

1 Introduction
The coordination chemistry and biochemistry of vanadium compounds have long been of great interest and have been studied from different points of view [1,2]. Vanadium compounds are extensively used not only in industry but also they have been proven to play a very limited role in biology. Vanadium nitrides or carbides are used for increasing the strength of steel [3] and vanadium pentoxide is used as a catalyst in manufacturing sulfuric acid [4] and as an oxidizer in maleic anhydride production [5] and also used to make ceramics [6]. On the other hand, vanadium participates in many enzymatic reactions such as halogenation of organic substrates by haloperoxidases [7], nitrogen fixation by vanadium nitrogeneses [8]. Many studies have shown that the complexes of vanadium lower glucose levels both in vitro and in vivo [9–12]. In other words, oxovanadium(IV) complexes have been extensively used as catalyst in the oxidation of a variety organic compounds [13–21].
In other words, selective oxidations of alkenes to the epoxides is one of the most important and fundamental reaction in organic chemistry both at the laboratory and industrial scale, and numerous catalysts such as Schiff base complexes [22–25] or metalloporphyrins [26–30] have been developed using a variety of reagents to achieve this purpose.
We report here the preparation and partial characterization of two oxo-vanadium(IV) Schiff base complexes and their application as catalyst on the oxidation of cyclooctene and styrene with tert-butyl hydroperoxide. The influence of the electronic and structural requirements of the ligand on the catalytic activity of the catalyst has also been investigated.
2 Experimental
2.1 Instruments and reagents
Infrared spectra were recorded as KBr pellets using Unicam Matson 1000 FT-IR. Elemental analysis (C, H, N) were performed using a Heraeus Elemental Analyzer CHN-O-Rapid (Elemental-Analysesysteme KBr pellets, Gmbh, West Germany). 1H and 13CNMR spectra were obtained in CDCl3 solutions with a Bruker FT-NMR 250 (250 MHz) spectrophotometer. The residual CHCl3 in conventional 99.8 atom% CDCl3 gives a signal at δ = 7.26 ppm, which was used for calibration of the chemical shift scale, and 13C{1H}NMR chemical shifts are reported relative to CDCl3 (77.0 ppm). A digital melting point measuring device (Electrotermal 9100) was used. A double beam spectrophotometer (Shimadzu, UV-240) was used for the UV-Vis absorption determination. A varian (AA220) flame atomic absorption spectrometer was used for vanadium determination. A Metrohm 757 VA Computrace was employed to evaluate electrochemical measurements at room temperature (25 °C) under nitrogen with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte. The reaction products of oxidation were determined and analyzed by HP Agilent 6890 gas chromatograph equipped with a HP-5 capillary column (phenyl methyl siloxane 30 m × 320 μm × 0.25 μm) and flame-ionization detector.
1,2-Diaminopropane (Merck, ≥ 98%), 1,3-diaminopropane (Merck, ≥ 98%) 2-hydroxy-5-chloroacetophenone (Merck, ≥ 98%), 2-hydroxy-3-methoxybenzaldehyde (Merck, ≥ 98%) naphtaldehyde, oxobis(pentane-2,4-dionato)vanadium (IV) (Merck, ≥ 98%) and tert-butylhydroperoxide (Merck, solution 80% in di-tert-butylperoxide) were used as received. Solvents were dried and distilled by standard methods before use [25]. Cyclooctene (Fluka, ≥ 90%) and styrene (Fluka, ≥ 99%) purity was checked by gas chromatographic analysis before using. Solvents for electrochemical experiments were re-distilled and passed through a column of activated alumina.
2.2 Preparation of the ligands
2.2.1 Preparation of H2{saln-1,2-pn(5-Cl)2}
To a stirred ethanolic solution (30 mL) of 1,2-diaminopropane (0.074 g, 1 mmol), 2-hydroxy-5-chloroacetophenone (0.341 g, 2 mmol) was added. The bright yellow solution was stirred and heated to reflux for 2 h. A yellow precipitate was obtained that was filtered off, washed with diethyl ether. Yield (95%), Mp 115 °C. Anal. Calc. for C19H20N2Cl2 (379.28): C, 60.16; H, 5.31; N, 7.38. Found: C, 60.21; H, 5.16; N, 7.49%. Selected FT-IR data, υ (cm−1): 2870–2900 (CH), 1615 (CN), 1492 (CC), 1138 (CO). 1H NMR (δ): 1.33, 1.35 (d, 3H, NCH2CH(CH3)N), 2.20, 2.30 (s, 6H, (CH3)CN), 3.67, 3.70 (d, 2H, NCH2CH(CH3)N), 4.17–4.24 (m, 1H, NCH2CH(CH3)N), 6.76–7.59 (m, 6H, ArH), 15.73, 15.98 (s, 2H, OH). 13C{1H}NMR (δ): 19.82 NCH2CH(CH3)N), 54.99 (NCH2CH(CH3)N), 56.44 (NCH2CH(CH3)N), 19.82 (CH3CN), 119.85–161.90 (aromatic C), 170.10, 171.77 (CN).
2.2.2 Synthesis of H2{saln-1,3-pn(3-OMe)2}
To a stirred ethanolic solution (10 mL) of 1,3-diaminopropane (0.074 g, 1 mmol), 2-hydroxy-3-methoxybenzaldehyde (0.308 g, 2 mmol) was added. The brown solution was stirred and heated to reflux for 1 h. A brown precipitate was obtained that was filtered off, washed with diethyl ether. Yield (90%), Mp 125 °C. Anal. Calc. for C19H22N2O4 (342.39): C, 66.65; H, 6.47; N, 8.18. Found: C, 66.72; H, 6.53; N, 8.22%. Selected FT-IR data, υ (cm−1): 2830–2961 (CH), 1630 (CN), 1476 (CO), 1092 (CO). 1HNMR (δ): 2.07–2.15 (q, 2H, NCH2CH2CH2N), 3.70–3.75 (t, 4H, NCH2CH2CH2N), 3.93 (s, 6H, OMe), 6.77–6.94 (m, 6H, ArH), 8.36 (s, 2H, HCN), 14.93 (s, 2H, OH). 13C{1H}NMR (δ): 31.60 NCH2CH2CH2N), 36.13 (NCH2CH2CH2N), 56.07 (OCH3), 113.92–151.82 (aromatic C), 165.55 (CN).
2.3 Preparation of oxo-vanadium(IV) complexes
Both complexes were prepared by a general procedure: the ligand, H2{saln-1,2-pn(5-Cl)2} (0.38 g, 1 mmol) or H2{saln-1,3-pn(3-OMe)2} (0.34 g, 1 mmol) was dissolved in 20 mL of ethanol. An ethanolic solution of oxobis(pentane-2,4-dionato)vanadium(IV) (0.265 g, 1 mmol) was added to above solution and the reaction mixture was refluxed for 3 h. The colored solution was concentrated to yield colored powders. The products were washed with warm ethanol. General structure of oxo-vanadium (IV) complexes is shown in Fig. 1 and analytical and physico-chemical data of the complexes are presented in Table 1.

