

1 Introduction
The development of environmentally benign methods in synthetic organic chemistry is of great interest, as ecological concerns increase. One of the most important goals in “green chemistry” is the elimination of volatile organic solvents [1]. Solvent-free organic reactions have captured great interest not only because of their immense ecological importance, but also because they offer many synthetic advantages in terms of high efficiency, yield, selectivity, simplicity of the reaction procedure, mild conditions, reduction of waste [2], as well as their safety, economy and low cost. The various successful solvent-free organic reactions based on grinding two reactants together at room temperature has been previously reported for aldol condensations [3], Dieckmann condensations [4], Biginelli reactions [5], Grignard reactions [6], Knoevenagel condensations [7,8], Reformatsky reactions [9], cyclopropanation reactions [10], oxidations [11], reductions [12], oximation of carbonyls [13], and rearrangement reactions [14]. Although, the potential of solvent-free methodologies based on grinding reactants together has started to emerge, there is still great need for exploring it [15].
On the other hand, imines have enormous application in organic and medicinal chemistry. Thus, they are important intermediates in reactions, such as the synthesis of secondary amines (by hydrogenation) [16], the formation of optically active α-alkyl aldehydes [17], nucleophilic additions of organometallic reagents [18], enantioselective oxidations (chiral oxaziridines) [19], cycloadditions [20,21], as well as in the synthesis of pharmaceutically significant compounds, like β-lactams [22–27]. Moreover, it is well known that they possess a wide spectrum of biological activities; they act as lipoxygenase inhibitors, anti-inflammatory agents [28] and anti-cancer agents [29–31]. Due to their fascinating pharmacological and therapeutic properties, as well as their great use in organic synthesis, the development of simple, efficient and environmentally friendly synthesis of imines is of great importance.
Imines can be prepared by various methods, but the most common one is probably the reaction of aldehydes and ketones with amines, discovered by Schiff [32]. It includes two general steps; the first one, nucleophilic attack at the carbonyl carbon by an amine, is reversible, and it is followed by the second step – removal of water – which determines feasibility of imine formation [33]. Removal of water can be accomplished by azeotropic distillation [34] (the first, traditional synthesis of imines, originally reported by Schiff et al. [35]), using molecular sieves [36], or dehydrating solvents such as tetramethyl orthosilicate [37] and trimethyl orthoformate (in situ dehydration strategy) [38,39]. Alternative approaches include condensation reactions in the presence of Lewis acids (ZnCl2 [40], TiCl4 [41–44], MgSO4–PPTS [45], alumina [46], K-10 clay [39,47] and CaO under microwave irradiation [48], Ti(OR)4 [49,50], CuSO4 [50], Mg(ClO4)2 [33]), which on, one hand, catalyze the amine nucleophilic attack on the carbonyl group and, on the other, serve as dehydrating agents in the final step. Microwave-assisted solvent-free synthesis of N-substituted aldimines has been recently reported by Paquin et al. [51].
There are several publications on the synthesis of N-methyl imines. The classical methods involve the use of aqueous solution of methylamine (which can be carried out either in water [52,53] or in the mixture of water and an organic solvent [54,55]), or the use of methylamine hydrochloride as the methylamine source [55]. An alternative way includes reaction of the appropriate aldehyde with N,N-bis(trimethylsilyl)methylamine in the presence of trimethylsilyl trifluoromethanesulfonate in dichloromethane or dichloroethane [56]. Solvent-free syntheses of N-methyl imines of aromatic aldehydes have already been reported, but these procedures include the use of either methylamine gas (overnight, at room temperature) [57] or 1,3-dimethylurea dispersed on montmorillonite K10 under microwave irradiation as the methylamine precursors [51]. Therefore, these methods have several inherent disadvantages, such as gas handling, the requirement of organic solvents, long reaction times and use of a catalyst. Herein, for the first time, we wish to report the use of methylamine hydrochloride under mild conditions as an efficient reagent in a simple and inexpensive one-pot solvent-free synthesis of N-methyl imines from aromatic aldehydes.
2 Results and discussion
The data accumulated from the reactions conducted with finely ground mixtures of 2-nitrobenzaldehyde and methylamine hydrochloride with or without a base were used as a learning set to determine the optimal conditions of the solvent-free synthesis of N-methyl aromatic imines (Scheme 1). These parameter-ranging experiments included variation of reaction time, reagent stoichiometry and the type of base used.

One-pot solvent-free synthesis of N-methyl imines of aromatic aldehydes.
We began our study by evaluating the influence of the reaction time and reagent ratio on the yield of N-(2-nitrobenzylidene)methanamine. Initially, equimolar amounts of o-nitrobenzaldehyde, CH3NH2·HCl and NaHCO3 were ground together for 10 min with a pestle and the reaction mixture was left to stand at room temperature for 30 min. The crude product turned out to be a mixture of the aldehyde and imine in the ratio 36:64 (based on a gas chromatography–mass spectrometry analysis), whereas the pure imine was isolated after column chromatography (SiO2) in the 56% yield (entry 1, Table 1). In order to improve the yield, the reaction was carried out with a 2- and 5-fold excess of CH3NH2·HCl and NaHCO3 and/or the reaction time was prolonged (to 1 and 2 h). As it can be seen from the data listed in Table 1, both of these parameters considerably affected the yield. Thus, by prolonging the reaction time to 1 and 2 h the yield increased to 66 and 69%, respectively (entries 2 and 3, Table 1), whereas the use of double amounts of CH3NH2·HCl and NaHCO3 in the reaction showed almost the same effect (entry 4, Table 1). However, the use of a 5-fold excess of these reactants did not increase the yield comparably to the preceding experiment (compare entries 4 and 5, Table 1). The best results (90%; see entry 6, Table 1) were achieved when the reaction was performed by grinding the reactants in the molar ratio aldehyde/CH3NH2·HCl/NaHCO3 = 1:2:2 and allowing the reaction mixture to stand for one additional hour at room temperature.
