1 Introduction and background
Matrix metalloproteinases (MMPs) are a family of about 30 zinc-dependent endopeptidases, which collectively have the capacity to degrade all the major components of the extracellular matrix [1]. Although the MMPs play crucial roles in physiological tissue remodelling and repair, their over-expression has been linked to a variety of chronic diseases including cancer, arthritis [2], osteoporosis [3], multiple sclerosis [4], arteriosclerosis [5], restenosis [5], meningitis [6], congestive heart failure [7], chronic obstructive pulmonary disease [8], chronic wounds [7a], liver cirrhosis [9], cerebral ischemia [10] and others. Efficient inhibition of MMPs is, therefore, an important scientific and therapeutic goal, which has attracted considerable attention within academia and industry for the last two decades or so. After more than 20 years of worldwide research, the scientific goals seem to have been met, and great advances have been made in the clarification of the mechanism of inhibition, however, the therapeutic targets are still waiting for their magic bullets.
This review does not include the various classifications of MMPs, for example, by their involvement in physiological and pathological processes, their substrate specificity or by their domain structures. Such information can be found in the reviews cited and the many earlier reviews cited in them.
The currently accepted mechanism of the proteolytic action of MMPs involves coordination of the catalytic Zn2+ cation by a water molecule. This interaction reduces the pKa of the solvent and facilitates its deprotonation by a glutamate residue, which acts as a general base/acid during catalysis, to produce an OH− nucleophile. This is the chemical species that attacks the carbonyl of the scissile peptide bond, which is also coordinated and thus polarized by the zinc ion. This is then followed by proton transfer to the peptide-bond nitrogen atom, also facilitated by the adjacent glutamate residue, leading to rupture of the peptide bond [11]. One crucial feature of all zinc proteinases is the presence of the catalytic Zn2+ cation in the active site, and following this recognition, most inhibitors have been designed to include a ZBG.
The desirable requirements for a potential transition state inhibitor for a metalloproteinase are: (i) tetrahedral structure to mimic the hydrolytic intermediate; (ii) the ability to form hydrogen bonds to bind to the protein; and (iii) disable the catalytic zinc ion by chelation. Phosphorus acid derivatives are the most obvious and natural candidates as they can fulfil all three of these requirements. Thus, the goal of the present review is to review MMP inhibitors (MMPIs) in which phosphorus-based functional groups have a central role in the inhibition (Fig. 1).
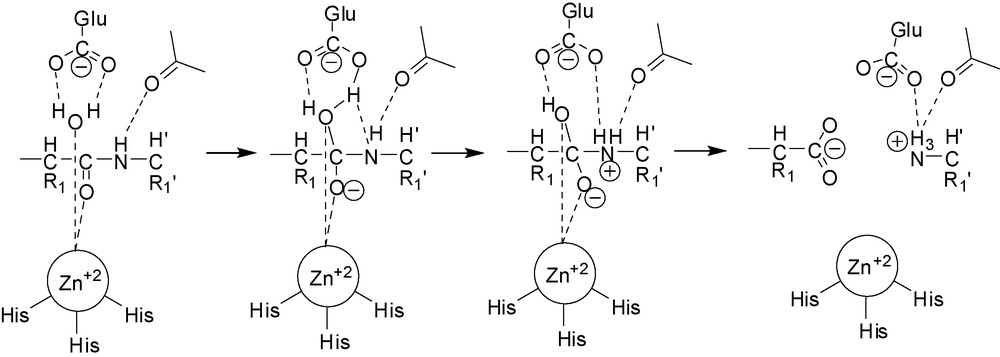
Reaction mechanism of proteolysis by MMPs.
1.1 Thermolysin, carboxypeptidase A and angiotensin converting enzyme
The earliest discovered zinc proteinases, thermolysin, (TLN) [12] and carboxypeptidase A (CPA) [13] had been crystallized prior to the MMPs and to the other medically significant angiotensin converting enzyme (ACE) [14].
The mode of binding of the naturally occurring TLN inhibitor phosphoramidon (Fig. 2) to the enzyme has been confirmed by crystallographic refinement [14]. Phosphoramidon binds to the zinc ion of TLN through a single oxygen of the phosphoramidate moiety. Together with three ligands from the protein, the resultant zinc complex has approximately tetrahedral geometry. However, in the case of P–Leu–NH2, two of the phosphoramidate oxygens interact with the zinc to form a complex that tends towards a pentacoordinate structure [15].
Structure of phosphoramidon.
Kim and Lipscomb determined the structures of three CPA-phosphinate complexes by X-ray crystallography [16]. The phosphinyl-zinc interactions in all three complexes are similar to those seen in complexes of thermolysin with phosphonamidate transition-state analogs. The knowledge gained from the inhibition and structural studies of TLN and CPA served as important guide for the development of medically useful ACE inhibitors as antihypertensive drugs even before ACE had been crystallized.
A practical consequence of these studies is the design and synthesis of Fosinopril (Fig. 3) (in addition to earlier non-phosphorus inhibitors), a clinically useful phosphinic acid drug to treat hypertension by inhibiting ACE [17]. Fosinoprilat is the active form of the drug, but as it is ionic at physiological pH, it has very low oral bioavailability. In order to increase its oral bioavailability it has been converted into the lipophilic prodrug, Fosinopril, by masking its acidic P–OH group as a bioreversible acyloxyalkyl ester.
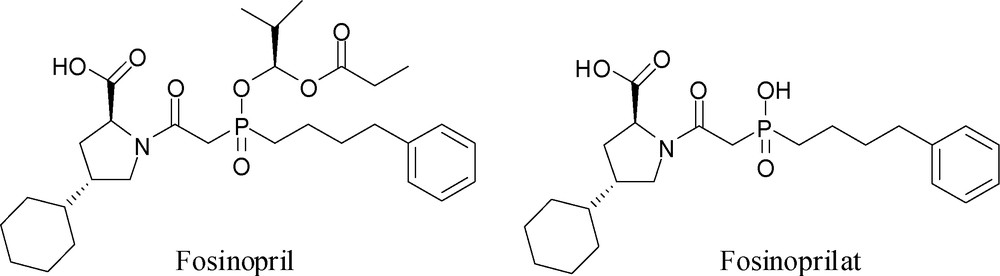
Structures of Fosinopril and Fosinoprilat.
