1 Introduction
The synthesis of π-conjugated 1,3,5-triarylbenzenes has received considerable attention in recent years due to their wide range of applications in the field of electrode devices [1], resisting materials [2], conducting polymers [3] or light-emitting diodes [4]. In addition, these molecules have also been used as potential building blocks for the synthesis of buckminsterfullerenes [5], polycyclic aromatic hydrocarbons (PAHs) [6], dendrimers [7] and highly conjugated polyaromatics [8]. Consequently, a number of synthetic methods have been developed previously to construct 1,3,5-triarylbenzenes. The most common strategies for their synthesis include the metal catalyzed cyclotrimerization of alkynes [9], cross coupling reactions of aryl halides with organometallic species [10] and triple self-condensation of aryl methyl ketones in the presence of an acidic catalyst such as PTSA [11], TiCl3(OTf) [12], TiCl4 [13], Bi(OTf)3.4H2O [14], phosphomolybdic acid [15], Nafion-H [16], potassium pyrosulfate and sulfuric acid [17], Amberlyst-15 [18], H3PW12O40@nano-SiO2 [19], zirconocene bis(perfluoroctanesulfonate)s [20], ethylenediamine and trifluoroacetic acid [21]. Although these processes have proved useful for the preparation of these compounds, there are some limitations including low yields, long reaction times, harsh reaction conditions, use of hazardous organic solvents, expensive metal catalysts, tedious workup procedures or requirement of special apparatus. Therefore, the development of an efficient and green methodology for the synthesis of 1,3,5-triarylbenzenes is still in high demand.
In context to develop green synthetic strategies for the sustainable development of the environment [22], there has been a growing interest to perform chemical reactions either in environmentally benign solvents such as water, ionic liquids or under solvent-free conditions. Moreover, high atom economy, use of preferred green reagents or solvents and generation of non-toxic by-products during the reaction also contribute to a clean and eco-friendly protocol [23].
Recently, DBSA has emerged as a cheap, non-hygroscopic, non-volatile and air-stable Brønsted acid surfactant-combined catalyst for carrying out various organic transformations in aqueous medium [24]. According to the literature, 1,3,5-triarylbenzenes can be efficiently obtained in high yields from acetophenones under the influence of acidic catalysts. Hence, we thought to investigate the synthesis of these molecules from aryl methyl ketones using DBSA as an acidic catalyst.
To continue our efforts on the development of one-pot syntheses [25] for various biologically important compounds and owing to the material importance of 1,3,5-triarylbenzenes, we wish to present herein a facile and economical procedure for the synthesis of 1,3,5-triarylbenzenes via triple self-condensation of aryl methyl ketones in the presence of DBSA as an efficient acidic catalyst under solvent-free conditions (Scheme 1).

DBSA catalyzed synthesis of 1,3,5-triarylbenzenes.
2 Results and discussion
In our initial study, we evaluated the scope of Yb(OTf)3, Ln(OTf)3 and DBSA as acidic catalysts for the cyclotrimerization of acetophenone at 130 °C under solvent-free conditions (Table 1, entries 1, 5 and 6). Surprisingly, the reaction catalyzed by 10 mol% Yb(OTf)3 gave the desired 1,3,5-triphenylbenzene (2a) as a white solid in only 16% isolated yield after 18 hours (Table 1, entry 1). Further attempts were unsuccessful to improve the yield of desired product (2a) by increasing the reaction time, temperature or catalytic load of Yb(OTf)3 (Table 1, entries 2–4). In contrast, Ln(OTf)3 failed completely to catalyze this transformation even after prolonged heating at 130 °C (Table 1, entry 5). On the other hand, when 10 mol% DBSA were employed to carry out this reaction under identical conditions, the desired product, 1,3,5-triphenylbenzene (2a) was obtained in 61% yield within 6 hours (Table 1, entry 6). Interestingly, the yield of the desired product (2a) did not improve significantly by enhancing either the reaction temperature or time (Table 1, entries 7 and 8); however, considerable increase in the yield of 2a was observed with higher loads of the catalyst (Table 1, entries 9 and 10). Thus, 20 mol% DBSA was