

1 Introduction
Metal phosphides MxPy are an interesting class of compounds which have been successfully used as catalysts and in materials science [1]. One of the possible routes for the preparation of these compounds is the decomposition of phosphorus-rich metal complexes, which can be synthesized by transmetalation reactions of alkali metal oligophosphanides and other metal salts [2]. Thus, the preparation and reactivity of linear alkali metal oligophosphanides has been extensively studied, and many complexes with potential applications as precursors for the preparation of metal phosphides could be obtained [3].
Alternatively, less is known about cyclic oligophosphanide anions than their open-chain counterparts. Although cyclo-(PPh)5 was described as early as 1877 [4], the first cyclooligophosphanide anions cyclo-(PnRn−1)−, were described only ca. 100 years later [5]. Thus, three-membered rings (in M[cyclo-(P3But2)] with M = Li, K), four-membered rings (in M[cyclo-(P4But2R)] with R = SiMe3, But) and five-membered rings (in M[cyclo-(P5R4)] with M = Li, R = But; M = K, R = Ph) have been reported [5,6]. However, these compounds have only been obtained as inseparable mixtures, and some of them have been only detected in solution by 31P NMR spectroscopy.
About 15 years after the investigations on these alkali metal cyclooligophosphanides, we reported the high-yield synthesis and full characterization of the sodium salt Na[cyclo-(P5But4)] [7] and the potassium salt [K(pmedta)(thf)][cyclo-(P5But4)] [2 h] (PMEDTA = Me2NCH2CH2NMeCH2CH2NMe2), which were used for the preparation of a wide variety of main group [8] and transition metal complexes [7,9]. However, some of the target compounds were obtained in low yield only, and others were difficult to synthesize, such as as-yet unknown rare-earth metal complexes, which were inaccessible in our hands starting from the highly reactive Na or K salts. In this context, lithium tetra-tert-butylcyclopentaphosphanide seemed to be a more promising starting material for the preparation of new cyclic oligophosphanide metal complexes. We now report the synthesis and structural characterization of [Li(tmeda)2][cyclo-(P5But4)] (1b) which has an unusual solid-state structure consisting of separated [Li(tmeda)2]+ cations and “naked” [cyclo-(P5But4)]− anions.
2 Results and discussion
2.1 Synthesis and spectroscopic studies
We have previously shown that the reaction of sodium with ButPCl2 and PCl3 in the ratio 12:4:1 in THF gives a mixture of Na[cyclo-(P3But2)] [5a,6b], Na[cyclo-(P4But3)] [6b], cyclo-(P4But4) [10], Na2(P4But4) [5a] and sodium tetra-tert-butylcyclopentaphosphanide Na[cyclo-(P5But4)]. The last complex was obtained in reasonable yield after optimization of this reaction [7]. Analogously, K[cyclo-(P5But4)] was prepared, but it was very difficult to separate from K2(P4But4) and thus always obtained in low yield [2f]. When lithium is employed, a mixture of cyclo-(P4But4) [10], Li2(P4But4), and lithium tetra-tert-butylcyclopentaphosphanide Li[cyclo-(P5But4)] (1) is formed after 6 hours at 80 °C; keeping the reaction mixture at this temperature increased the amount of 1 (to 90% according to 31P{1H} NMR after 1 d, or 95% after 2 d) (Scheme 1). Isolation and recrystallization from THF/TMEDA gave [Li(tmeda)2][cyclo-(P5But4)] (1b) in good yield. The 31P{1H} NMR spectrum of 1b shows three sets of signals which correspond to an ABB’CC’ spin system. Chemical shifts and coupling constants were successfully calculated by using the program SPINWORKS [11]. The coupling constants are similar to those observed for the analogous Na and K compounds (Table 1)1.
According to the spectroscopic data and based on the steric requirements of the tert-butyl groups, only one of the four conformations, the all-trans isomer, is preferred as was previously observed for related compounds [7,8,9] and confirmed by X-ray diffraction studies on 1b (Section 2.2). Additionally, the 1H NMR and 13C{1H} NMR spectra of 1b showed the expected signals.