The influence of reaction conditions and the base on the solvent-free imination of 2-nitrobenzaldehyde with CH3NH2·HCl at room temperature.
| Entry | Base | Reagents ratio (aldehyde:CH3NH3Cl:base) | t (h) | Ratioa aldehyde:imine | Yieldb(%) |
| 1 | NaHCO3 | 1:1:1 | 0.5 | 36:64 | 56 |
| 2 | 1:1:1 | 1 | 23:77 | 66 | |
| 3 | 1:1:1 | 2 | 22:78 | 69 | |
| 4 | 1:2:2 | 0.5 | 24:76 | 68 | |
| 5 | 1:5:5 | 0.5 | 29:71 | 66 | |
| 6 | 1:2:2 | 1 | 2:98 | 90 | |
| 7 | None | 1:5:0 | 2 | 100:0 | 0 |
| 8 | 1:5:0 | Overnight | 100:0 | 0 | |
| 9 | Et3N | 1:2:2 | 1 | 53:47 | 40 |
| 10 | NaOH | 1:2:2 | 1 | 3:97 | 77 |
| 11 | K2CO3 | 1:2:2 | 1 | 0:100 | 91 |
a GC–MS ratio between peak areas.
b Isolated yields of N-methyl imines.
The reaction completely failed when carried out without a base, even when CH3NH2·HCl was used in a 5-fold excess and the reaction mixture was allowed to stand overnight at room temperature (in order to exclude a very slow reaction; see entries 7 and 8, Table 1). In these particular cases, no imine was formed and the total amount of the starting aldehyde was recovered. Therefore, the base plays a crucial role, apparently through liberating methylamine from its salt (by neutralizing HCl), and we decided to examine the influence of the type of the applied base on the yield of this reaction, keeping in mind the conditions achieving the highest yield when NaHCO3 was used as the base (2-fold excess of base and CH3NH2·HCl and a 1 h run). Triethylamine, sodium hydroxide and potassium carbonate have been chosen for this purpose. The obtained results are listed in Table 1 (entries 9, 10 and 11), and show that the best yield was obtained in the case of K2CO3 (91%, entry 11, Table 1), and NaHCO3 (90%, entry 6, Table 1), probably because CO2, as one of the products of their reactions with methylamine hydrochloride, leaves the reaction mixture and drives the equilibrium of this reaction in the desired direction. The poor reaction yield when triethylamine was used (entry 9, Table 1), could be explained by the less favourable thermodynamics of this process (the driving force of the liberation of CO2 is lacking). However, the same rationalization cannot be applied to the reaction in which NaOH was used as the base (entry 10, Table 1), since there is no thermodynamic impediment for the completion of this reaction. The lower yield than expected in this case could be explained by the fact that it readily liberates a large amount of gaseous CH3NH2, which leaves the reaction mixture before having a chance to react with the aldehyde. These findings prompted us to continue our study with the less expensive base, NaHCO3.
To investigate the scope of this reaction, various aromatic aldehydes (with electron-withdrawing and/or electron-donating substituents) were subjected to the above mentioned reaction conditions. The reactivity of the aldehydes followed the expected general trend of electrophilicity (Table 2), i.e. it has been observed that the aldehydes bearing electron-withdrawing groups gave better yields in shorter reaction times and with lower aldehyde to amine ratios than the ones with electron-donating substituents. Specifically, among ortho-substituted aldehydes, o-nitrobenzaldehyde gave the highest yield (entry 6, Table 1), whereas the yields of the aldehydes with electron-releasing substituents in the ortho position, such as 2-hydroxy- (entries 47–52, Table 2), 2-alkyloxy- (entries 25, 32–34, 44–46, Table 2) or 2-acetoxybenzaldehydes (entry 43, Table 2) were lower but still better or comparable than those of the corresponding meta- and para-isomers (which is perhaps a consequence of the higher electrophilicity of the ortho-isomers compared to meta- and para- due to the displacement of the aldehyde group from the plane of the aromatic core by the ortho-substituent). Among the o-substituted aldehydes, the poorest outcome was noted for salicylaldehyde (entries 47–49, Table 2), but a gas chromatography–mass spectrometry GC–MS) analysis revealed that the resulting reaction mixture contains no starting aldehyde. This could be explained by acidic properties of the phenolic group (pKa = 8.37). During the grinding of salicylaldehyde with CH3NH2·HCl and NaHCO3, the sodium salt of the aldehyde is immediately formed as apparent from the yellow coloration of the mixture. Deprotonation is expected to slow down the reaction (–O− is a much more powerful electron-donating substituent compared to –OH) and make the work-up not as straightforward (a significant share of the imine could, as well, be in the form of a salt and, hence, that part would remain insoluble in ether). This becomes more apparent for more acidic derivatives of salicylaldehyde (3,5-dibromosalicylaldehyde and 5-nitrosalicylaldehyde) that either give much lower yields of the appropriate N-methyl imines or no imine at all (20 and 0%, respectively) (entries 50–52, Table 2). The GC–MS analysis revealed that in the case of 3,5-dibromosalicylaldehyde, the resulting reaction mixture contained no starting aldehyde, whereas in the case of 5-nitrosalicylaldehyde, the reaction failed completely (aldehyde:imine ratio was 100:0, according to the corresponding peak areas ratio in GC–MS chromatograms). On the other hand, the resulting ortho-hydroxy imines are additionally stabilized by intramolecular hydrogen bonding (perhaps this is the fact that can provide some insight into the formation of the imine of salicylaldehyde in general).
