

1 Introduction
A milestone in the chemistry of low-coordinated phosphorus compounds was set with the first successful preparation of λ3-2,4,6-triphenylphosphinine (1) by Märkl in 1966 and the parent phosphinine C5H5P (2) by Ashe, III in 1971 (Fig. 1) [1,2]. The stabilization of reactive PC double bonds by incorporation into aromatic systems opened up the access to formally sp2-hybridized phosphorus systems with significantly different electronic and steric properties compared to classical ligands based on trivalent phosphorus [3]. Long regarded as “chemical curiosities”, state-of-the-art synthetic methodologies developed mainly by the groups of Mathey and Le Floch allow nowadays specific derivatization and functionalization, including the introduction of additional donor-functionalities (3) [4,5]. This is an important aspect for potential applications, because it can lead to polydentate systems with interesting coordination and functional properties, not accessible by monodentate phosphinines [6]. In the last few years, we have focused on the design of functionalized and chiral [7] 2,4,6-triaryl-substituted phosphinines, which turned out to be accessible via the original pyrylium salt route. These derivatives often show a significant kinetic stability and are considerably inert towards water, oxygen and many acids and bases, which often facilitates their preparation and functionalization.
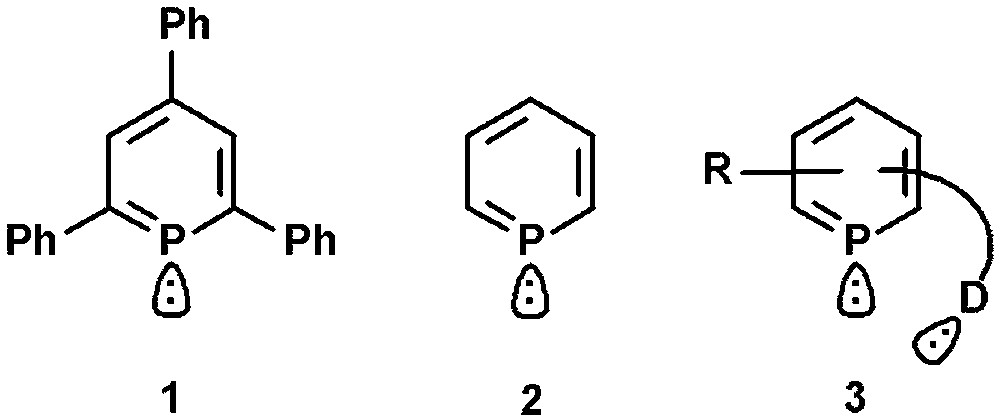
First reported phosphinines and donor functionalized system.
2 Phosphinines – electronic properties
Phosphinines are planar, aromatic systems. Recent calculations employing bond separation and homodesmic reaction energies suggest their aromaticity to be as high as 88% of benzene [8]. The electronic properties of phosphinines differ significantly from pyridines, as shown by photoelectron and electron transmission spectroscopy as well as by theoretical calculations [9]. The HOMO−2 orbital has a large coefficient at the phosphorus atom and represents essentially the lone-pair at the heteroatom. The phosphorus lone-pair occupies a more diffuse, partly delocalized, and less directional orbital than that of pyridine. While the HOMO−1 and HOMO orbitals contribute to π-donation, the LUMO orbital, with a large coefficient at the phosphorus atom, enables the heterocycle to act as a π-acceptor ligand, once coordinated to the metal center via the phosphorus atom (Fig. 2). In pyridines, the LUMO is located higher in energy compared to phosphinines, while the HOMO represents the lone pair located at the nitrogen atom. Consequently, phosphinines are much better π-acceptor ligands, but less good σ-donors compared to pyridines. The phosphorus atom in λ3-phosphinines has a strong 3s-orbital character, which is about 63.8%, versus 29.1% found for the nitrogen atom in pyridines [10]. This reflects the poor hybridization ability of phosphorus, which leads to a very low basicity of the phosphorus center (pKa (C5H6P+) = −16.1 ± 1.0 in aqueous solution), and consequently, phosphinines are relatively inert towards electrophilic attack [11]. Based on Tolman's electronic parameter χ, a comparison of phosphinines with classical phosphorus-based ligands, such as phosphines and phosphites, suggests that phosphinines are electron-withdrawing ligands with electronic properties similar to phosphites as already anticipated from the corresponding MO scheme (Fig. 2).
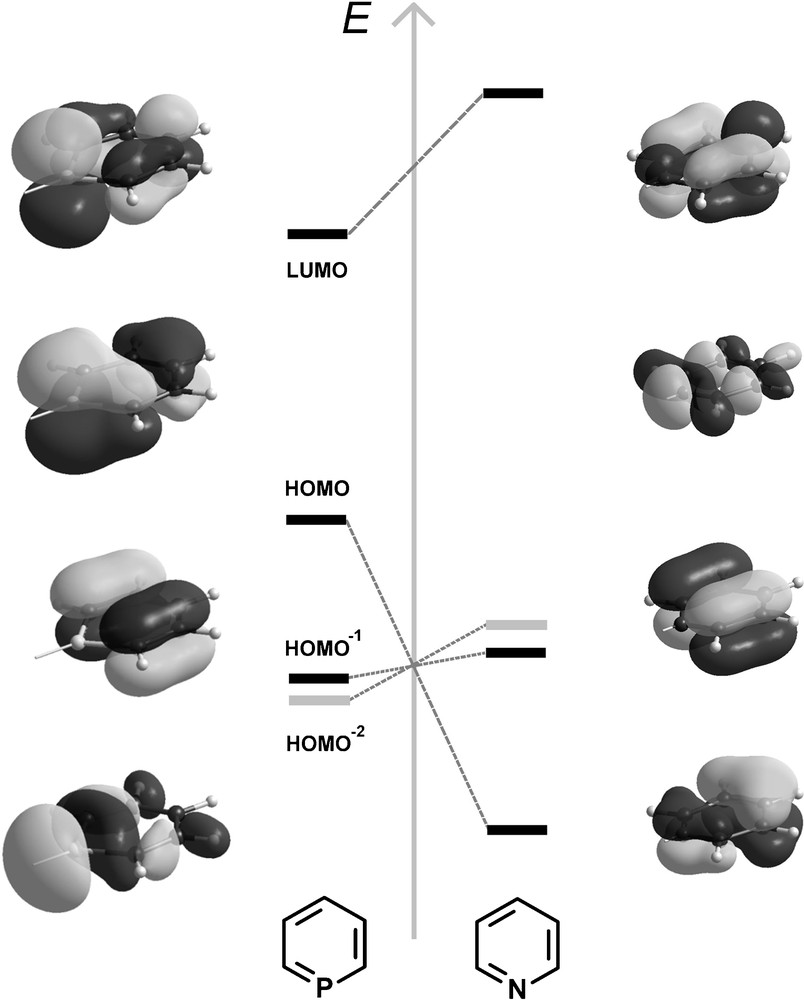
Qualitative MO-diagram of the frontier orbitals of phosphinine and pyridine. Light grey: MO representing the lone-pair.
Due to their particular electronic properties, phosphinines are especially suitable for the stabilization of late transition metal centers in low oxidation states. Moreover, phosphinines can be oxidized more easily than pyridines and form in fact rather stable radical monocations [12]. Phosphinines can also undergo stepwise electron transfer reactions to form the radical anion and the diamagnetic dianion, respectively upon reduction and can thus be regarded as “non-innocent” ligands [12]. Phosphinines show a typical downfield shift of about δ = +200 ppm in the 31P{1H} NMR spectrum, and all peripheral protons show chemical shift values downfield from benzene in the 1H NMR spectrum. These phenomena can be attributed to the presence of a diamagnetic ring current typical for aromatic systems. Nucleus-independent chemical shift values (NICS) of phosphinines lead to the same conclusion [10].
3 Phosphinines – structural characteristics and steric properties
Up to now, several λ3-phosphinines have been characterized crystallographically [13]. The phosphorus heterocycle is essentially planar and can best be described as a distorted hexagon due to the larger size of the phosphorus atom in comparison to a carbon atom (see also section S-functionalized phosphinines). The P–Cα bond lengths lie in-between a P–C single bond (triphenylphosphine: 1.83 Å [14]) and a P–C double bond (diphenylmethylenephosphaalkene: 1.66 Å [15]). There is no bond alternation between the C–C (∼1.39 Å) and the P–C (∼1.75 Å) bond lengths, which indicates delocalization of the π-system and the presence of an aromatic system. Interestingly, the internal C–P–C angle of approximately 100° is somewhat smaller compared to the C–N–C angle in pyridine (117°) [16]. This phenomenon is attributed mainly to the poor ability of phosphorus orbitals to hybridize, rather than to the lengthening of the heteroatom-carbon bond.
From the planarity of the P-heterocycle, it immediately becomes obvious that the steric demand of such ligand systems is significantly different compared to trivalent phosphines. Tolman's steric parameter θ (theta, cone angle), which provides a measure of the volume around a metal center occupied by a ligand, is not appropriate to describe the steric properties of phosphinines [17]. As a matter of fact, the occupancy angles α and ß along the two orthogonal planes x and y, show that the steric demand in the y plane is relatively small, while very large in the x plane. This leads to a flattened cone rather than a symmetric cone and the steric demand of phosphinines can thus be better described by the occupancy angles α and ß rather than by the cone angle θ (Fig. 3) [18].
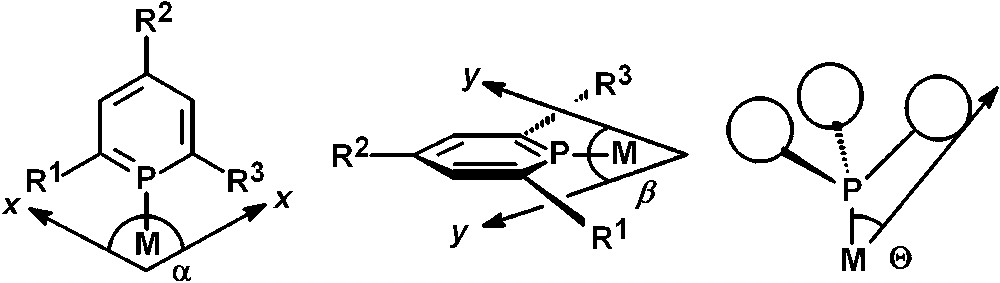
Occupancy angles α and β of a phosphinine in comparison to Tolman's cone-angle θ.
In this respect, phosphinines are much more similar to their pyridine-analogues and to N-heterocyclic carbenes, which have found nowadays many applications as ligands in homogeneous catalysis. On the basis of representative conformations and calculated structures of a 2,4,6-triphenylphosphinine-metal complex, values of α = 216°and β = 54° have been determined, which corresponds to an average parameter θ = 135° in comparison to θ = 145° for PPh3.
