

1 Introduction
The discovery in 1971 of the AyMo6Q8 (Q = chalcogen, A = cation) Chevrel phases in the Solid State Chemistry Laboratory of Rennes University [1,2] was the starting point in this laboratory for a new research field on Mo6 and Re6 chalcohalide cluster chemistry isolated at high temperature (1000–1200 °C) by solid state routes [3,4]. Notice that before the 1970s, the molybdenum and rhenium chalcohalides were obtained at moderate temperature – usually below 500 °C – and they were built from single cations, two-atom or triangular clusters, frequently with polysulfide groups located perpendicularly to the metal-metal bond, as in MoS2Cl3 or Mo3Cl4S7 [5–11]. The high-critical field superconducting properties of Chevrel phases stimulated an extensive research in order to explain and to improve their superconducting behaviour [12]. Their electronic structure has been the subject of many theoretical studies, which clearly evidenced that the number of valence electrons per octahedral cluster (VEC) in the HOMO level plays the essential role in their superconducting properties considering that these phases, built up from Mo6Q14 building blocks, exhibit some molecular character [13]. A simple way to modify the VEC was to change the Ay cationic stoichiometry, as shown by a great number of examples [12]. Two other original routes to change the VEC were to replace divalent chalcogen by monovalent halogen, for instance in Mo6S6X2 (X = Br, I) isotypical with Mo6S8 [14,15] or to replace a part of molybdenum (d5s1) in the Mo6 cluster by rhenium (d6s1), as in the mixed cluster compound Mo2Re4S8 [16]. The two last possibilities were investigated for the first time in the Rennes labs and they were the starting point of an important development of Mo6 and Re6 chalcohalide solid state chemistry presented below.
In this review, we will describe the crystal chemistry of Mo6 and Re6 chalcohalides synthesized at high temperature by solid state routes and structurally characterized by single crystal X-ray diffraction, starting from the 3D Chevrel phases in which the cluster building blocks are interconnected in the three directions, and decreasing the dimensionality to 2D, 1D architectures, down to 0D compounds built from discrete cluster units. In a second step we will show how the latter discrete units are, for several years, the object of an intensive use as individual nano-sized molecular precursors in soft chemistry for rebuilding 3D, 2D or 1D hybrid organic/inorganic cluster compounds and for the elaboration of nanocluster materials with potential applications in nanotechnology owing to their specific optical, redox and magnetic properties. As mentioned above, this review focuses essentially on chalcohalides. Nevertheless, some examples of pure halides and chalcogenides will be included when pertinent for comparison.
An aim of this article is to complete and update several previous accounts [17–25], and to remind us of a 50 year story of Mo6 and Re6 cluster chalcohalides in solid state chemistry. We have chosen to systematically refer to original papers, even when very old, in order to get a memory of them, because they are often forgotten by the timely used computer-assisted bibliographic tools and their users. Indeed, “if nobody remembers something, it did not ever exist” [26].
2 The M6Li8La6 building block
All the M6 (M = Mo, Re) chalcohalides are built from the nanometer-sized [(M6Li8)La6] building block in which the octahedral cluster, consisting in six metal atoms in d4 electronic configuration (MoII or ReIII) linked together by metal-metal bonds, is always bonded to 14 ligands. Indeed, this metal cluster (in the sense of the restricted definition of Cotton [27]) is face-capped by eight inner ligands (noted Li), while six apical ligands (noted La) are in apical positions that build a square-pyramidal environment around each metal atom (Fig. 1). A ligand shared between two units is noted La-a, Li-i or Li-a depending on the shared positions. This notation, which is very useful for describing schematically all the compounds built from the M6L14 units, was proposed by Schäfer and von Schnering [28].

The M6Li8La6 cluster unit.
Interestingly, for mixed chalcogen/halogen M6Li8La6 clusters, it is worth mentioning that, owing to the difference of valence and electronegativity between halogen and chalcogen, La and La-a positions are occupied preferentially by halogens, while Li-i and Li-a are occupied in priority by chalcogens. The non-shared Li positions (μ3-Li) can be occupied by either halogens or chalcogens leading almost systematically in the averaged structural model to a random occupancy of the inner sites by both of them, a situation often described as a “rotational disorder” (in the following, when both Qi and Xi appear in a developed formula it follows a Qi/Xi disorder on the corresponding inner positions). Indeed, formal metal (donor)-halogen/chalcogen (acceptor) interactions occur to satisfy the electronic demands of the latter (Q2− and X− to obey the octet rule). Consequently, the more negatively charged chalcogens will seek to bind a larger number of positively charged metal atoms than the less negatively charged halogens [29,30]. Notice that no exception to this rule has been observed up to now in the great number of Mo6 and Re6 chalcohalides. Of course, in the case of pure M6 chalcogenide chemistry, the chalcogens occupy all the ligand type positions, for instance in the Re6 ternary chalcogenides [31]
The molecular orbital diagram of these [(M6Li8)La6] units can theoretically be built from the frontier orbitals of six square-pyramidal ML5 fragments. The 12 highest molecular orbital occupied by electrons are metal-metal bonding in character with the HOMO level formed by a doubly degenerated orbital of eg symmetry considering an Oh symmetry for the unit. These metal-metal bonding orbitals are filled for 24 valence electrons per cluster (VEC) that constitutes the “magic number” in Mo6 and Re6 cluster chalcohalide chemistry [13,32–37]. Indeed, the M6 chalcohalides obtained up to now by solid state chemistry exhibit usually a VEC value of 24, with the exception of Chevrel phases for which the VEC value can vary from 20 to 24. In the latter case the interactions between cluster units lead to a band structure giving the possibility of metallic conduction when the bands are partly filled, while the other chalcohalides with a VEC value of 24 are generally semiconducting or insulating. Notice that an extremely rare example of a compound in which coexist discrete units with VEC = 24 and 23 has been isolated in solid state chemistry (Section 3.3.2).
3 Solid state compounds
3.1 Three-dimensional M6 chalcohalides
3.1.1 Three-dimensional Qi-a/Qa-i connections
The first discovered and structurally characterized 3D M6 chalcohalides were Mo6Q8-xXx (X = halogen, Q = chalcogen and 0 < x < 2) [14,15], isotypic with AyMo6Q8 (A = cations with various charges) Chevrel phases built from the [(Mo6Qi2Qi-a6/2)Qa-i6/2] three-dimensional framework [2]. In these chalcohalides the units are interconnected in the three directions by inner-apical chalcogen double bridges (Qi-a/Qa-i) in the same way as in Chevrel phases, while inner halogen atoms (Xi) are located on the threefold axis of the unit that builds channels developing in the three directions of the space.
When Q = S, definite compounds with x = 2 have been obtained: Mo6S6Br2 and Mo6S6I2 [(Mo6Xi2Qi-a6/2)Qa-i6/2] (Fig. 2). A slight defect on Q and X positions was observed in Mo6I1.76S5.96 [38]. While Mo6S8 (VEC = 20) is superconducting only at very low temperature (less than 2 K), Mo6S6X2 (VEC = 22) is superconducting at Tc = 13.8 K and 14 K for X = Br and I, respectively, and with high-critical field like for PbMo6S8 (VEC = 22) [14]. This VEC value of 22 corresponds to a maximum in the density of states near the Fermi level that should explain the optimal superconducting properties [39]. Notice that Mo6S8 (VEC = 20) is not thermodynamically stable and cannot be obtained by direct synthesis at high temperature, while Mo6S6X2 are prepared around 1200 °C like the other AyMo6Q8 phases and they are very stable. It was assumed that this stability is due to firstly, the addition of electrons in the metal-metal bonding orbitals and secondly, the location of two X halogens close to each other on the threefold axis leading to a van der Waals bond, partly filling the distorted vacancy at the origin of the unit-cell, that is occupied by the big A cations in AyMo6Q8 [12]. In Mo6S8 the corresponding two sulfur atoms are too small to be in contact in this vacancy, leading to a poor stability of this compound. Note that, in agreement with the latter assumption, “Mo6S6Cl2” (VEC = 22) has never been stabilized because the origin site should be too strongly distorted to get the small chlorine atoms in contact to each other [40]. Recently, the difference between the Mo6S8 and Mo6S6Br2 stability was discussed by bond valence analysis showing that lattice strain caused by anion polarization leads to instability [30]. The magnetic field penetration in nanometer-sized particles of Mo6S6I2 has been investigated in relation with decreasing the particle size [41].
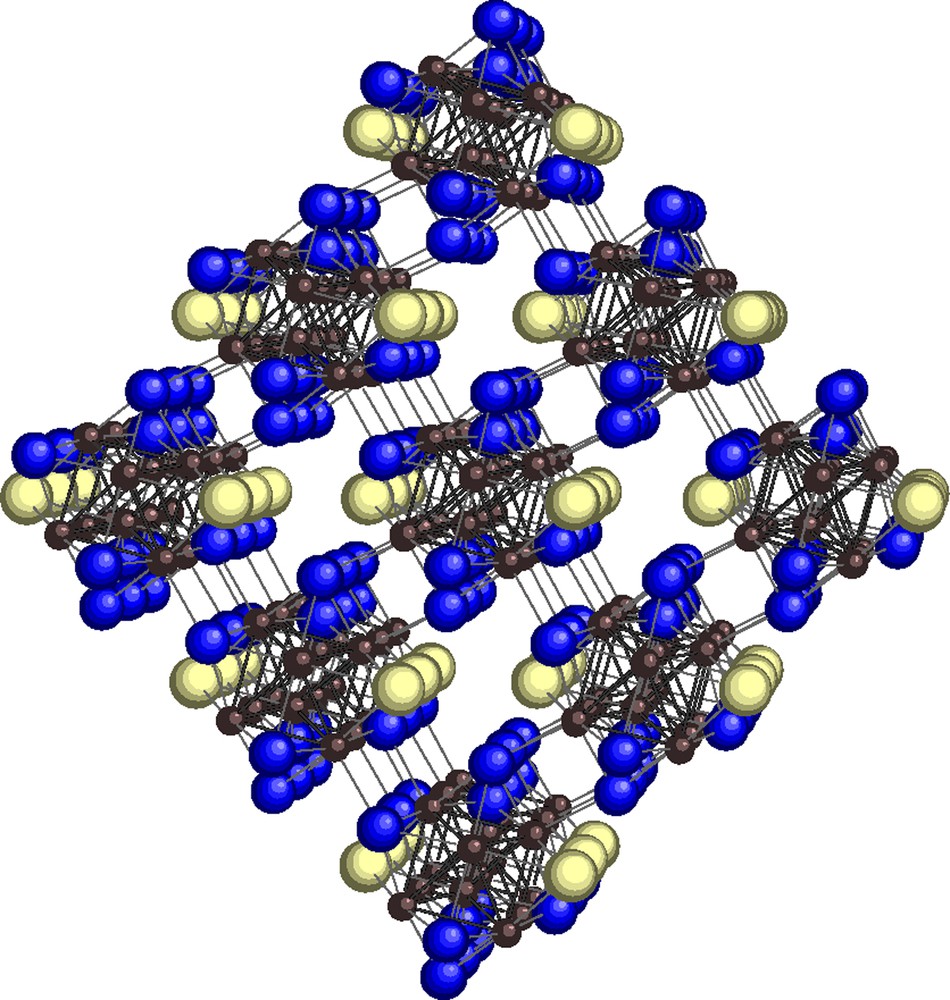
Crystal structure of Mo6S6Br2. S in blue (dark grey) Br in yellow (light grey) [15].
When Q = Se or Te, solid solutions Mo6Q8-xXx have been evidenced (X = Cl, Br, I and 0 < x < 2) with Tc increasing correlatively with the VEC value (Fig. 3) up to 7.0 K, 7.1 K, and 7.6 K for Q = Se and X = Cl, Br and I, respectively. Mo6Te8, not superconducting above 1 K, becomes superconducting after iodine substitution with a maximum Tc = 2.6 K for Mo6Te6I2 (Fig. 3). Note that analyses of X-ray powder diffraction data and superconducting critical temperature Tc performed for x > 2 evidenced a saturation of both the lattice constants and Tc, indicating that the halogen should only occupy the 2c site on threefold axis, as in Mo6S6Q2 with a maximum value x = 2. Thus, the various compositions of the solid solution are written as [(Mo6Qi2-xXixQi-a6/2)Qa-i6/2]. Indeed, as stated above, the halogen atoms should occupy only the inner sites and not the inner-apical ones.

Critical temperature Tc versus x stoichiometry of the superconductors Mo6Se8-xBrx, Mo6Se8-xIx and Mo6Te8-xIx [14] (top). Critical temperature Tc versus x stoichiometry and initial slope of the critical field of the superconductors Mo6Se8-xClx [40] (bottom).
The diffusion of small cations like Cu into the Mo6S6X2 host structure has been carried out at low temperature (450 °C) up to the limit of 1.5 Cu for X = Br [15] and 1.2 for X = I [42] with a decrease of the Tc down to 4.8 K for Cu1.2Mo6S6I2, related to the increase of the VEC above the ideal value of 22. Intercalation of sodium was performed by electrochemical reactions using Na/Mo6Se8-xIx (x = 0, 0.5, 1, 2) test cells. Cycling data indicate that intercalation process is initially irreversible and results in compounds of formula NayMo6Se8-xIx with y decreasing from 1 to 0 as x increases from 0 to 2, while after the first cycle the cells are readily reversible over several cycles [43].