chosen as the optimal catalyst load for carrying out the cyclotrimerization of acetophenone under solvent-free conditions. Since then, the synthesis of 1,3,5-triphenylbenzene (2a) has been recently accomplished [11] in 91% yield by heating acetophenone with 10 mol% p-toluenesulfonic acid (PTSA) at 130 °C for 10 hours. It was expected that increasing the load of PTSA from 10 to 20 mol% might reduce the reaction time as it is the case with DBSA. However, when the experiment was performed with 20 mol% PTSA, only 60% yield of 2a was obtained after 3 hours in contrast to 94% yield with 20 mol% DBSA under identical conditions (Table 1, entries 10 and 11). Thus, it may be concluded that DBSA is a better choice for the synthesis of 1,3,5-triarylbenzenes. Indeed, a control experiment was performed in the absence of DBSA under the same reaction conditions but no product formation was observed, even after 24 hours (Table 1, entry 12), illustrating the catalytic role of DBSA in the formation of 1,3,5-triphenylbenzene (2a). Encouraged by these results and to optimize the reaction conditions, the effect of temperature on the formation of 1,3,5-triphenylbenzene (2a) was examined. The reaction at lower temperatures such as 80 °C and 100 °C was found to be sluggish and produced poor yield of the desired product (2a) and the best results were obtained when the reaction was carried out at 130 °C (Table 1, entries 10, 13 and 14). Thus, the optimal condition for a model reaction was found to be 20 mol% DBSA at 130 °C temperature to generate 1,3,5-triphenylbenzene (2a) in 94% yield (Table 1, entry 10).
Optimization of the reaction conditions for the formation of 1,3,5-triphenylbenzene (2a) under solvent-free conditions.
| Entry | Catalysts | Catalyst load (mol%) | Temp (°C) | Time (h) | Yield (%)a |
| 1 | Yb(OTf)3 | 10 | 130 | 18 | 16 |
| 2 | Yb(OTf)3 | 10 | 130 | 24 | 25 |
| 3 | Yb(OTf)3 | 10 | 150 | 18 | 28 |
| 4 | Yb(OTf)3 | 20 | 130 | 18 | 30 |
| 5 | Ln(OTf)3 | 10 | 130 | 24 | b |
| 6 | DBSA | 10 | 130 | 6 | 61 |
| 7 | DBSA | 10 | 150 | 6 | 62 |
| 8 | DBSA | 10 | 130 | 24 | 64 |
| 9 | DBSA | 15 | 130 | 3 | 71 |
| 10 | DBSA | 20 | 130 | 3 | 94 |
| 11 | PTSA | 20 | 130 | 3 | 60 |
| 12 | – | – | 130 | 24 | b |
| 13 | DBSA | 20 | 100 | 3 | 22 |
| 14 | DBSA | 20 | 80 | 3 | c |
a Isolated yield.
b No product formation.
c Trace amount of product.
Under optimized reaction conditions, the cyclotrimerization of acetophenones was further explored in order to investigate the scope and limitations of the present methodology by heating various aryl methyl ketones at 130 °C in the presence of 20 mol% DBSA under solvent-free conditions. In general, the reaction proceeded smoothly with a wide range of aryl methyl ketones carrying either electron-donating or electron-withdrawing substituents on the aromatic ring and provided the corresponding 1,3,5-triarylbenzenes (2a–n) in good to excellent yields (Table 2). The acetophenones bearing electron-withdrawing substituents, particularly halogens on the aromatic ring, cyclotrimerized with high rates in short reaction times and afforded products (2b–h) in 72–95% yields (Table 2, entries 2–8). In contrast, the acetophenones having electron-donating substituents such as CH3 either at para- or meta-position of the aromatic ring reacted sluggishly and produced corresponding 1,3,5-triarylbenzene derivatives (2i and 2j) in 76–88% yields after 8 hours (Table 2, entries 9 and 10). In the case of acetophenones containing halogens at para-position of the aromatic ring, the yield of the cyclotrimerized products was significantly reduced, with decrease in the electronegativity of the halogen atoms (Table 2, entries 2–5). Further, the yields of triarylbenzenes varied with respect to the position of the substituents attached to acetophenone. For example, m-haloacetophenones provided high yields of the target 1,3,5-triarylbenzene products than the p-haloacetophenones (Table 2, entries 3 and 6; 4 and 7). On the other hand, m-methylacetophenone afforded the desired product (2i) in lower yield than p-methylacetophenone (Table 2, entries 9 and 10). In addition, the bulky aryl methyl ketones provided lower yields of the corresponding products (2k, 2l and 2n) when compared to acetophenone (Table 2, entries 1 and 11; 1 and 12; 1 and 14).