4 Donor-functionalized phosphinines
In contrast to arenes and pyridines, the direct functionalization of phosphinines is challenging because reagents for electrophilic substitution and metalation reactions usually attack at the phosphorus atom. The incorporation of additional donor-functionalities within the heterocyclic framework is, however, an important aspect in the design of phosphinines, because it can lead to polydentate systems with interesting properties. The chelate effect is an essential feature especially for the stabilization of metal complexes in higher oxidation states, preventing otherwise fast ligand dissociation from the metal center. The combination of “soft” and “hard” donor-sites within the same molecule (hybrid ligands) leads to electronically different binding sites in the corresponding metal complexes, which can influence significantly their reactivities and selectivities in catalytic reactions. Depending on the position of the additional donor group, the preparation of polynuclear species can be envisaged. Hemilability, often associated with hybrid ligands, is an important concept in the design of homogeneous catalysts, functional molecular materials, or activation of small molecules [19]. The generation of secondary interactions through the additional donor group is a key aspect in supramolecular chemistry. Additional reactive groups within the phosphorus heterocycle can also be used for further functionalizations. The incorporation of electron-rich or electron-poor aromatic systems, such as thiophene and pyridine groups (push-pull effects) is essential for the design of phosphorus-containing molecular materials for optoelectronic applications [20].
Since the first report on the synthesis of 2,4,6-triphenylphosphinine by O+/P exchange in pyrylium-salts and the synthesis of C5H5P by Sn/P exchange, ring expansion reactions in phospholes, metal-mediated functionalizations of 2-halogeno-derivatives and the use of (di)azaphosphinines as precursors are important synthetic procedures for the preparation of donor-functionalized phosphinines. We have recently shown that such systems are also accessible by the original pyrylium-salt route, expanding their scope significantly. In fact, starting from functionalized benzaldehydes and acetophenones, the stepwise assembly of the phosphorus containing heterocycle allows the incorporation of additional donor-functionalities into specific positions of the 2,4,6-triarylphosphinine core. A feature, which has largely been neglected for the design of functionalized phosphinines so far. The synthetic strategies mentioned here will be outlined in the following sections.
5 S-substituted phosphinines
One of the first thienyl-functionalized phosphinines (4) was reported by Le Floch et al. and prepared in good yields via Pd-catalyzed Stille-cross-coupling reaction of bromophosphinines with thienyl-trimethyltin derivatives (Scheme 1a) [21].
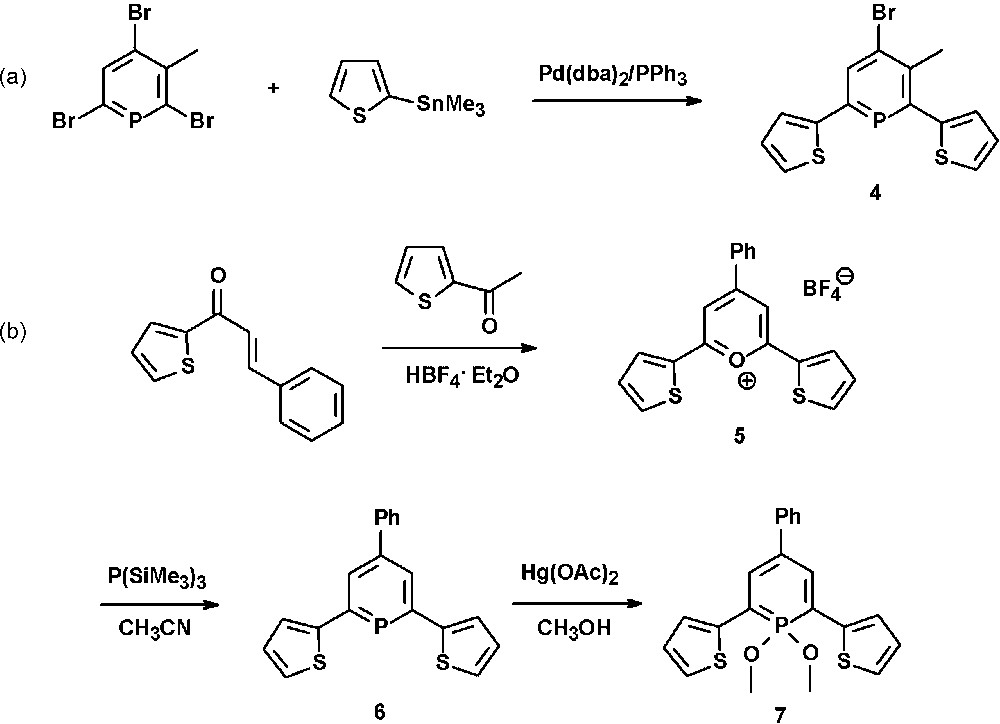
Synthetic procedures for the preparation of thienyl-functionalized λ3- and λ5-phosphinines.
We could show that similar systems can easily be prepared from the pyrylium salt 5 and P(SiMe3)3 (Scheme 1b) [22]. The strongly fluorescent pyrylium salt 5 is synthesized from benzal-α-acetothienone and 2-acetylthiophene in the presence of HBF4·Et2O. Fig. 4 shows the molecular structures of both the thienyl-substituted pyrylium salt 5 and phosphinine 6. The direct comparison nicely illustrates the effect of the larger phosphorus atom on the heterocyclic ring size in comparison to oxygen.
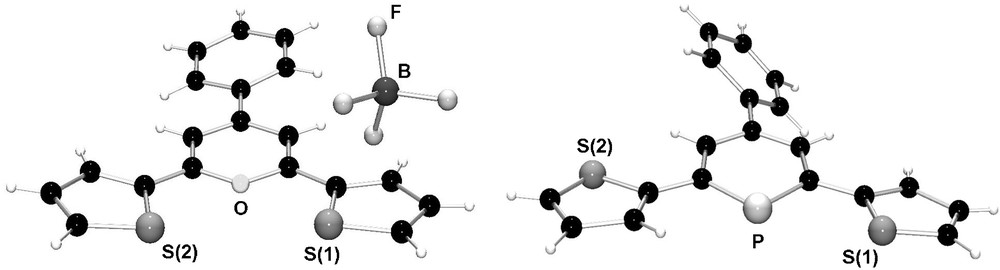
Molecular structures of thienyl-functionalized pyrylium salt 5 and phosphinine 6 in the crystal.
Due to the increasing interest in phosphorus-containing π-conjugated molecular materials, we started to explore the photophysical properties of λ3- and λ5-phosphinines. The λ3-phosphinine 6 was therefore converted quantitatively into the corresponding λ5-phosphinine 7 by reaction with Hg(OAc)2 and methanol (Scheme 1) [21]. While 6 shows only little fluorescence in solution, the fluorescence of 7 is significant (quantum yield 20%) and only slightly red-shifted (by ∼50 nm) compared to the maximum of the lowest energy (red-edge) absorption band, from which it seems to arise (Fig. 5). From theoretical calculations, we identified the lowest energy (red-edge) absorption bands as π → π* transition (mainly HOMO → LUMO), which are strongly allowed since they are accompanied by an apparent change of parity. Compound 7 also shows readily observable phosphorescence in solution (τ in the range of milliseconds), which was determined by time-gated detection at low temperature [22].
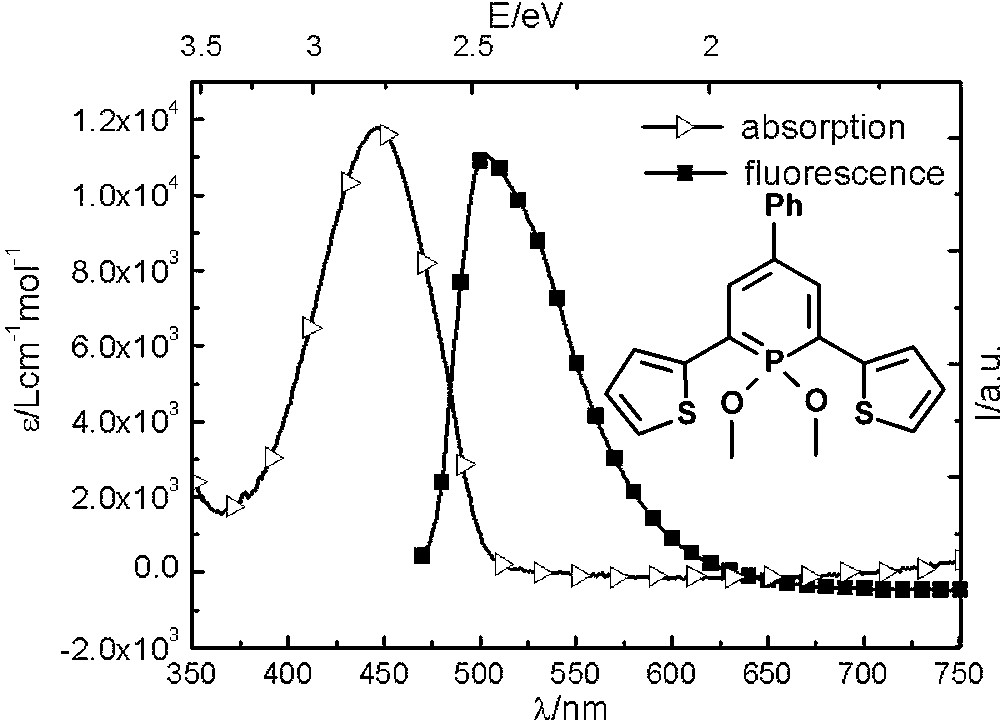
Absorption and Fluorescence spectrum of thienyl-functionalized λ5-phosphinine 7.
In view of potential applications of phosphorus-modified oligothiophenes in optoelectronic materials, Huy et al. recently reported on the synthesis of dithienophosphinines, such as 8, via ring-expansion reactions starting from dithienophospholes (Scheme 2) [23].
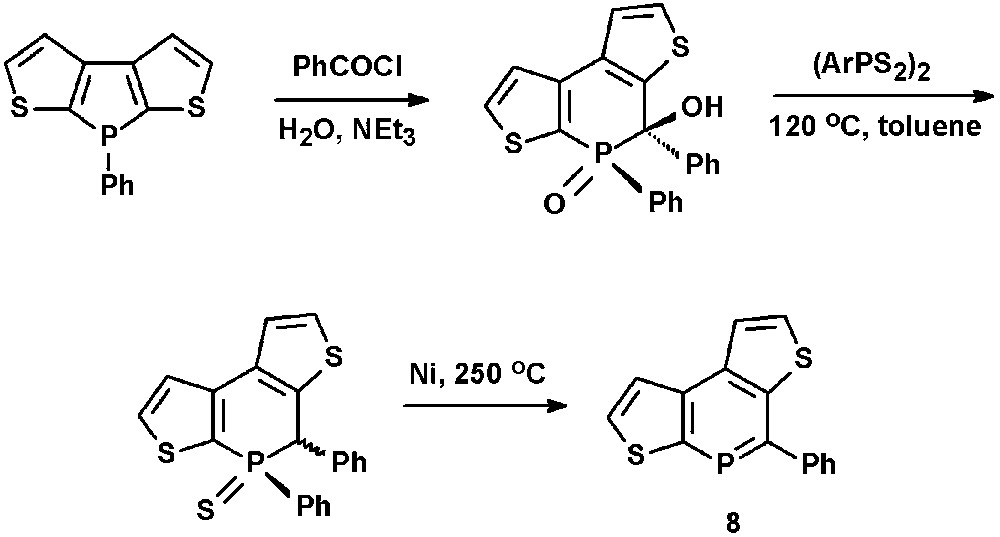
Synthetic procedure for the preparation of dithienophosphinines.
The mono-thienyl-functionalized phosphinines 9 and 10 can either be prepared via ring-expansion methods starting from a phosphole and 2-thiophenecarbonyl chloride [24] (see section N-functionalized phosphinines) or, alternatively, from the corresponding pyrylium salts and P(SiMe3)3 (Scheme 3) [25].