The first Chevrel phase discovered with rhenium was Mo2Re4S8 built from a mixed octahedral cluster with 24 valence electrons [16]. This compound is a rare example of a semiconducting Chevrel phase, owing to the complete filling of the energy bands near the Fermi level by 24 valence electrons. Indeed, changing the Mo/Re ratio affects the VEC and, for instance, the Mo4Re2Te8 compound (VEC = 22) turns out to be superconducting with Tc = 3.55 K [16]. It is also possible to change the Mo/Re ratio as well as the nature of the chalcogen and then several solid solutions were studied:
- • Mo2Re4Se8 is much easier to synthesize than the sulfur homologue, and a solid solution Mo2Re4S8-xSex extends over the full range 0 ≤ x ≤ 8 while Mo2Re4Se8-xTex is severely limited to the range 0 ≤ x ≤ 1.2 [16]. All these diamagnetic compounds are semiconducting, as expected (Fig. 4), but they do not follow an Arrhenius behaviour, exhibiting hopping conduction;
- • as a consequence the synthesis of Mo2Re4Te8 failed. In place Mo4Re2Te8, with VEC = 22 was obtained [16]. This compound is, with Mo6Te6I2, a unique example of superconducting telluride with Chevrel phase structure;
- • attempts of synthesis of a solid solution like Mo6Se8–Mo2Re4Se8 failed although the two extremes exhibit exactly the same structure, even if slight deviations from the 1:2 Mo:Re ratio were detected by EPMA [44] or XRD data [45]. This would be due essentially to electronic factors. In the example of the similar Mo-Ru heteroclusters, two domains of homogeneity were clearly identified, a very narrow semiconducting one close to Mo4Ru2Se8 and a more extended metallic one at the Mo6Se8 border [46]. Also, this reminds us that, in Chevrel phases, the 24 VEC limit (and then semiconducting compounds) is very difficult to reach actually by cationic filling of the cavities by solid state chemistry: the unique example of truly semiconducting compound seems to be Ti0.9Mo6Se8 [47].

Temperature dependence of the resistivity of Mo2Re4S4Se4, Mo2Re4Se8, Mo2Re4Se7Te and Mo4Re2Te8 [16].
The heterometallic cluster compound Mo2Re4Se8, as well as Mo4Ru2Se8, was suggested as a potential material for thermoelectronic applications [44,48,49]. These compounds were also studied as catalysts for two important electrochemical reaction processes, hydrogen evolution and molecular oxygen reduction in acid medium, an important electrocatalytic reaction in polymer electrolyte membrane fuel cells, and showed significant improvement in comparison to Mo metal and Mo6Se8 [50]. The Mo-Ru heterometallic cluster was shown to present a catalytic behaviour comparable to platinum [51].
The attempts to obtain pure Re6 chalcohalide Chevrel phases similar to Mo6 ones was unsuccessful because they should imply a VEC value higher than 24 (Re: d6s1 instead of Mo: d5s1) corresponding to fill antibonding metal-metal orbitals. However, the possibility to obtain mixed Mo2Re4 and Mo4Re2 octahedral clusters stimulated an intensive research to stabilize Re6 clusters that should be obtained by replacing a part of chalcogen by halogen in Re6L14 unit. It was the starting point of the development of the Re6 chalcohalide chemistry. Note that at the same period the first example of a mixed Mo2Re2 tetrahedral cluster was reported [52] that reinforced our purpose of isolating a homometallic cluster with rhenium.
3.1.2 Three-dimensional Xa-a connections
In M6 chalcohalide chemistry, apical-apical interunit connections are only obtained with halogen (Xa-a) as expected. Some examples are found with Mo6 and Re6 clusters. For instance, the compounds Mo6X10Q (X = Cl, Br and Q = S, Se, Te; X = I and Q = Se, Te) (Pccn space group) [53], isotypical with Nb6I11 [54], are built from [(Mo6Xi7Qi)Xa-a6/2] units. The six Xa-a bridges that develop in the three directions of the space are different: for instance, four Mo-Cl-Mo bridges of 122.8° and two of 152.9° are found in Mo6Cl10Se. The thioiodide member has never been isolated due to the strong difference between the ionic radii of iodine and sulfur that should distort the cluster unit too much.
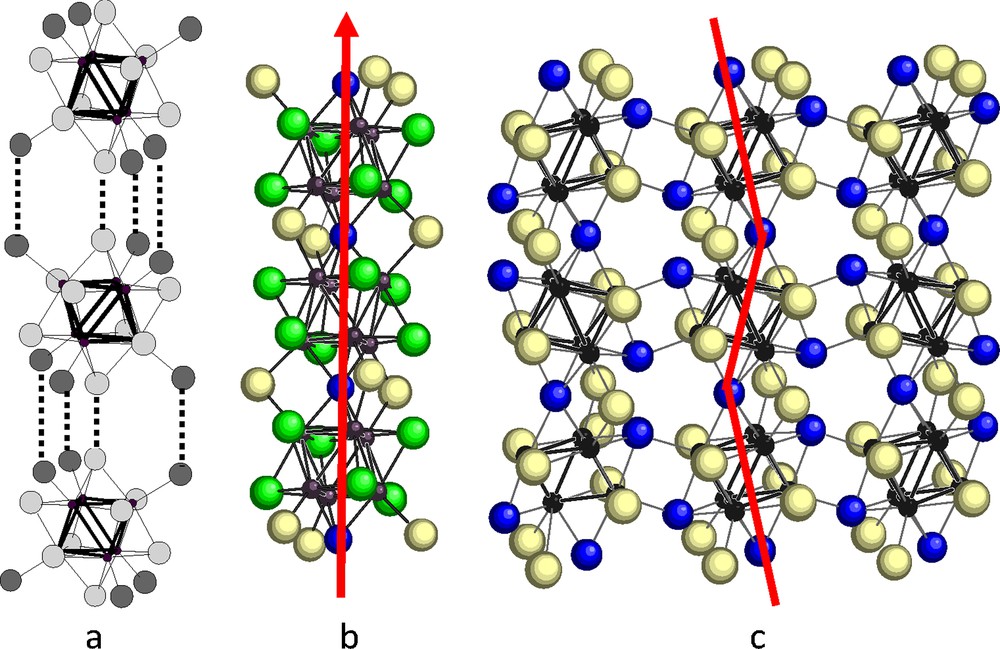
A similar stacking, but more symmetrical (R-3c space group), is observed in the rhenium chalcobromides Re6Q7Br4 (Q = S, Se) built from [(Re6Qi7Bri)Bra-a6/2] units [55,56]. Each cluster unit is here octahedrally surrounded by six vertex-sharing identical groups. The structure packing then derives from the ReO3 structure but a rotation of the cluster units leads to six identical bent halogen bridges of 115°. The angle of these bridges, much smaller than in Mo6X10Y leads to a more compact stacking (Fig. 5). Note that the corresponding selenochloride has a more distorted monoclinic structure [57], maybe closely related to the above mentioned molybdenum one. Unfortunately, its exact structure was not solved, due to severe twinning of the crystals.

Connection of a M6L14 cluster unit with the six adjacent units by 3D Xa-a bridges in the projection of Mo6X10Q (top) [53] and Re6Q7Br4 (bottom) [55] along the pseudo threefold axis or true threefold axis, respectively.
Finally, let us mention the presence of comparable 3D Qa-a connections in several compounds belonging to a rich series of Re6 cluster-based pure chalcogenides, where Q can be the usual chalcogen, but also a dichalcogen group [58–62]. Illustrating examples are found in the structures of Li4Re6S11 and K4Re6S12, with the structural formulas Li4[(Re6Si8)Sa-a6/2] and K4[(Re6Si8)Sa-a4/2(S-S)a-a2/2], respectively. Such compounds were studied for their catalytic properties, for instance hydrogenation and dehydrogenation of cyclohexanone [63] or condensation of primary amines, dehydrogenation of secondary amines and dealkylation of ternary amines [64]. Related to this series is the structure of the neutral Re2Te5, namely Re6Te15 [65–67]. Here the clusters are linked in the three directions by butterfly Te7 molecules, each of them being surrounded by 6 clusters, leading to the unit [(Re6Tei8)(Te7)a-a6/6].
In these 3D chalcohalides, the halogen/chalcogen distribution on the inner positions leads to a VEC value of 24 for all these series of chalcohalides and, in consequence, these compounds are insulating. The studies of dielectric permittivity performed for Mo6X10Q evidence dielectric relaxations. Indeed, the cluster units act locally as electrical dipoles due to the uneven halogen/chalcogen distribution around the Mo6 cluster. This random distribution of 1Q and 7X on the eight inner positions of the cluster unit gives eight possible orientations for the dipoles with two different orientations for each of them due to the symmetry of the unit-cell and, as a result, a strong disorder of dipoles in the framework. The dielectric permittivity is sensitive to temperature and frequency of the applied field [53,68,69]. Namely, the maximum of permittivity versus temperature shifts towards a lower temperature as the frequency decreases (Fig. 6), that is specific of a dipole glass, the electrical analogue of spin glasses observed in disordered systems [70]. Note that in the same conditions the isotypical Nb6I11 [(Nb6Ii8)Ia-a6/2] does not give any relaxation, due to the symmetrical arrangement of eight iodine atoms around the Nb6 cluster that prevents the presence of permanent dipoles in the structure [40]. It was reported that Re6S7Br4 exhibited a dielectric behaviour very close to that of Mo6Br10S [69], although the detail of the dielectric relaxations were not published.

Evolution of the dielectric constants ɛ′ and ɛ″ versus temperature at various frequencies for Mo6Cl10Q (Q = S, Se, Te) (top) and for Mo6I10Te (bottom) [53].
Photoelectrochemical investigations on Re6Se7Br4 single-crystals in aqueous electrolytes [71] indicated that this compound behaves as a p-type semiconductor photoelectrode. In the visible region, it exhibits considerable cathodic photocurrents. Unfortunately, this electrode was not stable under applied potentials; the band gap (indirect) is 1.78 eV.
3.1.3 Three-dimensional Xa-a and Qi-a/Qa-i mixed connections in Re6 chalcohalides
An illustrative example is given by the compounds Re6S8Cl2, Re6S8Br2 and Re6Se8Br2. The crystal structures, determined in the cases of Re6Se8Br2 [72] and Re6S8Br2 [73] show that the cluster units are here linked in two directions of the space by bent halogen Xa-a bridges, similar to those encountered in Re6Q7Br4, and in the third one by pairs of chalcogen Qi-a/Qa-i double bridges, similar to those present in Chevrel phases. Note that the latter were forgotten in [72], leading erroneously to some Re atoms deprived of their apical ligand [73]. Then, these compounds are in fact actually 3D and are written [(Re6Qi6Qi-a2/2)Qa-i2/2 Bra-a4/2]. The thiochloride Re6S8Cl2 also presents this structure [74,75]. The photoelectrochemical behaviour of Re6Q8Br2 (Q = S, Se) compounds was reported. Both of them act as p-type semiconductors and, assuming an indirect optical transition, have energy gaps of 0.84 (in good agreement with the value derived from optical absorption [72]) and 1.65 eV for the selenide and the sulphide, respectively, and appear stable in the photocathodic regime [73].
Note that the selenochloride parent Re6Se8Cl2 exhibits a very different structure, being strongly bidimentional (Section 3.2.3).
3.2 Two-dimensional and one-dimensional M6 chalcohalides
These compounds reviewed recently in detail [76], will be reported very briefly here.
3.2.1 Qi-i connections in 2D and 1D Mo6 chalcohalides
The inner-inner connection between two adjacent M6L14 cluster units, observed in Mo6 chalcohalides, has never been obtained with Re6 chalcohalides. Such a type of connection corresponds to condensate the M6L14 cluster units along one threefold axis of the L8 cube to form a polymeric chain of units in which the Li-i ligand is in prismatic environment of metal. Because the cluster units are rigid entities, three pairs of La condensate simultaneously with one pair of Li, giving a Li-i ligand surrounded by three La-a ligands (Fig. 7a).
a: schematic representation of the M6L14 cluster unit condensation along the threefold axis. The central cluster unit is 180° rotated around the threefold axis with respect to the two others leading to the formation of Li-i and La-a ligands that builds a prismatic environment of metal atoms around the Li-i ligand; b: the resulting 1D chain built from [(Mo6Xi5QiQi-i2/2)Xa-a6/2] cluster units in Mo6X8Q2 [77]. Q in blue (dark grey) Br in yellow (light grey), inner site randomly occupied by X and Q in green (middle grey); c: in Mo6Br6S3 [81], representation of three 1D chains built from [(Mo6Bri4Si-i2/2Si-a2/2)Sa-i2/2Bra-a4/2] cluster units, slightly tilted in order to locate two inner ligands in apical position for clusters of adjacent chains giving a 2D structure. S in blue (dark grey) Br in yellow (light grey). Masquer
a: schematic representation of the M6L14 cluster unit condensation along the threefold axis. The central cluster unit is 180° rotated around the threefold axis with respect to the two others leading to the formation of Li-i and La-a ligands that ... Lire la suite
These chains are observed in 1D Mo6Br8S2, Mo6I8S2 and Mo6I8Se2 i.e. [(Mo6Xi5QiQi-i2/2)Xa-a6/2] [77,78] (Fig. 7b). They are similar to the chains found in Nb6I9S reported later by J.D. Corbett et al., a rare example of Nb6L14 chalcohalide [79]. Van der Waals contacts between the chains ensure the (weak) cohesion of the structure. These compounds with 24 valence electrons per Mo6 cluster are insulating. Single crystals of Mo6I8Se2 oriented along or perpendicularly to the applied field exhibit a dielectric anisotropy with ɛ′///ɛ′⊥ ∼ 80 at room temperature [80].
In 2D Mo6Br6S3 [(Mo6Bri4Si-i2/2Si-a2/2)Sa-i2/2Bra-a4/2] similar chains are observed, but the units are now slightly tilted along the chain axis in order to locate two inner chalcogen atoms in apical position for two clusters of two adjacent chains, building layers of chains (Fig. 7c) [81]. Between adjacent layers exist van der Waals contacts. This 24 VEC compound is semiconducting with an activation energy of 0.12 eV calculated from single crystal measurement along the growing axis of the crystal [40].