General structure of oxovanadium(IV) complexes.
Physical and analytical data of the complexes.
| Compound formula | Formula weight | Yield (%) | Color | Founded (calculated) | |||
| %C | %H | %N | %V | ||||
| VOL1 | 444.21 | 90 | Green | 51.93 (51.37) | 4.42 (4.08) | 6.02 (6.31) | 11.53 (11.47) |
| VOL2 | 407.31 | 85 | Orange | 56.32 (56.03) | 4.98 (4.95) | 6.32 (6.88) | 12.63 (12.51) |
2.4 General oxidation procedure
Catalytic experiments were carried out in a 50 mL glass reaction flask fitted with a water condenser. In a typical procedure, 0.032 mmol vanadyl complexes were dissolved in 10 mL chloroform. Then 10 mmol alkene (cyclooctene or styrene) was added to the reaction mixture and 30 mmol TBHP was added. The reaction mixture was refluxed for 6 h. The reaction products were monitored at periodic time intervals using gas chromatography. The oxidation products were identified by comparison with authentic samples (retention times in GC).
3 Results and discussion
3.1 Characterization of the ligands and oxo-vanadium(IV) complexes
3.1.1 IR spectral studies
A practical list of IR spectral data is presented in Table 2. Comparison of the spectra of the complexes with the ligands provides evidence for the coordination mode of ligand in catalysts. A sharp band appearing at 1615–1630 cm−1 due to υ(CN) (azomethine), shifts to lower wave number by 7–15 cm−1 and appears at 1600–1623 cm−1. This indicates the involvement of azomethine nitrogen in coordination. IR spectra of 1 shows υ(VO) at 992 cm−1 and for 2 at 861 cm−1. Tetradentate Schiff base oxovanadium (IV) complexes generally show υ(VO) around 860 cm−1 for polymeric form and around 970 cm−1 for monomeric form. Thus, VO{saln-1,2-pn(5-Cl)2} and VO{saln-1,3-pn(3-OMe)2} are assigned to be polymeric (Fig. 2) and monomeric structure, respectively [31,32].
IR spectral data of ligands and vanadium complexes.
| Compound | ν(VO) | ν(CC) | ν(CN) | ν(CH) |
| H2{saln-1,2-pn(5-Cl)2} | – | 1492 | 1615 | 2870–2900 |
| H2{saln-1,3-pn(3-OMe)2} | – | 1476 | 1630 | 2830–2961 |
| VO{saln-1,2-pn(5-Cl)2} | 992 | 1530 | 1600 | 2890–3000 |
| VO{saln-1,3-pn(3-OMe)2} | 861 | 1461 | 1623 | 2938–2815 |
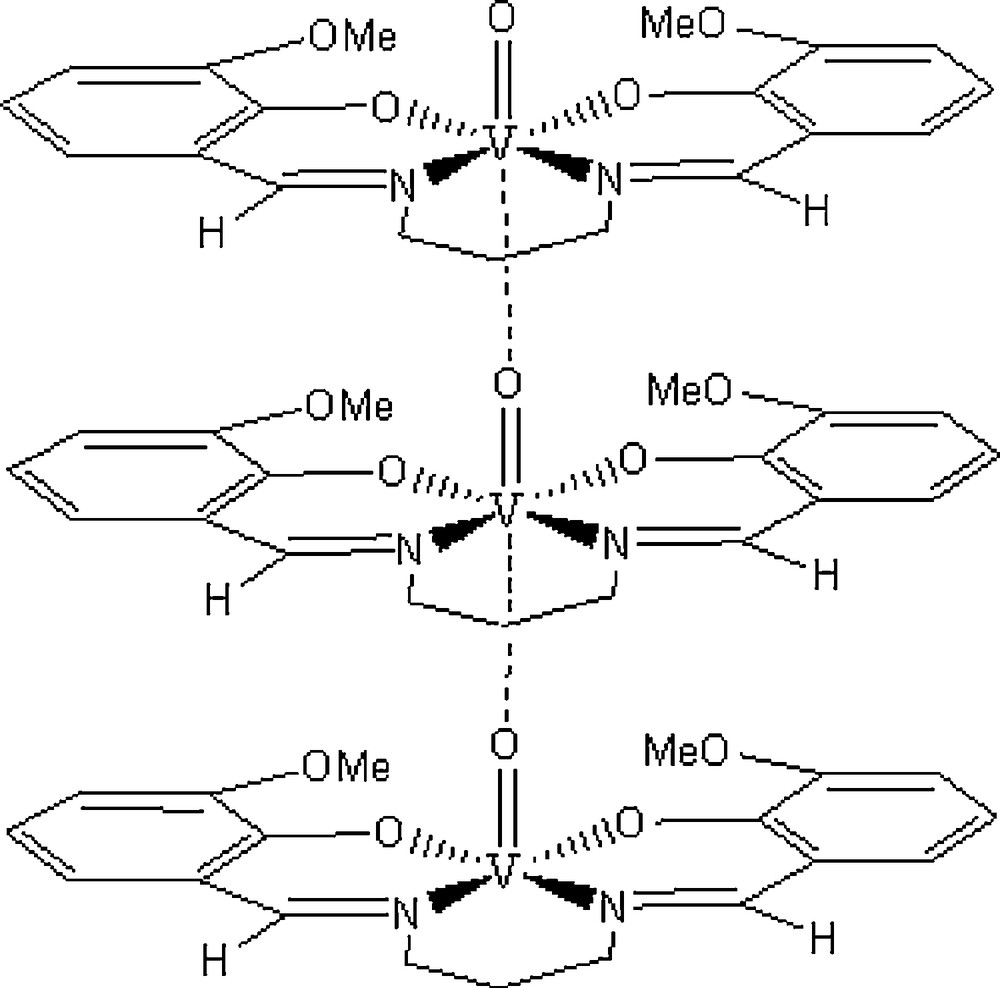
Polymeric structure of 2.
3.1.2 Electronic spectral studies
Table 3 provides electronic spectral data of the ligands and complexes along with their assignments. The electronic spectra of the ligands recorded in CHCl3 exhibit two bands between 253–420 nm that is due to π→π* and n→π* ligand transitions. Appearance of a lower intensity band at 368 nm (in 1) and 383 nm (in 2) are due to ligand to metal charge transfer (LMCT) transition [18,33]. In addition, both complexes display a weak broad band 575 for 1 and 528 for 2 nm due to expected d–d transition.