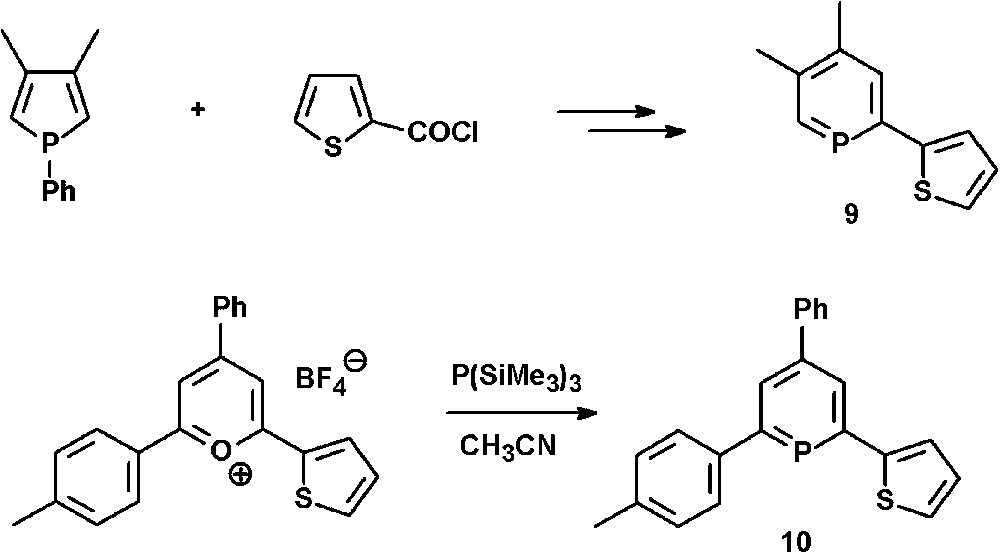
Synthetic procedures for the preparation of mono-thienyl-functionalized phosphinines.
Methylthio-functionalized phosphinines were recently prepared by us via the pyrylium-salt route, starting from para-methylthio benzaldehyde [26]. Interestingly, compound 11 is the first 2,4,6-triarylphosphinine which was characterized crystallographically and which resembles the first phosphinine-derivative reported by Märkl. Fig. 6 illustrates the molecular structure of 11 in the crystal.
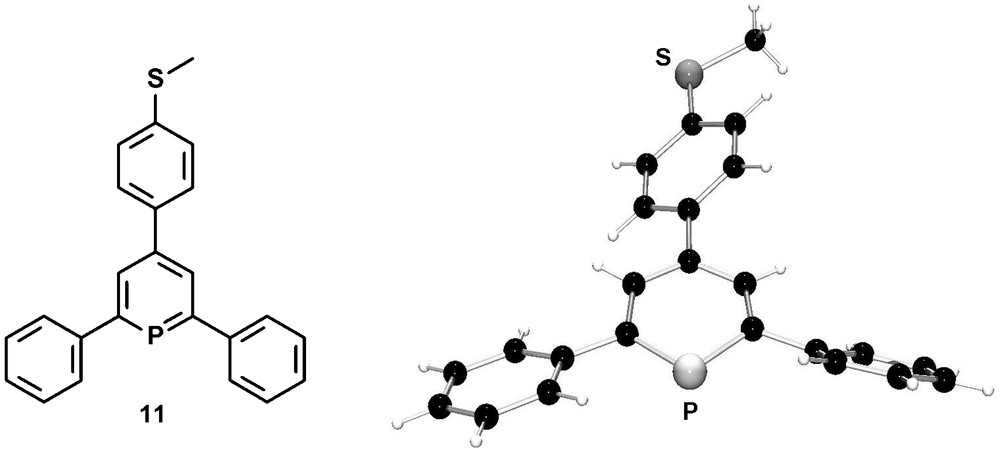
Molecular structure of the methylthio-functionalized phosphinine 11 in the crystal.
Thiol-functionalized phosphinines (12) were reported by Rosa et al. and can be prepared by reaction of 2,3-dehydrophosphinine-zirconocene complexes with triphenylphosphine sulfide and subsequent hydrolysis [27]. The intermediate η2-phosphabenzyne complexes are formed by oxidative addition of 2-bromophosphinines [27,28] with in situ generated Cp2Zr, Br/CH3 exchange and subsequent thermally promoted β-abstraction of methane in the presence of PMe3 (Scheme 4) [29].
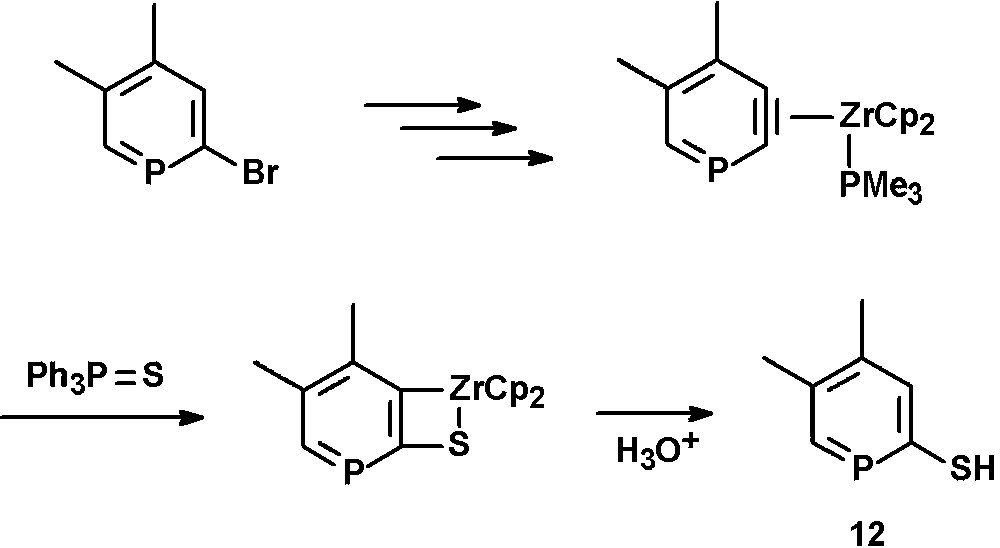
Synthetic procedure for the preparation of 2-thiol-functionalized phosphinines.
6 O-functionalized phosphinines
The furyl-functionalized phosphinine 13 is accessible in good yields by Pd-catalyzed Stille cross-coupling reactions of 2,6-dibromophosphinines with organotin derivatives containing furyl-residues (Scheme 5) [21]. Apparently, the 4-position cannot be converted under these reaction conditions. The mono-functionalized phosphinine 14 is available by ring-expansion reaction starting from a phosphole and 2-furoyl chloride (see section N-functionalized phosphinines) [22].
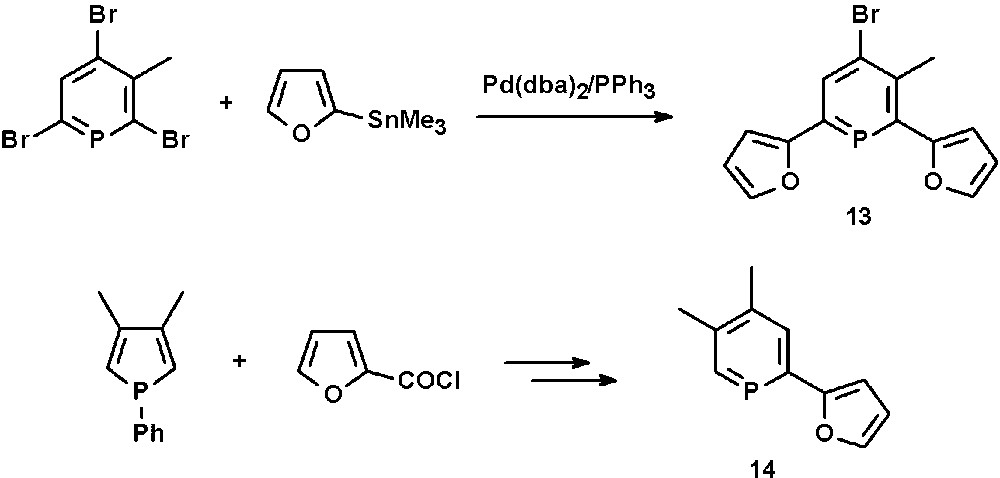
Synthesis of 2,6-difuryl- and 2-furyl-functionalized phosphinines.
The chiral (2-hydroxybicyclo[2.2.1]hept-2-yl)phosphinine 15 is accessible by reacting phosphinine-zirconium(IV) complexes as organometallic reagents with the carbonyl-functionality of (+)-camphor and subsequent hydrolysis (Scheme 6) [27].
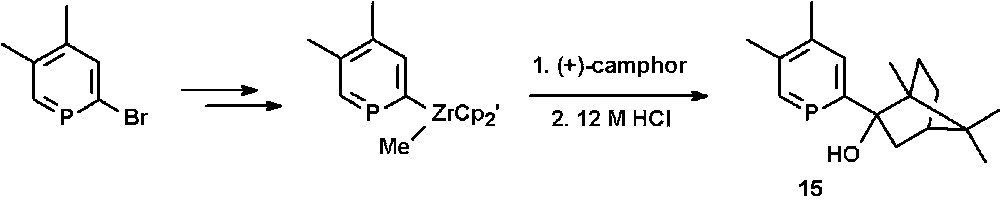
Preparation of the chiral hydroxy-functionalized phosphinine 15.
Reaction of the η2-phosphabenzyne zirconium complex with acetone yields the corresponding metallacycle, which can be converted to the hydroxy-functionalized phosphinine 16 by hydrolysis (Scheme 7) [27,29].
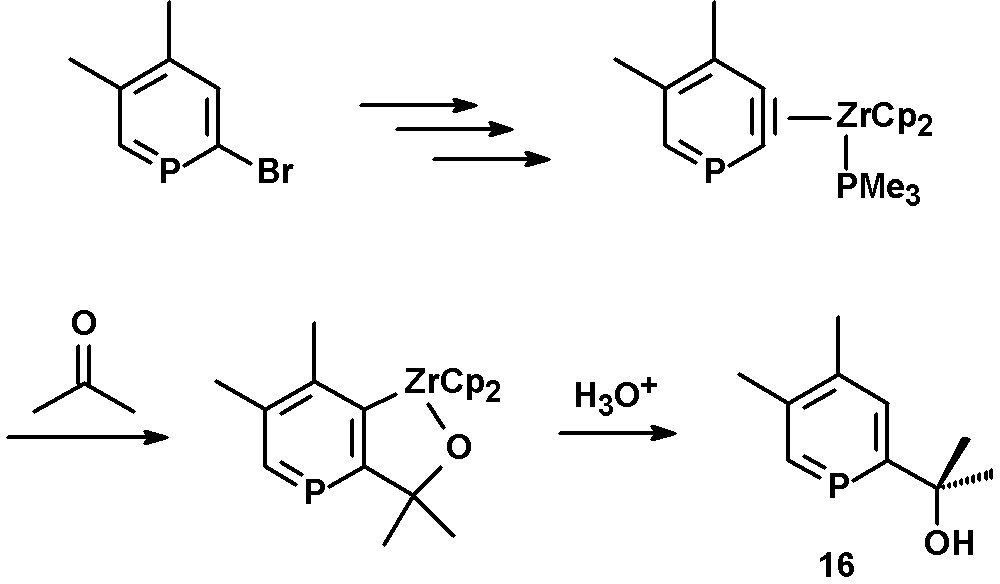
Preparation of hydroxyl-functionalized phosphinines.