No such luck was the MMPs’ share. The first report on the purification of collagenase (later called MMP-1) described the use of a hydroxamic acid affinity column [18]. This paper had crucial influence on future MMPI research, because it reported that only hydroxamate containing compounds showed “meaningful inhibition”, as opposed to other zinc enzyme inhibitors that included the angiotensin converting enzyme (ACE) inhibitor captopril, having an SH function as a ZBG, and other non-hydroxamate inhibitors: benzyl succinate, amastatin and phosphoramidon. Conversely, the only hydroxamate-based ACE inhibitor, idrapril [19] seems not to have entered into clinical use. To this date, the majority of MMP inhibitors (MMPIs) known are hydroxamate derivatives. While many of these yielded crystallizable enzyme-inhibitor complexes, providing invaluable structural information relevant to the mechanism of inhibition, none of them, nor other types of MMP inhibitors, got approved for clinical use to this date, apart from a tetracyline derivative, as a constituent of a mouthwash for treating periodontal disease [20].
2 Phosphorus based MMP inhibitors
2.1 Bisphosphonates
Geminal bisphosphonates (BPs) are P–C–P type synthetic analogs of the natural compound, diphosphoric acid (pyrophosphoric acid), having P–O–P type structure (Fig. 4). BPs are clinically used drugs for the treatment of calcium-related disorders and especially for bone diseases [21]. Their structure endows BPs with hydrolytic stability as opposed to diphosphoric acidate. In addition, because carbon is tetravalent, it can carry two additional substituents, thus opening the way to a wide variety of BP derivatives (Fig. 5).
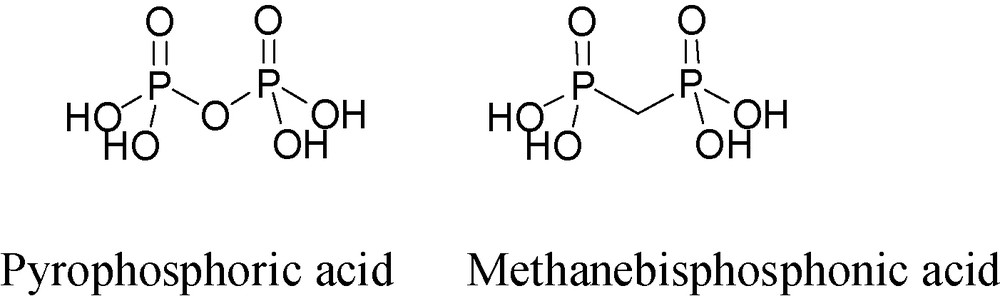
Structures of diphosphoric (pyrophosphoric) acid and methanebisphosphonic acid.
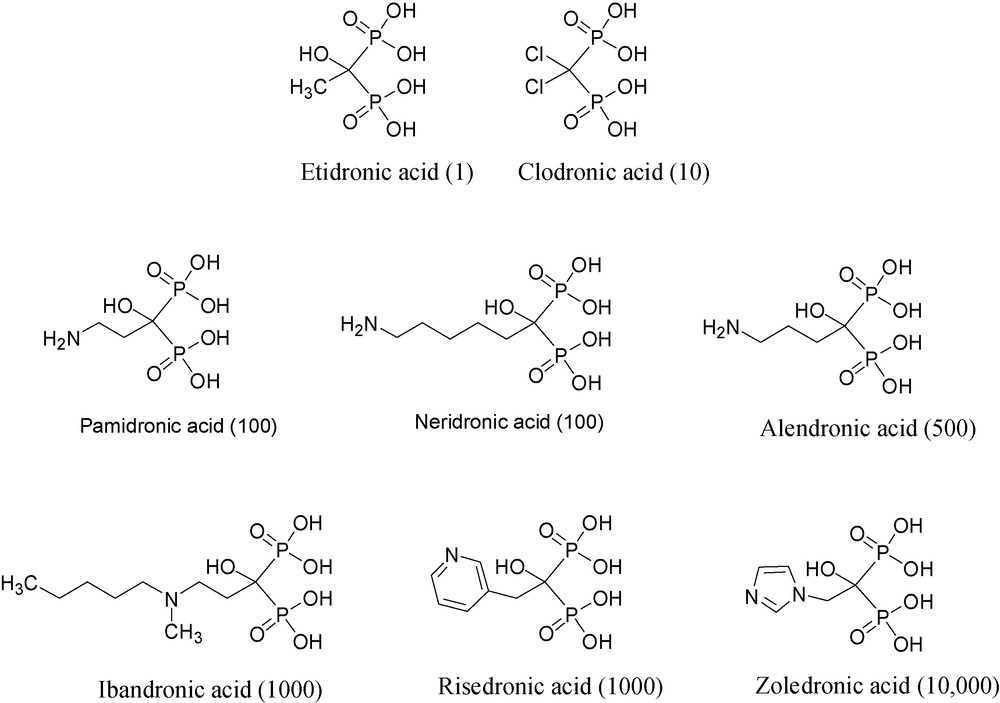
Bisphosphonates that inhibit MMPs or matrix metalloproteinases or Bisphosphonate MMP inhibitors.
Over the last three decades or so, a number of bisphosphonates have been shown to be useful for treating various bone diseases, among them hypercalcaemia of malignancy and metastatic bone disease [22]. The structures and the relative potencies in bone resorption of selected BPs are shown in Fig. 5.
In the course of studying the anticancer effects of BPs, the effect on the formation of bone metastases was noted [23], and was rapidly followed up by the discovery of the inhibitory effect of BPs on MMPs [24].
Using purified recombinant enzymes, several bisphosphonates, namely, clodronate, pamidronate, neridronate, alendronate and zoledronate, have been shown to inhibit the following enzymes quite non-selectively: MMP-1, -2, -3, -7, -8, -9, -12, -13, and -14, but with rather weak inhibition constants, i.e. IC50's ranging from 50 to 150 μM [24].
The common structural motif of BPs, the P–C–P moiety, is also called “bone-hook” (Fig. 6) because it binds to calcium, the bone mineral cation through chelation. The BP bone-hook is a nonselective chelator of heavy metal cations such as iron, zinc and others [25]. Because MMPs are zinc-dependent endopeptidases, it seemed reasonable to assume that BPs may inhibit the proteolytic activity of MMPs through chelation or, more likely, by abstraction of the divalent zinc cation. This conclusion is based on a number of findings. (a) Despite the differences in their potency against osteoporosis spanning over several orders of magnitude, all BPs used here were equipotent in inhibiting the proteolytic activity of MMP-2, -9, and -12. (b) An excess of zinc reversed the inhibitory effect of BPs on the proteolytic activity of MMPs [24].
Bone hook.