Solventless imination of aromatic amines by CH3NH2·HCl in the presence of NaHCO3 as the base at room temperature.
| Entry | Aldehyde | Reagents ratioa | Time (h) | Imine | Yield (%)b |
| 1 | 1a | 1:2:2 | 1 | 80 | |
| 2 | 1b | 1:2:2 | 0.5 | 38 | |
| 3 | 1:5:5 | 1 | 79 | ||
| 4 | o.n | 89 | |||
| 5 | 1c | 1:5:5 | 1 | 20 | |
| 6 | o.n | 94 | |||
| 7 | 1d | 1:5:5 | 1 | 22 | |
| 8 | o.n | 87 | |||
| 9 | 1e | 1:5:5 | 1 | 20 | |
| 10 | o.n | 78 | |||
| 11 | 1af | 1:5:5 | 1 | 85 | |
| 12 | 1f | 1:5:5 | 1 | 83 | |
| 13 | 1g | 1:5:5 | 1 | 78 | |
| 14 | 1h | 1:5:5 | 1 | 18 | |
| 15 | o.n | 79 | |||
| 16 | 1i | 1:5:5 | 1 | 32 | |
| 17 | o.n | 80 | |||
| 18 | 1j | 1:5:5 | 2 | 86 | |
| 19 | 1k | 1:5:5 | 1 | 41 | |
| 20 | 1:5:5 | o.n | 60 | ||
| 21 | 1l | 1:5:5 | 1 | 47 | |
| 22 | o.n | 56 | |||
| 23 | 1m | 1:5:5 | 1 | 43 | |
| 24 | o.n | 51 | |||
| 25 | 1n | 1:5:5 | 1 | 77 | |
| 26 | 1o | 1:5:5 | 1 | 48 | |
| 27 | o.n | 84 | |||
| 28 | 1p | 1:2:2 | 1 | 33 | |
| 29 | 1:5:5 | 1 | 51 | ||
| 30 | 1:5:5 | 3 | 65 | ||
| 31 | 1:5:5 | o.n | 75 | ||
| 32 | 1q | 1:5:5 | 2 | 73 | |
| 33 | 1r | 1:5:5 | 2 | 25 | |
| 34 | o.n | 58 | |||
| 35 | 1s | 1:2:2 | 1 | 3 | |
| 36 | 1:5:5 | o.n | 71 | ||
| 37 | 1t | 1:2:2 | 1 | 1 | |
| 38 | 1:5:5 | o.n | 1 | ||
| 39 | 1:10:10 | o.n | 80 | ||
| 40 | 1u | 1:5:5 | 2 | 48 | |
| 41 | o.n | 82 | |||
| 42 | 1v | 1:5:5 | 2 | 78 | |
| 43 | 1w | 1:5:5 | 2 | 81 | |
| 44 | 1z | 1:5:5 | 1 | 82 | |
| 45 | 1x | 1:5:5 | 1 | 38 | |
| 46 | o.n | 79 | |||
| 47 | 1y | 1:3:0 | 0.75 | 0 | |
| 48 | 1:5:5 | 1 | 55 | ||
| 49 | 1:2:2 | 2 | 61 | ||
| 50 | 1aa | 1:5:5 | 1 | 20 | |
| 51 | 1ab | 1:5:0 | o.n | 0 | |
| 52 | 1:5:5 | o.n | 0 | ||
| 53 | 1ac | 1:5:5 | 2 | 78 | |
| 54 | 1ad | 1:5:0 | 2 | 0 | |
| 55 | 1:5:5c | 4 | 0 | ||
| 56 | 1:5:1d | 2 | 26 | ||
| 57 | 1:5:5 | 2 | 89 | ||
| 58 | 1ae | 1:5:0 | 2 | 0 | |
| 59 | o.n | 0 | |||
| 60 | 1:5:5 | 2 | 0 |
a Aldehyde/CH3NH3Cl/base
b Isolated yields of N-methyl imines.
c NaH2PO4 was used as a base.
d Et3N was used as a base.
The yield of the reaction seems to be dependent on the steric nature of the ortho-substituent as well. The derivative with the sterically demanding ortho-hexyloxy-group gave noteworthy amounts of N-(2-(hexyloxy)benzylidene)methanamine only after the reaction mixture was left to stand overnight (entry 46, Table 2).
As expected, the aldehydes with para- positioned electron-releasing substituents gave the poorest results in this reaction. In the case of p-dimethylaminobenzaldehyde, the reaction failed and the total amount of the starting aldehyde was recovered (entries 58–60, Table 2). A quantitative synthesis of N,N-dimethyl-4-((methylimino)methyl)aniline from the corresponding aldehyde and gaseous methylamine was, however, previously reported [57]. This led us to consider the possibility that differing rates of the reaction between p-dimethylaminobenzaldehyde and methylamine and the loss of methylamine gas from the reaction mixture by evaporation might be the reason why no imine was detected.
A satisfying yield of N-methyl imine of 4-methoxybenzaldehyde (65%) was obtained when employing the reactants’ ratio (aldehyde/CH3NH2·HCl/NaHCO3) 1:5:5, already after 3 h, after grinding (entry 30, Table 2). In order to reach a comparable yield of N-(3,4-dimethoxybenzylidene)methanamine, the reaction mixture was required to stand overnight (entry 36, Table 2), whereas the conversion of 3,4,5-trimethoxybenzaldehyde to the corresponding imine, besides the overnight standing, necessitated also a 10-fold excess of CH3NH2 (entry 39, Table 2).
These results motivated us to study the kinetics of this reaction further. We decided to investigate the time dependence of the formation of the N-methyl imines of 4-metoxy- and 4-nitrobenzaldehyde, by 1H NMR spectroscopy, utilizing the reactants’ ratio (aldehyde/CH3NH2·HCl/NaHCO3) 1:5:5, by tracking the yield of the reactions left to stand after grinding from 10 min to 48 h. A plot (a) of the yields vs time for the two aldehydes and stacked 1H NMR spectra (b) of the reaction mixtures after 0, 0.5, 3 and 48 h of contact of 4-nitrobenzaldehyde, CH3NH2·HCl and NaHCO3 in the ratio 1:5:5 are presented in Fig. 1.