The synthetic access to the hydroxyl-functionalized phosphinine 17 via the pyrylium-salt route was recently reported by us [30]. This and related compounds are prepared in a straightforward manner from the corresponding methoxy-derivatives by reaction with BBr3 in CH2Cl2 and subsequent aqueous workup (Scheme 8). The products can generally be obtained in very high yields despite the drastic reaction conditions.
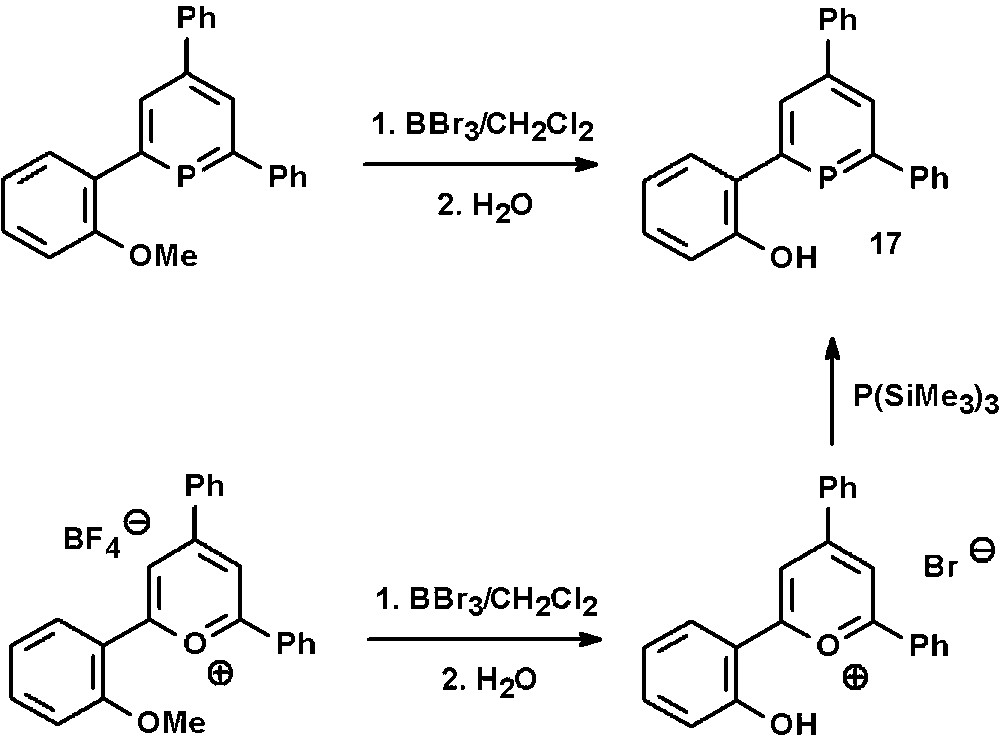
Preparation of hydroxyl-functionalized phosphinines via pyrylium salts.
Alternatively, the cleavage of the –OMe group can already be performed on the level of the pyrylium salt, leading to hydroxyl-functionalized pyrylium salts, which are converted into the corresponding phosphinines by reaction with P(SiMe3)3. Fig. 7 shows the molecular structure of the hydroxyfunctionalized triaryl-pyrylium bromide in the crystal.
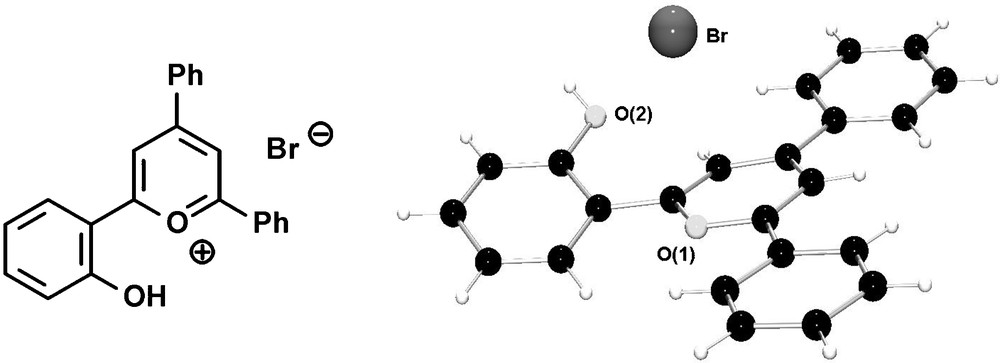
Molecular structure of hydroxyl-functionalized pyrylium bromide in the crystal.
Due to the presence of a reactive group within the phosphinine-heterocycle, hydroxy-functionalized phosphinines can easily be converted into the corresponding chrial bidentate phosphinine-phosphites (Section 7).
7 P-functionalized phosphinines
A 2,3-bis(diphenylphosphino)-6-phenyl-phosphinine has first been reported by Holah et al. starting from 1,2,5-triphenylphosphole and bis(diphenylphosphino)acetylene [31]. Alternatively, 2-phosphinophosphinines can be prepared by Pd-catalyzed coupling reactions of 2-bromophosphinines with PPh2SiMe3 [21]. An interesting method is a multistep synthesis reported by Le Floch, Mathey, and coworkers starting from 2-bromophosphinines and PBr3, as a wide variety of bidentate ligands can be prepared in this way. The intermediate λ5-phosphinine is reduced by P(CH2CH2CN)3 to yield the PBr2-substituted phosphinine 18. Subsequent reaction with organometallic reagents gives the corresponding 2-phosphinophosphinines of the type 19, including phosphole-phosphinine derivatives (Scheme 9) [32].
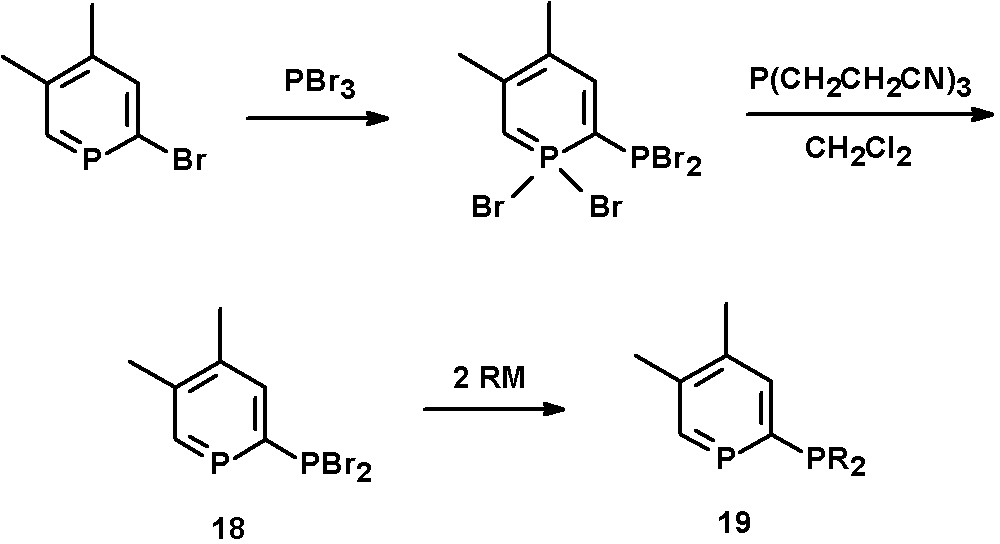
Synthetic procedure for the preparation of 2-phosphinophosphinines.
In contrast to the analogous 2-phosphinopyridines, 2-phosphinophosphinines allow the stabilization of various electron-rich dinuclear complexes due to the π-acceptor properties of the phosphinine subunit. Fig. 8 shows examples of Mo(I), Ni(0), and Cu(I) complexes containing a 2-phosphinophosphinine ligand [33]. In electrochemical studies, the Ni(0) complex 21 shows a reversible reduction to the Ni(0)-Ni(−1) anion radical, which can be attributed to the specific properties of phosphinines. The molecular structure of the Cu(I) complex 22 reveals an unusual η4 face-bridging mode of the iodine ligand [34a].
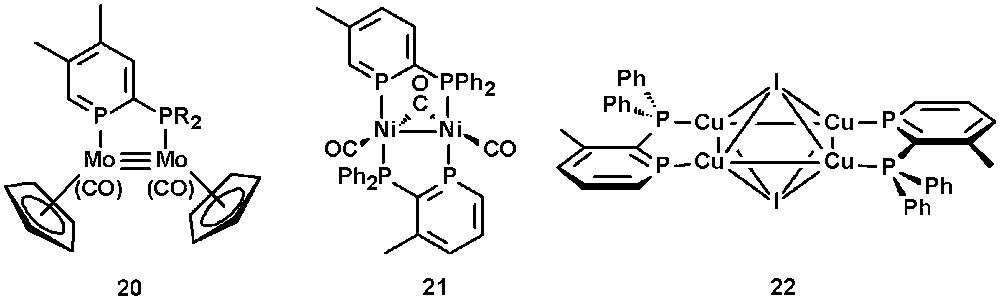
Selected examples of 2-phosphinophosphinine-metal complexes.
The synthesis of bidentate chiral phosphinine-phosphines were reported by Breit [34]. Reaction of the chiral R,R-diol with one equivalent ortho-diphenylphosphinobenzoic acid gave the corresponding monoester, which was converted into a pyrone by reaction with α-pyrone carboxylic acid chloride. Subsequent reaction with a phosphaalkyne under release of CO2 afforded the target compound 23 (Scheme 10).
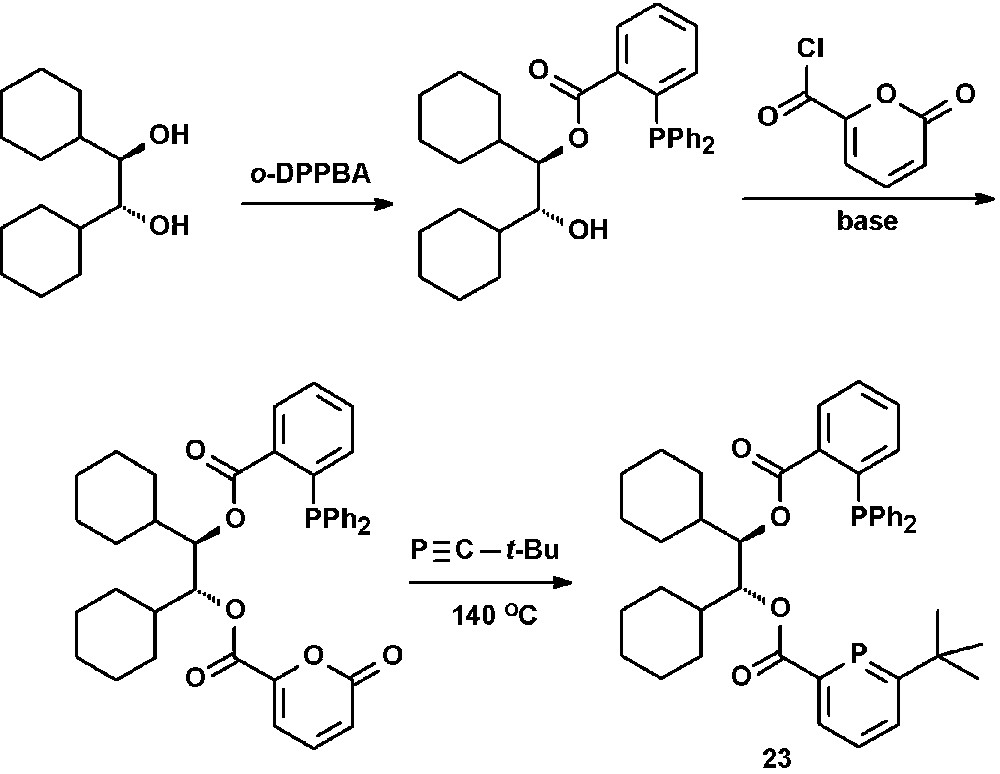
Preparation of chiral bidentate phosphino-phosphinines.