It was also shown that clodronate can downregulate the expression of MT1-MMP protein and mRNA in several cell lines. Additionally, several BPs have been shown in vitro to inhibit the invasion of malignant melanoma (C8161) and fibrosarcoma (HT1080) cells through reconstituted basement membrane (Matrigel) in cell cultures at IC50s of 50–150 μM and below [26].
Having low toxicity and proven to be well tolerated after several years in human use, it might appear that BPs have the potential to rapidly become widely useful inhibitors for MMP-related human soft and hard tissue–destructive diseases. However, it is necessary to consider the BP's unique pharmacokinetic characteristics.
The fate of BPs in the body is illustrated by the following in vivo results: upon intravenous administration of 14C-alendronate (1 mg/kg) to rats, the drug was distributed widely throughout the body, including calcified and noncalcified tissues. With the exception of kidney, the noncalcified tissue/plasma concentration ratio was less than 1%, ranging from about 0.05–0.7%. The kidney/plasma ratio varied from 3 to 6%. The kidney is the only eliminating organ, which may explain the high concentration ratio observed in this tissue. The drug level in all noncalcified tissues declined rapidly with time, parallel to the decline in plasma concentration. The amount of the drug in the noncalcified tissues decreased from 63% of the dose at 5 min to 5% at 1 h. Meanwhile, the drug concentration in the bone continuously increased and reached its peak at 1 h, suggesting that a significant redistribution of drug from noncalcified tissues into bone tissue occurred. The rapid redistribution means that the noncalcified tissues are exposed to bisphosphonates for only short periods [27].
Similar results were reported by other laboratories when other bisphosphonates had been given intravenously at high doses [28].
From these pharmacokinetic data, it appears that there is little likelihood to achieve any significant concentration for more than a short time of any BP in the extracellular fluid, where the disease causing MMP enzymes are present, even when the BP is administered systemically. Of orally administered bisphosphonates, only a minute fraction of the dose (< 1%) is absorbed.
In summary, zoledronic acid, the most potent BP known, inhibits tumor cell adhesion to the extracellular matrix and invasion through Matrigel and also has antiangiogenic activity. A growing body of evidence from animal models demonstrates that zoledronic acid and other bisphosphonates can reduce skeletal tumor burden and prevent metastasis to bone [29]. BPs fight certain cancers by various mechanisms and the inhibition of MMPs is one of them. But because of BPs exceptional pharmacokinetic behavior dictated by their strong affinity to bone, they are likely to be of value only to bone-related diseases.
2.2 Phosphoramides and phosphonamides
Phosphonamides are structurally the closest transition state analogs of carboxamides (Fig. 7). Therefore, among the first MMP (and other zinc peptidase) inhibitors there were phosphapeptide analogs of the natural substrates in which the scissile amide carbonyl was replaced by the tetrahedral phosphorus amide. A characteristic of the phosphorus amidates (and phosphinates, see next section) is that is their structure makes it possible the design of “combined inhibitors” [30].1
2.2.1 Phosphoramides
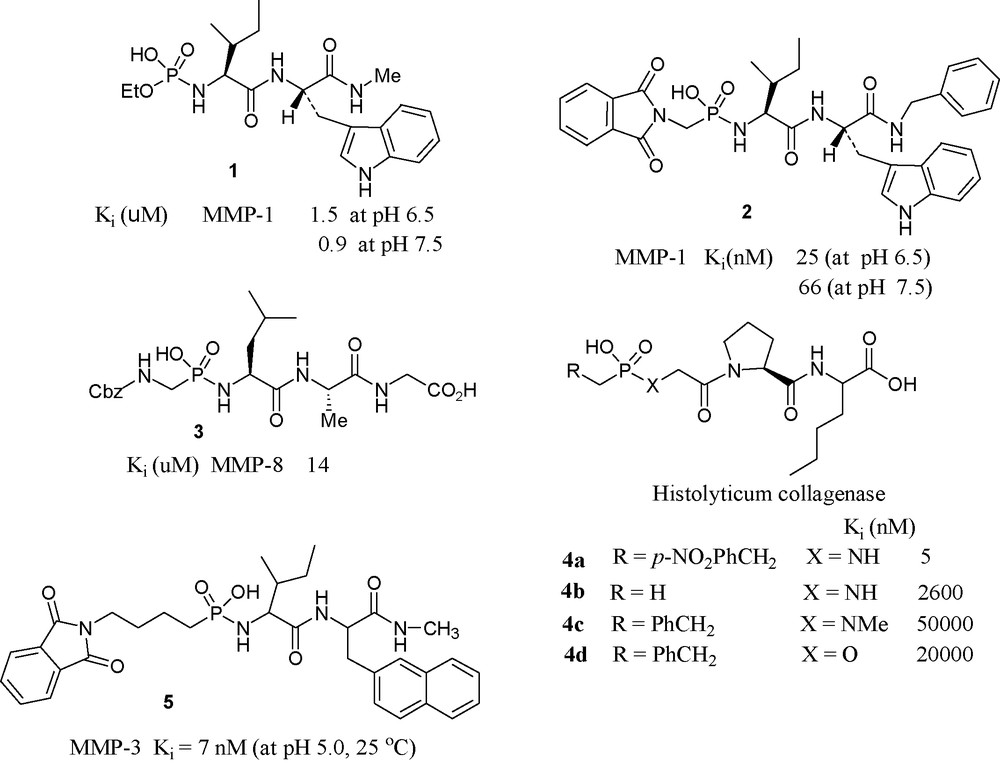
Scientists from the University of Kentucky have synthesized a series of N-(monoethylphosphoryl) peptides and examined their inhibition potency towards MMP-1. Among them, the most potent compound is 1, which inhibits MMP-1 with the Ki value of 1.5 μM [31].
2.2.2 Phosphonamides
In phosphonamidate 2, pH dependence of Ki has been noted. The compound was found approximately 3-fold more potent at pH 6.5 than at pH 7.5 [32].
A collaborative effort from two American Universities (Tallahassee, FL and Berkeley, CA) produced a series of phosphonamidates and evaluated their inhibitory potencies towards MMP-8. Compound 3 was the most potent in the series [33].
A Greek–French collaboration has produced and tested phosphonamide peptides having general formula R–PO(OH)–Xaa–Yaa–Zaa for clostridium collagenase. The most potent one found to be the compound 4a, with a Ki value of 5 nM [34].
Scientists at Merck USA have investigated the inhibition of stromelysin by phosphonamidate 5 that was found to be a slow binding potent inhibitor showing interesting pH dependence [35].
