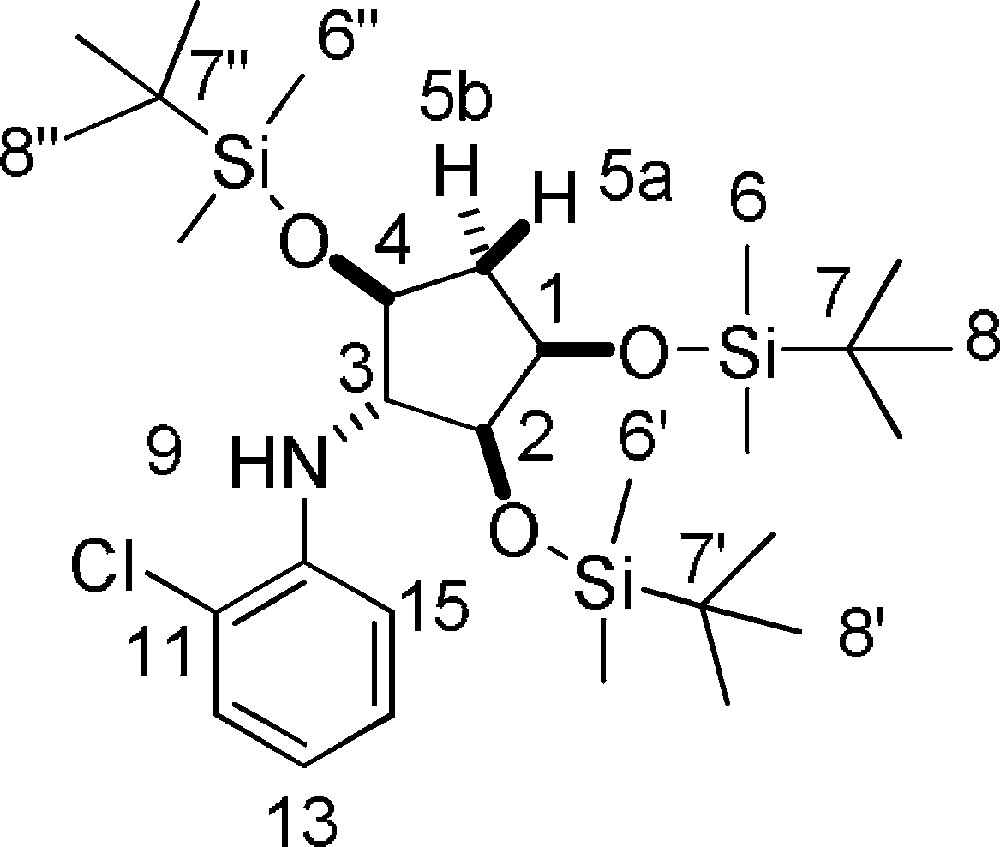
5.30 (±)-2-chloro-N-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl)aniline 19 (Fig. 2. – S36)
TBDMSOTf (302 μL, 1.314 mmol) was added at 0 °C to a solution of 2,6-lutidine (218 μl, 1.877 mmol), 7b (224 mg, 0.626 mmol) and 8b (224 mg, 0.626 mmol) in dry THF (4 mL). The solution was stirred for 2 h, the solvent was removed under reduced pressure and the residue was taken up in Et2O (4 mL). The organic layer was washed two times with water, dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel (n-hexane/CH2Cl2 from 100/0 to 85/15) to give pure 19 (493 mg, 68%) as a white solid. Rf = 0.5 (Hexane/DCM 9/1). 1H-NMR (400 MHz, CDCl3): −0.09 + −0.01 + −0.01 + 0.05 + 0.08 (5 s, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.85 + 0.86 + 0.93 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.75 (dt, 1 H, 5a-H, Jgem = 13.8 Hz, J = 4.4 Hz); 2.15 (ddd, 1 H, 5b-H, Jgem = 13.8 Hz, J = 8.3 Hz, J = 5.3 Hz); 3.65 (dd, 1 H, 2-H, J = 7.2 Hz, J = 3.7 Hz); 3.84–3.95 (m, 2 H, 3-H + 4-H); 3.98 (dd, 1 H, 1-H, J = 9.0 Hz, J = 4.1 Hz); 6.59 (ddd, 1 H, 13-H, J = 7.9 Hz, J = 7.4 Hz, J = 1.5 Hz); 7.00 (dd, 1 H, 15-H, J = 8.3 Hz, J = 1.5 Hz); 7.05–7.10 (m, 1 H, 14-H); 7.20 (dd, 1 H, 12-H, J = 7.9 Hz, J = 1.5 Hz). 13C-NMR (100.61 MHz, CDCl3): −4.7 + −4.5 + −4.2 + −4.1 + −4.0 + −3.9 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 18.2 + 18.47 + 18.52 (7-C + 7’-C + 7”-C); 26.1 + 26.2 + 26.3 (8-C + 8’-C + 8”-C); 41.0 (5-C); 66.5 (3-C); 72.8 (1-C); 77.4 (4-C); 78.7 (2-C); 113.6 (11-C); 117.3 (13-C); 119.0 (15-C); 127.9 (12-C); 129.0 (14-C); 144.5 (10-C). MS (ESI+): 586 [M + H]+.