Although 23 was applied as ligand in the Rh-catalyzed hydroformylation of styrene, no enantiomeric access of the chiral 2-phenyl propionaldehyde was observed.
Very intriguing P-functionalized phosphinines are the phosphorus analogues of 2,2′-bipyridine, as oligopyridines have attracted considerable interest as mediators for solar energy conversion. The replacement of nitrogen by phosphorus in similar structures can lead to extraordinary properties which might differ significantly due to the distinct electronic differences that exist between phosphorus and nitrogen. Especially the π-acceptor ability of phosphinines with respect to pyridines allows the exploration of a coordination chemistry, which is directed to the stabilization of electron-rich metal centers.
The synthetic strategy reported by Le Floch, Mathey, and coworkers involves a preliminary masking of the PC double bond with sulfur and a 1,3-diene, followed by a NiCl2-catalyzed coupling reaction of the di-lithio derivative. Subsequent reductive thermolysis affords 4,4′,5,5′-tetramethyl-2,2′-biphosphinine 24 (tmbp) (Scheme 11) [35].
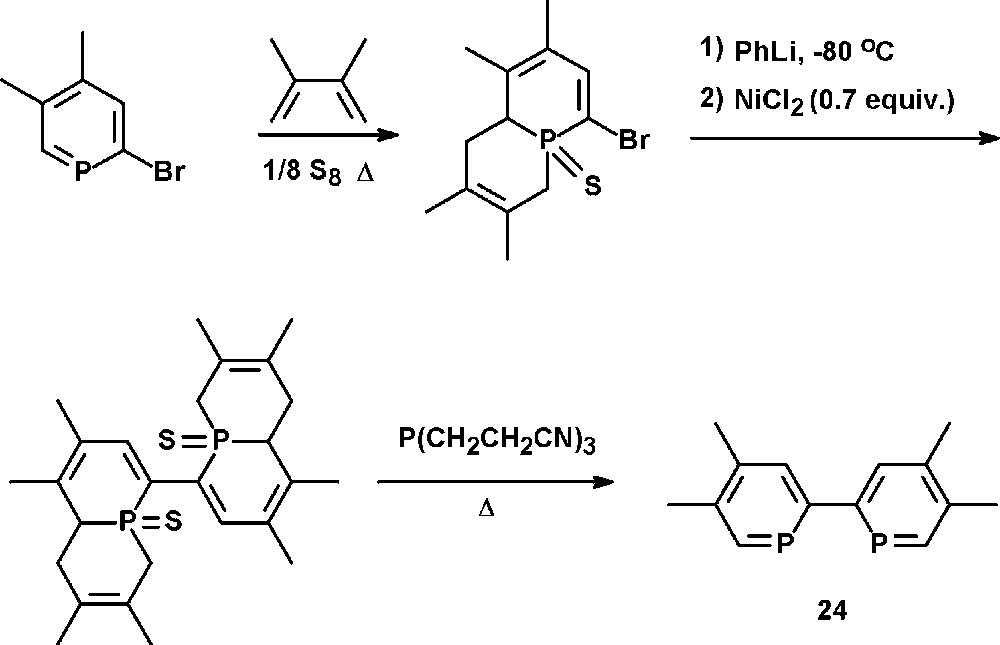
Preparation of 4,4′,5,5′-tetramethyl-2,2′-biphosphinine 24 (tmbp).
Electrochemical studies [36] on tmbp have been performed and especially the coordination chemistry of tmbp has been investigated. Selected examples of metal complexes are depicted in Fig. 9. Among those are phosphorus analogs of [Ru(bipy)n]2+, such as 25 [37] and classical M(CO)4 complexes of the type 26 (M = Cr, M, W) [35]. Interestingly, recent studies have shown that tmbp can also efficiently stabilize highly reduced species, as observed in the Ru(−2) complex 27 and the Co(−1) complex 28 [38]. The same was investigated for Zr(−2), Ti(−2), Hf(−2), Mn(−1), Fe(−2), Rh(−1) and Ni(−1) species [39]. These properties can again be attributed to the strong π-acceptor properties of phosphinines.
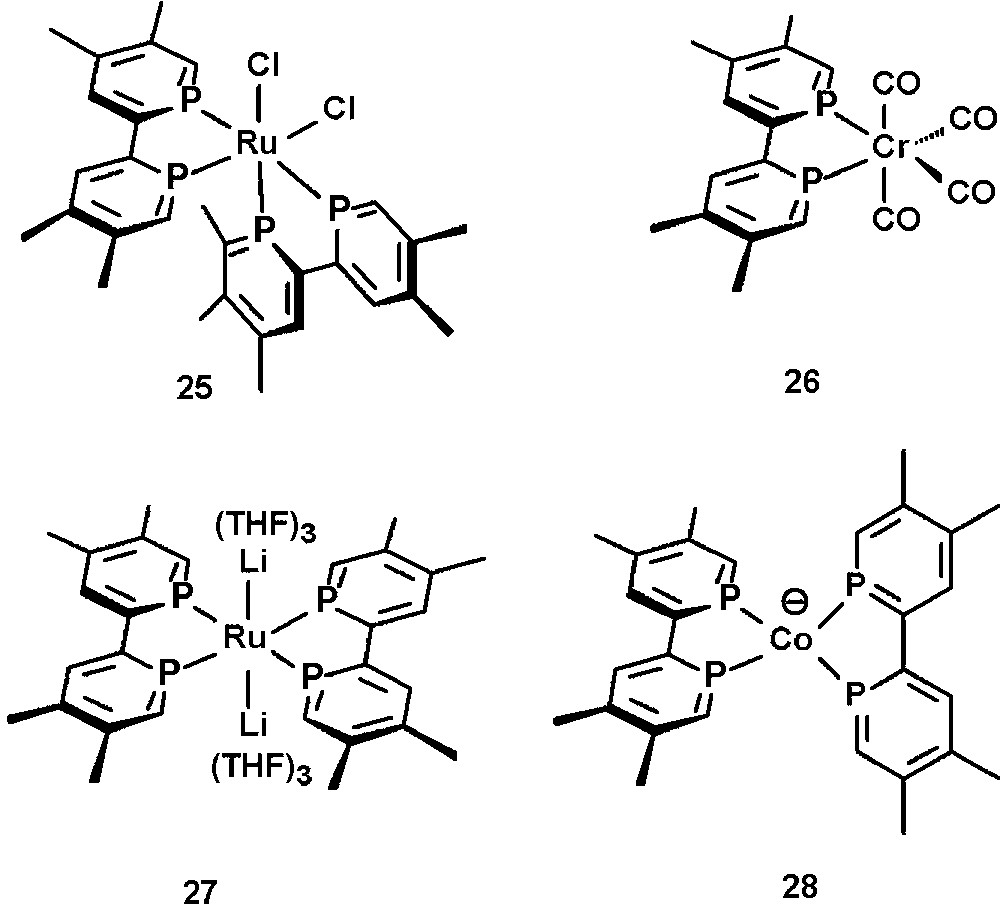
Selected examples of tmbp-transition metal complexes.
The phosphorus analogue of 4,4′-bipyridine, 4,4′-bis-2,6-diphenylphosphinine 29, was reported by Märkl et al. and prepared by thermolysis of 1,1′-dibenzyl-1,1′-diphospha-4,4′-pyrylene at T = 350 °C (Scheme 12) [40]. The direct synthesis starting from 4,4′-bis-2,6,2′6′-tetraphenylpyrylium tetrafluoroborate and P(CH2OH)3 was unsuccessful.
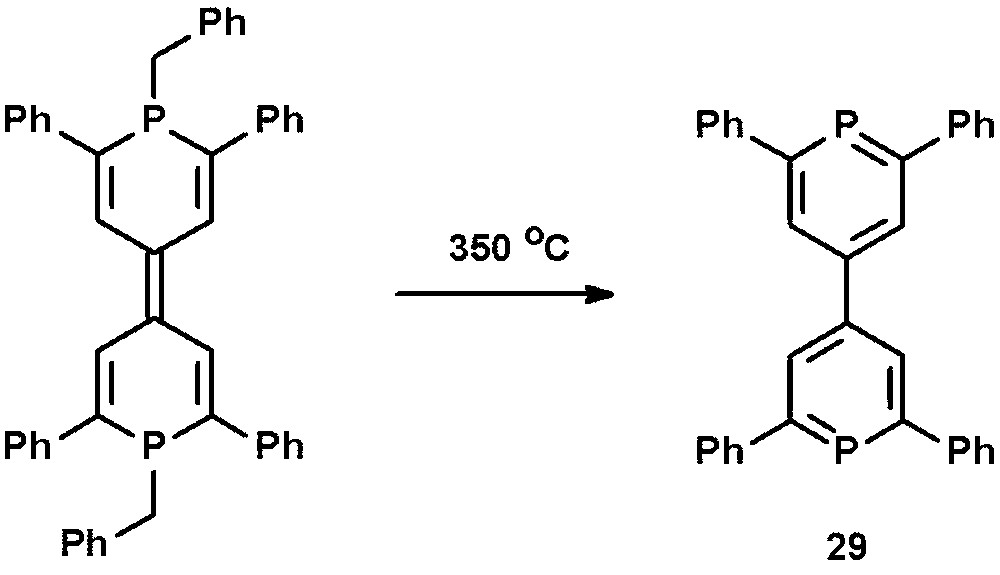
Preparation of 4,4′-bis-2,6-diphenylphosphinine 29 by thermolysis.
The development and preparation of silacalix-[4]-phosphinines, such as 30, has been reported by Le Floch, Mathey, and coworkers [41]. These compounds are the first examples of phosphinine-based macrocycles and thus related to pyrrol-based porphyrines. The crucial step in their synthesis is the reaction of the phosphinine-bridged diyne with the precursor under dilute conditions in order to close the cavity and to prevent the formation of oligomers (Scheme 13).