2.2.3 Hydroxamate based inhibitors containing phosphorus amides
The publication by Novartis scientists of CGS 27023A (Fig. 8), at that time a novel structure among MMP inhibitors stimulated research in other laboratories. CGS 27023A is a hydroxamate inhibitor having a sulfonamide moiety in the molecule [36]. This moiety was crucial for potency, since its replacement by a carboxamide function caused the loss of all activity, either because the sulfonamide oxygens make strong hydrogen-bonds to the enzyme or because the geometry of the tetrahedral sulfonamide's role in the enzyme cannot be fulfilled by the planar amide.
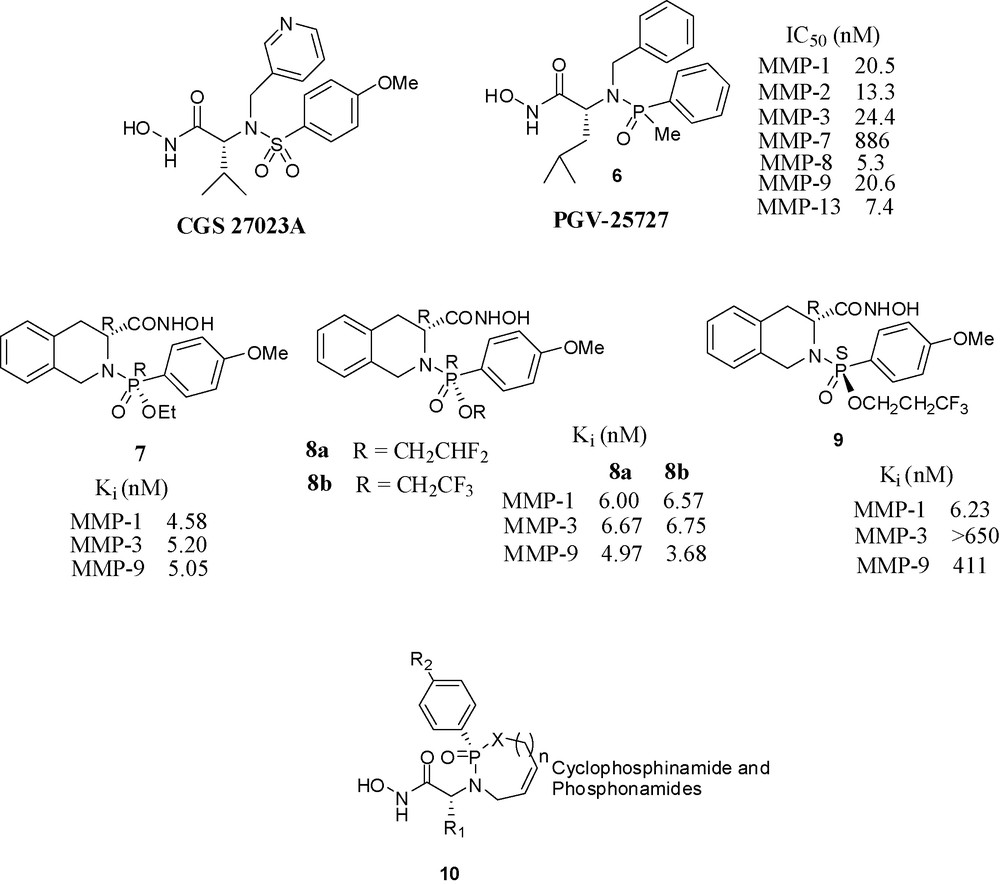
Hydroxamate based inhibitors containing phosphorus.
In this section, we present analogs of CGS 27023A, having hydroxamate ZBG, in which the original sulfonamide has been replaced by a phosphorus amide (phosphinamide or phosphonamide) moiety. These phosphorus amides do not compete with the hydroxamate on zinc binding but fulfil the roles of hydrogen-bond acceptor and other interactions with the enzymes.
Scientists at Procter and Gamble have synthesized phosphinamide based hydroxamic acids and found the molecules with (R) configuration at phosphorus were potent inhibitors, whereas the molecules with (S) configuration were almost completely inactive. Compound 6 inhibited MMP-1 and MMP-3 with an IC50 in the nanomolar range. Its binding mode in its stromelysin complex was clarified by X-ray crystallography [37]. The crystal structure of 6 with truncated stromelysin was also reported [38].
Scientists of Nippon Organon have prepared phosphonamide-based hydroxamate derivatives and evaluated their inhibitory potencies towards various MMPs. These compounds also have a chiral center at the phosphorus atom which is absent in the corresponding sulfonamide derivatives. Among the four diastereomers, the (R,R) isomer 7 has shown potency in nanomolar range towards MMP-1, 3, 9. The other three (S,S), (R,S), (S,R) isomers showed no or moderate activities. When the methoxy group of 7 was replaced with OPh, the potency towards MMP-9 was greatly increased (Ki = 0.08 nM) [39]. The next paper from the same laboratory described the potential applicability of difluoro and trifluoroethyl esters 8a and 8b as antedrugs for psoriasis [40]. In a third paper from the same laboratory, the activity profiles of various fluoroalkyl esters was checked. Interestingly, the 3,3,3-trifluoropropyl ester of the isomer (S)-9 was highly active and selective towards MMP-1 vs. MMP-3 and MMP-9 [41]. Scientists from LEO Pharma in Denmark have reported a series of potent cyclic phosphinamide or phosphonamide containing hydroxamates. A representative structure 10 is shown [42].
2.3 Phosphinic acids
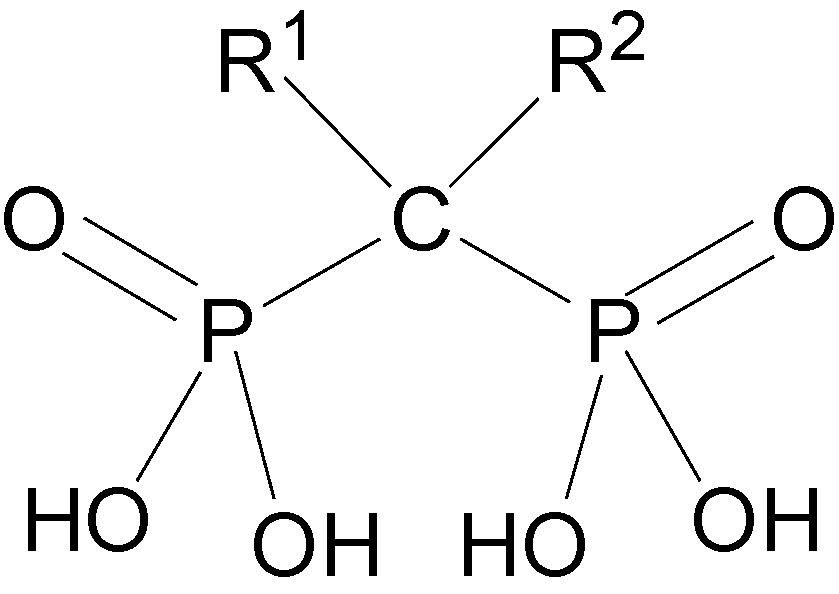
Phosphinic acids have evolved from phosphonamides by replacing the hydrolytically unstable P–N by the stable P–C bond, giving compounds in which the phosphorus in the tetrahedral O=P–O− group is bound to two carbon atoms (Fig. 9). The clinically successful phosphinate ACE inhibitor Fosinopril [17] also might have served as motivation for imitation. The conceptual transfer from phosphonamides to phosphinates involves the replacement of the NH by a CH2 group and loss of a hydrogen bond, which might cause decrease in inhibitory potency [43]. However, as shown in the following paragraphs, no such decrease was observed.