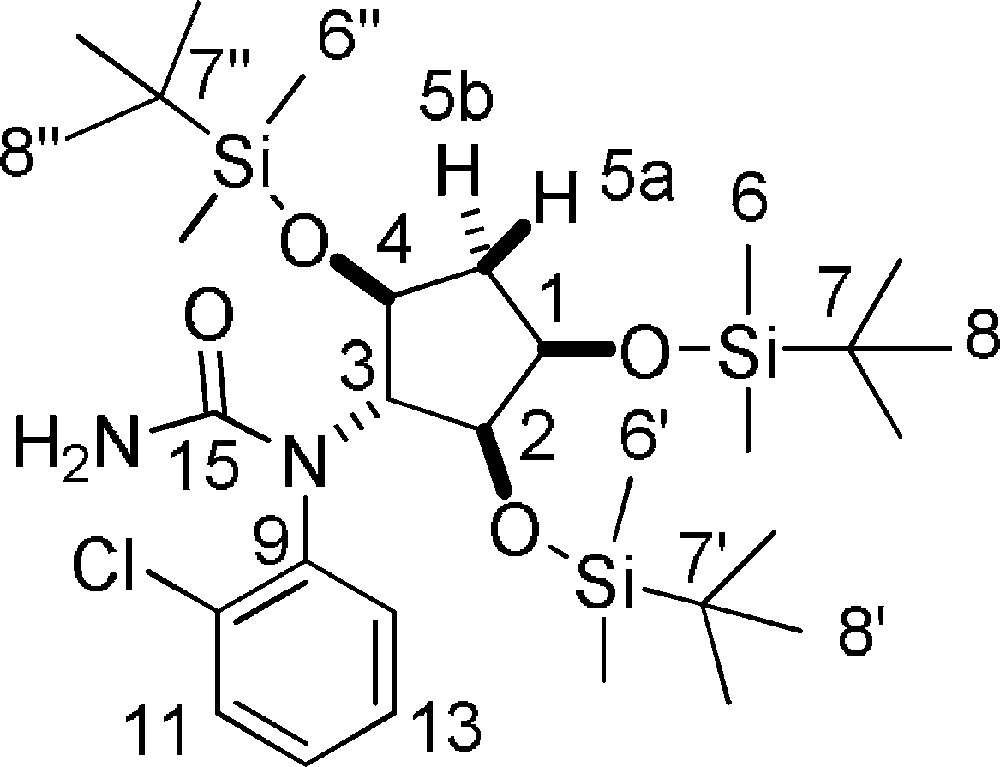
5.31 (±)-2-chloro-N-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl)aniline 20
A solution of 19 (363 mg, 0.619 mmol) in THF (2 mL) was slowly added to a stirred solution of Chlorosulfonyl isocyanate (0.081 mL, 0.928 mmol) in THF (1 mL) at −10 °C. The reaction mixture was stirred 10 min at −10 °C, then quenched with water and stirred for 30 min, then NaOH (3 M) was added. The organic layer was diluted with AcOEt and washed 2 times with brine, dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was purified with Biotage SP1™ (n-hexane/AcOEt from 100/0 to 60/40, SNAP 10 g column) to give pure 20 (362 mg, 93%) as a white solid. Rf = 0.5 (Hexane/EtOAc 7/3). 1H-NMR (400 MHz, C6D6): 0.09 + 0.11 + 0.12 + 0.14 + 0.17 + 0.18 + 0.21 + 0.22 + 0.26 + 0.27 (10 s for 2 rotamers, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.93 + 0.95 + 0.99 + 1.02 + 1.11 (5 s for 2 rotamers, 3 × 9 H, 9 × 8-H + 9 × 8-H’ + 9 × 8”-H); 1.70–1.81 (m, 0.6 H for 1 rotamer, 5a-H); 2.12–2.37 (m, 1.4H, 1 × 5b-H + 0.4 × 5a-H); 3.92 (d, 0.4H, 3-H, J = 3.9 Hz); 4.07 (bs, 2 H, NH2); 4.14 (dd, 0.6 H, 3-H, J = 8.5 Hz, J = 5.7 Hz); 4.76–4.89 (bs, 1 H, 1-H); 4.94–5.05 (m, 1 H, 2-H); 5.12–5.24 (m, 1 H, 4-H); 6.56–6.71 (m, 1 H, Ar-H); 6.78–6.93 (m, 1 H, Ar-H); 7.05 (d, 1 H, Ar-H, J = 8.0 Hz); 7.65 (d, 0.4H, Ar-H, J = 7.8 Hz); 7.70 (d, 0.6 H, Ar-H, J = 7.8 Hz). 13C-NMR (100.61 MHz, C6D6): −4.6 + −4.4 + −4.3 + −4.0 + −3.9 + −3.8 + −3.3 (2 × 6-C + 2 × 6’-C + 2 × 6”-C for the 2 rotamers); 18.1 + 18.4 + 18.5 (7-C + 7’-C + 7”-C); 26.1 + 26.2 + 26.3 + 26.5 + 26.6 (8-C + 8’-C + 8”-C for the 2 rotamers); 40.5 + 40.6 (5-C for the 2 rotamers); 71.5 + 72.0 + 72.1 + 72.6 + 73.7 + 78.3 + 78.4 + 80.9 (3-C + 4-C + 1-C + 2-C for the 2 rotamers); 128.2 (Ar-C); 128.4 (Ar-C); 130.5 (Ar-C); 131.0 (Ar-C); 133.1 (Ar-C); 142.8 (Ar-C); 156.4 (15-C). MS (ESI+): 629 [M + H]+.