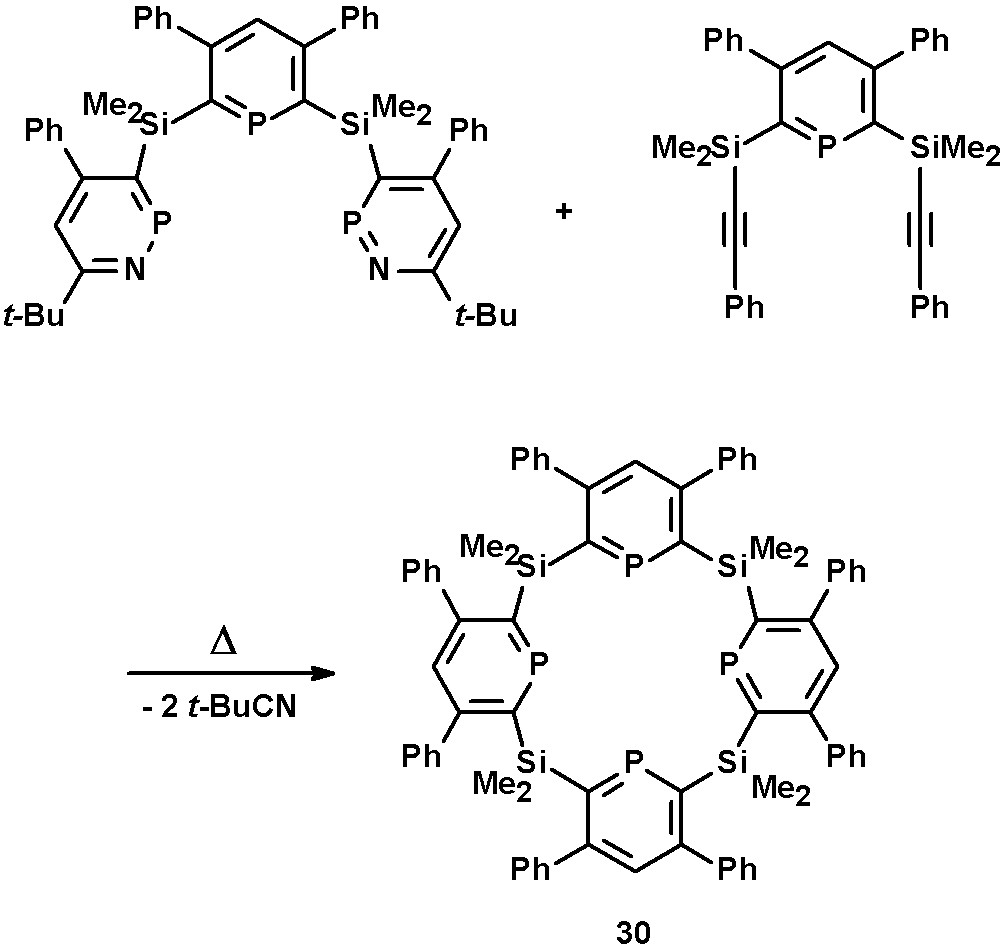
Preparation of silacalix-[4]-phosphinine macrocycle 30.
The coordination chemistry of 30 towards square-planar cationic Rh(I) and Ir(I), as well as towards Au(I) has been investigated. Electrochemical studies on the Rh(I) complex revealed the formation of reduced Rh(0) and Rh(−1) species, stabilized by the π-accepting phosphinine-units [6b]. The paramagnetic Au(0) complex formed by single-electron reduction of the silacalix[4]phosphinine-Au(I) complex could be characterized by EPR spectroscopy in solution [42].
We have recently designed diphosphinines, which might be suitable candidates for the synthesis of exclusively trans-coordinating ligands towards a transition metal center [43]. Due to the remarkable structural (as well as electronic) features of phosphinines, the orientation of the lone-pair electrons, responsible for σ-coordination, is significantly different compared to the situation in classical phosphines; while a pyramidal geometry is present in the latter ones, the formally sp2-hybridized phosphorus atom in phosphinines shows an angle of approximately 120° with the α carbon atoms. This structural element of phosphinines in comparison to phosphines should in principal allow a simpler design of trans chelating ligands. In combination with an appropriate aryl-based ligand backbone that locates the phosphorous atoms at the appropriate distance, the two lone-pairs are expected to point in a linear way to one another, thus resulting in the ideal case in a true trans-coordination with a P–M–P angle of 180° in which both P–P distance and lone-pair orientation cope for this coordination mode. We could synthesize the diphosphinine 31 based on a terphenyl-backbone via the classical pyrylium salt route and the corresponding Rh(I) complex was characterized cyrstallographically (Fig. 10).
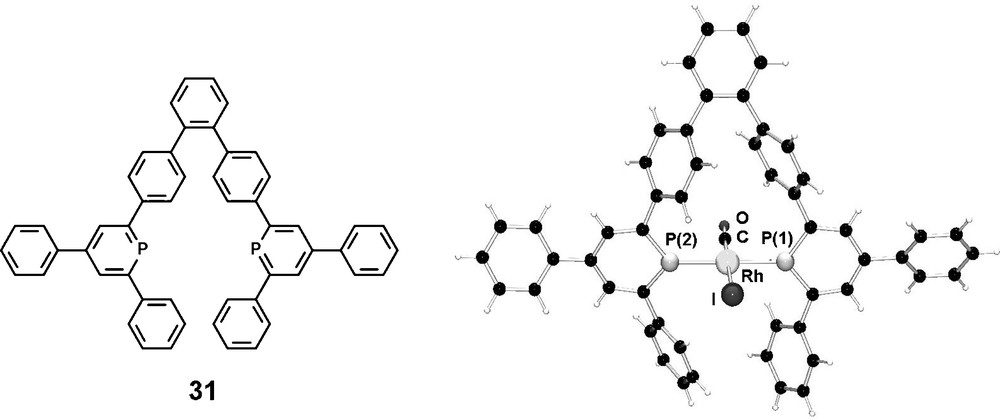
Molecular structure of the trans-(31)Rh(CO)I in the crystal.
The chiral BINOL- or TADDOL-substituted phosphinine-phosphites 32–34 were obtained quantitatively from the corresponding hydroxy-substituted phosphinines (vide supra) by reaction with phosphorchloridites in the presence of an amine (Fig. 11) [30].
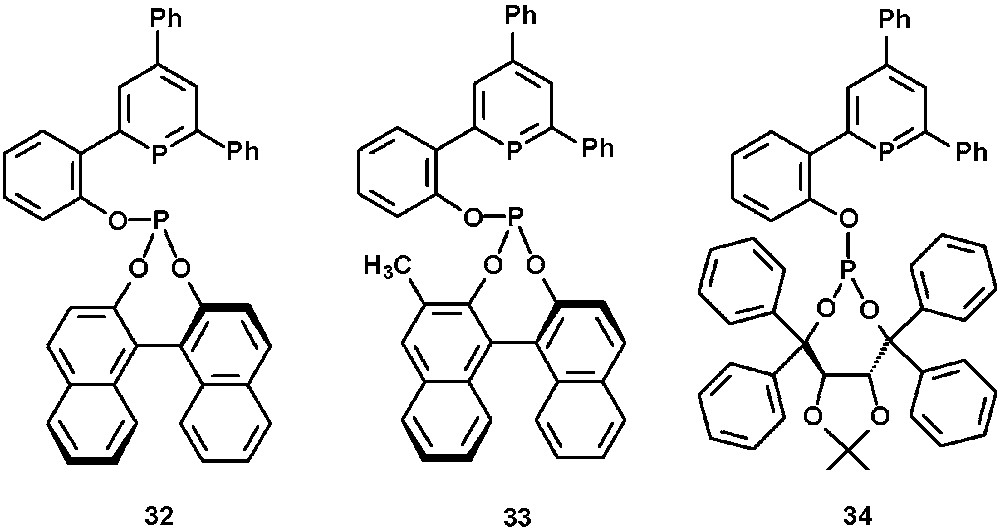
Chiral phosphinine-phosphites 32–34.
We studied the coordination behavior of this ligand class towards Rh(I) by reacting 32 with one equivalent of Rh(cod)2BF4. The loss of cyclooctadiene can be observed by NMR spectroscopy and the corresponding chiral rhodium complexes (P1P2)Rh(cod)BF4 (P1: phosphinine-P; P2: phosphite-P) were formed quantitatively. Fig. 12 shows the 31P{1H} NMR spectrum of [(32)Rh(cod)]BF4 and reveals that the phosphinine-phosphite ligand coordinates to the metal center in a bidentate fashion: a doublet of doublets (JRh–P1 = 172.9 Hz, JP1–P2 = 68.8 Hz) at δ = 160.5 ppm is observed in the phosphinine region P1 as well as in the phosphite region P2 at δ = 136.5 ppm (JRh–P2 = 243.2 Hz, JP2–P1 = 68.8 Hz).
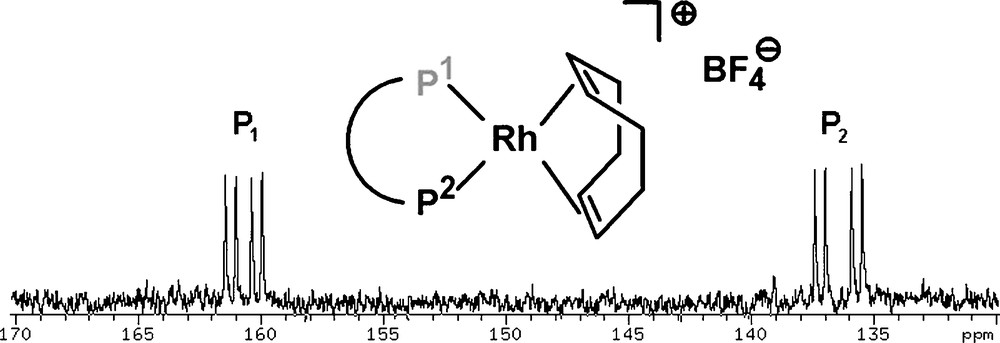
31P NMR spectrum (CD2Cl2) of (32)Rh(cod)BF4. P1: phosphinine-P; P2: phosphite-P.
Cationic Rh(I) complexes based on chiral phophinine-phosphites were further applied in the asymmetric hydrogenation of dimethyl itaconate and methyl 2-(N-acetylamino)cinnamate, showing turnover frequencies of up to 2500 h−1 and enantioselectivities of up 79% at T = 25 °C and a H2 pressure of p = 10 bar (0.1 mol%, cRh = 1.25 mM, CH2Cl2) [30]. As a matter of fact, these are the first reported example of a chiral phosphinines showing asymmetric induction in a transition-metal catalyzed reaction. However, it should be pointed out that the chirality is exclusively located in the phosphite parts, rather than in the phosphinine units. The latter ones solely permit the chelating behavior and contribute to the steric and electronic properties of the bidentate ligand. It should also be mentioned that although hydrogenation of the phosphinine ligand was not observed in the above-mentioned catalytic transformations, it cannot be excluded especially under more drastic hydrogenation conditions.
8 N-functionalized phosphinines
Chiral oxazoline functionalities as hard σ-donors were used by Breit to prepare bidentate phosphinine-based ligands with two electronically rather different coordination sites [34]. The synthesis was achieved starting from L-valinol and α-pyrone-carboxylic acid chloride. The phosphinine-moiety in 35 was formed by [4 + 2] cycloaddition reaction of the oxazoline-pyrone derivative with a phosphaalkyne under subsequent release of CO2 (Scheme 14). Derivatives of these ligands and the corresponding cationic Ir(I) complexes were investigated by Pfaltz and Neumann [44]. Attempts to apply these chiral ligands in the Rh-catalyzed asymmetric hydroformylation of styrene showed good conversions towards the branched 2-phenylpropionaldehyde but no enantiomeric excess was reported.