DBSA catalyzed cyclotrimerization of aryl methyl ketones to form 1,3,5-triarylbenzenes under solvent-free conditionsa.
| Entry | Ar | Products | Time (h) | Yield (%)b |
| 1 | C6H5 | 2a | 3 | 94 |
| 2 | p-FC6H4 | 2b | 4 | 95 |
| 3 | p-ClC6H4 | 2c | 4 | 86 |
| 4 | p-BrC6H4 | 2d | 4 | 76 |
| 5 | p-IC6H4 | 2e | 4 | 72 |
| 6 | m-ClC6H4 | 2f | 4 | 90 |
| 7 | m-BrC6H4 | 2g | 4 | 79 |
| 8 | 3,4-Cl2C6H3 | 2h | 3 | 88 |
| 9 | m-CH3C6H4 | 2i | 8 | 76 |
| 10 | p-CH3C6H4 | 2j | 8 | 88 |
| 11 | p-PhC6H4 | 2k | 3 | 92 |
| 12 | 4-cyclohexylphenyl | 2l | 10 | 74 |
| 13 | m-CF3C6H4 | 2m | 4 | 70 |
| 14 | Fluoren-2-yl | 2n | 8 | 70 |
a Reaction conditions: aryl methyl ketone (3 mmol), DBSA (0.6 mmol), 130 °C temperature.
b Isolated yields of pure products.
The proposed mechanism for the formation of 1,3,5-triarylbenzenes is depicted in Scheme 2. The reaction possibly proceeds through the protonation of aryl methyl ketone to form intermediate (I) and (II) in the presence of DBSA as an acidic catalyst. The reaction of these intermediates in the next step affords β-hydroxy ketone (III). After subsequent dehydration and addition steps, it forms an enol intermediate (VI) which on intramolecular 6-π-electrocyclization followed by dehydration affords the desired 1,3,5-triarylbenzenes.

Plausible reaction mechanism for the synthesis of 1,3,5-triarylbenzenes.
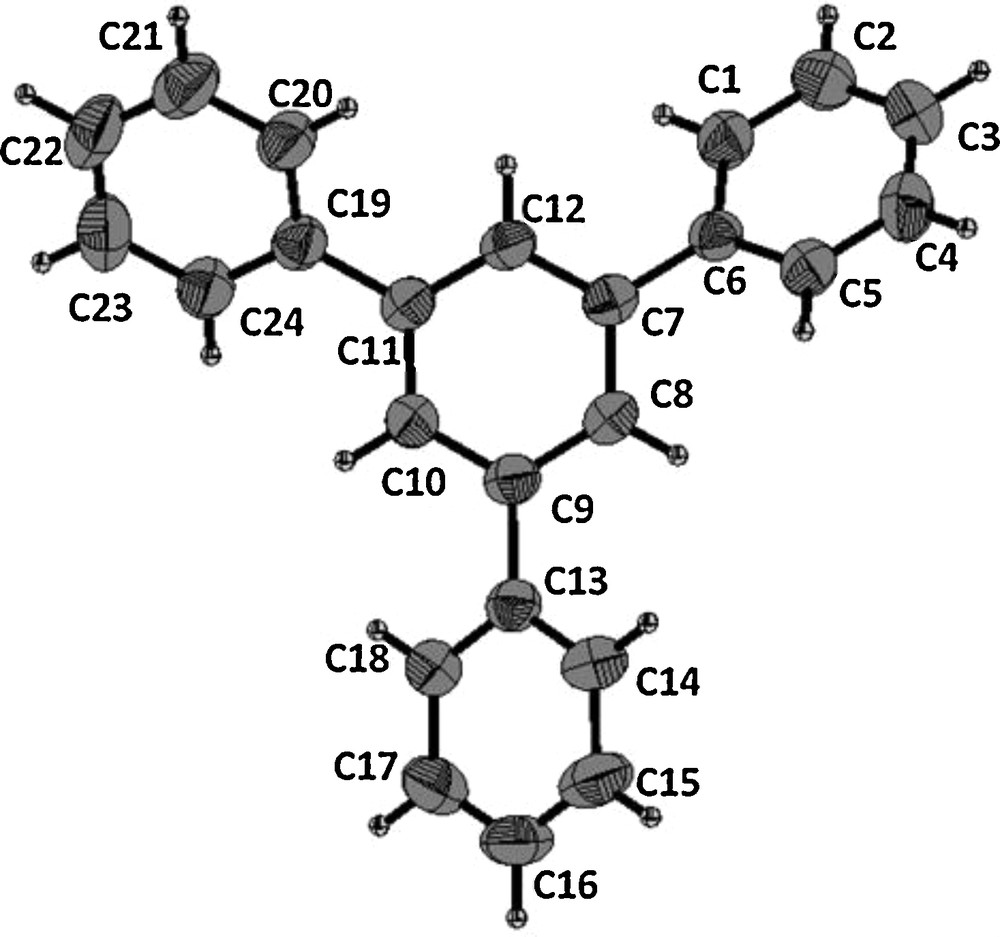
The structures of all the synthesized compounds were established on the basis of spectral data. In addition, X-ray analysis of compound 2a further proved its structure to be that of 1,3,5-triphenylbenzene (Fig. 1)1.
Furthermore, the catalytic efficiency of DBSA was compared with that of other reported catalysts for the formation of 1,3,5-tris(4′-fluorophenyl)benzene, 2b (Table 3). Though, catalysts such as PTSA, Bi(OTf)3·4H2O and phosphomolybdic acid are capable to generate the desired product (2b) in 79–90% yields by using comparatively lower catalyst loads but these procedures require either prolonged reaction time, elevated temperature or inflammable organic solvents