2.2 Structural studies
Orange crystals of 1b were obtained from THF/TMEDA solution at –28 °C. Compound 1b crystallizes in the noncentrosymmetric monoclinic space group Cc with four molecules in the unit cell. Selected bond lengths and angles for 1b are given in Table 2.
Selected bond lengths (pm) and angles (°) for 1b.
| 1b | |
| P(1)–P(2) | 220.7 (2) |
| P(1)–P(5) | 212.5 (2) |
| P(2)–P(3) | 222.6 (2) |
| P(3)–P(4) | 221.5 (2) |
| P(4)–P(5) | 221.6 (2) |
| P(2)–C(1) | 191.6 (4) |
| P(3)–C(5) | 190.9 (5) |
| P(4)–C(9) | 189.8 (5) |
| P(5)–C(13) | 188.1 (5) |
| Li(1)–N(1) | 211.3 (8) |
| Li(1)–N(2) | 210.4 (8) |
| Li(1)–N(3) | 214.4 (8) |
| Li(1)–N(4) | 213.3 (8) |
| P(1)–P(2)–P(3) | 102.45 (6) |
| P(1)–P(5)–P(4) | 106.35 (6) |
| P(2)–P(3)–P(4) | 103.16 (6) |
| P(2)–P(1)–P(5) | 104.70 (6) |
| P(3)–P(4)–P(5) | 106.63 (6) |
| C(1)–P(2)–P(1) | 98.5 (2) |
| C(1)–P(2)–P(3) | 100.8 (2) |
| C(5)–P(3)–P(2) | 103.4 (2) |
| C(5)–P(3)–P(4) | 102.0 (2) |
| C(9)–P(4)–P(3) | 101.1 (2) |
| C(9)–P(4)–P(5) | 99.7 (2) |
| C(13)–P(5)–P(1) | 112.0 (2) |
| C(13)–P(5)–P(4) | 102.3 (2) |
| N(1)–Li(1)–N(3) | 122.3 (4) |
| N(1)–Li(1)–N(4) | 121.4 (4) |
| N(2)–Li(1)–N(3) | 118.5 (4) |
| N(2)–Li(1)–N(4) | 121.6 (4) |
Compound 1b consists of separated [Li(tmeda)2]+ cations and [cyclo-(P5But4)]− anions (Fig. 1a). The lithium cation is coordinated in an usual distorted tetrahedral fashion by four nitrogen atoms of two TMEDA molecules (Li–N 210.4(8) to 214.4(8) pm, see for example [12]), which prevents interaction of the cation with the anion (Fig. 1b) and thus results in a “naked” [cyclo-(P5But4)]− anion (Li(1)⋯P(1) 494.0(6) pm). This type of anionic two-coordinate phosphorus atom is typical in metal complexes of white phosphorus (see for example [13] and references therein), but rare for linear or cyclic oligophosphanides [2,3,7,9,14]. Only a few examples display an ion-separated structure, such as [K(18-crown-6)(thf)2][P4HMes4] (Mes = 2,4,6-Me3C6H2), in which the potassium cation is coordinated by 18-crown-6 and two THF molecules [3a], and the lithium phosphanides [Li(12-crown-4)2][PPh2] and [Li(12-crown-4)2][PMes(BMes2)] [14]. In known lithium oligophosphanides the coordination of only one TMEDA molecule and a phosphorus atom was observed [2i], rather than the coordination of two TMEDA molecules as in 1b.