UV-Vis data for ligands and complexes.
| Compound | λmax (nm) (ɛ, M−1 cm−1) | Assignment |
| H2L1 | 337 (11 690) | π→π* |
| 420 (8020) | n→π* | |
| H2L2 | 253 (22 830) | π→π* |
| 417 (2870) | n→π* | |
| VOL1 | – | π→π* |
| 263 (30 880) | n→π* | |
| 368 (7590) | LMCT | |
| 575 (110) | d–d | |
| VOL2 | – | π→π* |
| 288 (28 460) | n→π* | |
| 383 (6870) | LMCT | |
| 528 (100) | d–d |
The electronic spectra of 1 and 2 in various solvents have been recorded and the absorption spectra in the d–d region are presented in Table 4. The results show that d–d transition band shifts to higher energy upon going from chloroform to coordinating solvents. Both complexes exhibit a different d–d spectrum in coordinating solvents. From IR and electronic spectra of the 2, we conclude that this complex has a six-coordinate structure in the solid state (achieved by V-O-V bridging) and a six-coordinate structure in donor solvents (achieved by coordination of a solvent molecule) but exhibit a five-coordinate square-pyramidal structure in non-donor solvents, while VO{(salnptn(3-OMe)2} has a five-coordinate, square-pyramidal structure in the solid state and six-coordinate structure in donor solvents.
d–d transition band for the 1 and 2 in various solvents.
| Solvent | Chloroform | Acetonitrile | Acetone | Ethanol | Methanol |
| Donor number | – | 14.1 | 17 | 19 | 20 |
| λ (nm) 1 | 590 | 588 | 584 | 579 | 575 |
| λ (nm) 2 | 550 | 547 | 540 | 534 | 525 |
3.2 Electrochemical studies
Electrochemical studies were carried out in anhydrous acetonitrile by cyclic voltammetry at room temperature. Fig. 3 shows the cyclic voltammograms of 0.01 mmol of 1 and 2 complexs in acetonitrile at various scan rates to show peak separation. As can be seen in Fig. 3, the redox processes were reversible, and the cathodic peak current of the complexes were increased and the peak shifted to more positive potentials direction with increasing the scan rates, which is a good evidence for high redox electroactivity of these complexes. Redox potentials for two complexes are given in Table 5. The redox potential for the metal center of 1 shifts in the cathodic direction. It seems that the electronic effects due to the presence of two methyl substituents at imine bonds and the two chlorine atoms at the aryl groups of 1 cause the decreased redox potential of 1 with respect to that of 2. It should be noted that OCH3 and Cl groups are considered as the π electron-donating groups in electrophilic substitution reactions of substituted benzenes [34]. However, the result of electrochemical studies suggests that the neat electron-donating effects of the two methyl and two chlorine substituents of 1 was larger than that of the OCH3 groups of 2.
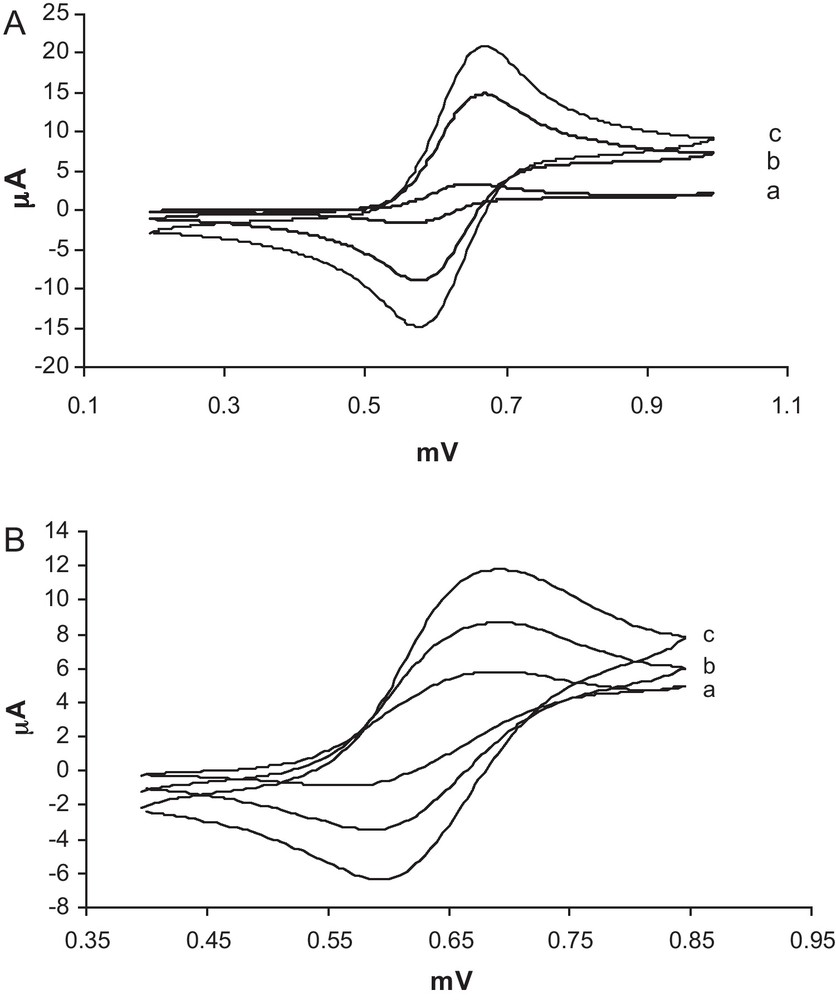
Cyclic voltammograms of 0.01 mmol 1 (A) and/or 2 (B) in acetonitrile solution containing 0.1 M TBAPF6 as a supporting electrolyte. Scan rates were (in mV/s) (a) 20, (b) 50 and (c) 100, respectively.
Electrochemical data for the oxidation of oxovanadium(IV) complexes in CH3CN.
| Complex | Eap (V) | Ecp (V) | ΔEp (V) | E1/2 (V) |
| 1 | 0.675 | 0.570 | 0.105 | 0.622 |
| 2 | 0.685 | 0.590 | 0.095 | 0.637 |
3.3 Catalytic activity studies
3.3.1 Oxidation of cyclooctene
Oxidation of cyclooctene with tert-butyl hydroperoxide as oxidizing agent in the presence of catalytic amount of 1 and 2 was carried out.
Different reaction parameter such as reaction solvent, temperature, reaction time, oxidant concentration and also catalyst concentration that may affect the conversion and selectivity of the reaction has been optimized.
Figs. 4 and 5 illustrate the influence of the solvent nature and also reaction time in the catalytic epoxidation of cyclooctene by 1 and 2. Ethanol, acetonitrile, chloroform and dichloromethane were used as solvents. It is clear from the plots that maximum cyclooctene conversion (98% for 1 and 93% for 2) was obtained in chloroform as a non-coordination solvent. It seems that coordinating solvent may weakly coordinate to the vacant site of VO species and prevent the activation of the TBHP. Excellent yield of the epoxide (100%) obtained using these catalytic method display the superior catalytic activity of the present oxo-V(IV) complexes. It was observed that oxidation of cyclooctene required 6 h for completion.