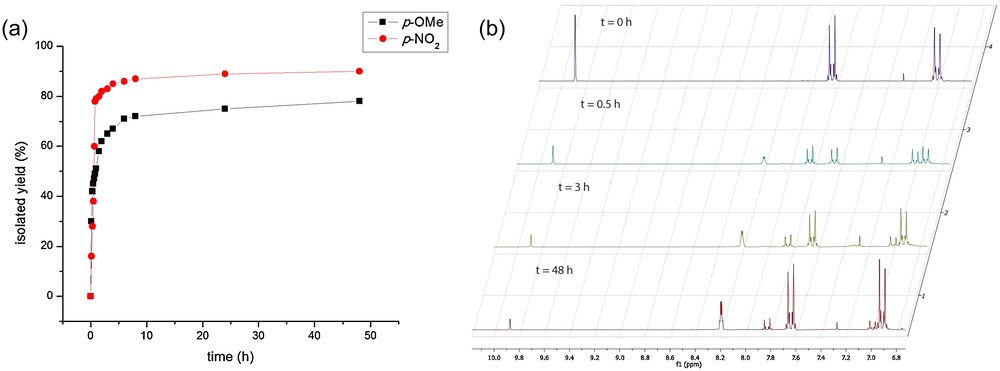
(a) Time dependence of the reaction yield of N-methyl imines of 4-metoxy- and 4-nitrobenzaldehyde; (b) Stacked 1H NMR spectra of the reaction mixtures after 0, 0.5, 3 and 48 h of contact of 4-nitrobenzaldehyde, CH3NH2·HCl and NaHCO3 in the ratio 1:5:5.
The reaction was expectedly slower in the case of the electron-rich aldehyde. After 1 h, the isolated yield of N-(4-methoxybenzylidene)methanamine was 51% (entry 29, Table 2), whereas the same of N-(4-nitrobenzylidene)methanamine was 79% (entry 3, Table 2). However, N-(4-methoxybenzylidene)methanamine could still be obtained in a good yield but only after leaving the reaction mixture to stand overnight.
Mechanistically, the formation of an imine must involve two steps. First, the amine nitrogen acts as a nucleophile attacking the carbonyl carbon and a tetrahedral intermediate is formed. Dehydration of the intermediate gives an imine [58]. The data given in Table 2 and in Fig. 1 are in agreement with the fact that electron-withdrawing substituents promote the addition of CH3NH2 to an aldehyde, but also that the electron-donating ones favour the dehydration step. It can be concluded that the first step is the rate-limiting one, whereas the second step is faster. As the nucleophilic attack by the amine is reversible, the feasibility of imine formation largely depends on the removal of water in the second step [33]. An acid is required to protonate the aminal intermediate, but too much acid would protonate all of the amine and prevent it from acting as a nucleophile. Thus, imine formation is sensitive to the amount of acid and it is fastest at about pH 4–6 [58], making NaHCO3, with pKb ca. 7 and a CO2 driving force, almost an ideal choice for the base utilized to liberate methylamine from its hydrochloride.
Following the procedure described herein, we prepared 31 N-methyl imines out of which eight (2r, 2s, 2 v, 2w, 2x, 2y, 2z, 2ag, Scheme 1) were synthesized for the first time ever and their structures were elucidated by spectral means (1H- and 13C-NMR, IR, MS) (Section 4.6). N-methyl imines, of some of the aldehydes with electron-donating substituents, were synthesized in the amounts insufficient for detailed spectral characterization, due to their lower electrophilicity (2k, 2w, 2x, 2ab, 2ae, Scheme 1).
In order to evaluate the preparative value of the present methodology, we carried out this reaction on a gram-scale. Salicylaldehyde (5 g, 41 mmol) or 4-chlorobenzaldehyde (2 g, 14 mmol), when reacted with methylamine hydrochloride and NaHCO3 (5 mol eq), gave the corresponding imine in 72 and 84% yield, respectively. The initial product of salicylaldehyde required no further purification according to a GC–MS analysis.
Finally, we tried to perform a solvent-free synthesis of N-methyl imines of aliphatic aldehydes and aromatic and aliphatic ketones, but in all of these cases, the reactions failed. As it is well known, primary aliphatic aldehydes, in general, give polymeric materials with amines, due to the ease with which the imines initially formed, undergo subsequent aldol condensations [32]. On the other side, the reactions of ketones (both aromatic and aliphatic ones) were unsuccessful because of the lower electrophilicity, in part from the greater steric hindrance, of carbonyl groups, in comparison to those of the aldehydes.
3 Conclusion
In conclusion, we have established a convenient one-pot solvent-free synthesis of N-methyl imines from aromatic aldehydes, using methylamine hydrochloride as the methylamine source. This method not only provides good to high yields, but also eliminates the use of hazardous solvents, more or less expensive catalysts and the necessity of work/handling with an anhydrous gas in pressurized containers. Additionally, the advantages of this method include mild reaction conditions, the simplicity of the procedure, easy work-up, as well as the fact that this is an environmentally benign and inexpensive approach. Methylamine hydrochloride is easy and safe to handle, and it could be either commercially obtained or prepared by a very simple procedure from inexpensive formaldehyde solution and ammonium chloride. This synthesis provides high yields of imines of aldehydes with electron-withdrawing substituents in short reaction times, whereas in the case of aldehydes with electron-releasing substituents, yields can be enhanced with an excess of methylamine hydrochloride and base and/or by prolonged reaction times. All the before mentioned facts make this method the simplest one for the synthesis of N-methyl imines of aromatic aldehydes.
4 Experimental
4.1 General
Preparative medium-pressure liquid chromatography (MPLC) was performed with a pump module C-601 and a pump controller C-610 Work-21 pump (Büchi, Switzerland) and was carried out on pre-packed column cartridges (40 × 75 mm, Silica-gel 60, particle size distribution 40–63 μm, Büchi). Silica-gel 60 on Al plates, layer thickness 0.2 mm (Kieselgel 60 F254, Merck) was used for thin layer chromatography (TLC). The spots on TLC were visualized by UV light (254 nm) and by spraying with 50% (v/v) aqueous H2SO4 followed by heating. The GC-MS analyses were performed on a Hewlett-Packard 6890 N gas chromatograph equipped with fused silica capillary column HP-5MS (5% phenylmethylsiloxane, 30 m × 0.25 mm, film thickness 0.25 μm, Agilent Technologies, USA) and coupled with a 5975B mass selective detector from the same company as detailed below. The IR measurements (ATR-attenuated total reflectance) were carried out using a Thermo Nicolet model 6700 FTIR instrument (Waltham, USA). The NMR spectra were recorded on a Varian Gemini 200 (1H at 200 MHz, 13C at 50 MHz) spectrometer, using CDCl3 as the solvent. Chemical shifts are expressed in δ (ppm) using TMS (Me4Si) as an internal standard. Microanalysis of carbon, hydrogen and nitrogen were carried out with a Carlo Erba 1106 microanalyzer; their results agreed favorably with the calculated values.