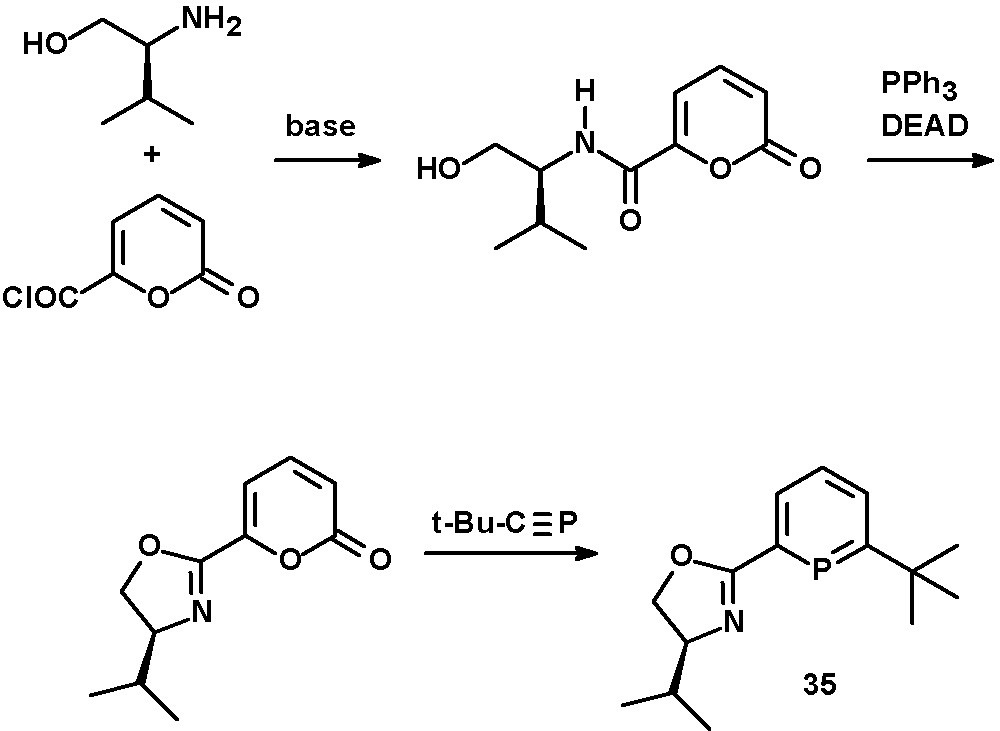
Synthesis of chiral oxazoline-phosphinines.
N-methyl-pyrrol-functionalized phosphinines, such as 36, are accessible via Pd-catalyzed Stille cross-coupling reactions of 2,6-dibromophosphinines with organotin derivatives containing N-methyl-pyrrol-residues (Scheme 15) [21].
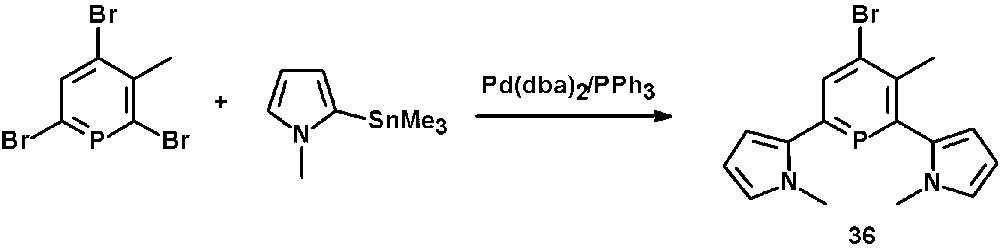
Preparation of pyrrol-functionalized phosphinines via Pd-catalyzed Stille cross-coupling reaction.
A special class of donor-substituted phosphinines are pyridyl-functionalized phosphinines. These compounds represent phosphorus-analogues of well-studied oligopyridines, such as bipyridines and terpyridines. Latter compounds possess a rich coordination chemistry that has often been exploited for the development of molecular devices, homogeneous catalytic systems or modern materials with interesting photophysical properties. The replacement of a pyridine unit by a π-accepting phosphinine entity leads to 2-(2′-pyridyl)phosphinine, a semi equivalent of 2,2′-bipyridine containing a low-coordinated “soft” phosphorus and a “hard” nitrogen heteroatom. Such chelates are intriguing bidentate hybrid ligands, which have first been described by Mathey and coworkers in 1982 with the synthesis of 2-(2′-pyridyl)-4,5-dimethylphosphinine 37 (NIPHOS, Scheme 16) [45].
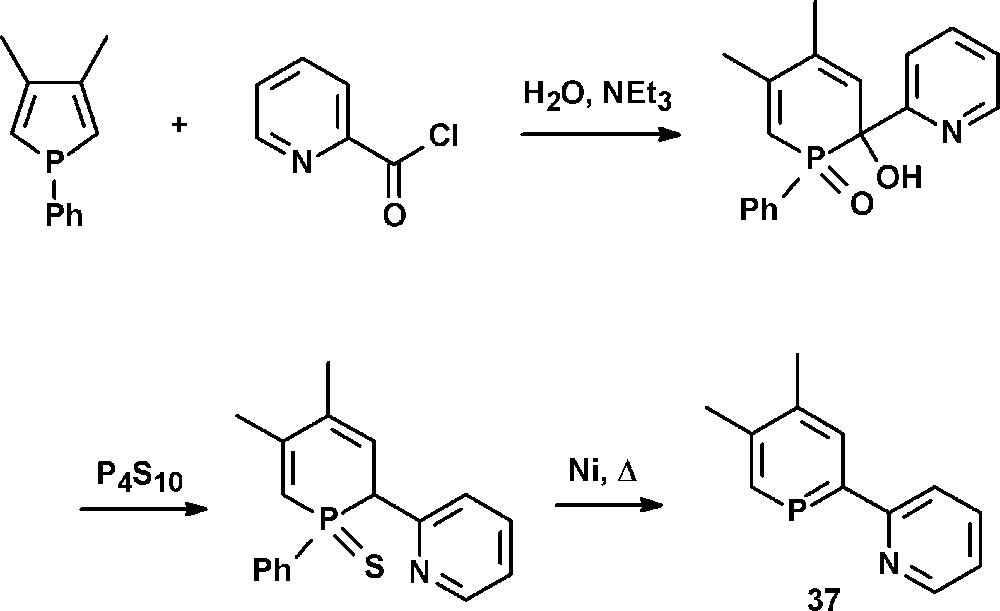
Synthesis of 2-(2′-pyridyl)-4,5-dimethylphosphinine 37 (NIPHOS).
A few examples on the coordination chemistry of NIPHOS have been reported in literature by the groups of Mathey and Venanzi. The P,N ligand forms complexes of the type [M(NIPHOS)(CO)4] (M = Cr, Mo, W) (38) [46], and Rh(I) and Ir(I) dimers of the type [Ir2(cod)2(NIPHOS)2][X]2 and [Rh2(nbd)2(NIPHOS)2][X]2 (X = SbF6, cod = 1,5-cyclooctadiene, nbd = norbornadiene) (39), in which the phosphinine ligand adopts an η2-bonding mode (Fig. 13) [47]. Interestingly, studies have shown that NIPHOS binds stronger to Ir(I) and Rh(I) than 2,2′-bipyridine. The cationic Pd(II) and Pt(II) complexes [MCl(L)(NIPHOS)][MCl3(L)] (L = tertiary phosphine) (40) could not be isolated and are highly sensitive towards nucleophilic attack. They easily add ROH groups to one of the PC double bonds under formation of 41 [48].
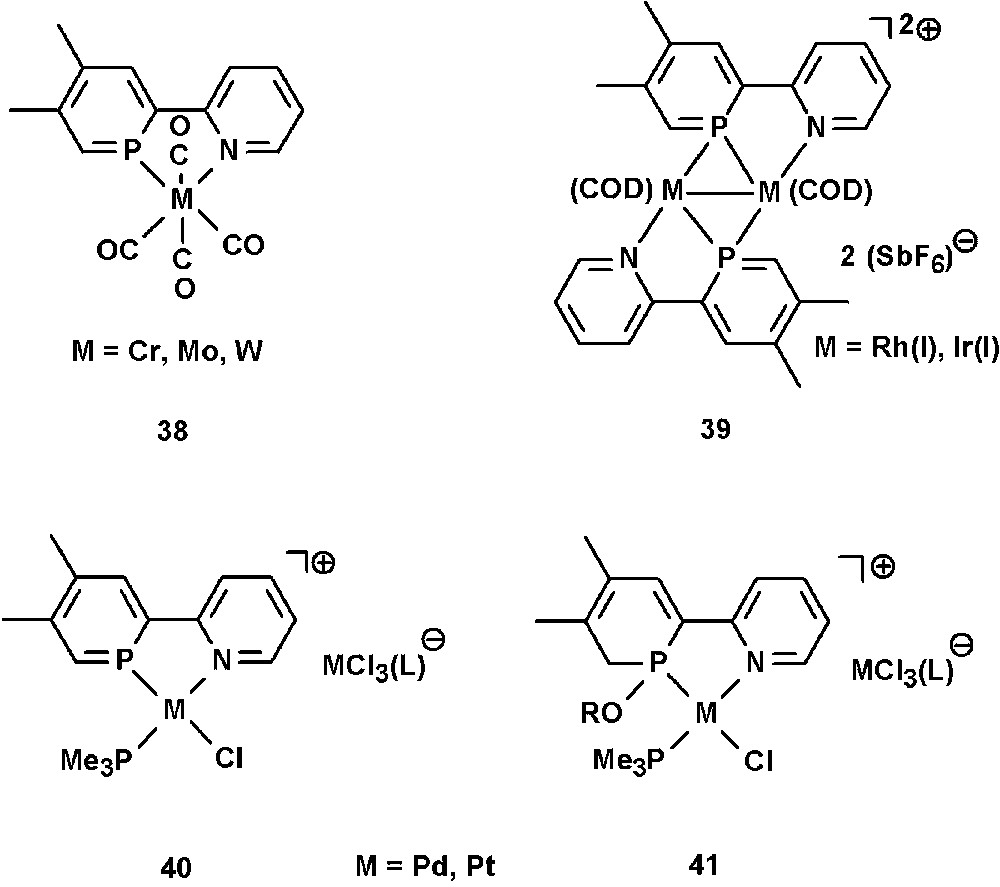
Transition-metal complexes containing the NIPHOS ligand.
We have demonstrated that the classical λ3-phosphinine synthesis via pyrylium salts is also suitable for the incorporation of pyridyl-functionalities within the phosphinine-framework. Thus, the 2,2′-bipyridine derivative 2-(2′-pyridyl)-4,6-diphenyl-phosphinine 42 is readily available from the corresponding pyridyl-functionalized pyrylium salt and P(SiMe3)3 (Scheme 17) [22].
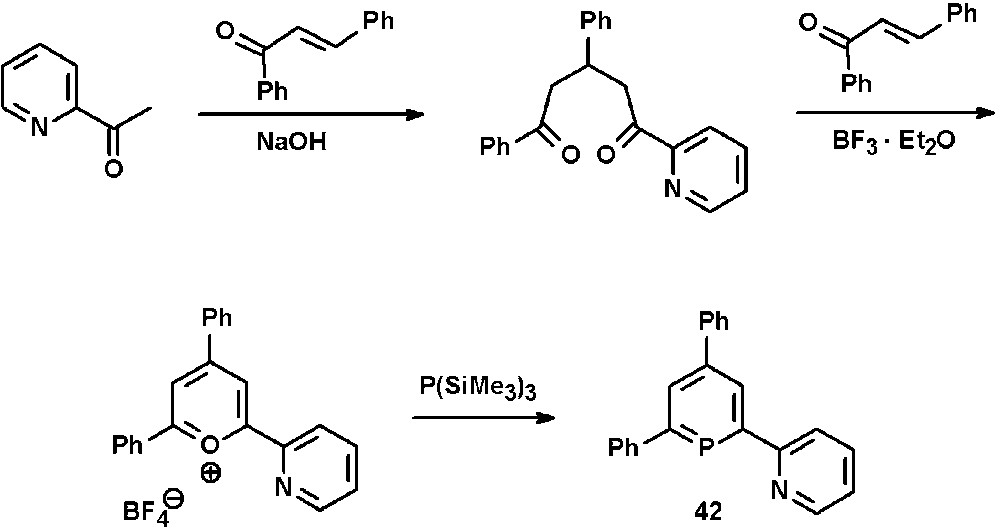
Preparation of 2-(2′-pyridyl)-4,6-diphenyl-phosphinine 42 via the pyrylium salt route.