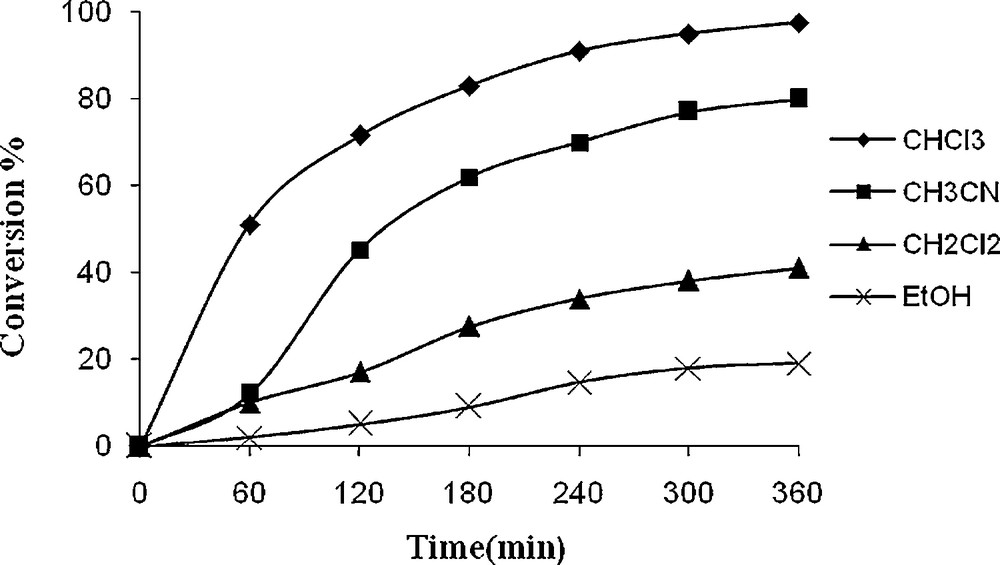
Effect of different solvent on the oxidation of cyclooctene as a function of time. Reaction conditions: cyclooctene (110 mg, 10 mmol), 1 (14.4 mg, 0.032 mmol), TBHP (336 mg, 30 mmol), solvent (10 mL) and reflux.
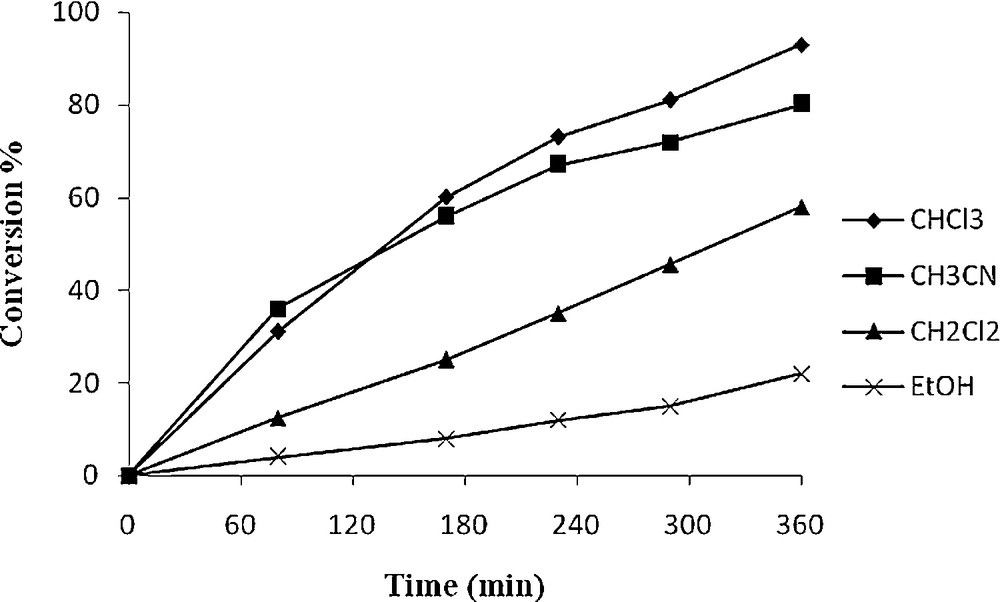
Effect of different solvent on the oxidation of cyclooctene as a function of time. Reaction conditions: cyclooctene (110 mg, 10 mmol), 2 (13 mg, 0.032 mmol), TBHP (336 mg, 30 mmol), solvent (10 mL) and reflux.
In order to find suitable reaction temperature, reaction mixture was stirred at three different temperatures (viz. 40, 50 and 60 °C) and the obtained results are presented in Table 6. It is clear from the Table 6 that increment of temperature increases the conversion and at 60 °C, maximum conversions were achieved.
Oxidation of cyclooctene with TBHP in the presence of 1 and 2 in different temperatures after 6 h.
| Temperature (°C) | Conversion (%) 1 | Conversion (%) 2 | Selectivity (%) |
| 60 | 98 | 93 | 100 |
| 50 | 95 | 90 | 100 |
| 40 | 73 | 82 | 100 |
In order to optimize the oxidant concentration, four different cyclooctene to TBHP molar ratios, viz. 1:0.5, 1:1, 1:2 and 1:3 were taken in chloroform and the reaction was carried out at reflux condition (Figs. 6 and 7). The formation of cyclooctene oxide was regularly analyzed at time intervals. As illustrated in Fig. 6, the oxidation of cyclooctene improved from 43 to 91% upon increasing the cyclooctene to TBHP molar ratio from 1:0.5 to 1:2. The oxidation remained nearly constant upon further increasing this ratio to 1:3, which suggested that a large amount of oxidant is not an essential condition to improve the oxidation of cyclooctene. This observation may be due to some differences between the kinetics of reaction performed in the presence of the two catalysts. The rate-determining step of the reaction seems to be difference for the reaction catalyzed by 1 and 2. It should be noted that, the steric hindrance around the reaction center of the active oxidation intermediate is difference for 1 and 2.