(a) Phosphinic acid based inhibitors I. (b) Phosphinic acid based inhibitors II.
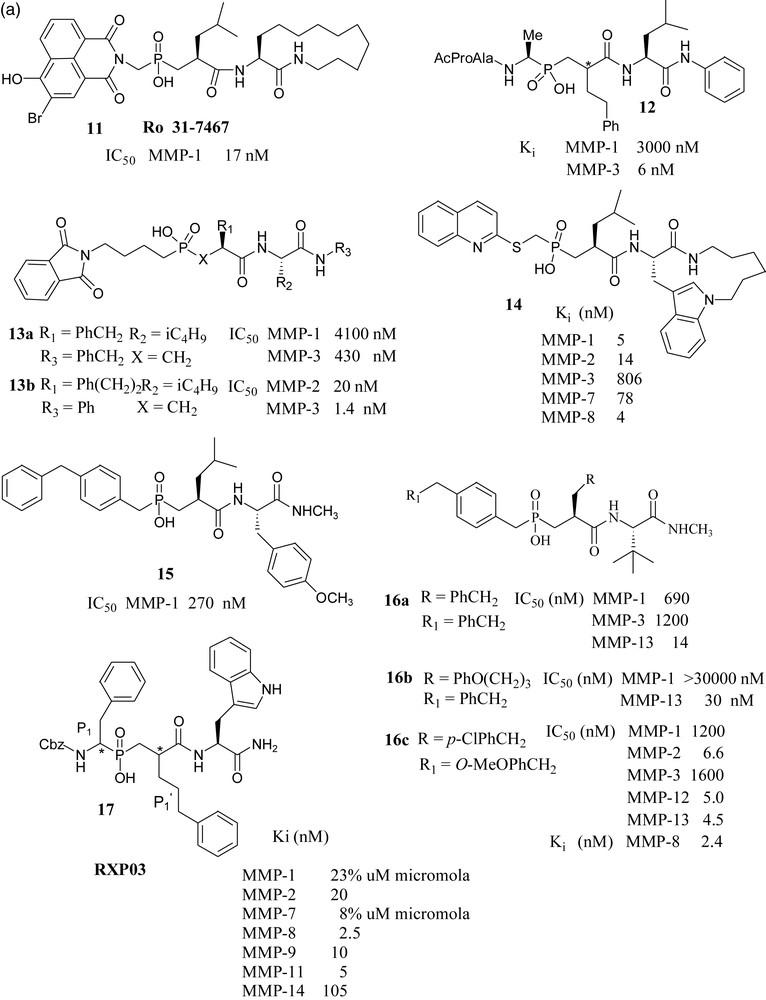
A British industry-academia collaboration reported that phosphinate MMP inhibitor Ro 31-7467 (11) has been shown to inhibit smooth muscle cell proliferation, [44] and bone resorption through gelatinase inhibition [45].
Merck USA scientists synthesized and tested a series of phosphapeptide inhibitors. They have shown that potent and selective inhibition of MMP-3 with these peptidylphosphinic acids requires extension of substitution into the P3 position. Thus, compound 12 was found to be approximately 500 fold more potent against MMP-3 vs. MMP-1 [46]. In a later paper from the same laboratory, it has been shown that phosphonic acid 13a inhibited MMP-3 more effectively than the corresponding phosphonamidate and phosphonate analogs. Its potency has further increased when benzyl group at P1’ was replaced by phenylethyl group (compound 13b). It has been noted that 13b becomes practically inactive towards MMP-3 when the phthalimidobutyl substituent in the P1–P3 region was replaced by a methyl group [47].
Syntex scientists have reported that placement of the quinolylthiomethyl group in the P1–P2 region and a macrocyclic indolactam moiety on the primed side of the phosphinic acid endowed the molecules 14 with significant potency against MMP-1, 2, 3, 7 and 8, with little selectivity [48].
Pfizer scientists have studied the binding mode of compound 15 with MMP-1, in which a P2 diphenylmethane group effectively occupied the S2 pocket and the isobutyl side chain occupied the S1’ pocket. Loss of activity was observed when the group at P2 was replaced by other substituents like biphenyl analogs, isopropyl or cyclohexyl groups, which cannot effectively occupy S2 pocket. Compounds of this series have shown poor absorption and very low oral activity. This group has also reported interesting compounds 16a, 16b and 16c, which showed considerable selectivity towards MMP-13 over MMP-1 and MMP-3 [49,50].
Collaboration of French and Greek groups made significant contributions to the chemistry and biology of phosphinates as MMP (and ACE - not shown) inhibitors.
The present paper describes a detailed study of a large number of phosphinate MMP inhibitor; among them, several molecules with nanomolar Ki constants. An interesting finding is the potentiating influence of phenylpropyl group in the P1’ position, e.g 17, combined with more conventional side chains [51]. The crystal structure of MMP-11 complexed with phosphinic TS inhibitor, RXP03 has been determined [52]. From the results of the evaluation of this phosphinic acid MMP inhibitor in a rat model of liver ischemia/reperfusion, it was concluded that MMP inhibitors might be of clinical relevance in liver-associated ischemic diseases or after liver transplantation [53].