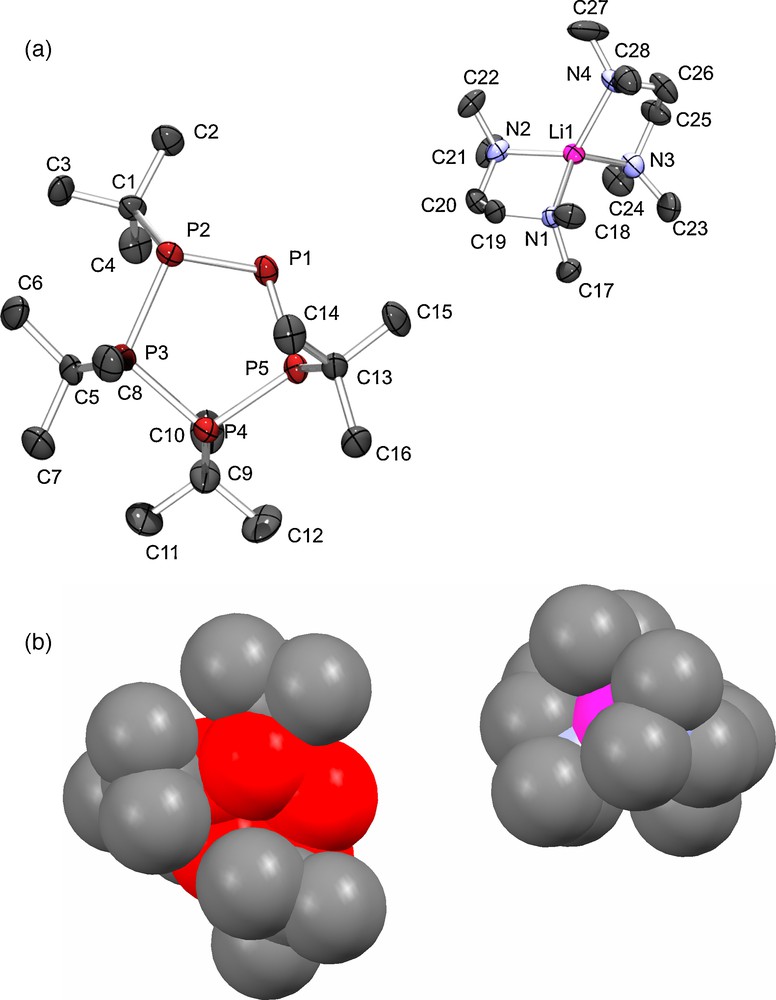
a) Molecular structure and atom labeling scheme for 1b with thermal ellipsoids at 50% probability (hydrogen atoms are omitted for clarity) and b) space-filling diagram of 1b (color code: P red; Li pink; N light blue; C gray).
The [cyclo-(P5But4)]− anions show an all-trans arrangement of the tert-butyl groups on the phosphorus atoms with an envelope conformation of the chiral anionic P5 ring (best plane is defined by P(1)–P(3)–P(4)–P(5), deviation of P(2) from this plane is 87.5 pm (P(1) is the anionic phosphorus atom). This conformation is different from those found in [Na(thf)4][cyclo-(P5But4)] [7] and [K(pmedta)(thf)][cyclo-(P5But4)] [2g], in which the neutral phosphorus atoms are almost coplanar, while the anionic phosphorus atom lies about 80 pm above this plane. The P(1)–P(5) bond length of 212.5(2) pm in 1b is about 8 pm shorter than the other P–P bonds, which have values ranging from 220.7(2) to 222.6(2) pm (Table 2), validating the larger coupling constant 1J(PA−PB) = 1J(PA−PB’) of −394.9(1) Hz observed in the 31P{1H} NMR spectrum. A comparison of selected structural parameters of alkali and coinage metal complexes containing the cyclo-P5But4− ligand is given in Table 3.
Comparison of selected structural parameters for alkali and coinage metal complexes of (cyclo-P5But4)−.
| Compound | P(1)–P(5) | Other P–P bonds | M(1)–P(1) | Reference |
| 1b | 212.5(2) | 220.7(2) 221.5(2) 221.6(2) 222.6(2) | 490.4(6) | This work |
| [Na(thf)4][cyclo-(P5But4)] | 213.2(2) | 220.4(2) 220.8(1) 221.1(2) 222.9(1) | 293.8(2) | [7] |
| [K(thf)(pmedta)][cyclo-(P5But4)]a | 212.9(3) | 219.5(4) 220.8(6) 221.4(3) 222.6(5) | 325.3(3) | [2h] |
| [Cu{cyclo-(P5But4)}(PPh3)2] | 214.6(2) | 221.0(2) 221.1(2) 221.9(2) 222.2(2) | 228.6(2) | [9c] |
| [Au{cyclo-(P5But4)}(PCyp3)]b | 217.3(2) | 220.7(2) 221.6(2) 222.5(2) 223.0(2) | 233.3(2) | [9c] |
a Data of only one of the three crystallographically independent molecules of the asymmetric unit are given.
b Cyp = cyclo-C5H9.