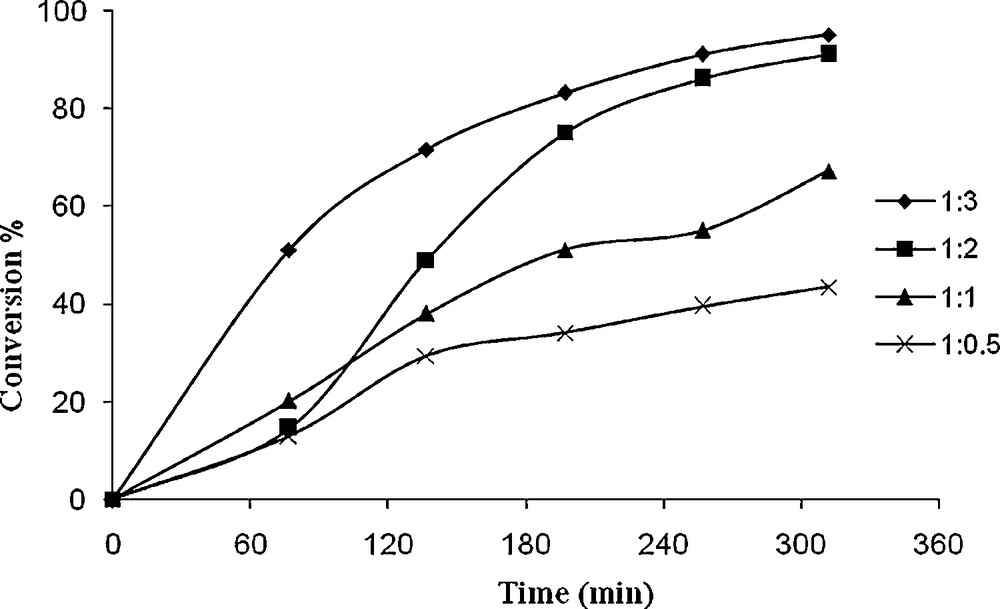
Effect of amount of TBHP on the oxidation of cyclooctene as a function of time. Reaction conditions: cyclooctene (110 mg, 10 mmol), 1 (14.4 mg, 0.032 mmol), solvent (10 mL) and reflux.
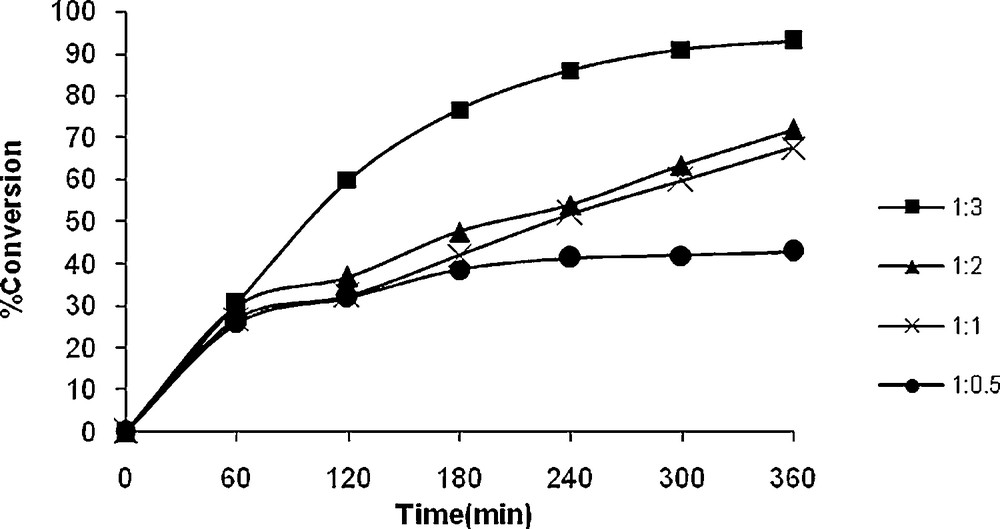
Effect of amount of TBHP on the oxidation of cyclooctene as a function of time. Reaction conditions: cyclooctene (110 mg, 10 mmol), 2 (13 mg, 0.032 mmol), solvent (10 mL) and reflux.
The effect of catalyst concentration on the performance of catalyst was studied and the results are presented in Table 7.
Effect of amount of catalyst on the cyclooctene oxidation.a
| Amount of catalyst (mg) | Conversion % for 1a | Amount of catalyst (mg) | Conversion % for 2 | Selectivity (%) |
| Without catalyst | 8 | Without catalyst | 8 | 100 |
| 11 | 89 | 10 | 90 | 100 |
| 14.4 | 98 | 13 | 93 | 100 |
| 22 | 92 | 20 | 92 | 100 |
a Reaction conditions: cyclooctene (110 mg, 10 mmol), solvent (10 mL), oxidant (30 mmol) and reflux. Reaction time: 6 h.
The highest conversion was obtained with 14.4 mg of catalyst (1) and 13 mg of catalyst (2) and further increments of catalyst hardly improve the conversion. This may be due to the higher degradation of TBHP at higher concentration of catalysts [35] or may interpreted in terms of thermodynamic and mass transfer limitations at higher reaction rates. [36].
3.3.2 Oxidation of styrene
Oxidation of styrene with TBHP in presence of catalytic amounts of 1 and 2 gave styrene oxide and benzaldehyde as major products. However, the formation of at least five products has been reported [37]. Production of benzaldehyde is possibly due to further oxidation of styrene oxide by TBHP [38].
To get optimized reaction conditions for the maximum conversion of styrene, the effect of various parameters viz. amount of oxidant (moles of TBHP per mole of styrene), catalyst, temperature of the reaction mixture and reaction solvent were studied. The influence of the solvent nature in the catalytic oxidation of styrene by 1 and 2 were studied. Acetonitrile, chloroform and dichloromethane have been used as solvent and results illustrated in Tables 8 and 9. Higher conversion was observed in acetonitrile than in chloroform or dichloromethane may be due to the higher polarity of acetonitrile (relative dielectric constants, ɛ/ɛ0 = 37.5) with respect to the chloroform (ɛ/ɛ0 = 4.9) or dichloromethane (ɛ/ɛ0 = 9.1) [39].
Effect of various solvent on the oxidation of styrene in the presence of 1.a
| Solvent | Conversion (%) | TON (Total) | Product selectivity (%) | |
| Styrene oxide | Benzaldehyde | |||
| Acetonitrile | 98.8 | 309 | 48 | 52 |
| Chloroform | 67.5 | 211 | 57 | 43 |
| Dichloromethane | 38 | 119 | 53 | 47 |
a Reaction conditions: cyclooctene (110 mg, 10 mmol), 1 (14.4 mg, 0.032 mmol), solvent (10 mL) and reflux. Reaction time: 6 h.
Effect of various solvent on the oxidation of styrene in the presence of 2.a
| Solvent | Conversion (%) | TON | Product selectivity (%) | |
| Styrene oxide | Benzaldehyde | |||
| Acetonitrile | 98.5 | 308 | 57.7 | 42.3 |
| Chloroform | 22 | 62 | 58 | 42 |
| Dichloromethane | 20 | 62.5 | 44 | 56 |
a Reaction conditions: cyclooctene (110 mg, 10 mmol), 2 (13 mg, 0.032 mmol), solvent (10 mL) and reflux. Reaction time: 6 h.
In order to find the effect of TBHP concentration in the oxidation of styrene, different TBHP to styrene molar ratios were taken in acetonitrile and the reaction was carried out at reflux. The oxidation of styrene improved from 24 to 98.8% (in 1 after 6 h) (Fig. 8) and 27 to 98.5% (in 2 after 6 h) (Fig. 9) upon increasing the styrene to TBHP molar ratio from 1:0.5 to 1:3.