The electronic properties of the parent compound 2-(2′-pyridyl)phosphinine were evaluated by means of DFT calculations and a selection of relevant frontier orbitals is illustrated in Fig. 14 [49].
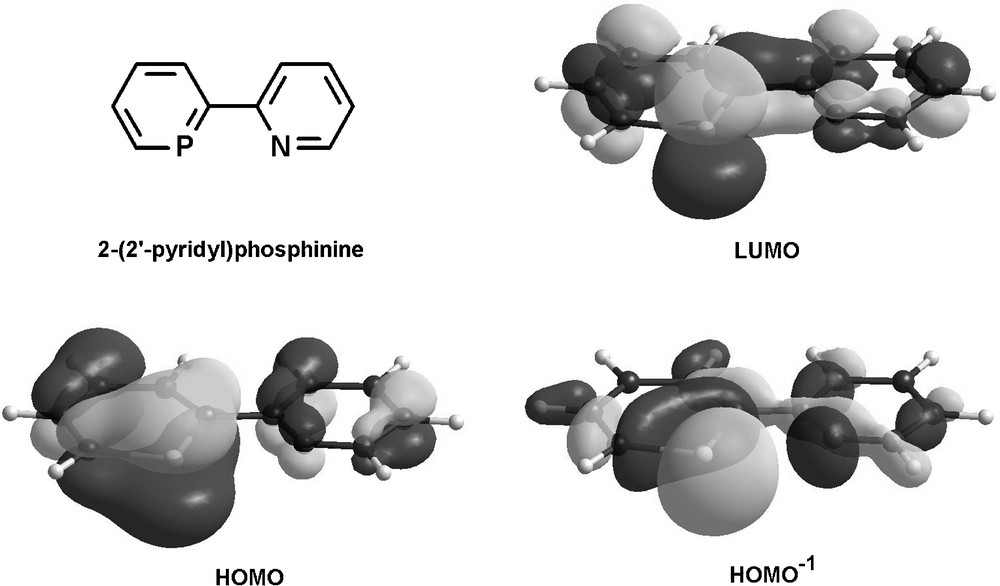
Frontier orbitals of 2-(2′-pyridyl)phosphinine.
As evident from the distribution and size of coefficients, the LUMO enables the phosphinine-moiety to act as a strong π-acceptor upon coordination of the phosphorus atom to a metal center. In contrast, the most important coefficients of the pyridine moiety are localized on the carbocyclic part, demonstrating the weak π-acceptor properties of this subunit. Furthermore, the HOMO can participate in π-donation from the phosphorus atom, while the HOMO−1 represents the lone-pairs at the heteroatoms for participation in σ-bonding with a metal center. The pronounced spherical character and larger coefficient of the phosphorus lone-pair compared to nitrogen reflects its less-directional and more diffuse nature.
We are currently studying the coordination properties of 42 and could prepare the cationic Rh(I) complex 43, which was characterized crystallographically [49], the neutral Pd(II) and Pt(II) complexes 44 and 45, as well as the neutral and cationic Ru(II) complexes 46 and 47 (Fig. 15) [25]. Apparently, this particular P,N ligand is suitable for the stabilization of late transition metal centers in both low and high oxidation states.
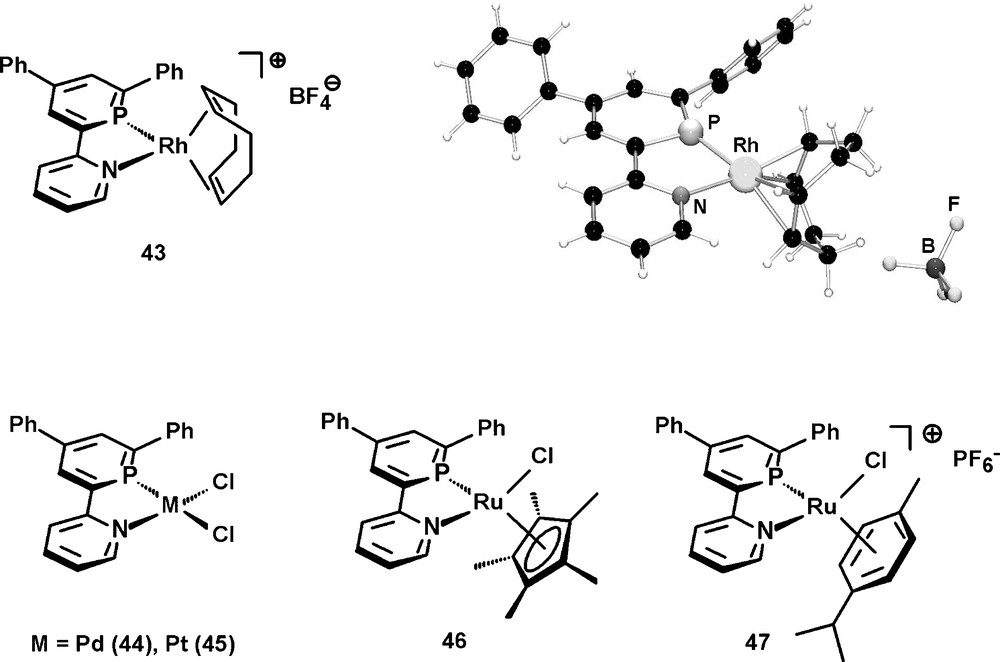
Selected examples of transition-metal complexes containing the 2-(2′-pyridyl)-4,6-diphenyl-phosphinine ligand 42 and molecular structure of (42)Rh(cod)BF4 in the crystal.
The additional phenyl-substituents in 4- and 6-position of the heterocyclic framework might also contribute to a kinetic stabilization of the metal complexes, as no particularly high sensitivity towards moisture could be observed, in contrast to some reported NIPHOS-metal complexes (vide supra). The synthetic procedure can be extended to the preparation of the 2,2′:6′,2″-terpyridine derivative 48, in which two of the three pyridine moieties have been substituted by the homologous aromatic phosphinine heterocycle (Scheme 18) [50].
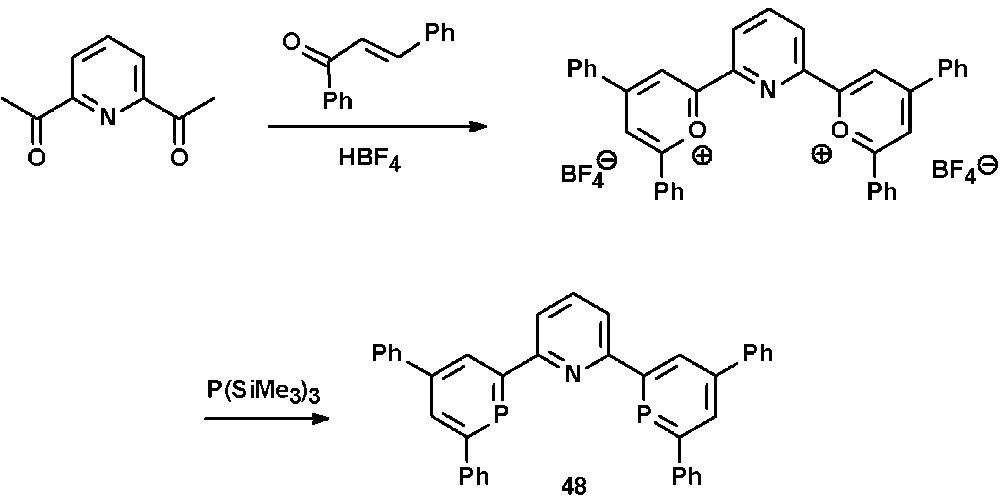
Synthesis of a diphosphinine-analogue of terpyridine via the pyrylium salt route.
In contrast to the well studied neutral PNP-pincer ligands containing predominantly σ-donative donor groups, the tridentate ligand 48 consists both of a σ-donative pyridine group as well as two π-accepting phosphinine donors. Due to the presence of electronically rather inequivalent donor atoms, these compounds represent therefore a new class of π-accepting PNP-pincer ligands. The corresponding CuIBr complex 49 was recently characterized crystallographically and revealed a distorted tetrahedral coordination geometry of the metal center as a result of an unusual coordination mode of the two phosphinine ligands. The molecular structure in the crystal is shown in Fig. 16.
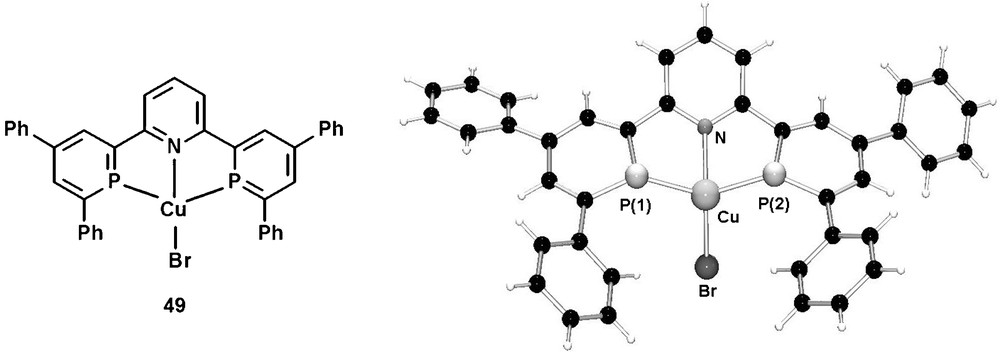
Molecular structure of the (48)Cu(I) complex 49 in the crystal.
9 Summary
State-of-the-art synthetic methodologies allow nowadays specific derivatization and functionalization of phosphinines, including the introduction of additional donor-functionalities into certain positions of the heterocyclic framework. In their function as phosphorus ligands with particular electronic and steric properties, they often show interesting and unusual coordination properties in combination with transition metal centers. The design of polydentate phosphinines with tailored properties can be envisaged, which will stimulate even further their exploitation in more applied projects in the future. In fact, very promising examples have already appeared in recent years in literature, such as their use as ligands in homogeneous catalysis. Moreover, the investigation of the photophysical properties of transition metal complexes containing phosphorus analogues of the well studied 2,2′-bipyridine and 2,2′:6′,2″-terpyridine derivatives is obvious, as they might lead to systems with interesting properties with respect to the development of optoelectronic devices. The history of phosphinines nicely demonstrates that these intriguing phosphorus heterocycles can still lead to unexpected discoveries and their further investigation remains an exciting, challenging and rewarding research field.
Acknowledgements
C.M. thanks The Netherlands Organization for Scientific Research (NWO-CW) for financial support. The European COST action CM0802 (PhoSciNet) is gratefully acknowledged.