Note that such Li-i connections, never observed in other M6L14 chalcohalides, were already found in Mo6 and Re6 chalcogenides like Ba4Mo12S18 [82] and K8[Re12CS17(CN)6] [83] built from dimeric cluster units, the Li-i position being occupied by S or C, respectively (Fig. 8). These dimeric units correspond to one link of the infinite chains observed in Mo6X8Q2 fibers. However, these compounds do not grow like fibers because in Ba4Mo12S18 [((Mo6)2Si8Si-iSi-a6/2)Sa-i6/2Sa-a3] these dimeric units are interconnected in the three directions by sharing six Si-a/Sa-i ligands with adjacent dimeric units building large channels, while they are discrete in K8[Re12CS17CN6] i.e. [((Re6)2Si14Ci-i)Sa-a3(CNa)6]. In Cs∼1Mo12S14 [84] the [(Mo6Si-i2/2Si-a6/2)Sa-i6/2] units are also interconnected by Si-i ligands, but the cluster units are now rotated around the threefold axis building an antiprismatic environment of molybdenum around Si-i that prevents any condensation of the Sa ligands in the form of Sa-a one (Fig. 9), in contrast to the compounds reported above, while Si-a ligands are now building a 3D architecture with large channels.
a: the dimeric [((Mo6)2Si8Si-iSi-a6/2)Sa-i6/2Sa-a3] cluster unit in Ba4Mo12S18. The six Sa ligands represented on the figure are in fact Sa-i in the structure, and the six Si ligand located above and below Si-i are in fact Si-a in the structure. From Gougeon et al. [82]; b: the dimeric [((Re6)2Si14Ci-i)Sa-a3(CNa)6]6− discrete unit in Cs6[Re12CS17CN6]. From Fedorov et al. [83].

Schematic representation of the cluster units condensation along the threefold axis building an antiprismatic environment of metal atoms around the Li-i ligand (left). Comparing with Fig. 7a, remark that all three clusters have now the same orientation. The 1D chain built from [(Mo6Si-i2/2Si-a6/2)Sa-i6/2] cluster units in Cs∼1Mo12S14.(right). Redrawn from Gougeon et al. [84]. All the Si and Sa ligands represented on the figure are in fact Si-a and Sa-i respectively in the structure.
3.2.2 Xa-a connections in two-dimensional and one-dimensional Re6 chalcohalides
The needle-shaped 1D Re6Se5Cl8, exhibits a crystal structure based on trans Cla-a bridged [(Re6Sei5Cli3)Cla-a2/2Cla4] cluster units. The zigzag chains are linked together only by van der Waals interactions [57]. The corresponding thiobromide was reported to present a related structure, although not isotypical, presumably related to a change in halogen bridge angle [85].
In the example of the 2D Re6Se6Cl6, two pairs of Cla-a bridges develop in a plane, while the remaining apical ligands are terminal and point towards the third direction [55,57]. Such interunit connections build layers of units interpenetrating perpendicularly to the layer plane. This compound, that can be written [(Re6Sei6Cli2)Cla-a4/2Cla2], is in fact strictly isostructural with the binary halides M6X12 (M = Mo, W), the first examples of octahedral clusters, discovered early by C. Brosset [86] and structurally characterized by H. Schäfer et al. [87]. Note that attempts to synthesize the analogous Re6S6Br6 have failed [85] presumably for steric reasons.
3.2.3 Qi-a/Qa-i connections in two-dimensional and one-dimensional Re6 chalcohalides
Accurate structural data were obtained for the fibrous 1D Cs2Re6S8Br4 [88], confirming previous reports [29,85,89,90]. The cluster units are linked by trans Qi-a/Qa-i double bridges, leading to linear chains [(Re6Qi6Qi-a2/2)Qa-i2/2Xa4]2− interacting together by coulombic forces via Cs+ cation.
In the structure of the 2D Re6Se8Cl2, trans Sei-a/Sea-i double bridges define a plane where the clusters are strongly interconnected, while in the third direction are terminal apical chlorine ligands. This compound, written as [(Re6Sei4Sei-a4/2)Sea-i4/2Cla2], then exhibits a specially strong bidimensional character, the cohesion of the layers being due only to weak van der Walls contacts [91]. Transport measurements confirmed an anisotropy of the resistivity of about 102 [74] for this n-type semiconductor with a direct band gap of 1.42 eV and photoconductive properties [92]. A solid solution around Re6Q8Cl2 was evidenced for a partial substitution of Se by S or Te [74], but a complete substitution by sulfur and/or substitution of chlorine by bromine led to the completely different three-dimensional Re6Q8Br2 structure, described in Section 3.1.3 above. Re6Se8Cl2 is easily exfoliated by, for instance, hydrazine [93].
3.2.4 Mixed Qi-a/a-i and Xa-a connections in two-dimensional Re6 chalcohalides
The two-dimensional CsRe6S8Br3 is built from rigid chains based on trans Si-a/Sa-i double bridges (refer to 1D Cs2Re6S8Br4 above), linked together by Bra-a bridges, building layers of cluster units. Then the framework is written [(Re6Si6Si-a2/2)Sa-i2/2Bra-a2/2Bra2]− [94] The structural cohesion is ensured by electrostatic interactions with Cs+ countercations sandwiched between the layers. This compound is isostructural with the iodine representative CsRe6S8I3 [29].
3.3 Discrete M6 chalcohalides
In such chalcohalides, the cluster units are not interconnected, implying a number of 14 ligands per M6 cluster in the formula of the compound. They have been isolated long ago with Re6 clusters, but more recently with Mo6 ones due to the recent interest of such compounds in solution chemistry. Indeed, they act essentially as salts and, because they are soluble in many polar solvents, they constitute the best M6 precursors for the elaboration of hybrid organic/inorganic M6 cluster compounds.
From a general point of view, these chalcohalide compounds lie near the halogen-rich border of the phase diagrams, because they can be written as [(M6Xi8-xQix)Xa6]n-, the six apical ligands being in priority halogens. Many of them present structures related to the cubic A2+Mo6X14 [95–98], to the trigonal Cs2Mo6X14 [99,100] or to the orthorhombic Cu2Mo6I14 [98] prototypes, depending on the size of the counteraction. Note an exception, illustrated by the structure of Cs10Re6S14 [101]. In this unique example, implying a very important anionic charge, sulfur occupies both the inner and apical positions because there is no choice, as mentioned above (in this system and related ones were reported a number of compounds, based on various organizations of apical sulfur bridges and even disulfur ones, not detailed in this report).
3.3.1 Ternary discrete M6 chalcohalides
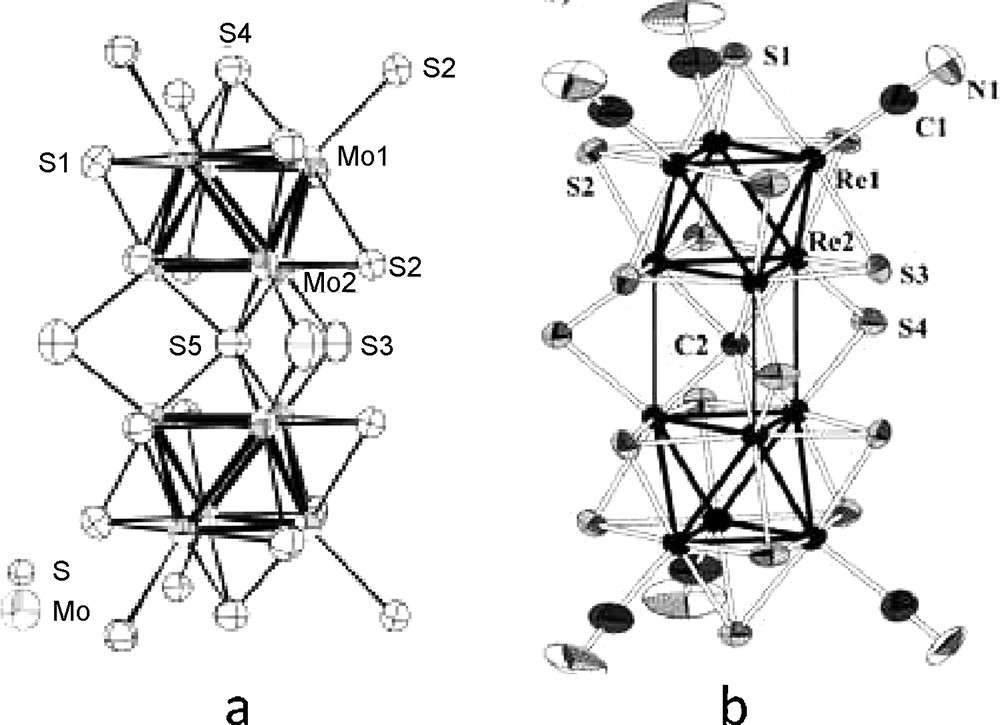
The family Re6Q4X10 is a unique example of discrete neutral clusters in this chemistry. Re6Se4X10 (X = Cl, Br) and Re6Te4Br10 were synthesised very early in a pioneering work by Opalovslii et al. [102,103] but logically formulated at that time as Re3Q2X5, in the absence of any structural data. Crystal structures were first determined independently in the example of Re6Se4Cl10 (Fig. 10) [55,104–106]. Complete structural data were reported subsequently for several parent compounds: Re6S4Br10 [85], Re6Se4Br10 [107], Re6Te4Cl10 [108] and Re6Te4Br10 [109]. The structure of Re6S4Cl10 was also solved: unit-cell and main distances were reported in the original paper [110] and structural data are given as Supplementary Material.
Crystal structure of Re6Se4Cl10 [106]. The inner ligands are omitted for clarity. One of the empty distorted octahedral site formed by the apical ligands of six adjacent cluster units is represented in blue (light grey).
Briefly, the structure is built from two independent [(Re6Qi4Xi4)Xa6] units, where the eight inner positions are randomly occupied by halogens and chalcogens, with a regular distribution in the examples of Re6Se4Cl10 [106] and Re6Te4Cl10 [108], whereas an irregular distribution was reported for Re6Te4Br10 [109]. Because the cluster units are neutral, the cohesion of the structure is done only by Q…Q or Q…X van der Walls contacts [106].
Dielectric behaviour of some of these compounds was studied in detail. Re6Se4Cl10 exhibits around 120 K a phase transition (step in ɛ′(T) and λ point in ɛ′′(T)) strongly sensitive to applied field and frequency, suggesting the coexistence of two close energy states. Single-crystals of Re6S4Br10 also show a phase transition and dielectric relaxations with a gaussian distribution of relaxation times, agreeing well with the statistical occupancy of the inner sites [106].
Note a second example of neutral cluster unit, but with some complex apical ligands, [(Re6Tei6Cli2)(TeCl2)a2Cla4]; this compound was synthesized at quite low temperature, 450 °C [111].
3.3.2 Quaternary discrete M6 chalcohalides
For this class of compounds, only a few examples are known in the case of molybdenum, while, in contrast, it is very rich in rhenium chemistry. This is obviously related to the oxidation state of the metal (Mo II vs Re III). Indeed, even the pure molybdenum halides (for instance, A2+[Mo6Cl14]2− [96]) are yet dianions, and any halogen-chalcogen substitution will result in increasing anionic charge in order to maintain the ideal 24 VEC value. In contrast, in rhenium chemistry, there is much space to play with such substitutions, as exemplified in the following.
With Mo6 clusters, only quaternary 0D compounds have been obtained with the general formula Ay[(Mo6Xi8-xQix)Xa6] with 0 ≤ x ≤ 2. Note that the Xi/Qi distribution on the eight inner positions is often but not systematically random, and such a distribution will be specified for the following Mo6 compounds. Up to now, the syntheses have been performed with X = Br and I, Q = S, Se and Te and with alkalin used as countercation. Definite compounds and solid solutions have been isolated and structurally characterized.
The two Cs4Mo6Br12Q2 (Q = S, Se) chalcobromides are definite compounds with a VEC value of 24 [112]. They are built from [(Mo6Bri6Qi2)Bra6]4− discrete units in which the eight inner positions are randomly occupied by six bromine atoms and two chalcogen atoms. Cesium cations, in complex coordination of inner and apical ligands, ensure the neutrality of the compounds.
With a rubidium cation a continuous solid solution is obtained in the Mo-Br-Se system, Rb2+x[(Mo6Bri8-xSeix)Bra6] (0.25 < x < 0.7) where coexist, in various ratios depending on the x value, [(Mo6Bri8)Bra6]2− and [(Mo6Bri7Sei)Bra6]3− cluster units, both of them with VEC = 24. In the latter unit deduced from the average structure, the eight inner positions are randomly occupied by seven bromine atoms and one selenium atom [113]. In contrast, in the Mo-Br-Te system a definite compound, Rb2.5Mo6Br13.5Te0.5, is obtained, built from the 24 VEC cluster units [(Mo6Bri8)Bra6]2− and [(Mo6Bri7Tei)Bra6]3− in 1:1 ratio. From the average structure one can deduce that in the latter unit only six of the eight inner positions are randomly occupied by bromine and tellurium atoms, while the two other inner positions located on the threefold axis of the unit are fully occupied by bromine atoms [113].