4.2 Gas chromatography–mass spectrometry analyses
The reaction progress was monitored and the reaction mixture composition preliminary were determined by GC–MS. The GC–MS analyses (three repetitions for each sample) were performed on a Hewlett-Packard 6890 N gas chromatograph equipped with fused silica capillary column HP-5MS (5% phenylmethylsiloxane, 30 m × 0.25 mm, film thickness 0.25 μm, Agilent Technologies, USA) and coupled with a 5975B mass selective detector from the same company. The injector and interface were operated at 250 and 290 °C, respectively. Oven temperature was raised from 70 to 290 °C at a heating rate of 5 °C/min and then isothermally held for 20 min. As a carrier gas, He at 1.0 mL/min was used. The samples, 1 μL of the solutions in Et2O (ca. 1 mg in 1 mL of Et2O), were injected in a pulsed split mode (the flow was 1.5 mL/min for the first 0.5 min and then set to 1.0 mL/min throughout the remainder of the analysis; split ratio 40:1). Mass selective detector was operated at the ionization energy of 70 eV, in the 35–650 amu range and scanning speed of 0.34 s. The percentage composition was computed from the total ion chromatogram peak areas without the use of correction factors. Qualitative analyses of the reaction mixture constituents were based on the analysis of the fragmentation patterns from their MS.
4.3 Materials
All of the commercially obtained reagents (Aldrich, USA; Merck, Germany; Roth, Germany; Carlo Erba, Italia; B.D.H laboratory reagents, England) were used as received, except that the solvents were purified by distillation. Methylamine hydrochloride, utilized in the syntheses, was prepared by the procedure reported by Marvel and Jenkins [59]. Some of the starting aldehydes were commercial, while the others were synthetized by the previously described procedures: 2-acetoxybenzaldehyde and 4-acetoxy-3-methoxybenzaldehyde [60]; 2-hydroxy-5-nitrobenzaldehyde [61]; 3-nitrobenzaldehyde [62]; 3-iodovanilin [63]. Alkoxybenzaldehydes (2-methoxy-, 2-methoxy-5-nitro-, 3,5-dibromo-2-methoxy-, 2-isopropoxy-, 2-(hexyloxy)benzaldehyde and 3-iodo-4-methoxybenzaldehyde) were prepared from corresponding hydroxybenzaldehydes and alkyl halides [64]. The structures of all of the synthesized aldehydes were confirmed by spectral means (1H- and 13C-NMR, IR, MS), and spectral data, which were not available in the literature, are listed below (the literature survey of the CAS database was accomplished through the use of SciFinder Scholar 2007 [65]).
4.4 Spectral data not available in the literature of the employed aldehydes
4.4.1 2-methoxy-5-nitrobenzaldehyde (1r)
Colorless oil; FTIR (neat): 3081, 2984, 2948, 2850, 1678 (CO), 1584, 1521, 1484, 1468, 1427, 1399, 1338, 1279, 1248, 1163, 1140, 1073, 1016, 940, 906, 831, 746, 634 cm−1; EIMS m/z 181 [M]+ (100.0), 180 [M–H]+ (30.7), 166 [M–CH3]+ (11.2), 164 (27.8), 163 (41.4), 150 [M–OCH3]+ (6.4), 149 [M–CH3OH]+ (9.3), 135 [M–NO2] (18.7), 134 [M–H–NO2] (34.9), 120 [M–CH3–NO2]+ (29.4), 118 (13.8), 106 [M–CHO–NO2]+ (4.9), 103 (19.9), 92 (33.6), 77 (47.2), 75 (32.4), 74 (17.1), 64 (22.1), 63 (35.4), 51 (11.7); Calcd for C8H7NO4: C 53.04, H 3.89, N 7.73, O 35.33%; found: C 53.06, H 3.88, N 7.80, O 35.26%; RI (HP-5MS) 1714.
4.4.2 3,5-dibromo-2-methoxybenzaldehyde (1s)
White amorphous solid; FTIR (neat): 3067, 2878, 1683 (CO), 1572, 1556, 1457, 1414, 1380, 1243, 1217 (C–O–C, asym.), 1144, 975, 890, 870, 811, 746, 717, 628 cm−1; EIMS m/z 296 [M81Br2]+ (48.0), 295 [M81Br2–H]+ (53.0), 294 [M81Br79Br]+ (9.6), 293 [M81Br79Br–H]+ (25.3), 292 [M79Br2]+ (2.2), 291 [M79Br2–H]+ (12.9), 281 [M81Br2–CH3]+ (25.7), 280 [M81Br2–H–CH3]+ (11.5), 279 [M81Br79Br–CH3]+ (66.3), 278 [M81Br79Br–H–CH3]+ (50.1), 277 [M79Br2–CH3]+ (55.0), 276 [M79Br2–H–CH3]+ (71.4), 267 (10.2), 265 (24.4), 264 [M81Br2–CH3OH]+ (11.9), 263 (22.5), 262 [M81Br79Br–CH3OH]+ (19.1), 261 (6.9), 260 [M79Br2–CH3OH]+ (9.4), 252 [M81Br2–CHO–CH3]+ (16.1), 250 [M81Br79Br–CHO–CH3]+ (36.9), 248 [M79Br2–CHO–CH3]+ (47.4), 236 [M81Br2–CHO–OCH3]+ (10.3), 234 [M81Br79Br–CHO–OCH3]+ (16.5), 232 [M79Br2–CHO–OCH3]+ (6.4), 225 (20.6), 223 (42.9), 221 (22.2), 172 (33.3), 170 (33.8), 156 (24.5), 155, (28.9), 143 (32.5), 141 (34.3), 77 (41.5), 75 (53.7), 74 (59.7), 63 (55.9), 62 (64.2), 61 (33.0); 13C NMR (CDCl3, 50 MHz): 63.5, 118.2, 119.2, 130.5, 131.5, 141.3, 159.1, 187.5; 1H NMR (CDCl3, 200 MHz): 3.98 (s, 3H, OCH3), 7.89 (d, J = 2.5 Hz, 1H, H6), 7.93 (d, J = 2.5 Hz, 1H, H4), 10.28 (s, 1H, H7); Calcd for C8H6Br2O2: C 32.69, H 2.06, Br 54.37, O 10.89%; found: C 32.63, H 2.03%; RI (HP-5MS) 1624.