3 Conclusions
While the reaction of sodium with ButPCl2 and PCl3 in the ratio 12:4:1 in THF gave Na[cyclo-(P5But4)] after five days at 80 °C, and the analogous K[cyclo-(P5But4)] was obtained only in low yield and with K2(P4But4) as impurity, the preparation of the corresponding lithium compound Li[cyclo-(P5But4)] (1) is achieved in two days at 80 °C. Recrystallization of 1 from THF/TMEDA gave crystals of [Li(tmeda)2][cyclo-(P5But4)] (1b), which consists of separated [Li(tmeda)2]+ cations and [cyclo-(P5But4)]−anions, representing the first example of a “naked” [cyclo-(P5But4)]− anion.
We expect that lithium salts 1 and 1b may facilitate transmetalation reactions with transition metal or rare-earth metal complexes which were unsuccessful starting from the analogous sodium or potassium salts.
4 Experimental section
4.1 General methods
All experiments were performed under an atmosphere of dry argon using standard Schlenk techniques. The NMR spectra were recorded at 25 °C on a Bruker AVANCE-DRX-400 spectrometer. 1H NMR (400.13 MHz) and 13C NMR (100.16 MHz): internal standard solvent, external standard TMS; 31P NMR (161.9 MHz): external standard 85% H3PO4. FAB mass spectra were recorded in a MASPEC II spectrometer with 3-nitrobenzyl alcohol as matrix. IR spectrum: a KBr pellet containing 1b was prepared in a nitrogen-filled glove box and the spectrum was recorded on a Perkin-Elmer System 2000 FTIR spectrometer in the range 350–4000 cm−1. All solvents were purified by distillation, dried, saturated with argon, and stored over potassium mirror. Elemental analyses were performed on a VARIO EL (Heraeus). The melting point was determined in a sealed capillary under argon and is uncorrected. ButPCl2 was prepared according to the literature procedure [15].
4.2 Data collection and structure refinement of 1b
The data of 1b were collected on a CCD Oxford Xcalibur S (λ(MoKα) = 71.073 pm) using ω and φ scans mode. Semi-empirical from equivalents absorption corrections were carried out with SCALE3 ABSPACK [16]. The structures were solved with direct methods [17]. Structure refinement was carried out with SHELXL-97 [17]. All non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed in calculated positions and refined with calculated isotropic displacement parameters. Table 4 lists crystallographic details. CCDC 768441 (1b) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Crystallographic data for 1b.
| 1b | |
| Empirical formula | C28H68LiN4P5 |
| Formula mass (g mol−1) | 622.65 |
| Collection T (K) | 130(2) |
| Crystal system | Monoclinic |
| Space group | Cc |
| a (pm) | 1939.3(2) |
| b (pm) | 1135.45(3) |
| c (pm) | 1982.1(2) |
| α (deg) | 90 |
| β (deg) | 115.689(1) |
| γ (deg) | 90 |
| V (nm3) | 3.9332(6) |
| Z | 4 |
| ρcalcd (Mg m−3) | 1.052 |
| Absorption coefficient (mm−1) | 0.254 |
| F(000) | 1368 |
| Crystal size (mm3) | 0.6 × 0.4 × 0.3 |
| θ range (°) | 2.65 to 25.51 |
| Range h, k, l | −23 ≤ h ≤ 23 |
| −13 ≤ k ≤ 13 | |
| −24 ≤ l ≤ 24 | |