5.32 (±)-1-((1R,2S,3R,5S)-2,3,5-tris(tert-butyldimethylsilyloxy)cyclopentyl)1H-benzo[d]imidazol-2(3H)-one 22
DIPEA (0.195 mL, 1.122 mmol), palladium(II) acetate (8 mg, 0.036 mmol) and 2-dicyclohexylphosphino-2’,4’,6’-triisopropylbiphenyl (178 mg, 0.373 mmol) were added to a stirred solution of 20 (235 mg, 0.373 mmol) in 2-propanol (3 mL) under N2 atmosphere at room temperature. The mixture was stirred at reflux overnight, and then the crude product was taken up in CH2Cl2 and filtered on a celite pad. The filtrate was washed twice with water, dried over Na2SO4, filtered and concentrated under reduced pressure. The crude was purified with Biotage SP1™ (n-hexane/EtOAc from 90/10 to 50/50) to give pure 22 (187 mg, 69%) as a yellow oil. Rf = 0.5 (Hexane/EtOAc 8/2). 1H-NMR (400 MHz, CDCl3): −0.34 + −0.14 + −0.09 + 0.11 + 0.12 (6 s, 6 × 3 H, 6 × 6-H + 6 × 6’-H + 6 × 6”-H); 0.65 + 0.73 + 0.98 (3 s, 3 × 9 H, 9 × 8-H + 9 × 8’-H + 9 × 8”-H); 1.72 (ddd, Jgem = 14.2 Hz, J4–5a = 2.6 Hz, J1–5a = 2.0 Hz, 1 H, 5a-H); 2.34 (ddd, Jgem = 14.2 Hz, J4–5b = 8.6 Hz, J1–5b = 4.4 Hz, 1 H, 5b-H); 4.08 (bs, 1 H, 1-H); 4.56 (dd, J2–3 = 9.6 Hz, J3–4 = 6.0 Hz, 1 H, 3-H); 4.61 (dd, J2–3 = 9.6 Hz, J1–2 = 4.1 Hz, 1 H, 2-H); 4.90 (ddd, J4–5b = 8.6 Hz, J3–4 = 6.0 Hz, J4–5a = 2.6 Hz, 1 H, 4-H); 6.99–7.09 (m, 4H, 10-H + 11-H + 12-H + 13-H); 8.96 (bs, 1 H, 15-H). 13C-NMR (100.61 MHz, CDCl3): −5.5 + −5.3 + −5.1 + −4.7 + −4.6 + −4.4 (2 × 6-C + 2 × 6’-C + 2 × 6”-C); 17.7 + 17.8 + 18.1 (7-C + 7’-C + 7”-C); 25.5 + 25.6 + 25.9 (8-C + 8’-C + 8”-C); 40.5 (5-C); 67.6 (3-C); 69.1 (4-C); 72.3 (1-C); 73.3 (2-C); 108.4 (13-C); 108.9 (12-C); 121.0 (10-C); 121.3 (11-C); 127.2 (14-C); 132.1 (9-C); 154.8 (16-C). MS (ESI+): 593 [M + H]+; 615 [M + Na]+.