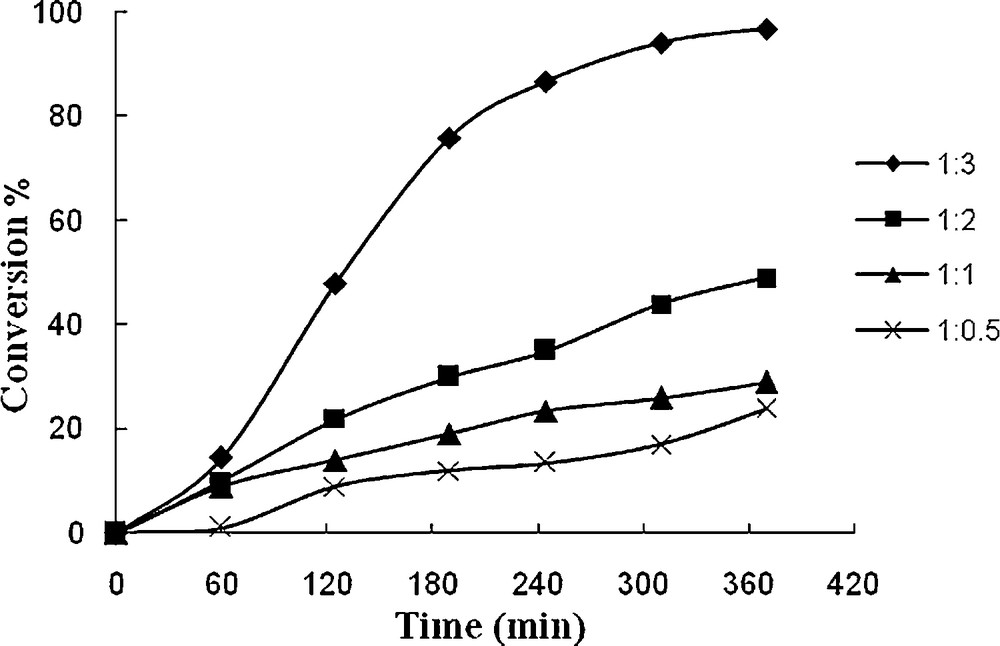
Effect of amount of TBHP on the oxidation of styrene as a function of time. Reaction conditions: styrene (104 mg, 10 mmol), 1 (14.4 mg, 0.032 mmol), solvent (10 mL) and reflux.
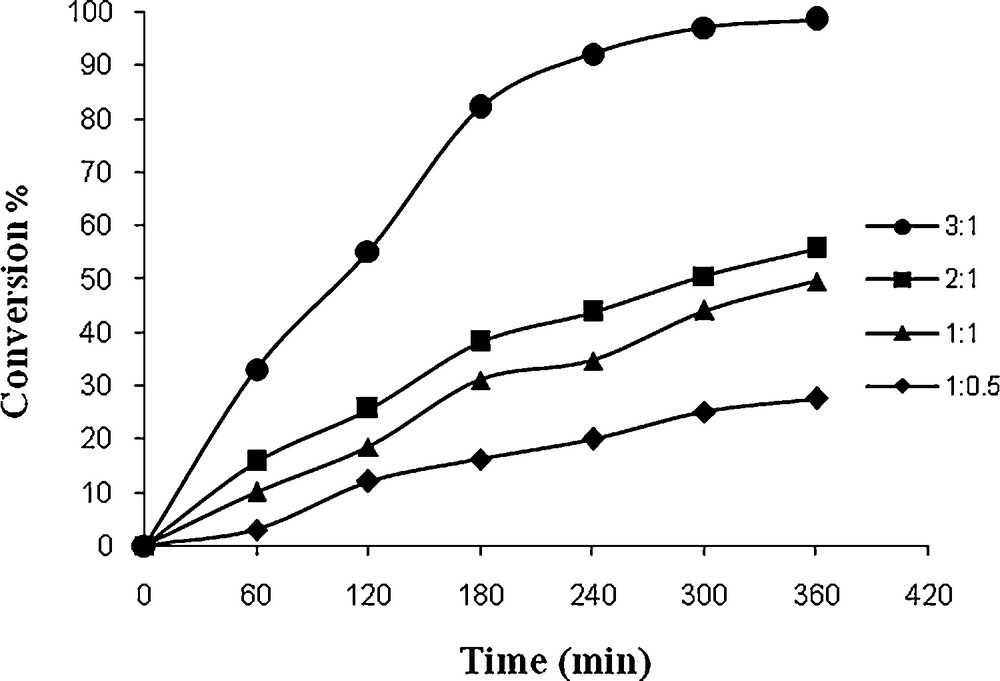
Effect of amount of TBHP on the oxidation of styrene as a function of time. Reaction conditions: styrene (10 mg, 10 mmol), 2 (13 mg, 0.032 mmol), solvent (10 mL) and reflux.
It could be found from Figs. 10 and 11 that only 10% styrene could be converted in the absence of catalyst, indicating that the catalyst is crucial for the oxidation reaction. As depicted in Figs. 10 and 11, it was observed that the yield is considerably enhanced when the catalyst was used. Besides, 14.4 mg catalyst (1) and/or 13 mg catalyst (2) was optimum amount of catalyst for oxidation of styrene.
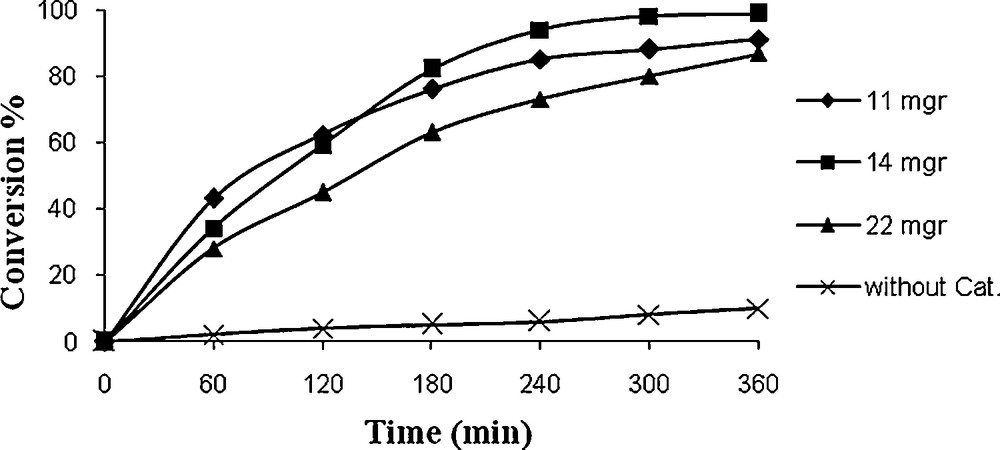
Effect of catalyst (2) weight on oxidation of styrene. Reaction conditions: styrene (10 mg, 10 mmol), TBHP (336 mg, 30 mmol), solvent (10 mL) and reflux.