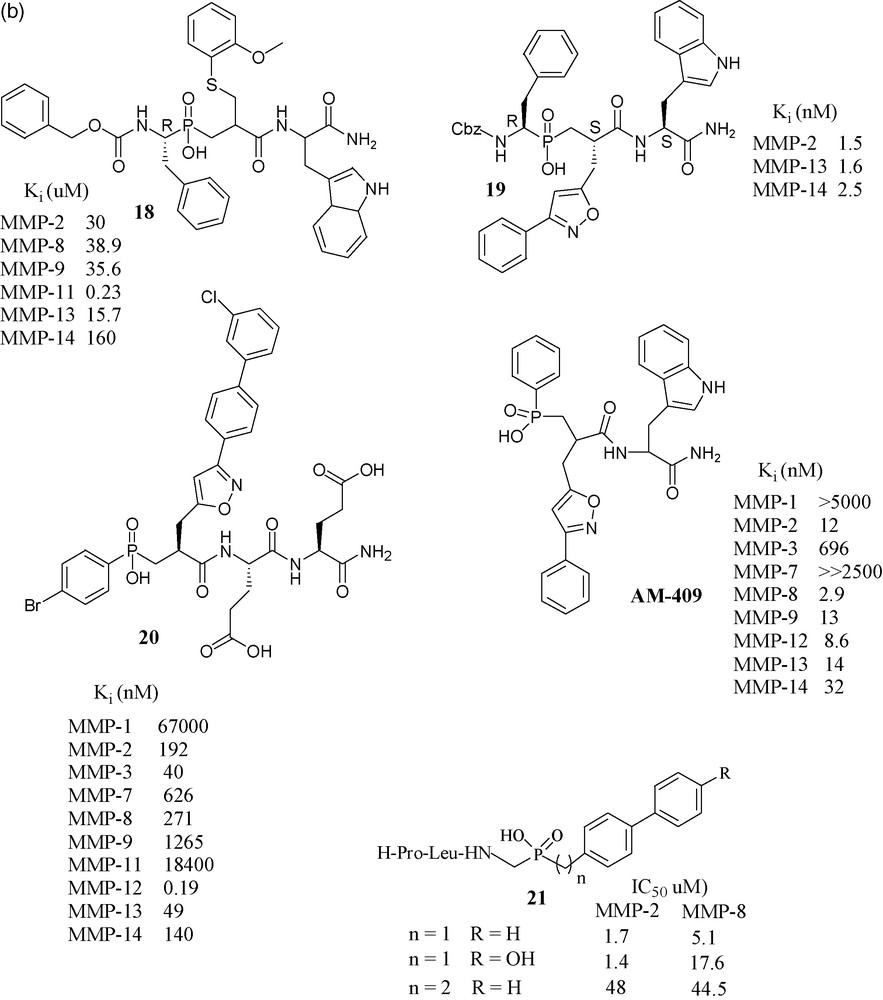
In an effort toward selective MMP-11 inhibitors, a matrixin over expressed in breast cancer, compound 18 was found to be the most selective phosphapeptide inhibitor of MMP-11 (Ki is 0.23 μM), while its potency toward the other MMPs was two orders of magnitude lower [54]. Considering the promising results exhibited by RXP03, this structure was further developed by focusing on P1’ position, as it is well known that this position is one of the key side chains for the control of inhibitor potency and selectivity. The introduction of an isoxazole group into the P1’ position gave compound 19, which was 36-fold more potent than RXP03 (R,S,S) towards MMP-14, than that having an aralkyl group at P1’ position. There was found also stereospecificity among the isomers of 19. Diastereoisomer (R,S,S) is 100-fold more potent than its (R,R,S) isomer, which shows that orientation of the P1’ group is critical for this series of isoxazole compounds [55]. This project was further developed with the synthesis of four additional phosphinic inhibitors having the isoxazole side chains in the P1’ position, to probe the S1’ cavity. Among them, compound 20 has shown subnanomolar potency towards MMP-12 [56]. Another compound, AM-409, reported by the French–Greek group formed a crystalline complex with MMP-9, the crystal structure of which was reported by the Max Planck Institute's protein structure group [57].
Collaboration of Italian laboratories have synthesized three novel peptidomimetic phosphinate inhibitors 21 and evaluated them for MMP-2 and MMP-8. Their IC50 values were in the micromolar range [58].
2.4 Phosphonic acids
Relative to phosphinate-based inhibitors which have side chains on both sides of the ZBG, most phosphonates had side chains only on one side. Therefore, in the latter class lower selectivity and potency may be expected relative to phosphinates.
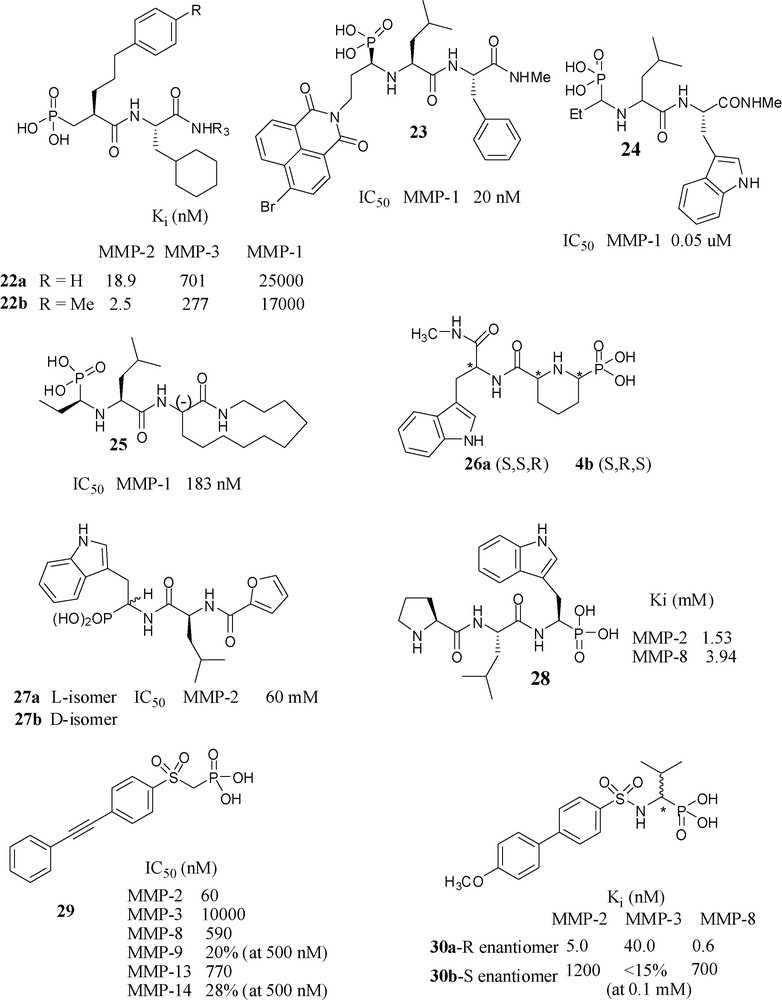
CELLTECH UK scientists have prepared phosphonic acid peptides of structures 22a and 22b (Fig. 10), which had nanomolar potency towards MMP-2 and had selectivity relative to MMP-3 and MMP-1. The potency and selectivity was increased almost 10-fold by methyl substitution: 22b [59].