4.4.3 2-isopropoxybenzaldehyde (1y)
Colorless oil; FTIR (neat): 2978, 2930, 2862, 2361, 1673 (CO), 1597, 1581, 1479, 1457, 1385, 1283, 1237 (C–O–C, asym.), 1192, 1160, 1112, 1092, 1041, 949, 865, 825, 752 cm−1; 13C NMR (CDCl3, 50 MHz): 21.9 (2 C), 71.0, 113.9, 120.3, 125.6, 128.2, 135.7, 160.5, 190.2; Calcd for C10H12O2: C 73.15, H 7.37, O 19.49%; found: C 73.09, H 7.40, O 19.51%; RI (HP-5MS) 1330.
4.5 General procedure for the solvent-free synthesis of N-methyl imines of aromatic aldehydes
The reactants – corresponding aldehyde (entries 1–60, Tables 1 and 2) (1.5 mmol), CH3NH3Cl (2–10 mol eq, Tables 1 and 2) and the base (2–10 mol eq, Tables 1 and 2) (the amount of CH3NH3Cl and NaHCO3 were equimolar) – were grinded in a mortar with a pestle for 10 min and then left to stand at room temperature from 1 h to overnight (Tables 1 and 2). The reaction was monitored by TLC, GC-MS and 1H NMR. The reaction mixture was taken up with Et2O and the resultant suspension was filtrated through a layer of anhydrous MgSO4. The solvent was evaporated in vacuo to give the crude product. Some of the obtained azometines were pure enough for spectral characterization, while the others were purified by MPLC. The products were characterized by IR, 1H, and 13C NMR, MS, whenever they were synthesized in the sufficient amounts. The spectral data of the known compound were compared with those from the literature and showed favorable agreement.
4.6 Spectral data of the newly synthesized imines
4.6.1 N-(2-nitrobenzylidene)methanamine (2a)
Colorless oil; FTIR (neat): 3099, 3073, 3034, 2972, 2953, 2900, 2850, 2763, 1646 (CN), 1608, 1574, 1514 (C–NO2, sym.), 1454, 1444, 1394, 1372, 1348 (C–NO2, sym.), 1213, 999, 965, 938, 843, 779, 742, 699, 685 cm−1; EIMS m/z 163 [M–H]+ (0.5), 147 (39.2), 135 [M–NCH3]+ (16.5), 121 (17.0), 118 [M–NO2]+ (9.1), 117 [M–H–NO2]+ (100.0), 105 (4.4), 102 (5.5), 90 (17.4), 89 [M–NCH3–NO2]+ (13.7), 77 (30.0), 76 [M–CHNCH3–NO2]+ (16.6), 65 (7.6), 63 (8.7), 51 (11.0), 50 (10.4), 42 [M–C6H4NO2]+ (17.2); 13C NMR (CDCl3, 50 MHz): 48.2, 124.1, 129.3, 130.4, 130.9, 133.2, 148.6, 158.0; 1H NMR (CDCl3, 200 MHz): 3.57 (d, J = 1.6 Hz, 3H, NCH3), 7.57 (dt, J = 7.6, 1.6 Hz, 1H, H4), 7.66 (dt, J = 7.6, 1.2 Hz, 1H, H5), 7.98 (dd, J = 7.6, 1.2 Hz, 1H, H3), 8.02 (dd, J = 7.6, 1.6 Hz, 1H, H6), 8.68 (q, J = 1.6 Hz, 1H, NCH); Calcd for C8H8N2O2: C 58.53, H 4.91, N 17.06, O 19.49%; found: C 58.45, H 4.86, N 17.14, O 19.55%; RI (HP-5MS) 1377.
1H NMR spectroscopic data are in accordance with [54].
4.6.2 N-(3-nitrobenzylidene)methanamine (2b)
Colorless oil; FTIR (neat): 3084, 2952, 2875, 2768, 1650 (CN), 1614, 1521 (C–NO2, asym.), 1474, 1455, 1438, 1395, 1371, 1347 (C–NO2, sym.), 1216, 1124, 999, 954, 829, 807, 732, 673 cm−1; EIMS m/z 164 [M]+ (73.4), 163 [M–H]+ (100), 147 (13.6), 118 [M–NO2]+ (23.3), 117 [M–H–NO2]+ (56.5), 105 (3.4), 90 (11.1), 89 [M–NCH3–NO2]+ (13.1), 77 (21.6), 76 [M–CHNCH3–NO2]+ (11.5), 65 (3.6), 63 (8.9), 51 (9.1), 50 (9.8), 42 [M–C6H4NO2]+ (56.4); 13C NMR (CDCl3, 50 MHz): 48.1, 122.6, 124.8, 129.5, 133.2, 137.8, 148.5, 159.8; 1H NMR (CDCl3, 200 MHz): 3.58 (d, J = 1.6 Hz, 3H, NCH3), 7.60 (t, J = 7.8 Hz, 1H, H5), 8.06 (dt, J = 7.8, 1.2 Hz, 1H, H6), 8.26 (ddd, J = 7.8, 2.0, 1.2 Hz, 1H, H4), 8.37 (q, J = 1.6 Hz, 1H, N = CH), 8.54 (t, J = 2.0 Hz, 1H, H2); Calcd for C8H8N2O2: C 58.53, H 4.91, N 17.06, O 19.49%; found: C 58.48, H 4.89, N 17.11, O 19.52%; RI (HP-5MS) 1460.
13C NMR and 1H NMR spectroscopic data are in accordance with [55].