to carry out the reaction (Table 3, entries 1–3). In contrast, the present methodology requires 20 mol% DBSA to generate the corresponding product (2b) in 95% yield at 130 °C within 4 hours under solvent-free conditions (Table 3, entry 5). Hence, DBSA can be considered as an efficient catalyst for the synthesis of 1,3,5-triarylbenzenes.
Comparative study of catalytic efficiency of DBSA with other reported catalysts for the formation of 1,3,5-tris(4′-fluorophenyl)benzene (2b).
| Entry | Catalysts (mol%) | Solvents | Time (h) | Temp (°C) | Yield (%) [References] |
| 1 | PTSA (10) | – | 24 | 143 | 90 [11] |
| 2 | Bi(OTf)3.4H2O (2) | Toluene | 18 | Reflux | 79 [14] |
| 3 | Phosphomolybdic acid (2) | Ethanol | 9 | Reflux | 80 [15] |
| 4 | Amberlyst-15 (25–30) | Toluene | 10 | Reflux | 75 [18] |
| 5 | DBSA (20) | – | 4 | 130 | 95a |
a Present work.
3 Conclusion
In summary, we have developed an efficient method for the synthesis of various 1,3,5-triarylbenzenes via cyclotrimerization of aryl methyl ketones. The reaction worked well with acetophenones bearing either electron-donating or electron-withdrawing substituents. The notable features of the present protocol are operational simplicity, high atom economy, short reaction times, good to excellent yields of the target products and most importantly, water as the only by-product generated in the reaction.
4 Experimental
All the chemicals were purchased from Sigma-Aldrich and used without further purification. The progress of the reactions was monitored by thin layer chromatography (TLC) using silica gel 60 F254 (pre-coated aluminium sheets) from Merck. TLC spots were visualized by UV-light irradiation followed by iodine. NMR spectra were obtained in CDCl3 or DMSO-d6 on a Jeol ECX 400 MHz NMR spectrometer and chemical shifts are expressed in parts per million (ppm). Infrared spectra were recorded on a Perkin Elmer IR spectrometer and absorption maxima (vmax) are given in cm−1. The melting points were determined in open capillary tubes on Buchi M-560 melting point apparatus and are uncorrected.