| Data/restraints/parameters | 7248/2/363 |
| Goodness-of-fit on F2 | 1.069 |
| Final R indices [I > 2σ(I)] | R1 = 0.0592 |
| wR2 = 0.1557 | |
| R indices (all data) | R1 = 0.0666 |
| wR2 = 0.1591 | |
| Largest diff. peak and hole (e·Å−3) | 0.551 and −0.326 |
| Absolute structure parameter (Flack parameter) | 0.00(13) |
4.3 Synthesis of [Li(tmeda)2][cyclo-(P5But4)] (1b)
A solution of PCl3 (2.89 g, 0.021 mol) and ButPCl2 (13.35 g, 0.084 mol) in THF (50 mL) was carefully added at 0 °C to lithium sand prepared by trituration of Li (1.75 g, 0.25 mol) in THF (300 mL). The reaction mixture was then warmed to room temperature and heated under reflux for 2 d. After 1 h, the initially colorless solution had turned yellow-green, and it changed to dark brown after one day. THF was then removed under reduced pressure and the brown residue extracted with n-hexane (450 mL) to give a green-brown solution. The solvent was removed under reduced pressure to give a brown oily solid (Li[cyclo-(P5But4)] (1), 8.20 g, 85%). Recrystallization of this solid from THF/TMEDA (10/1) gave orange needles of [Li(tmeda)2][cyclo-(P5But4)] (1b; m.p. 139.3–141.6 °C) at −27 °C. Yield of 1b 7.32 g (56%); IR (KBr): 2951 (s), 2889 (s), 2857 (s), 2368 (w), 2287 (w), 1908 (w), 1648 (w), 1458 (s), 1386 (m), 1359 (s), 1261 (s), 1169 (s), 1046 (s), 897 (m), 806 (s), 667 (w), 559 (m), 500 (w), 481 (m), 440 (w), 406 (w); 1H NMR (400 MHz, d8-THF, 25 °C): δ = 1.21 (d, 3J(P,H) = 3.0 Hz, 9H; But), 1.26 (d, 3J(P,H) = 3.0 Hz, 9H; But), 1.29 (d, 3J(P,H) = 3.0 Hz, 18H; But), 2.16 (s, 24H, Me of TMEDA), 2.31 (s, 8H, CH2 of TMEDA); 13C{1H} NMR (100.6 MHz, d8-THF, 25 °C): δ = 30.4 (m, C(CH3)3), 31.1 (m, C(CH3)3), 32.6 (br, C(CH3)3), 33.4 (br, C(CH3)3), 41.8 (s, Me of TMEDA); 56.7 (s, CH2 of TMEDA). 31P{1H} NMR (Table 1); FAB MS, matrix: 3-NBA; m/z (%): 383.1 (19.7) [M+ − Li(tmeda)2], 327.1 (100.0) [M+ − Li(tmeda)2 − But + H], 270.1 (18.5) [M+ − Li(tmeda)2 − 2 × But + H], 239.0 (14.1) [M+ − cyclo-(P5But4)]; elemental analysis (%) found (calcd for C28H68LiN4P5, M = 622.65 g/mol): C 54.01 (53.59); H 11.01 (10.77); N 9.00 (9.25).
Acknowledgements
We thank Ministerio de Educación y Ciencia (FPI fellowship for R.H.) and the Alexander von Humboldt-Stiftung (Humboldt-Fellowship for S.G.-R.), the Deutsche Forschungsgemeinschaft (He 1376/22-2) and the EU-COST Action CM0802 “PhoSciNet” for financial support.
1 The signs for the coupling constants 1J(P,P) were set as negative, and the remaining signs and all the other coupling constants were obtained with the program SPINWORKS.
| Compound | δA (ppm) | δBB’ (ppm) | δCC’ (ppm) | 1J(PA−PB) = 1J(PA−PB’) (Hz) | 1J(PB−PC) = 1J(PB’−PC’) (Hz) | 1J(PC−PC’) (Hz) | 2J(PA−PC) = 2J(PA−PC’) (Hz) | Reference |
| 1b | −92.5(1) | 86.2(1) | 73.8(1) | −394.9(1) | −328.7(1) | −313.4(1) | 0.8(1) | This work |
| Na[cyclo-(P5But4)] | −105.6(1) | 82.7(1) | 75.0(1) | −379.2(1) | −317.3(1) | −309.4(1) | 0.1(1) | 7 |
| K[cyclo-(P5But4)] | −104.4(1) | 86.9(1) | 79.8(1) | −376.9(1) | −319.4(2) | −312.3(1) | 0.9(1) | 2h |