4.6.3 N-(4-nitrobenzylidene)methanamine (2c)
Yellow-brown amorphous solid; FTIR (neat): 2967, 2901, 2834, 2774, 1648 (CN), 1601, 1511 (C–NO2, asym.), 1440, 1340 (C–NO2, sym.), 1291, 1219, 1192, 1101, 1001, 972, 953, 847, 832, 813, 745, 692, 682 cm−1; EIMS m/z 164 [M]+ (73.8), 163 [M–H]+ (100), 147 (7.0), 118 [M–NO2]+ (9.7), 117 [M–H–NO2]+ (70.9), 105 (6.8), 91 (9.7), 90 (11.9), 89 [M–NCH3–NO2]+ (12.7), 77 (15.0), 76 [M–CHNCH3–NO2]+ (11.7), 65 (5.9), 63 (10.6), 51 (9.5), 50 (11.3), 42 [M–C6H4NO2]+ (52.8); 13C NMR (CDCl3, 50 MHz): 48.1, 123.5 (2 C), 128.3 (2 C), 141.5, 148.6, 159.9; 1H NMR (CDCl3, 200 MHz): 3.58 (d, J = 1.6 Hz, 3H, NCH3), 7.87 (m, 2H, ArH (AA’)), 8.23 (m, 2H, ArH (BB’)), 8.36 (q, J = 1.6 Hz, 1H, NCH); Calcd for C8H8N2O2: C 58.53, H 4.91, N 17.06, O 19.49%; found: 58.50, H 4.90, N 17.09, O 19.51%; RI 1455 (HP-5MS).
1H NMR spectroscopic data are in accordance with [51,66].
4.6.4 3-((methylimino)methyl)benzonitrile (2d)
Colorless oil; FTIR (neat): 3062, 2943, 2854, 2771, 2222 (CN); 1651 (C = N), 1475, 1455, 1434, 1400, 1360, 1289, 1147, 1000, 955, 797, 738, 683 cm−1; EIMS m/z 144 [M]+ (48.0), 143 [M–H]+ (100.0), 128 [M–CH3–H]+ (1.5), 116 (13.9), 103 (5.1), 102 [M–CHNCH3]+ (12.9), 89 [M–NCH3–CN]+ (5.5), 76 [M–CHNCH3–CN]+ (6.6), 75 (7.2), 63 (4.0), 51 (4.4), 50 (4.6), 42 [M–C7H4N]+ (30.0); 13C NMR (CDCl3, 50 MHz): 48.0, 112.8, 118.2, 129.4, 131.2, 131.7, 133.4, 137.1, 159.8; 1H NMR (CDCl3, 200 MHz): 3.55 (d, J = 1.6 Hz, 3H, NCH3), 7.53 (t, J = 7.7 Hz, 1H, H5), 7.69 (dt, J = 7.7, 1.5 Hz, 1H, ArH), 7.94 (dt, J = 7.7, 1.5 Hz, 1H, ArH), 8.00 (t, 1.5, 1H, H2), 8.29 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H8N2: C 74.98, H 5.59, N 19.43%; found: C 74.95, H 5.60, N 19.44%; RI (HP-5MS) 1345.
4.6.5 4-((methylimino)methyl)benzonitrile (2e)
White amorphous solid; FTIR (neat): 2951, 2882, 2860, 2775, 2223 (CN), 1649 (C = N), 1606, 1495, 1454, 1394, 1363, 1301, 1290, 1220, 1195, 1161, 1108, 1002, 944, 863, 835, 643 cm−1; EIMS m/z 144 [M]+ (51.6), 143 [M–H]+ (100.0), 128 [M–CH3–H]+ (1.8), 116 (13.2), 103 (6.4), 102 [M–CHNCH3]+ (13.2), 89 [M–NCH3–CN]+ (5.0), 76 [M–CHNCH3–CN]+ (6.2), 75 (6.4), 63 (3.7), 51 (4.2), 50 (4.5), 42 [M–C7H4N]+ (24.1); 13C NMR (CDCl3, 50 MHz): 47.8, 113.2, 118.0, 127.8 (2 C), 131.9 (2 C), 139.6, 160.1; 1H NMR (CDCl3, 200 MHz): 3.55 (d, J = 1.6 Hz, 3H, NCH3), 7.67 (m, 2H, ArH (AA’)), 7.80 (m, 2H, ArH (BB’)), 8.30 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H8N2: C 74.98, H 5.59, N 19.43%; found: C 75.02, H 5.57, N 19.46%; RI (HP-5MS) 1349.
1H NMR spectroscopic data are in accordance with [67].
4.6.6 N,N’-(1,3-phenylenedimethylidyne)bis(methanamine) (2f)
Light yellow amorphous solid; FTIR (neat): 2939, 2880, 2847, 2772, 1647 (C = N), 1583, 1455, 1433, 1399, 1355, 1292, 1267, 1157, 1000, 957, 795, 690, 653, 639 cm−1; EIMS m/z 160 [M]+ (84.1), 159 [M–H]+ (100.0), 145 [M–CH3]+ (8.1), 144 [M–CH3–H]+ (3.7), 143 (13.6), 133 (6.4), 132 (7.2), 131 [M–NCH3]+ (4.9), 119.1 (8.0), 118 [M–CHNCH3]+ (44.1), 117 (20.8), 116 (7.4), 91 (9.1), 90 (6.2), 89 [M–CHNCH3–NCH3]+ (11.0), 77 (9.9), 76 [M–CHNCH3–CHNCH3]+ (5.0), 65 (3.2), 63 (6.6), 51 (5.4), 42 [M–C6H4CHNCH3]+ (32.1); 13C NMR (CDCl3, 50 MHz): 48.1 (2 C), 127.8, 128.8, 129.4 (2 C), 136.5 (2 C), 161.8 (2 C); 1H NMR (CDCl3, 200 MHz): 3.52 (d, J = 1.6 Hz, 6H, NCH3), 7.44 (t, J = 7.6 Hz, 1H, H5), 7.78 (dd, J = 7.6, 1.6 Hz, 2H, H4,6), 7.97 (bs, 1H, H2), 8.29 (q, J = 1.6 Hz, 2H, N = CH); Calcd for C10H12N2: C 74.97, H 7.55, N 17.48%; found: C 75.01, H 7.56, N 17.45%; RI 1425 (HP-5MS).