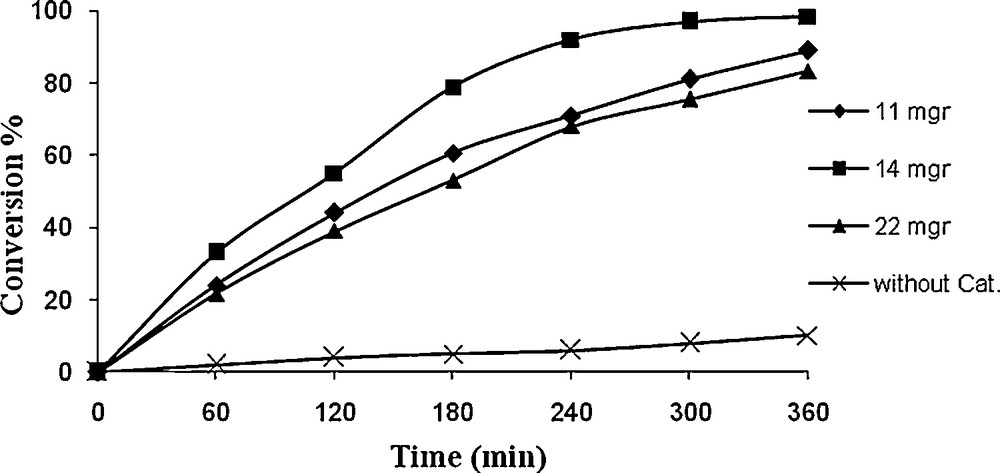
Effect of catalyst (2) weight on oxidation of styrene. Reaction conditions: styrene (10 mg, 10 mmol), TBHP (336 mg, 30 mmol), solvent (10 mL) and reflux.
Table 10 shows that oxidation of styrene depend upon temperature of the reaction. Therefore, lowering the temperature causes a decrease in styrene conversion.
Oxidation of styrene with TBHP in the presence of 1 and 2 in different temperatures after 6 h.
| Temperature (°C) | Conversion (%) 1 | Conversion (%) 2 | Selectivity (%) |
| 80 | 98.8 | 98.5 | 100 |
| 70 | 70 | 70 | 100 |
3.3.3 Proposed mechanism
In order to establish possible reaction pathway, titration of 1 and/or 2 (10−4 M in methanol) with methanolic solution of TBHP was monitored by electronic absorption spectroscopy.
As shown in Fig. 12, the intensity of the weak broad band due to d–d transition (363 nm for 1 and 554 nm for 2) decreases on drop wise addition of TBHP and the intensity of the band at 368 or 383 nm decreases on drop wise addition of TBHP to the methanolic solution of 1 or 2. The observed result confirms that initially, oxovanadium(IV) oxidized to oxovanadium(V) (Scheme 1, I) and then another TBHP will be activated by coordination to the oxovanadium(V) center. Consequently, an alkylperoxidic intermediate (Scheme 1, II) will be formed and then nucleophilic attack of olefin on the electrophilic oxygen atom covalently bonded to the metal leading to the formation of epoxide product [40–42].
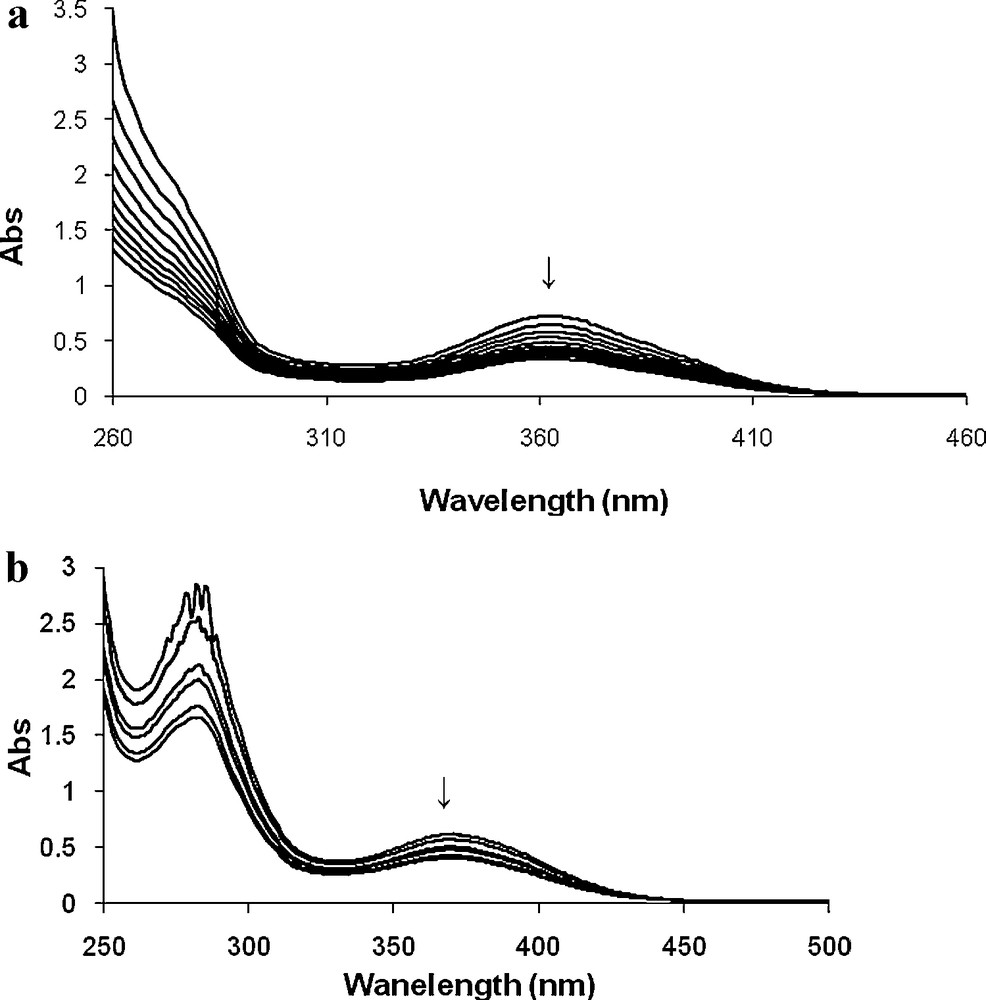
Titration of 1 (a) and 2 (b) with TBHP. Spectra were recorded after the successive addition of one drop portions of TBHP dissolved in methanol to 10 mL of ca. 10−4 M solution of 1 (a) or 2 (b).