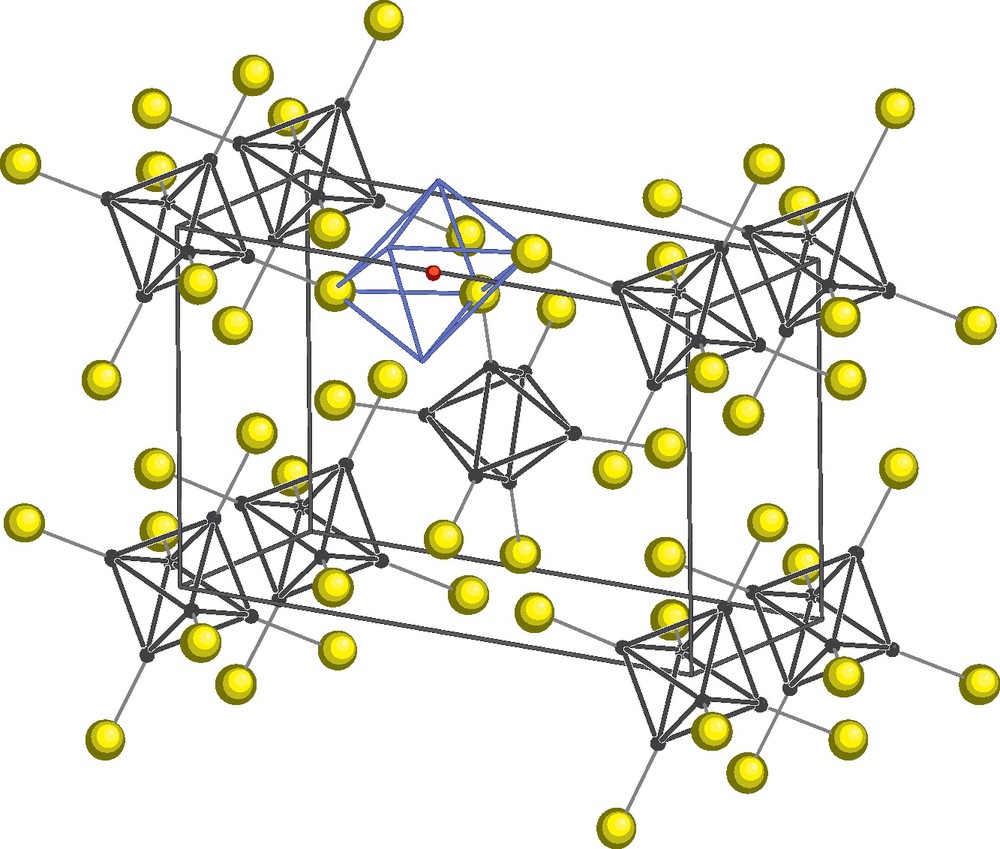
Increasing the rubidium ratio in the two Mo-Br-Q (Q = Se, Te) systems, Rb12Mo9Br31Q are obtained that can be written as double salts {Rb3[(Mo6Bri7Qi)Bra6]}[Rb3(MoBr6)]3. They are built from the coexistence of [MoIIIBr6]3− complex and [(Mo6Bri7Qi)Bra6]3− (VEC = 24) cluster units in which all the eight inner positions are randomly occupied by bromine and chalcogen atoms (Fig. 11). The magnetic susceptibility measured for Rb12Mo9Br31Te follows a Curie law with an experimental moment consistent with molybdenum at the three oxidation state in the [MoBr6]3− complex and a 24 VEC value for the cluster unit that is non magnetic [114].
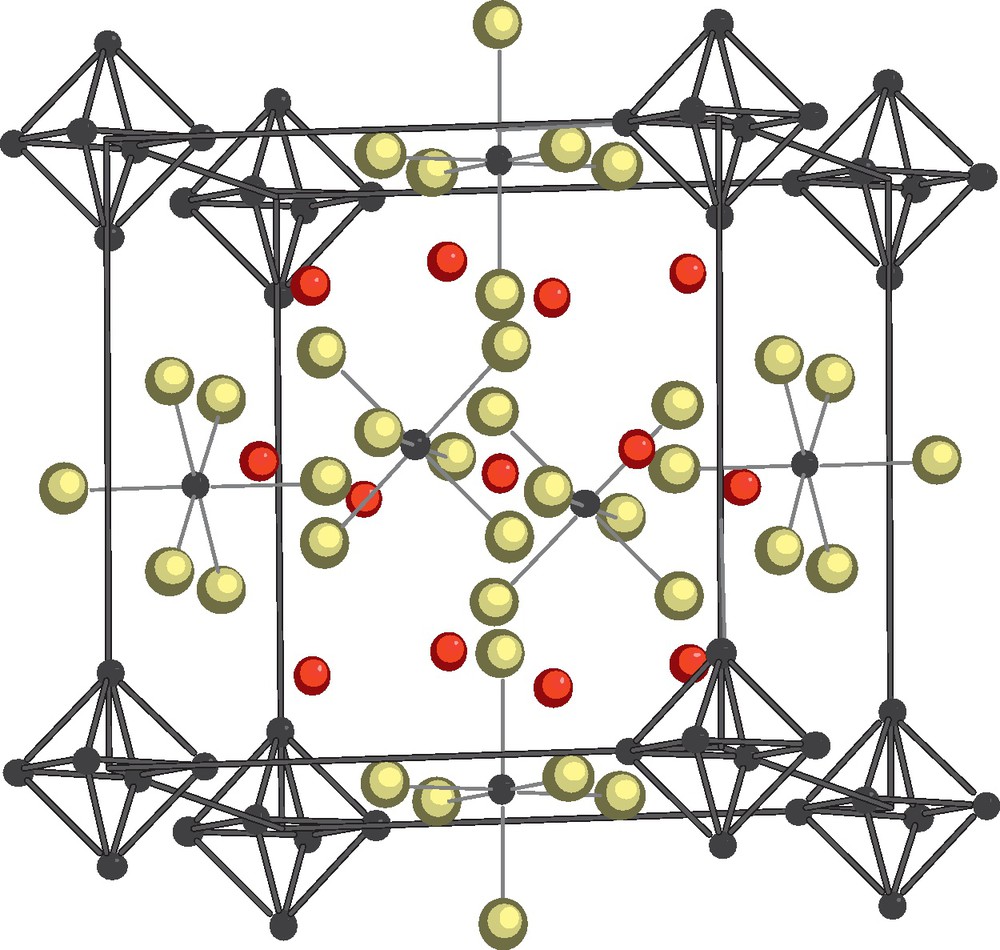
Crystal structure of the double salts {Rb3[(Mo6Bri7Qi)Bra6]}[Rb3(MoBr6)]3 [114]. The inner and apical ligands of the Mo6 clusters are omitted for clarity. Br in yellow (light grey) and Rb in red (dark grey).
In the Mo-I-Se system, with cesium atom as counter cation, the solid solution Cs3[(Mo6Ii6Ii2-xSeix)Ia6] (1.2 < x < 1.6) has been isolated [115]. It is based on a mixture of (2-x)[(Mo6Ii7Sei)Ia6]3− and (x-1)[(Mo6Ii6Sei2)Ia6]3− cluster units, in various ratios depending on x value, with VEC = 24 and 23, respectively. It constitutes an exceptional example where cluster units with two different VEC coexist in a same cluster compound. Two inner positions located on the threefold axis of the unit are randomly occupied by I and Se atoms. The 23 VEC cluster unit is magnetic due to one unpaired electron on the HOMO level of the Mo6L14 molecular orbital diagram and, as a consequence, the compound is paramagnetic as evidenced by a low temperature electron paramagnetic resonance (EPR) study. Note that a similar crystal structure has been encountered for Rb3Re6S7Br7 (see below) and for the Cs3Mo6Br13O oxybromide, with one oxygen atom randomly distributed on the two inner positions located on the threefold axis of the cluster unit [116]. However, in the latter compound, that was the first Mo6L14 compound with μ3-face-capping oxygen obtained by solid state chemistry, the structure is built only from one [(Mo6Bri7Oi)Bra6]3− unit in contrast to the chalcoiodides reported above which contain cluster units with two different VEC values.
In the case of Re6 clusters, the full series [(Re6Qi4+nXi4-n)Xa6]n− with 0 ≤ n ≤ 4 (including the neutral [(Re6Qi4Xi4)Xa6] Section 3.1.1) has been obtained, mainly with alkali countercations [85]. The n parameter is in most cases an integer although some non-integer values were encountered in a few examples of solid solutions and half-integer ones were reported in rare cases of definite compounds.
3.3.2.1 Mono-anionic cluster units
A number of compounds ARe6Se5Cl9 were early mentioned with A = Li, K, Rb, Cu, Ag [55] and completed by some additional compounds characterized by powder X-ray diffraction [117]. The prototype structure, solved in the example of KRe6Se5Cl9 [55,117], is cubic, isotypic with the ternary molybdenum halides like PbMo6Cl14 [96]. A close relationship with Re6Se4Cl10 (Fig. 10) was also evidenced: a twist of the cluster units in the latter opens octahedral sites where the countercations are located [19,55,107] (Fig. 12). The structure becomes slightly different with larger cations and/or larger apical ligands and, for instance:
- • KRe6S5Br9 is monoclinic, and the K+ ions are in prismatic sites instead of octahedral ones, the signature of steric constraints [118];
- • CsRe6Se5Cl9 is trigonal (Fig. 13) [55], related to Cs2Mo6Cl14 [99] as well as Cs(Rb)Re6S5Br9 [85];
- • RbRe6S5Cl9 is monoclinic [110].
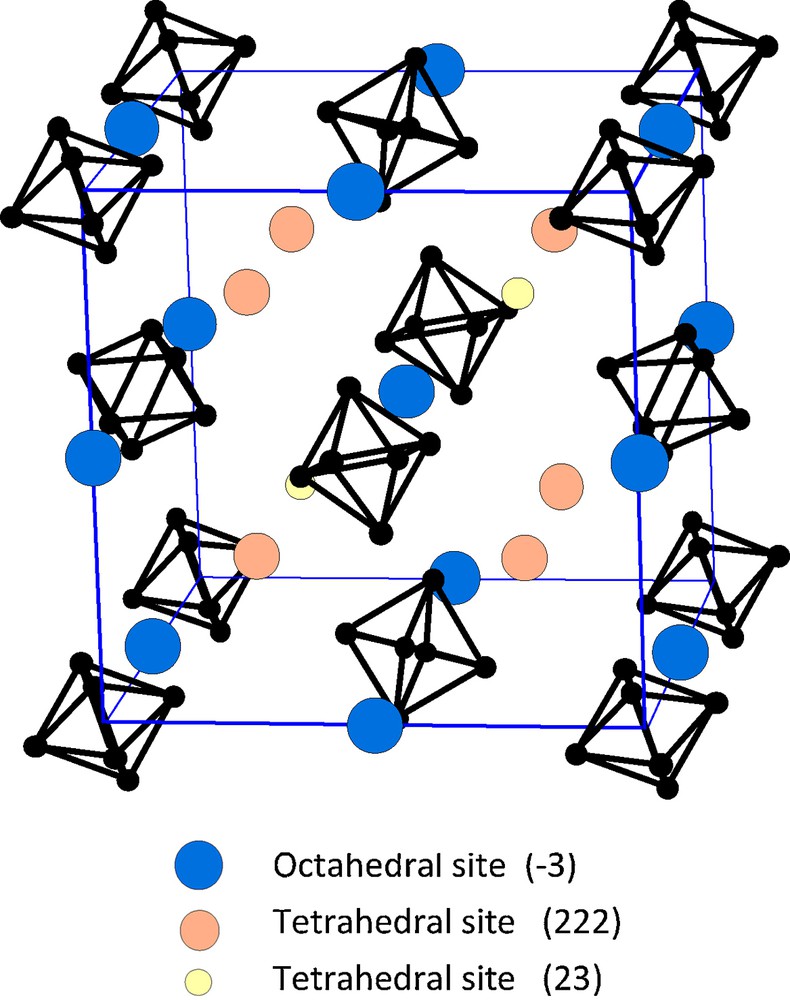
Schematic representation of the octahedral and tetrahedral sites arranged by the cluster units in the Pn-3 structure-type.

Crystal structure of Cs1+xRe6S5+xBr9-x (0 ≤ x ≤ 2). The inner positions are randomly occupied by either S or Br atom (both of them are represented on the figure). Br in yellow (light grey), S in blue (dark grey); Cs in red (large ball in dark grey) almost fills its site while the other Cs+ ions in pink (large balls in middle grey) randomly occupy several sites delocalized along the c axis (represented only on the 0z vector for clarity) [122]. Masquer
Crystal structure of Cs1+xRe6S5+xBr9-x (0 ≤ x ≤ 2). The inner positions are randomly occupied by either S or Br atom (both of them are represented on the figure). Br in yellow (light grey), S in blue (dark grey); Cs ... Lire la suite
3.3.2.2 Di-anionic cluster units
Substituting one Se for one Cl on the randomly occupied inner positions allows the charge balance without any change in the structure and various cubic compounds with di-anionic cluster units [Re6Se6Cl8]2-, in most cases isotypic with the previous ones, were reported [55].
Note that with bromine ligands, K2Re6S6Br8 is not isotypic with the monoclinic parent KRe6S5Br9, but turns again to be cubic as most of the compounds with small cations [119]. In fact, while the first potassium occupies the octahedral site like in KRe6Se5Cl9, the insertion of the second potassium ion occurs in a site which was very small in the monoclinic cell but opens as a genuine tetrahedral site (222) in the cubic one (Fig. 12) (in fact, K2Re6S6Br8 is a point of a solid solution, see below, and the determined structure corresponded actually to 1.7 K per cluster unit). The structure of Ag0.5K1.5Re6S6Br8 shows some cations ordering, as one potassium fully occupies the same tetrahedral site while 0.5 Ag and 0.5 K are distributed on the octahedral one [120]. However, Ag2Re6S6Br8 turns to exhibit a different triclinic structure, where the countercations are distributed on four distorted sites (in fact there is a slight excess of silver, Ag2.2) [120].
With divalent cations, the high temperature synthesis was reported for alkalin-earth salts A(II)Re6Q6Cl8 (A = Mg, Ca; Q = S, Se) [121] and for A(II)Re6Se6Cl8 (A = Cr, Mn, Fe, Co, Cd, Pb, Eu) [55] which are isotypic with the cubic prototype structure KRe6Se5Cl9. The thiobromide CdRe6S6Br8 also exhibits the cubic Pn-3 structure, but with a slight Cd deficiency, compensated by a related change in the (Se/Br)i ratio. The Cd ion occupies the octahedral site, while the tetrahedral one remains empty [120].