Phosphonic acid based inhibitors.
Scientists from SKB have investigated the effect of bulky substituents at P1 position. Having a bromonaphthalimidoethyl analog (R,S,S isomer) at P1 position in 23 was the most potent in the series, 80-fold more potent than the (S,S,S) isomer towards MMP-1. Incidentally, this molecule is an example of phosphonic acids that, similarly to phosphinates, have side chains of both the primed and the unprimed side of the molecule [60]. Another paper from SKB reported a series of N-phosphonoalkyl dipeptides and found 24 as the most potent molecule in this series, having the bicyclic aromatic tryptophan analogue as P2’ substituent [61]. Introduction of 3-(-)-aminoazacyclotridecane-2-one moiety to the P2’/P3’ position however, not surprisingly, 25 showed lower potency than the corresponding hydroxamate towards MMP-1 [62].
A collaboration of three French laboratories has produced conformationally constrained N-phosphonoalkyl dipeptides 26a (S,S,R) and 26b (S,R,S), by gathering P1 and P1’ side chains of molecule 24 to form a piperidine ring however neither of showed any significant activity at concentrations less than 10−4 M [63].
A collaboration of Italian laboratories have prepared two diastereomers 27a and 27b and evaluated their inhibitory potency towards MMP-2, MMP-3, MMP-8, MMP-9. They observed no inhibition even at 100 μM concentration (except for MMP-2 for which 27a shown IC50 of 60 μM). In fact, the corresponding carboxylic ZBG analog showed better potency [64].
An Italian–German collaborative project has reported the crystal structure of 28 complexed with MMP-8, where it occupies the unprimed region of active site [65]. Scientists from the same laboratory prepared a series of phosphonic acid analogs by replacing the terminal l-proline with other amino acid residues to yield compounds of increased affinity towards MMP-2 and MMP-8. Computer simulations were performed to explain the effects of substitutions on the preferred mode of binding to MMP-8 [66].
The collaboration of three Italian research groups has produced a series of α-sulfonylphosphonates, most of which had IC50 values in nanomolar range towards MMP-2, -8, -13 and -14. Among them, compound 29 found to be the most potent one towards MMP-2 (IC50 = 60 nM) and selective vs. MMP-3, MMP-8, MMP-9, MMP-13 and MMP-14 [67].
A collaboration of European groups has synthesized (R)-30a and (S)-30b α-arylsulfonylamino phosphonates, and studied their detailed mode of binding through the crystal structures of the complexes with MMP-8 [68]. Because of the favourable binding interactions, the (R)-enantiomer 30a is > 1000-fold more potent than its (S)-enantiomer towards MMP-8. Potency of the (R)-isomer towards MMP-2 and MMP-3 was also in the nanomolar range. In the case of corresponding hydroxamate analogs, the (R)-isomer is 100-fold potent than its (S)-isomer towards MMP-2 and MMP-3. However, in contrast among the corresponding carboxylate analogs, the S-isomer is 12-fold more potent towards MMP-3 than its R-isomer. When the methoxy group was substituted by ethoxy they obtained IC50 values in subnanomolar range 0.37–1.1 nM for MMP-2, -8, -9 and -13 [69]. The m-methyl substituted compound showed some degree of selectivity toward MMP-8. In order to clarify the involvement of MMP-8 in neuroimmunological disorders such as multiple sclerosis, a pharmacological inhibition of MMP-8 with this “selective” inhibitor (IC50 = 0.4 nM) was carried out. This study revealed that IP administration of the MMP-8 selective inhibitor to mice having experimental autoimmune encephalomyelitis (EAE) reduced the severity of the disease. Based on this finding, the authors concluded that MMP-8 plays an important role in EAE development and proposed that this enzyme may be a novel therapeutic target in human neuro-inflammatory diseases such as multiple sclerosis [70].
2.5 Carbamoylphosphonic acids
The MMP inhibitory action of carbamoylphosphonates (CPOs) was discovered in the screening of various acylphosphonic derivatives in an in vitro invasion assay. This assay determines the extent of invasion of HT1080 tumor cells across a reconstituted membrane in the absence or in the presence of varying concentrations of potential inhibitors. The assay evaluates the effect of the drug in an environment similar to that in vivo, and therefore it has a higher predictive value than measuring inhibitory potency on recombinant enzymes. All aroylphosphonic acids (ArCOPO3H2) showed modest but significant inhibitory potency. In comparison, an analogous benzylphosphonic (ArCH2PO3H2) acid and an aroylphosphonic acid monoester (RCOPO3HR) showed no activity at all in this assay. The breakthrough appeared when the first carbamoylphosphonic acid, N-cyclohexylcarbamoylphosphonic acid, was tested. The potency showed by this compound motivated to further study systematically the MMP inhibitory potency of various classes of carbamoylphosphonic acids [71,72].