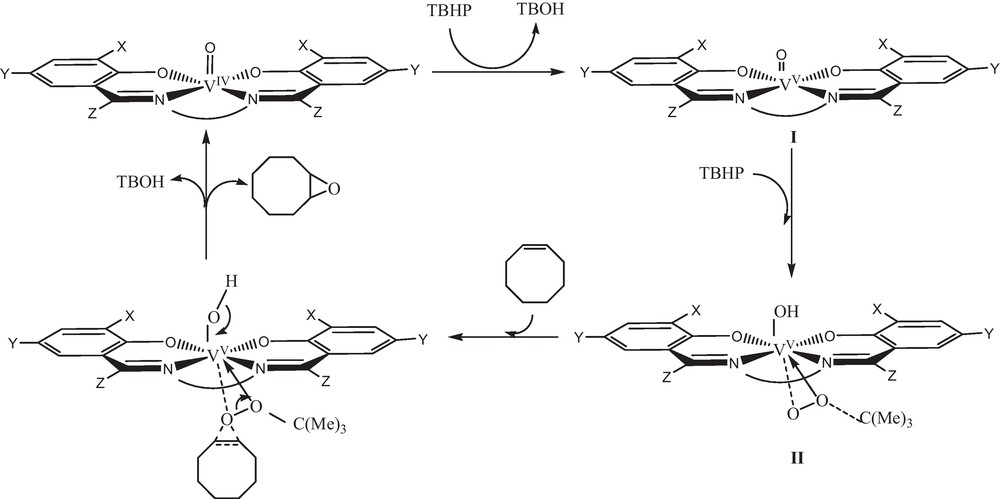
Proposed mechanism.
The influence of the electronic and structural requirements of the Schiff base ligand on the catalytic activity was investigated (Figs. 13 and 14). The order of catalytic activity for oxidation of cyclooctene was found to be 1 > 2 (Fig. 13). As expected, an electron-donating group (Cl) in the salicylidene ring (1) increases the effectiveness of the catalyst, resulting from the easier oxidation of the vanadium(IV) to the vanadium(V) and therefore, the activation of TBHP.
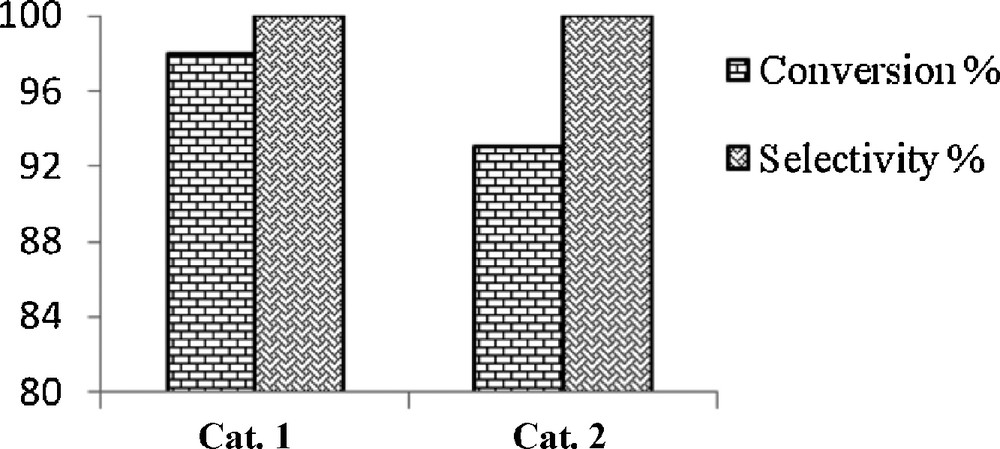
Conversion and selectivity of the 1 and 2 complexes in the epoxidation of cyclooctene by TBHP.
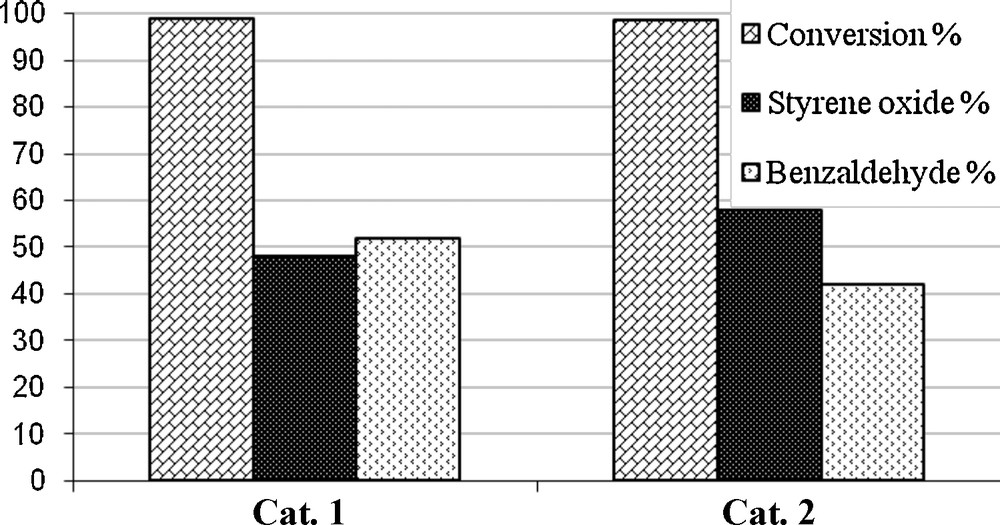
Conversion and product distribution of the 1 and 2 complexes in the oxidation of styrene by TBHP.
The higher selectivity of the used catalysts may be due to the both electronic effects as well as the steric effects of the substituents at different positions of the Schiff base complexes. We have discussed the point previously and compared the catalytic activity of the electron-rich Schiff base complexes compared to the electron-deficient ones [15,22]. Therefore, electron-rich Schiff base complexes are more efficient than electron-deficient ones.
A comparison of the results obtained for our present catalytic system with those reported in the literature which used TBHP as oxidizing agent [43–45] reveal that higher conversion and selectivity for epoxide formation will be achieved.
4 Conclusions
In summary, a highly efficient epoxidation method using TBHP activated by simple VO(VI)-Schiff base complexes has been developed. To investigate the influence of the electronic demands of the ligands on the catalytic activity of vanadium complexes, two VO(IV) complexes, as N2O2 tetradentate Schiff base ligands have been synthesized. It was observed that electron-donor groups on the salicylidene ring of ligand enhance the effectiveness of the catalyst. Moreover, the epoxide yields show that the efficiency of catalytic system is strongly dependent on the temperature and nature of solvent, and the best yields in the oxidation of cyclooctene were achieved in chloroform and at reflux while in the oxidation of styrene, 100% conversion was obtained in acetonitrile at reflux.
Acknowledgements
The financial support of this work by K.N. Toosi University of Technology research council is acknowledged.
Vous devez vous connecter pour continuer.
S'authentifier