Cs2Re6S6Br8 is trigonal (S.G. P31c) like CsRe6S5Br9 reported above and, in fact, they are the two ends of a continuous solid solution [85] (vide infra). One Cs+ is sandwiched between cluster units while the second one is randomly distributed on several sites aligned on the c axis of the structure. As usual, there is some rotational disorder on the inner positions, but the two Li ligands located on the threefold axis are ordered (one is pure Br, the second is mainly sulfur) breaking the centrosymmetry. Thanks to the acentric space group, and analysing a crystal belonging to the solid solution, it was possible, in this specific case, to determine unambiguously the actual isomers of the cluster cores (Re6Si6Bri2)4+ and (Re6Si5Bri3)5+ present in both Cs2Re6S6Br8 and CsRe6S5Br9 (Fig. 14) [122]. DFT calculations were carried out for all possible isomers in this Re-S-Br system [123] as well as in the Re-S-Cl one [124]. The 2a di-substituted and the 3b tri-substituted isomers were computed to be the most stable in Cs2Re6S6Br8 and CsRe6S5Br9, respectively [123], in full agreement with structural data [122]. In contrast, for the molybdenum compound Cs3Mo6Ii6Ii2-xSeixIa6 it is the 2c isomer which was found experimentally to be the most stable, again in agreement with the theoretical study by DFT which indicates that occupation of the inner position on the threefold axis by Se in this compound is energetically preferred to the other positions [115]. The structure of the rubidium parent (although less precise due to the poor quality of crystals) is similar [89].

Sketch of the possible isomers of the [Re6S6Br2] and [Re6S5Br3] cores (di- and tri-substituted isomers, respectively) in Cs1+xRe6S5+xBr9-x. Rhenium and bromine apical ligands were omitted for clarity. Light grey and black circles are sulfur and bromine inner ligands, respectively. From [123].
3.3.2.3 Tri-anionic cluster units
They have been observed as definite compounds mainly for large cations, rubidium and cesium. However Na3Re6S7Br7 was mentioned as the upper boundary of a cubic solid solution [85], and K3Re6S7Br7 is thought to have been formed (with a small yield and may be not crystallized) in a mild temperature sulfidation of Re6S4Br10 by KNCS [125]. This K3Re6S7Br7 was not detected in the systematic high temperature study of the K-Re-S-Br system [85], but was obtained later as single-crystals suitable for structure determination, after recrystallization in water, but no yield was given [126]. The most striking feature in this structure is the considerable disorder of K+ cations, which arrange as partially occupied (K4)4+ tetrahedral groups and (K3)3+ angular groups. In the same A-Re-S-Br system, only the major structural features of the monoclinic P21/n cesium counterpart were obtained, due to a severe twinning effect [89,120,127], while Rb3Re6S7Br7 (trigonal R-3c), which presents the same structure as Cs3[(Mo6Ii6Ii2-xSeix)Ia6] (see above), has been fully characterized as well as its tetrahydrate [127]. The main difference between the two rhenium salts is the alkalin environment, as Rb+ fully occupies one site, while Cs+ is distributed on three different sites. In the Cs-Re-S-Cl system was reported the compound Cs3Re6S7Cl7, with a space group (monoclinic, P21/c) different from the two above mentioned compounds [128].
3.3.2.4 Tetra-anionic cluster units
These compounds are well documented in the case of cesium and thallium countercations while some examples were reported with Rb+ [88] and K+ [126]. In the latter case, K4Re6S8Cl6, the potassium ions, distributed in the voids of the cluster units packing, present a considerable disordering. Cs4Re6Se8I6 [29] is monoclinic P21/c while Cs4Re6S8Br6 is trigonal P-6c2 [88]. In the latter, the cesium ions form a very regular cuboactahedron at the center of which lies the tetra-anionic cluster unit. Such an arrangement is reminiscent in the iodide, but severely distorted (Fig. 15 a and b). However, most of the examples of tetra-anionic cluster units appear in isotypical structures R-3c of general formula A5Re6Q8X7 (A = Rb, Cs, Tl; Q = S, Se; X = Cl, Br) [29,89,90,129]. As the structure contains an extra halogen (in fact half distributed on two very close positions), the compounds could be written M4Re6Q8X6.MX, but it happens that the A+ ions occupy two crystallographic positions corresponding to 3 and 2 cations per cluster, respectively and, in addition, it appears clearly a triangular bipyramidal complex cation [A5X]4+. Then it would be better to write this series as [A5X]4+[(Re6Qi8)Xa6]4-, leading to a very simple description of this large structure as a body centered rhombohedral stacking (Fig. 15 c and d). Note that the recrystallization of a water/HBr solution of what is thought to be Cs5Re6S8Br7 gives the hydrate Cs4Re6S8Br6,2H2O [130]. Finally, the even halogen richer Cs6Re6Se8I8 [29] is built from a cuboctahedral stacking of Cs+ ions, like in the example of Cs4Re6S8Br6, but now these polyhedra are apex-sharing in their equatorial plane instead of edge-sharing, opening octahedral sites were locate the extra iodine ions (Fig. 15e).
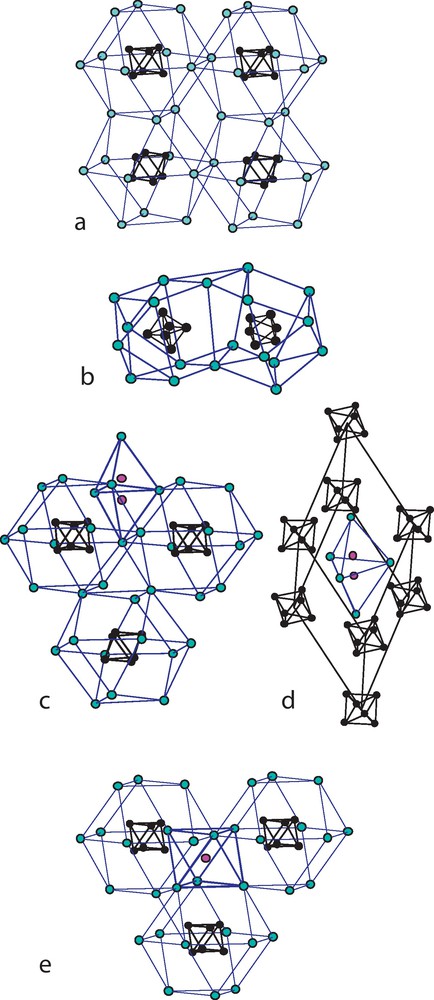
Alkalin ion polyhedra surrounding the Re6 cluster in various tetra-anionic compounds: a: in Cs4Re6S8Br6 [88]; b: in Cs4Re6Se8I6 (redrawn from data ref. [29]; c: in Cs5Re6S8Br7 (redrawn from [129]); d: the rhombohedral CsCl-type pseudo unit-cell in the Cs5Re6S8Br7 structure evidencing the (Cs5Br)4+ complex cation where bromine ion is splitted in two half occupied positions due to steric hindrance (redrawn from [129]); e: in Cs4Re6S8I8, the site of the extra iodine ions is hightlighted (redrawn from [29]). Masquer
Alkalin ion polyhedra surrounding the Re6 cluster in various tetra-anionic compounds: a: in Cs4Re6S8Br6 [88]; b: in Cs4Re6Se8I6 (redrawn from data ref. [29]; c: in Cs5Re6S8Br7 (redrawn from [129]); d: the rhombohedral CsCl-type pseudo unit-cell in the ... Lire la suite
3.3.2.5 Solid solutions and non-integer n
Most of the above mentioned structures correspond to definite compounds An[(Re6Qi4-nXi4+n)Xa6] (A = alkalin) where n is an integer. When n is non-integer, cluster units with different negative charges (but 24 VEC) coexist in the same structure as detailed above for Rb2+x[(Mo6Bri8-xSeix)Bra6] (0.25 < x < 0.7). Indeed, the systematic study of the quaternary A-Re-S-Br phase diagrams evidenced several solid solutions extending mainly between monoanions and dianions, but with some variations depending on the cation size [85]. For instance, the domain of homogeneity extends from Na1 to Na3 (cubic), K1.1 to K2.5 (cubic, while for K1.0 the structure is monoclinic), Rb1 to Rb2 as well as Cs1 to Cs2 (trigonal in both cases). In the latter systems, definite compounds (non-integer n) were evidenced for n = 0.6 (Rb) and 0.4 (Cs), with a different structure, expected to be related to the KRe6S5Br9 one [85]. The other systems were not so systematically studied. However note the structure determination of Rb2.5Re6S6.5Cl7.5 [110]: similarly to K2.5Re6S6.5Br7.5 [120] such composition is obtained by filling first the four octahedral sites of the Pn-3 unit-cell (one alkalin per cluster) and then the six tetrahedral 222 sites up to their maximum (Fig. 12). It corresponds then to the upper limit of the cubic solid solution. In fact, in the Pn-3 structure exist also smaller additional tetrahedral 23 sites, but they were always found to be vacant, even when a composition like Rb2.5Li0.5Re6S7Cl7 was attempted [131].
3.3.2.6 Cationic cluster units
This case is exceptional and one can mention the examples of [(Re6Tei8)(TeX2)a6]X2 (X = Br, I) [111,132] prepared at mild temperatures (400–450 °C). Here the (Re6Tei8) core is bounded to the tellurium of the six neutral apical ligands TeX2. It results large cationic units that form a distorted close packing in the voids of which are located the halogen counter-anions.
3.4 Summary of structure dimensionality and cluster interconnections
Table 1 reports the Mo6 and Re6 chalcohalides detailed in this review versus the dimensionality of the intercluster connections from 3D to 1D, giving the developed formula of the compounds and those for which the crystal structures were determined by single crystal X-ray diffraction.
Mo6 and Re6 chalcohalides reported with 3D, 2D and 1D interunit connections.
| Type of connections | General formula | Developed formula | Molybdenum | Rhenium | Crystal structures reported | Space group | Ref. |
| 3D | |||||||
| 3D Xi-a/Xa-i | M6Q8-xXx | [(M6Qi2-xXixQi-a6/2)Qa-i6/2] | Mo6S6X2 (X = Br, I) | – | Mo6S6Br2 | R-3 | [15] |
| Mo6Se8-xXx (0 < x < 2) (X = Cl, Br, I) | Mo6S5.96I1.76 | R-3 | [38] | ||||
| Mo6Te8-x(0 < x < 2) (X = Cl, Br, I) | |||||||
| 3D Xa-a | M6X10Q | [(M6Xi7Qi)Xa-a6/2] | Mo6X10Q (X = Cl, Br; Q = S, Se, Te) (X = I; Q = Se, Te) | Mo6Cl10Se | Pccn | [53] | |
| Re6Q7X4 (Q= S, Se; X = Cl, Br) | Re6S7Br4 | R-3c | [57] | ||||
| Re6Se7Br4 | R-3c | [56,57] | |||||
| 2D Xa-a + 1D Qi-a/Qa-i | M6Q8X2 | [(M6Qi6Qi-a2/2)Qa-i2/2Xa-a4/2] | Re6Q8X2 (Q = S, Se; X = Cl, Br) | Re6S8Cl2 | P21/n | [75] | |
| Re6S8Br2 | P21/n | [73] | |||||
| Re6Se8Br2 | P21/n | [72] | |||||
| 2D | |||||||
| 2D Xa-a | M6Q6X6 | [(M6Qi6Xi2)Xa2Xa-a4/2] | – | Re6Se6Cl6 | Re6Se6Cl6 | Cmca | [57] |
| 2D Qi-a/Qa-i | M6Q8X2 | [(M6Qi4Qi-a4/2)Qa-i4/2Xa2] | – | Re6Se8Cl2 | Re6Se8Cl2 | P-1 | [91] |
| Re6Se8-xQxCl2(Q = S, Te) | |||||||
| 1D Qi-i, Xa-a + 1D Qi-a/Qa-i | M6X6Q3 | [(M6Xi4Qi-i2/2Qi-a2/2)Qa-i2/2Xa-a4/2] | Mo6Br6S3 | – | Mo6Br6S3 | Cmcm | [81] |
| 1D Xa-a + 1D Qi-a/Qa-i | AM6Q8X3 | A[(M6Qi6Qi-a2/2)Qa-i2/2Xa-a2/2Xa2] | – | ARe6Q8X3 (Q = S, Se; X = Cl, Br, I) | TlRe6S8Cl3 | P-1 | [90] |
| CsRe6S8Br3 | P21/n | [94] | |||||
| CsRe6Se8I3 | P21/n | [29] | |||||
| 1D | |||||||
| 1D Xa-a | M6Q5X8 | [(M6Qi5Xi3)Xa-a2/2Xa4] | – | Re6Se5Cl8 | Re6Se5Cl8 | P-1 | [57] |
| Re6S5Br8 | Re6S5Br8 | P-1 | [85] | ||||
| 1D Qi-a/Qa-i | A2M6Q8X4 | A2[(M6Qi6Qi-a2/2)Qa-i2/2Xa4] | – | Tl2Re6S8Cl4 | Tl2Re6S8Cl4 | P-1 | [90] |
| Cs2Re6S8Br4 | Cs2Re6S8Br4 | P21/n | [88] | ||||
| 1D Qi-i, Xa-a | M6X8Q2 | [(M6Xi5QiQi-i2/2)Xa-a6/2] | Mo6Br8S2 | – | Mo6I8Se2 | P63 | [77,78] |
| Mo6I8Q2 (Q = S, Se) |
All the shared ligand types, La-a, Li-a, La-i and Li-i are involved in these connections. La-a or Li-a connections develop either in one, two or three directions without any other type of shared ligands or they can be associated together in a same compound. Only missing is (for 3D compounds) the “1D Xa-a + 2D Qi-a/Qa-i” combination, for instance the hypothetic 24 VEC compound “A[(Mo6Xi4Qi-a4/2)Qa-i4/2Xa-a2/2]” has never been obtained up to now. Li-i connections develop only in one direction, due to strong steric hindrance, accompanied by La-a connections when the Li-i ligand is in prismatic site of metal. One could imagine other arrangements involving several types of shared ligands and various counter cationic charges. A virtual library of all possible Re6 chalcohalides was already reported [24]. However, such hypothetic compounds are subject to the rule concerning the specific ligand sites that can be occupied preferentially by halogen and/or chalcogen (Section 2), to a 24 VEC value (or close to 24) depending on the halogen/chalcogen stoichiometry and on the cluster unit charge, and to a reasonable steric hindrance of the ligands. They have not been obtained up to now for these reasons or because the corresponding stoichiometries were never experimentally explored. Another reason is the possibility of several connectivities for the same chemical formula, one of them being the most stable. For instance M6Br8S2 can be written as 1D [(M6Bri5SiSi-i2/2)Bra-a6/2] or 3D [(M6Bri6Si-a2/2)Sa-i2/2Bra-a4/2], the former arrangement being the most stable in the case of molybdenum (Mo6Br8S2), while the latter one was found only with rhenium in Re6S8Br2. Other examples can be found in rhenium chemistry. For instance Re6S6Br6 can be written either as 2D [(Re6Si6Bri2)Bra-a4/2Bra2] or 1D [(Re6Si4Bri2Si-a2/2)Sa-i2/2Bra4], only the first case being encountered. Similarly for A2Re6S8Br4 i.e. 2D [(Re6Si8)Bra-a4/2Bra2]2− or 1D [(Re6Si6Si-a2/2)Sa-i2/2Bra4]2− or for ARe6S8Br3 i.e. 3D [(Re6Si8)Bra-a6/2] or 2D [(Re6Si6Si-a2/2)Sa-i2/2Bra-a2/2Bra2]− only one of the two possible structures was obtained (the 1D and 2D, respectively). Conversely, in the case of Re6Q8X2, the two possible connectivities 3D and 2D were known for the same general formula, but for different sets of halogen and chalcogens, probably in relation with steric constraints. Notice that the 3D Xa-a connection is obtained for both Mo6 and Re6 chalcohalides while the other combinations are specific of each element.