5.33 (±)-1-((1R,2S,3R,5S)-2,3,5-trihydroxycyclopentyl)1H-benzo[d]imidazol-2(3H)-one 23 (Fig. 2. – S39)
Hydrochloric acid (0.464 mmol, 23 μL, 20 M solution in MeOH) was added to a stirred solution of 22 (83.3 mg, 0.140 mmol) in MeOH (3 mL). The reaction was stirred for 3 h at room temperature, and then the solvent was removed under reduced pressure. The residue was taken up with distilled water and washed three times with CH2Cl2. The aqueous phase was evaporated under reduced pressure and pure 23 (35 mg, quantitative yield) was obtained as a white solid without further purification. 1H-NMR (400 MHz, CD3OD): 1.82 (ddd, Jgem = 14.4 Hz, J4-5a = 5.4 Hz, J1–5a = 3.4 Hz, 1 H, 5a-H); 2.55 (ddd, Jgem = 14.4 Hz, J1–5b = 8.5 Hz, J4–5b = 5.4 Hz, 1 H, 5b-H); 4.15–4.20 (m, 1 H, 1-H); 4.56 (dd, J2–3 = 8.9 Hz, J3–4 = 7.7 Hz, 1 H, 3-H); 4.62 (dd, J2–3 = 8.9 Hz, J1–2 = 5.0 Hz, 1 H, 2-H); 4.67–4.77 (m, 1 H, 4-H); 6.32–6.44 (m, 3 H, 3 × Ar-H); 6.98–7.06 (m, 1 H, Ar-H). 13C-NMR (100.61 MHz, CD3OD): 40.0 (5-C); 67.7 (3-C); 69.8 (4-C); 70.8 (1-C); 73.1 (2-C); 109.8 (13-C); 110.3 (12-C); 122.3 (10-C); 122.4 (11-C); 129.6 (14-C); 132.2 (9-C); 156.8 (16-C). MS (ESI+): 273 [M + Na]+; 523 [2M + Na]+. HRMS: C12H14N2O4Na: calcd. 273.08458; found 273.08455.
6 Conflicts of interest
There is no conflict of interest for all the authors.
Acknowledgements
This work was financially supported in part by the Italian “Ministero dell’Università e della Ricerca” (MiUR, Grant number RBPR05NWWC_004).