4.1 General procedure for the synthesis of 1,3,5-triarylbenzenes (2a–n)
A mixture of acetophenone (3 mmol) and DBSA (0.6 mmol) was heated at 130 °C in a preheated oil bath for 3–8 hours. After completion of the reaction as indicated by thin layer chromatography (TLC), the reaction mixture was cooled to room temperature and diluted with equal volumes of saturated solution of NaHCO3 and brine (5 mL + 5 mL). The resulting solution was extracted with ethyl acetate (10 mL × 3) and the organic layers were combined, dried over anhydrous Na2SO4 and evaporated under reduced pressure to dryness. The crude product obtained was purified by silica gel (60–120 mesh size) column chromatography using 1–2% ethyl acetate in heptane as the eluent to afford the desired products in pure form.
The compounds, 2a–g and 2i–l are known and their spectral and analytical data are found to be in agreement with the reported data [11,14,16,18] while the characterization data of unknown compounds (2h, 2m and 2n) are given below.
4.1.1 1,3,5-Tris(3′,4′-dichlorophenyl)benzene (2h)
White solid; mp 280 °C; yield: 94%. IR (Nujol): ν 1459, 1377, 1137, 1026, 860, 816, 718, 695 cm−1; 1H NMR (400 MHz, DMSO-d6): δ 8.28 (dd, J1 = 9.15 Hz, J2 = 2.20 Hz, 3H, ArH), 8.04 (s, 3H, ArH), 7.95 (dd, J1 = 8.05 Hz, J2 = 2.20 Hz, 3H, ArH), 7.74 (d, J = 8.79 Hz, 3H, ArH) ppm; ESI-MS: m/z = 514 [M + H]+; Anal. calcd for C24H12Cl6.H2O: C, 54.28; H, 2.66. Found: C, 54.05; H, 2.45.
4.1.2 1,3,5-Tris(3′-trifluoromethylphenyl)benzene (2m)
White solid; mp 280 °C; yield: 70%. IR (CHCl3): ν 2926, 1599, 1459, 1401, 1359, 1330, 1281, 1234, 1165, 1125, 1099, 1074, 1042, 911, 872, 801, 704, 674, 661, 621 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.92 (bs, 3H, ArH), 7.87 (d, J = 7.32 Hz, 3H, ArH), 7.79 (s, 3H, ArH), 7.69–7.61 (m, 6H, ArH) ppm; 13C NMR (100 MHz, CDCl3): δ 141.47 (d, 2JC−F = 24.92 Hz), 131.74 (d, 3JC−F = 32.59 Hz), 131.10 (d, 3JC−F = 31.63 Hz) 130.71, 129.49, 125.88, 124.60 (d, 4JC−F = 3.83 Hz), 124.14 (d, 4JC−F = 2.88 Hz), 124.09 (d, 1JC−F = 272.20 Hz) ppm; ESI-MS: m/z = 549 [M + K]+; Anal. calcd for C27H15F9.0.5H2O: C, 62.44; H, 3.10. Found: C, 62.71; H, 3.08.
4.1.3 1,3,5-Tris(9H-fluoren-2-yl)benzene (2n)
Pale yellow solid; mp 156 °C; yield: 70%. IR (Nujol): ν 1458, 1377, 1301, 1153, 824, 766, 732 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.93–7.89 (m, 9H, ArH), 7.84 (d, J = 7.32 Hz, 3H, ArH), 7.77 (d, J = 8.05 Hz, 3H, ArH), 7.58 (d, J = 7.32 Hz, 3H, ArH), 7.43–7.39 (m, 3H, ArH), 7.35–7.31 (m, 3H, ArH), 4.01 (s, 6H, CH2) ppm; 13C NMR (100 MHz, CDCl3): δ 143.95, 143.48, 142.58, 141.34, 141.16, 139.81, 126.82, 126.79, 126.20, 125.06, 125.03, 123.98, 120.17, 120.01, 37.00 ppm; ESI-MS: m/z = 593 [M + Na]+; Anal. calcd for C45H30: C, 94.70; H, 5.30. Found: C, 94.48; H, 5.30.
Acknowledgements
This work is supported by the University of Delhi, India under the scheme to strengthen R&D Doctoral Research Programme. We are thankful to Central Instrumentation Facility, University of Delhi, India for providing NMR and single crystal X-ray data. Amreeta Preetam is grateful to CSIR, New Delhi, India for providing JRF.
1 CCDC 867818 contains the supplementary crystallographic data for the deposited structure. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.