Table 2 reports the 0D compounds Ay[(M6Xi8-xQix)Xa6] versus the x stoichiometry in chalcogen. Note that this x stoichiometry concerns only the cluster core, because in these discrete units the apical positions are only occupied by halogens. As expected, the smaller x values (0 < x < 2) were obtained for the molybdenum compounds, that is required to maintain a 24 VEC value (or close to 24) due to the smaller valence of this element when compared to rhenium one. However, cyano-Mo6 chalcohalides with x > 2 have been obtained by solution chemistry ([156] Section 4.3). Many of these compounds are cubic Pn-3, or derived from this structure-type in relation to the size of the cluster unit and of the cationic sites arranged in the cluster packing (Section 3.3.2).
Details of crystal structures for 0D Ay[(M6Xi8-xQix)Xa6] compounds versus increasing x chalcogen stoichiometry.
| x value | Molybdenum, general formula | Rhenium, general formula | Building blocks (developed formula) | Crystal structures reported | Space group | Ref. |
| 0 | A2+Mo6X14 | [(Mo6Xi8)Xa6]2- | Rb2Mo6Br14a | C2/c | [113] | |
| 0.5 | Rb2.5Mo6Br13.5Te0.5 | 0.5 [(Mo6Bri8)Bra6]2- | Rb2.5Mo6Br13.5Te0.5 | Pn-3 | [113] | |
| +0.5 [(Mo6Bri7Tei)Bra6]3- | ||||||
| 0.25 < x < 0.7 | Rb2+xMo6Br8-xSexBr6 | (1-x) [(Mo6Bri8)Bra6]2- | x = 0.25, x = 0.5, x = 0.7 | Pn-3 | [113] | |
| + x [(Mo6Bri7Sei)Bra6]3- | ||||||
| 1 | [Rb3Mo6Br13Q][Rb3(MoBr6)3] | [(Mo6Bri7Qi)Bra6]3- | Rb12Mo9Br31Se | Pm-3 m | [114] | |
| (Q = Se, Te) | + 3 (MoBr6)3- | Rb12Mo9Br31Te | Pm-3 m | [114] | ||
| 1.2 < x < 1.6 | Cs3Mo6I6I2-xSexI6 | (2-x) [(Mo6Ii7Sei)Ia6]3- | x = 1.2, x = 1.5, x = 1.6 | R-3c | [115] | |
| + (x-1) [(Mo6Ii6Sei2)Ia6]3- | ||||||
| 2 | Cs4Mo6Br12Q2 (Q = S, Se) | [(Mo6Bri6Qi2)Bra6]4- | Cs4Mo6Br12S2 | Pbca | [112] | |
| Cs4Mo6Br12Se2 | Pbca | [112] | ||||
| 4 | Re6Q4X10 (Q = S, Se, Te; X = Cl, Br) | [(Re6Qi4Xi4)Xa6]0 | Re6S4Cl10 | P-1 | [110] | |
| Re6S4Br10 | P-1 | [85] | ||||
| Re6Se4Cl10 | P-1 | [105,106] | ||||
| Re6Se4Br10 | P-1 | [107] | ||||
| Re6Te4Cl10 | P-1 | [108] | ||||
| Re6Te4Br10 | P-1 | [109] | ||||
| 5 | AIRe6Q5X9 (A = Li, K, Rb, Cs, Cu, Ag; Q = S, Se; X = Cl, Br) | [(Re6Qi5Xi3)Xa6]- | KRe6S5Br9 KRe6Se5Cl9 | C2/c Pn-3 | [118] [117] | |
| 5 < x < 6 | A1+yRe6S5+yBr9-y | (1-y).[(Re6Si5Bri3)Bra6]- | Cs1.95Re6S5.82Br8.19 | P31c | [122] | |
| + y.[(Re6Si6Bri2)Bra6]2- | K1.7Re6S5.7Br8.3 | Pn-3 | [119] | |||
| Cd0.7Re6S5.5Br8.5 | Pn-3 | [120] | ||||
| 6 | A+2Re6Q6X8 | [(Re6Qi6Xi2)Xa6]2- | Ag2Re6S6Br8 | P-1 | [120] | |
| K1.45Ag0.55Re6S6Br8 | Pn-3 | [120] | ||||
| 6 < x < 7 | A+2.5Re6S6.5Cl7.5 | [(Re6Si6Cli2)Cla6]2- | Rb2.5Re6S6.5Cl7.5 | Pn-3 | [110] | |
| + [(Re6Si7Cli)Cla6]3- | K2.5Re6S6.5Br7.5 | Pn-3 | [120] | |||
| 7 | A+3Re6Q7X7 | [(Re6Qi7Xi)Xa6]3- | K3Re6S7Br7 | P21/c | [126] | |
| Rb3Re6S7Br7 | R-3c | [127] | ||||
| Cs3Re6S7Cl7 | P21/c | [128] | ||||
| Cs3Re6S7Br7 | P21/n | [120] | ||||
| 8 | A+4Re6Q8X6 | [(Re6Qi8)Xa6]4- | K4Re6S8Cl6 | C2/m | [126] | |
| Cs4Re6Se8I6 | P21/c | [29] | ||||
| Cs4Re6S8Br6 | P-6c2 | [88] | ||||
| A+5Re6Q8X7 (A = Rb, Cs, Tl; Q = S, Se; X = Cl, Br) | [(Re6Qi8)Xa6]4− [A5X]4+ | Cs5Re6S8Cl7 | R-3c | [90] | ||
| Cs5Re6S8Br7 | R-3c | [29] | ||||
| Tl5Re6S8Cl7 | R-3c | [90] | ||||
| Tl5Re6Se8Cl7 | R-3c | [90] | ||||
| A6Re6S8I8 | [(Re6Qi8)Xa6]4-(CsI)2 | Cs6Re6S8I8 | Fm-3 m | [29] |
a As an example. See also [95–100].
4 Solid state M6 chalcohalides as relevant precursors for soft chemistry
The [(Mo6Li8)La6] and [(Re6Li8)La6] cluster units reported above exhibit various intrinsic properties. For instance, as a rule, they are highly emissive in the red-near infrared region, the characteristics of the emission depending on the Q/X ratio and on the geometry of the unit [133–135]. They have 24 e-/23 e− oxidation process by electrochemistry or soft chemical routes [110,133,136]; they are magnetic when VEC = 23 that corresponds to the presence of one unpaired electron on the HOMO level of the molecular orbital diagram of the unit; they act as electrical dipole when the ligand charges are non symmetrically distributed around the M6 cluster [53]. As a consequence such discrete cluster units are relevant candidates for the elaboration of functional hybrid cluster materials that combine the properties of both the cluster units and functional ligands, provided that they can be extracted as an individual entity from the solid state precursors and then handled for ligand substitution by functional ligands in solution or by soft chemistry.
4.1 Intrinsically soluble cluster-based compounds
Due to the strength of intercluster bonds, the polymeric 3-D to 1-D compounds are obviously not directly soluble. In contrast, many discrete anionic units are soluble in common polar solvents. For instance, for rhenium compounds this solubility strongly depends on their charge and also on the elements present in the quaternary system. As a general rule, the most important is the anionic charge, the highest is the solubility and tetra-anionic units turn out to be readily soluble in water. Table 3 gives some typical examples as an illustration [137].
Example of solubility of M6 chalcohalides in usual solvents [137].
| Compound | H2O | EtOH | CH3CN | CH2Cl2 |
| Cs4Re6S8Br6, CsBr | Soluble | Weakly soluble | Insoluble | Insoluble |
| Cs4Re6S8Br6 | Insoluble | – | – | – |
| Cs3Re6S7Br7 | Soluble | Soluble | Weakly soluble | Insoluble |
| Cs2Re6S6Br8 | Insoluble | Insoluble | Soluble | Soluble |
| CsRe6S5Br9 | Weakly soluble | Soluble | Insoluble | Insoluble |
4.2 Lowering the dimensionality by chemical excision
Chemical excision is the cleavage of interunit bridges in extended solids, affording a reduction of the dimensionality. The first example in cluster chemistry is the replacement of Ia-a bridges in 3-D Nb6I11 (isostructural with Mo6Cl10S, Section 3.1.2) by alkyl-amines, yielding a 0-D compound [138]. The dimensional reduction formalism in solid state chemistry was extensively applied to cluster-based frameworks by the group of R.H. Holm [21,29,90,139,140]. This concept is illustrated by the following sequence of reactions where TlCl is incorporated in extended networks:
However, as emphasized by the authors, such reactions describe a ‘formalistic relationship between structures and not necessary a specific synthetic pathway’. Indeed, from an experimental point of view, they prepared intermediate and final compounds by direct synthesis and not by reaction of TlCl with a previously prepared extended solid.
4.3 Synthesis of cyano and hydroxo derivatives
In many examples (CN)− groups behave like halogen anions and cyano compounds of M6 clusters are now well documented. They are obtained in most cases by a genuine excision (associated to a substitution) process. The most usual route consists in the reaction of a presynthesized cluster compound, typically Re6Q8Br2, with an excess of molten KCN at medium temperatures (530–600 °C) [141]. The same reaction, but with a KCN deficiency, produces mixed apical ligands, like [(Re6Sei8)(CN)a4Bra2]4− [142]. It is also conveniently possible to simply substitute (CN)− for halogen in 0 D tetra-anionic species at around 635 °C [143].
An alternative preparation is the reaction of Re6Te15 (which already contains the [Re6Te8] core), with NaCN at 600 °C, affording the [(Re6Tei8)(CN)a6]4− ion [144,145]. When using in place thiocyanate as reagent, a core substitution (Si for Tei) occurs, leading to [(Re6Si8)(CN)a6)]4− ion, more precisely [Re6Si6(S1-xTex)i2(CN)a6]4−. In fact this approach afforded the first example of chalco-cyano compounds in this chemistry as early as 1995 (Fig. 16) [146,147]. The partial statistical disorder was studied in more detail later [148]. In fact, it appears not mandatory to start from a pre-existing octahedral cluster: examples are given by the reaction of the binaries ReS2 or ReSe2 with KCN to give K4[(Re6Si8)Sa-a4/2(CN)a2] (layered) or K4[(Re6Sei8)(Se2)a-a2/2(CN)a4] (chain) [149]. Adding an excess of CsCl in the starting mixture yields Cs4[(Re6Si8)Sa-a2/2(CN)a4] [150].

Projection of the structure of KCs3Re6S8(CN)6 [147]. The inner ligands were omitted for clarity. C in dark green (dark gray), N (light gray), K/Cs in red (large balls in dark grey).
Another route, not so explored, is the conversion of tetrahedral cluster to octahedral one: for instance, a mixture of [(Re5MoSi8)(CN)a6]5− and [(Re4Mo2Si8)(CN)a6]5− heterometallic cluster anions was obtained by reaction of Re3MoS4Te4 and KCN at 850 °C [151]. Considering the high temperature of reaction, one cannot exclude a full decomposition of the starting compound. However, an example of conversion of tetrahedral to octahedral cluster by a solvothermal route working at a temperature as low as 200 °C was reported, giving access to the [(Re6Sei8)(CN)6]4− ion [152].
Note that the isoelectronic technecium [(Tc6Qi8)(CN)a6]4− ion was recently reported: the structures, electronic structures and electronic transitions are very similar to those of the rhenium parents, although the energy gap is smaller in the technetium complexes [153].
The cyano derivatives of Mo6 clusters are less documented. The early exchange of bromine apical ligands by CN ones in Mo6Br12 via an aqueous solution route [154] should be mentioned. On the opposite side, it was shown that the octahedral cluster-based binary Mo6Se8 can be excised by molten KCN in a way similar to that used in rhenium chemistry, resulting in the formation of the [(Mo6Sei8)(CN)a6]7− and [(Mo6Sei8)(CN)a6]6− ions [155]. In fact the starting composition can be as well a mixture of MoSe2 and Mo, for instance, and starting from various mixtures, the above mentioned ions were obtained as well as K6[(Mo6Sei8)(CN)a4(CN)a-a2/2], which is a chain-based compound [156]. True Mo6 chalcohalides have also been converted to chalcohalocyanides, but again by exchange in aqueous medium, giving the ions [(Mo6Qi2Bri6)(CN)a6]4− [112] and the [(Mo6Sei3Xi5)(CN)a6]5−/[Mo6Sei4Xi4(CN)a6]5− solid solution although the intermediate chalcohalide implied in the reaction was not actually isolated [157].
A similar strategy was used to access the corresponding hexahydroxo complexes, treating as an example Re6Q8Br2 by molten KOH at 200-280 °C, leading to the [(Re6Qi8)(OH)a6]4− tetra-anion. Here, the hydroxo ligands are very labile, and easily back replaced by bromine ones for instance, when subjected to the action of HBr [158]. A related hexa-aqua [Re6Si8(H2O)a6]2+ dication was also reported: it was obtained by removing Cla apical ligands of [Re6S8Cl6]4− by Ag+ in perchloride medium [159].