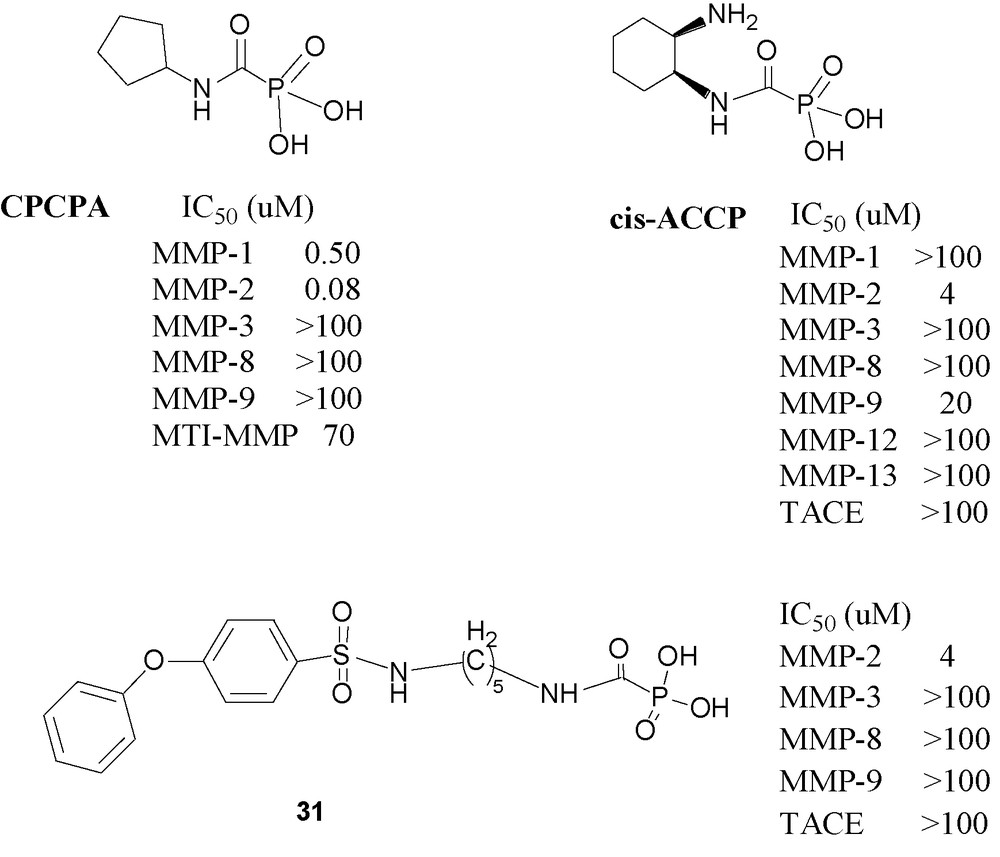
Following this preliminary report, it was of interest to examine in depth the most potent compound, namely cyclopentylcarbamoylphosphonic acid (CPCPA) (Fig. 11) an MMP-2 specific inhibitor [73]. The results of this study indicate that CPCPA significantly inhibited cellular invasion and capillary formation in vitro. Furthermore, i. p. or peroral administration of the CPCPA significantly reduced lung metastasis formation and s. c. tumor growth in a murine melanoma model. Simultaneously with its biological explorations of CPOs, it was of interest to explore the ability of the carbamoylphosphonic function to serve as zinc-binding-group (ZBG). It was also found that CPOs containing basic amino groups have enhanced zinc-binding potency in aqueous solutions [74].
Carbamoylphosphonic acid-based inhibitors.
A more recent publication describes cis-2- aminocyclohexylcarbamoylphosphonic acid (cis-ACCP) an MMP-2 specific CPO, which was evaluated in vitro and in two in vivo cancer metastasis models [75]]. It reduced metastasis formation in mice by ∼ 90% when administered by a repetitive once daily dosing regimen of 50 mg/kg via oral or intraperitoneal (IP) routes and was nontoxic up to 500 mg/kg, following IP administration daily for two weeks. Pharmacokinetic investigation of cis-ACCP in rats revealed distribution restricted into the extracellular fluid, which is the site of action for the antimetastatic activity. Sustained and prolonged absorption (t1/2 ∼ 126 min) occurred via paracellular mechanism along the small and large intestine with overall bioavailability of 0.3%. Yet, even with its poor oral bioavailability, cis-ACCP was found to have a sufficiently high quasi steady state concentration in the extracellular fluid, significantly above the observed minimal inhibitory concentration (MIC), for a prolonged period of time that enables once-a-day administration.
The results indicate that cis-ACCP is a novel MMP inhibitor characterized by rapid systemic elimination. Its limited permeability through biological membranes enforces slow and sustained absorption to the systemic circulation enabling it to maintain effective concentration in the extracellular fluids, where it exerts its antimetastatic activity, as the active compound does not permeate into cells to induce adverse effects. Although cis-ACCP seems an ideal drug candidate, it seemed that it would be desirable to improve its relatively high IC50 value (IC50 ≈ 4 μM) and low duration (≈ 20 min) of MMP-2 inhibition [75].
To achieve these goals new CPO inhibitors have designed recently [76]. The design of these molecules is based on the combination of the CPO group combined with a diphenyl ether moiety with a series of linker chains of varying lengths. The diaryl ether moiety has been shown to fit the S1’ pocket in a variety of molecules’ and it indeed endowed the resulting CPOs with strong affinity to MMP-2. The most useful molecule in the series (CH2)n, n = 2–6, is 31, which has five methylene groups in the linking chain. Compound 31 is a selective MMP-2 inhibitor. It inhibits the enzyme throughout the duration of the measurement, namely at least 3 h, without showing signs of weakening inhibition. It is orally active in the murine melanoma model showing a dose related reduction in metastasis formation: 50 mg/kg giving 80% and 150 mg/kg giving 86%, relative to control without any signs of toxic effect.
In conclusion, carbamoylphosphonates are medium potency, water-soluble MMP inhibitor, which because of their ideal pharmacological, toxicological, and pharmacokinetic profiles appears preferable over previous, much more in vitro potent MMP inhibitors that have reached phase 2 and phase 3 clinical trials in recent years and have failed.
CPOs are in a way reminiscent of bisphosphonates, the previously mentioned drugs for bone diseases, which have also very low (< 1%) oral bioavailability. This small fraction is sufficient for achieving their therapeutic action because they are selectively self-targeted only to the bone where they perform their therapeutic function. Similarly, CPOs self-target only to the extracellular fluid where they inhibit MMPs that are concentrated there. Because of their anionic polar nature, they are unable to cross membranes and to enter cells, thus exert no toxic side-effects.
3 Conclusion
The first efforts toward clinically useful MMP inhibitors started appearing in the literature, to the best of our knowledge in the 1980s. Since then enormous amounts of manpower and money were used up gathering immense amounts of scientific knowledge, but no MMPI drug. The famous saying of Niels Bohr about the difficulty of prediction about the future, which we cited five years ago [77], appears to be still valid for MMP inhibitors. However, it is safe to say that not necessarily the lowest nanomolar inhibitor will win the race toward the first MMPI drug. To us it seems (while remembering Niels Bohr's dictum) that the requirements for a successful inhibitor, in addition to affinity to the enzyme, are solubility in the extracellular fluid, staying out of cells and organs, and weak or no zinc binding, i.e. allosterism.
1 There exist three types of inhibitors depending on which side of the scissile amide bond lines the molecule up, relative to the ZBG. Molecules which extend on both sides of the ZBG are called “combined inhibitors”. Most inhibitors extend from the ZBG only to one direction to afford either right, or left hand side inhibitors, because they bind to the primed (right hand) subsites or to the unprimed (left hand) sites.