4.4 Quaternary ammonium salts
In order to increase the solubility in common polar organic solvents (for instance CH3CN) it was proposed very early to use a cationic metathesis to obtain, for instance the corresponding tetrabutylammonium salt [100,110,160,161] like as an example (TBA)2Mo6X14 or the whole series (Bu4N)n+[Re6(Si4+nCli4-n)Cla6]n− used in the following as starting material for solution chemistry [110]. Note that obtaining a homogeneous series enabled to evidence a cluster core contraction when the ratio S/Cl increases [110].
5 Solution chemistry of M6 clusters
5.1 Playing with organic countercations or ligands
5.1.1 Organic (organometallic) countercations
In addition to the pioneering work just above mentioned, the treatment of polymeric compounds like Re6Se5Cl8 or Re6Se6Cl6 by an excess of R4NCl in hot DMF or acetonitrile results in an excision reaction and the formation of R4N− salts of mono- or di-anionic cluster units, respectively [139]. Mesolamellar phases were reported with long chain (n = 14–18) alkyltrimethylammonium surfactants [162]. Several works involving tetraphenylphosphonium countercations [125], solvates or kryptates [163,164], metal complexes [165–168] or ferrocenium [169] should also be mentioned. Cluster anions [M6L14]n− were combined by electrocrystallisation, very early in some examples, with various cations derived from tetrathiafulvalene TTF (Fig. 17) [160,170–175].
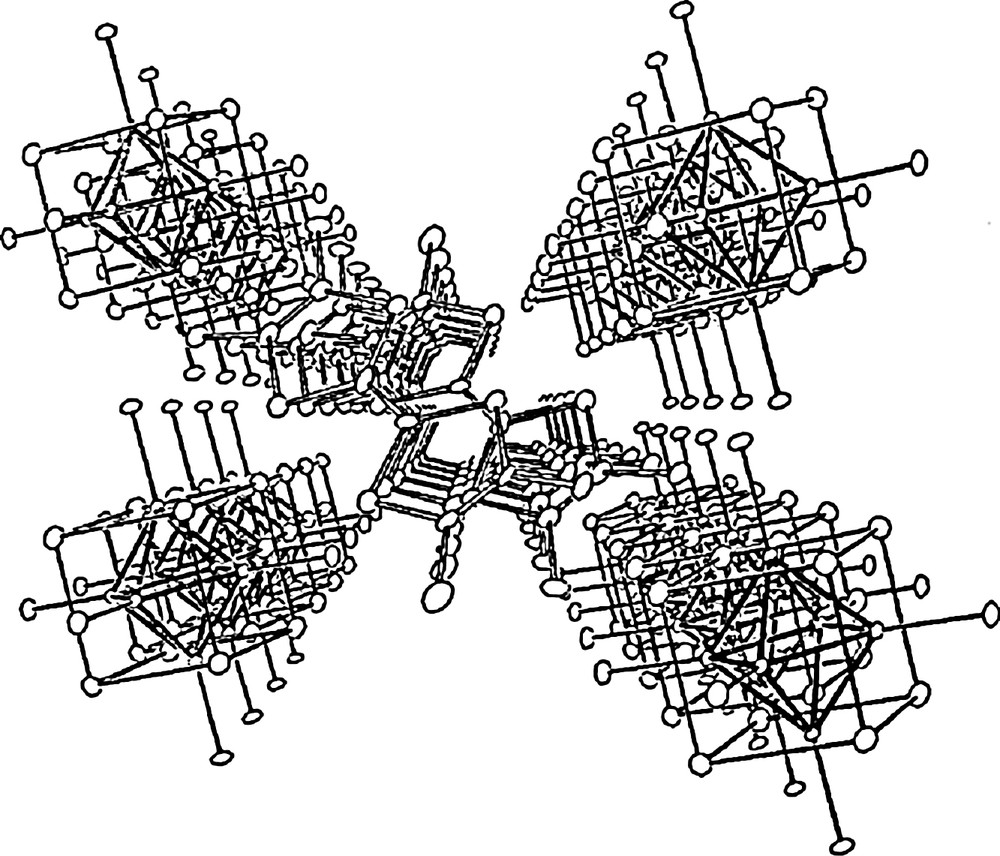
Structures of β-(TMTTF)2Re6Se5Cl9 and β-(TMTSF)2Re6Se5Cl9 [160].
5.1.2 Ligand exchanges
5.1.2.1 Apical substitutions
As mentioned above, hydroxo apical ligands are very labile, and easily replaced by halogen ions, cyano ions [176] but also organic anions for instance formates [177].
Refluxing (Bu4N)3-4Re6Q8X6 with PEt3 in DMF for instance affords the [(Re6Qi8)(PEt3)anXa6-n](n-4)+ ion (Q = Se, X = I, n = 3–6 [178]; Q = S, X = Br, n = 2–6 [179]; Q = S, X = Cl, n = 1–4 [180] (Note that n = 5 and 6 imply the formation of the rarely encountered cationic cluster units). Similar reactions were reported with PPh3, AsPh3 and SbPh3, although in quite stronger conditions (sealed tube, 200 °C) [181,182] and also diphosphines were used as monodentate ligands [183] as well as bidentate chelating ones [184]. Some molybdenum homologues were obtained some time ago, by an indirect route using the reductive dimerization of a trinuclear molybdenum thiochloride cluster, Mo3S7Cl4, after its reaction with triethylphosphine, to give Mo6S8(PEt3)6 [185]. The analogous seleno derivative was synthesized in a similar way [186].
Due to the strong Re-P (As, Sb) bond, only the (eventually) remaining apical halogens are subsequently removable by Ag salts, giving access to cluster carbonyl [187,188], triflates [189] and solvates like, for instance, in [(Re6Sei8)(PEt3)an(MeCN)a6-n]2+ (n = 0,4,5) [178]. The latter compounds open the way to a large range of site-differentiated cluster complexes, as very labile triflates and nitriles can be in turn substituted by other ligands [190]. Alcohol reaction with acetonitrile ligand(s) activated by the cluster core afforded imino ester ligands by nucleophilic addition [191]. N3− azide ion (as NaN3) does not substitute for acetonitrile, but in place undergoes an addition reaction across the nitrile triple bond, to give tetrazolato complexes [192] (note that, in contrast, N3− substitutes readily for halogen apical ligands, to give [(Re6Si8)(N3)a6]4− and [(Mo6Bri8)(N3)a6]2− anions [193]). On the other hand, organic azides unexpectedly form iminocomplexes with cluster-bound nitriles [194,195]. It was suggested the formation of a cluster-azido intermediate followed by a stereoselective rearrangement and the concerted photoelimination of dinitrogen [194]).
A number of examples are reported relative to the substitution of pyridine and its derivatives on the apical ligand positions [196–205]. These compounds form either by direct substitution (for a terminal halide), or via a presynthesized solvate or with the help of a silver salt, usually in reflux conditions. The hexaammine group was evidenced in the [Re6Se7Br(NH3)6][Re6Se7BrBr6].12H2O salt [206].
5.1.2.2 Core substitutions
Although the cluster core is very rigid, implying strong M-Li inner bonds, there are some restricted examples of inner ligands substitutions in “soft chemistry” conditions.
While Mo6 chalcohalides presented in the first part of this review have been prepared at high temperature by solid state routes, several examples of [(Mo6Xi8-xQix)Xa6]n− cluster units prepared in solution have been reported:
- • for x = 1, from reaction of Mo6X12 (X = Cl, Br) precursors with NaSH or NaSeH in pyridine [207–210];
- • for x = 1 or coexistence of x = 1 and 2 in a same compound, from reaction of (H7O3)2[Mo6Cl14] with H2Se generated in situ from ZnSe under hydrothermal condition [211];
- • for x = 2 from the refluxed reaction mixture of Mo6Cl12, NaSeH and pyridine [212].
In these reactions the apical halogen is not substituted and the corresponding compounds crystallize as hybrid salts with organic countercations. It is different from a reaction of Mo6X12 in aqueous solution of Cs2Se3 and then in aqueous solution of KCN, in which the apical halogens are replaced by CN− groups after the inner ligand substitution by chalcogen: the [(Mo6Sei3.6Cli4.4)(CN)a6]n− and [(Mo6Sei3.4Bri4.6)(CN)a6]n− cluster units are obtained and the two compounds crystallize with Cs cation [157]. A similar example is the conversion of the [(Re6Sei5Cli3)Cl6a]− anion to [(Re6Sei6Cli2)Cl6a]2− one by Li2Se in THF [140]. Exchange of two inner chlorines by hydroxo ions was evidenced by structure determination, associated with IR spectra and mass spectroscopy, in K[(Re6Sei5Cli3)Cla6] when recrystallized at room temperature from water/ethanol mixtures [213]. In contrast, the cluster core remained unchanged when the same experiment was carried out in pure ethanol [214]. Action of water in DMF at a slightly higher temperature (60 °C) on [(Re6Sei4Cli4)Cla6] afforded the oxo-chalcohalogeno [(Re6Sei4Oi2Cli2)Cl6a]2− anion [140]. Note that such hydrolysis was shown to work also in high temperature solid state synthesis: an example is the formation of the [(Re6S5OCl7)2O]4− oxo-bridged dicluster with oxo-chalcohalide core at 850 °C in a sealed silica tube [215]. An efficient approach to carry out such substitutions is the use of silylated reagents (Me3Si)2E and the [(Re6Qi5EiCli2)Cl6a]2− anion was early obtained at room temperature for E = O, S, Se, Te [216] and subsequently for E = As, P, NH [217].
A slightly different approach is the use in mild conditions of molten ligands or salts: for instance, reaction of Re6S4Br10 with KNCS in a quartz ampoule at 550 °C gives access to the [(Re6Si7Bri)Bra6]3− ion which was crystallized as the hybrid salt (PPh4)3[Re6S7Br7] [125] (but in contrast, it was reported that the action of molten KNCS at 200 °C on the [Re6S8Cl6]4− cluster anion resulted in the formation of the isocyanate complex [Re6S8(NCS)6]4− by apical substitution [218]). Similarly, action of molten 3,5-dimethylpyrazole on [(Re6Qi7Bri)Br6a]2− gave the cationic species [(Re6Qi7Oi)L6a]2+ (L = 3,5-Me2PzH) [219].
Finally, a substitution of an organic ligand on an inner position would require ligands not only tricoordinated but also of very small dimension because of severe steric hindrance. To the best of our knowledge the only one example reported concerns the Mo6 cluster with eight alkoxy ligands in inner positions in Na2[Mo6(OCH3)i8(OCH3)a6] [220] and Na2[Mo6(OCH3)i4(OC6H5)i4(OC6H5)a6] [221].
5.2 Rebuilding polymeric structures from isolated building blocks
5.2.1 Connecting clusters by coordination chemistry
Taking advantage of the bidentate behaviour of the cyano ligand a number of polymeric structures were rebuilt, creating for instance transition metal complex bridges between cluster units [143,222]. This approach is illustrated, for instance, by the formation of discrete dimers [{Cu(en)2}{Re6Te8(CN)6}2]6− (Fig. 18), 1-D [Ni(NH3)2(en)2]2[{Ni(NH3)4}Re6Se8(CN)6]Cl2,2H2O (linear chains) or 1-D [{Mn(H2O)(en2)}{Mn(en2)Re6Te8(CN)6], 3H2O (zigzag chains) and 2-D (NH4)2[{Ni(en)2}3{Re6Te8(CN)6}2], 6H2O (wavy layers) [223]. Three-dimensional frameworks are illustrated for instance by Na[Mn(salen)]3[Re6Se8(CN)6] [224] or (Me4N)4[{Mn(H2O)2}1.5Mo6Se6(CN)6], 8H2O [225]. The reaction of [Re6Tei8(CN)a6]4− ion with Fe3+ cations in aqueous solution results in the synthesis of the porous expanded Prussian Blue analogue Fe4[Re6Te8(CN)6] [226]. In many other examples the 3-D structures can be described as deriving from Prussian Blue [227–229]. A specific example with Mo6 chalcohalide core is A(Et4)11[(Mo6Br6S2)(CN)6]3,16H2O (A = mixed alkalins Cs/K), but here alkalin ions in octahedral environment of (CN) replace transition element complexes, leading to an even more porous framework [112]. This approach of cyano-based coordination chemistry was subsequently widely developed and more details can be found in a recent review [230]. The first coordination polymers based on hexahydroxo rhenium clusters anions and alkalin-earth metal cations were published more recently: they include discrete molecular structures, 1-D chains and two types of 2-D layered structures [231].
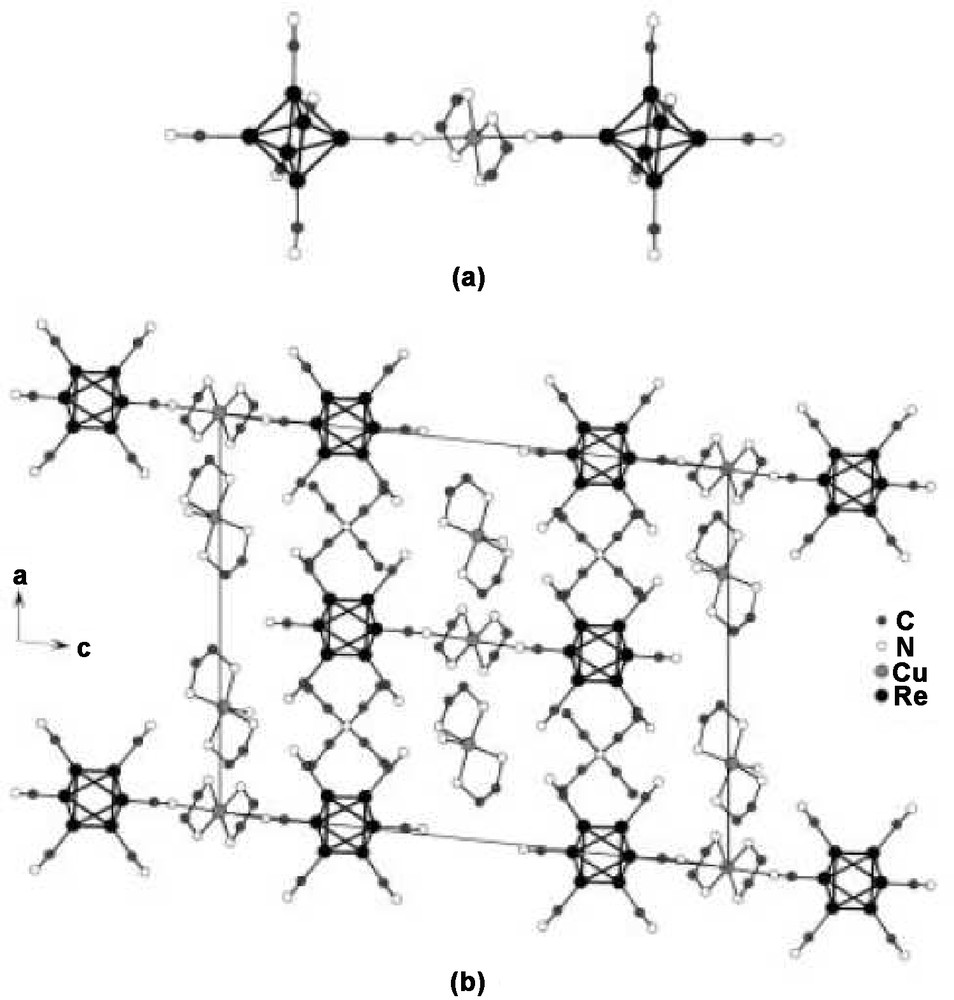
Structure of anionic dimer [{Cu(en)}2{Re6Te8(CN)6}2]6− (a) and view along [010] of the crystal packing in (Et4N)2[Cu(NH3)(en)2]2[{Cu(en2)}{Re6Te8(CN)6}2].2H2O (b). All hydrogen and tellurium atoms and water molecules have been omitted for clarity [223].
5.2.2 Creating self organized structures using organic spacers
Generating site-differentiated species [Re6Qi8(PEt3)a6-xXax]2-x and subsequently substituting labile MeCN for halogen offers the possibility of assembling new molecular species, provided that the number (and isomer geometry) of solvate ligands and the nature of grafted ligands are well chosen. For instance, assembling [Re6Qi8(PEt3)a5(MeCN)a]2+ with bipyridines gives discrete bridged diclusters, and even triclusters (with the use of trans-[Re6Qi8(PEt3)a4(MeCN)a2]2+) [197], while with tri and tetradentate pyridil-based ligands, star-shaped (three- and tetra armed) multiclusters were obtained [232]. Using the cis-[Re6Qi8(PEt3)a4(MeCN)a2]2+ isomer instead of the trans- one afforded the first examples of cluster-based molecular squares cyclo-[Re6Qi8(PEt3)a4La-a2/2]48+ with various lengths of the organic spacer [233].
Within this approach, supramolecular arrays were obtained via three routes:
- • the mediation of trans-[(Re6Qi8)(PEt3)a4(dipyridyl)a2]2+ with transition metal ions (Co2+, Cd2+, Zn2+ as nitrates) gave sinusoidal and zigzag chains and porous structures [234] while the use of the cis- isomer resulted in the formation of one-dimensional chain of corner-sharing squares [235];
- • the use of hydrogen bonding involving ligands like nicotinamides [236], giving again dimers, linear and zigzag chains;
- • the use of halogen bonding between the nitrogen of a cyano ligand and the iodine of a TTF derivative [237] or a bis iodoethynyl benzene [238].
6 Conclusion: recent trends in M6 cluster chemistry and perspectives
In conclusion, we would like to emphasize the recent use of the M6L14 cluster units – obtained by solid state chemistry – as building blocks for the elaboration of nano-sized materials. Indeed, they are ideal candidates for elaboration of such materials due to their molecular size, their various intrinsic properties (magnetism, luminescence, oxydo-redox, electrical dipolar) and the possibility of apical ligand exchange for functional one. Up to now, these cluster materials were obtained with (Mo6Xi8) or (Re6Qi8) cluster cores starting from Mo6 halides or Re6 chalcohalides. However, due to their potential applications in catalysis, in biology, or in optoelectronic it is worth thinking about their future extension towards chalcohalides (Mo6Xi8-xQix) or (Re6Qi8-xXix) cluster cores, because the presence of mixed Q/X ligands on the cluster core modifies the intrinsic properties of the cluster units that could be used for specific applications. In the following we briefly report some examples of such cluster nano assemblings or nanomaterials.
In [n-Bu4N]2[(Mo6Bri8)(CF3SO3)a6], the substitution of the six apical triflate ligands by the organometallic pyridine ligands [RuCp(PPh3)2(η1-C2-4-pyridinyl)], the tri-silylferrocenyl phenolate dendron p-NaO-C6H4C{CH2CH2CH2Si(Me)2Fc}3, or the dendronic pyridine 3,3′-{CH2O-p-C6H4C(CH2CH2CH2SiMe2C5H5FeCp)3}2Py, gives new cluster-dendrimer assemblings. The two latter ones with 18 or 36 ferrocenyl endings (Fig. 19), respectively, can recognize biologic molecules by cyclic voltametry. [239,240]. The hexa arm dendrimer based on the Mo6Cl8 core [241] should also be mentioned. In the solvated [(Re6Sei8)(CH3CN)a6](SbF6)2 compound the apical ligands can be replaced by a variety of functionality-bearing dendrons to give cluster-dendrimer assemblings [190,200,242,243], with luminescence properties [242]. In another approach, [(Re6Si8)(OH)a6]4− units have been encapsulated and released in maltose-decorated polypropylene amine dendrimer, showing the interest of such assemblings for therapeutic treatment [244].
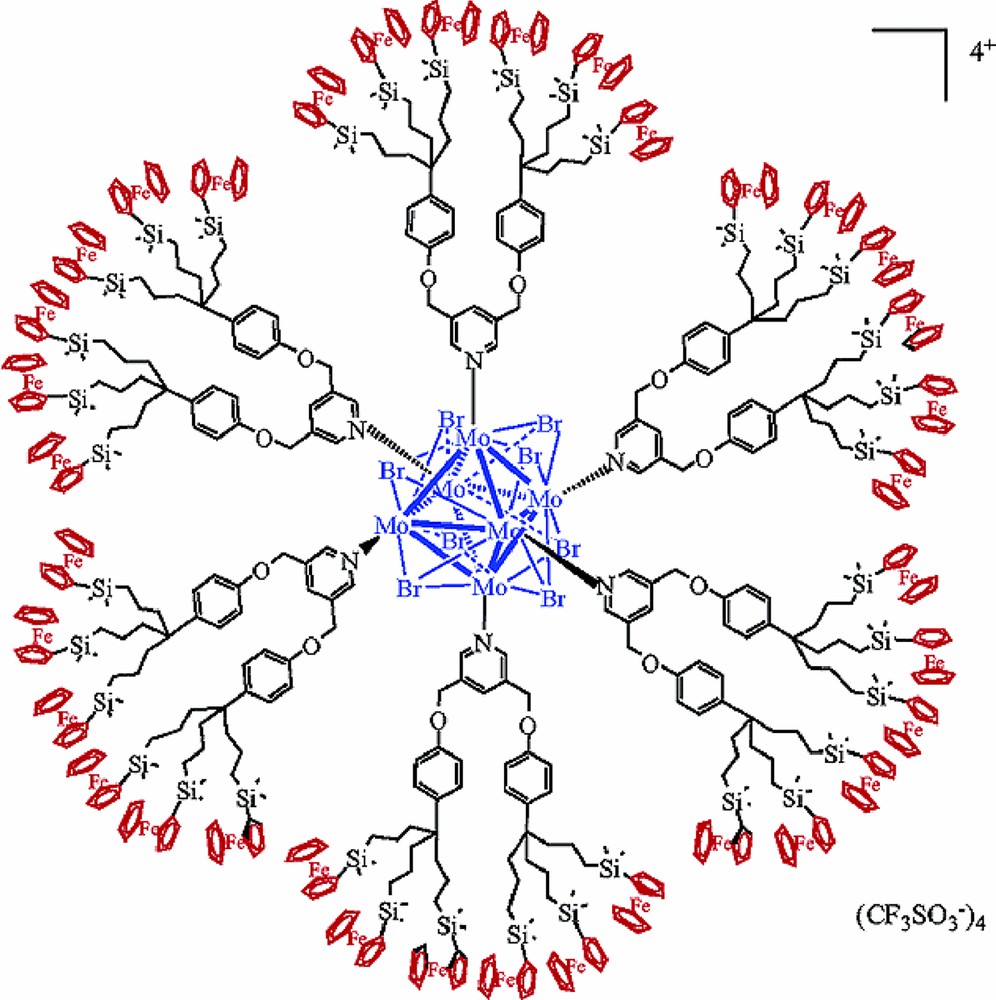
Mo6 cluster-dendrimer assembling with 36 ferrocenyl endings [239].
Other cluster nanomaterials have been obtained, like, for instance:
- • luminescent liquid crystals after the grafting of mesogenic ligands in apical position of (Mo6Bri8) [245,246] cluster cores or after using mesogenic countercations [247,248];
- • silica nanoparticles Cs2Mo6Br14@SiO2 [249], (nBu4N)2[Mo6I8(CF3CO2)6] [250] and γ-Fe2O3-Cs2Mo6Br14@SiO2 [251] in which the [Mo6L14]2− units are well-dispersed inside the silica matrix, while γ-Fe2O3 is located at the center of the particle. They are luminescent in the red and near infrared region under UV excitation and additionally magnetic for the latter bifunctional ones, and thus they are promising candidates for applications in biotechnology. Similar luminescent nanoparticles were elaborated from rhenium cluster compounds [252,253]. Characterization of photonic colloidal crystals from silica spheres with incorporated luminescent [Mo6Br14]2− cluster units was also reported [254];
- • nanocolloids and nanocomposites, [(n-C4H9)4N]2Mo6Br14@ZnO, with [Mo6Br14]2− cluster units immobilized on the ZnO nanocrystal surface. Depending on the excitation wavelength, the visible emission of either ZnO or the cluster units is observed, giving them properties of tunable emission [255];
- • [Mo6Ii8(CF3SO3)a6]2− units anchored in apical position on n- and p- type Si(111) surfaces through alkyl monolayers end-capped by pyridine, previously covalently bound to hydrogen terminated Si(111) [256,257] and trans-[(Re6Sei8](TBP)a4(OH)a2] cluster units (TBP = 4-terbutylpyridine) immobilized on Si(111) surfaces through alkyl spacers [258]. Such assemblings provide new types of molecular junctions the electronic properties of which being influenced by the presence of metal clusters;
- • [(Re6Sei8)(TBP)a4(MAC)a2] neutral cluster units embedded into polymeric matrices after copolymerization with methyl metacrylate to give cluster-polymer materials that combine the processability of the polymeric matrix with luminescent properties, keeping their properties even after several months of ageing and with a small amount of cluster units embedded in the polymer [259]. Another way was to copolymerize the cluster complex [(Re6Sei8)(PEt3)a5(4-vinylpyridine)a](SbF6)2 with styrene via the polymerizable 4-vinylpyridine ligand [199]. With molybdenum clusters, the properties of the photoactive composite materials of (nBu4N)2[Mo6I8(CF3CO2)6] in polyurethane nanofibers have been reported [250];
- • Mo6I8S2 [77,78] fibers dispersed in isopropanol after ultrasonic debundling and sedimentation [260] for use as an alternative to carbon nanotubes for applications in nanotechnology. These fibers were sulfurized under argon-H2/H2S flow in order to obtain MoS2 nanotubes with encapsulation of MoS2 fullerenes inside them [261] or, according to a similar process, using the Mo6S4I6 [262,263] fibers strongly related to Mo6I8S2 fibers [264]. These Mo6I8S2 nanofibers have been also used as precursors for the elaboration of new composite films based on MoO3-x nanowires aligned in a liquid single crystal elastomer matrix [265]. Other nanofibers in the Mo-S-I system have been used for elaboration of various electronic devices, but no precise crystal structure was reported for such fibers [266].
Finally, all the recent examples above mentioned evidence the great potentiality of cluster compounds as precursors in the new field of nanotechnology. This short review that covers the Mo6 and Re6 chalcohalides in solid state chemistry since the origin to recent period, evidences that a number of cluster compounds with chalcohalide cluster cores could be also used as precursors for elaboration of cluster nanomaterials, that opens in the future a promising and exciting way of wide investigations. Curiosity, perseverance and determination are required to develop such a research field, that were the qualities of the pioneer researchers who have started – sometimes in difficult conditions – the transition metal cluster chemistry a very long time ago [28,86].
Acknowledgements
Dr M. Sergent, who has initiated the solid state cluster chemistry in the Rennes Laboratory, is warmly acknowledged for helpful advice at the beginning of this work, many discussions and great dynamism. We would like to acknowledge our previous PhD students and coworkers of Rennes for their works and helpful discussions: Dr R. Chevrel, Dr S. Cordier, Prof R. Gautier, Dr P. Gougeon, Dr F. Grasset, Dr J.-F. Halet, Dr O. Hernandez, Dr K. Kirakci, Dr L. Leduc, Dr L. Le Pollès, Dr Y. Molard, Dr G. Pilet, Dr M. Potel and Dr A. Slougui. We would like also to mention Dr L. Ouahab and Dr P. Batail for their collaboration at the beginning of this research. The Novosibirsk group of Prof V.E. Fedorov is thanked for fruitful exchanges. The “Centre de Microscopie à Balayage” (CMEBA) with Drs J. Le Lannic, I. Peron and F. Goutefangeas, and the “Centre de Diffractométrie” of Rennes 1 University with Dr Thierry Roisnel are also thanked. Fondation Langlois is greatly acknowledged for financial support during years.