4.6.7 N-(2-chlorobenzylidene)methanamine (2g)
Colorless oil; FTIR (neat): 2942, 2891, 2772, 1642 (C = N), 1592, 1571, 1468, 1433, 1399, 1365, 1270, 1201, 1156, 1121, 1050, 1029, 1001, 957, 854, 827, 751, 709, 628 cm−1; EIMS m/z 155 [M37Cl]+ (18.9), 154 [M37Cl–H]+ (37.1), 153 [M35Cl]+ (57.7), 152 [M35Cl–H]+ (100.0), 137 (3.5), 125 (12.7), 118 [M–Cl]+ (4.6), 117 [M–Cl–H]+ (4.5), 113 (3.4), 112 (10.2), 111 (8.6), 102 (7.2), 89 [M–NCH3–Cl]+ (10.1), 77 (6.1), 76 [M–CHNCH3–Cl]+ (4.7), 75 (11.1), 63 (6.0), 51 (5.3), 50 (6.6), 42 [M–C6H4Cl]+ (22.1); 13C NMR (CDCl3, 50 MHz): 48.4, 127.0, 128.1, 129.7, 131.3, 133.2, 134.9, 159.2; 1H NMR (CDCl3, 200 MHz): 3.56 (d, J = 1.7 Hz, 3H, NCH3), 7.25-7.40 (m, 3H, ArH), 7.98 (dd, J = 7.3, 2.5 Hz, 1H, H6), 8.72 (q, J = 1.7 Hz, 1H, N = CH); Calcd for C8H8ClN: C 62.55, H 5.25, Cl 23.08, N 9.12%; found: C 62.58, H 5.26, N 9.13%; RI (HP-5MS) 1198.
NMR spectroscopic data are in accordance with literature (1H NMR [54,68]; 13C NMR [68]).
4.6.8 N-(3-chlorobenzylidene)methanamine (2h)
Colorless oil; FTIR (neat): 2943, 2892, 2855, 2771, 1652 (C = N), 1594, 1568, 1471, 1453, 1429, 1362, 1281, 1262, 1211, 1194, 1073, 1002, 952, 888, 782, 722, 702, 682 cm−1; EIMS m/z 155 [M37Cl]+ (14.2), 154 [M37Cl–H]+ (35.7), 153 [M35Cl]+ (44.2), 152 [M35Cl–H]+ (100.0), 137 (3.3), 127 (3.9), 125 (12.5), 118 [M–Cl]+ (15.8), 117 [M–Cl–H]+ (8.1), 113 (4.7), 112 (6.2), 111 (13.8), 102 (4.0), 91 (4.2), 89 [M–NCH3–Cl]+ (9.9), 76 [M–CHNCH3–Cl]+ (4.5), 77 (5.6), 75 (12.7), 63 (6.3), 51 (5.2), 50 (7.1), 42 [M–C6H4Cl]+ (29.3); 13C NMR (CDCl3, 50 MHz): 48.1, 126.1, 127.7, 129.8, 130.4, 134.8, 138.1, 160.9; 1H NMR (CDCl3, 200 MHz): 3.51 (d, J = 1.6 Hz, 3H, NCH3), 7.32 (dd, J = 7.9, 6.8 Hz, 1H, H5), 7.37 (dt, J = 7.9, 2 Hz, 1H, ArH), 7.54 (ddd, J = 6.8, 2, 1.8 Hz, 1H, ArH), 7.71 (t, J = 1.8 Hz, 1H, H2), 8.22 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C8H8ClN: C 62.55, H 5.25, Cl 23.08, N 9.12%; found: C 62.56, H 5.26, N 9.07%; RI (HP-5MS) 1219.
1H NMR spectroscopic data are in accordance with [54].
4.6.9 N-(4-chlorobenzylidene)methanamine (2i)
Colorless oil; FTIR (neat): 2944, 2892, 2846, 2769, 1649 (C = N), 1596, 1572, 1488, 1455, 1399, 1296, 1206, 1087, 1013, 1001, 959, 858, 817, 702 cm−1; EIMS m/z 155 [M37Cl]+ (17.4), 154 [M37Cl–H]+ (36.6), 153 [M35Cl]+ (52.9), 152 [M35Cl–H]+ (100.0), 137 (4.6), 127 (4.5), 125 (14.2), 118 [M–Cl] + (5.7), 117 [M–Cl–H]+ (5.5), 113 (4.7), 112 (6.3), 111 (12.9), 102 (5.1), 89 [M–NCH3–Cl]+ (9.7), 77 (4.9), 76 [M–CHNCH3–Cl]+ (3.9), 75 (11.4), 63 (5.7), 51 (4.6), 50 (6.5), 42 [M–C6H4Cl]+ (18.0); 13C NMR (CDCl3, 50 MHz): 48.1, 128.8 (2 C), 129.0 (2 C), 134.7, 136.4, 161.0; 1H NMR (CDCl3, 200 MHz): 3.51 (d, J = 1.6 Hz, 3H, NCH3), 7.38 (m, 2H, ArH (AA’)); 7.64 (m, 2H, ArH (BB’)), 8.24 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C8H8ClN: C 62.55, H 5.25, Cl 23.08, N 9.12%; found: C 62.47, H 5.25, N 9.15%; RI (HP-5MS) 1219.
1H NMR spectroscopic data are in accordance with [51,54].
4.6.10 N-(4-bromobenzylidene)methanamine (2j)
Light yellow amorphous solid; FTIR (neat): 2943, 2880, 2845, 2768, 1649 (C = N), 1588, 1570, 1487, 1452, 1433, 1400, 1365, 1295, 1066, 1009, 1001, 958, 935, 854, 814, 705, 679 cm−1; EIMS m/z 199 [M81Br]+ (52.6), 198 [M81Br–H]+ (100.0), 197 [M79Br]+ (55.1), 196 [M79Br–H]+ (99.5), 171 (9.7), 169 (10.2), 157 [M81Br–CHNCH3]+ (10.3), 155 [M79Br–CHNCH3]+ (10.3), 118 [M–Br]+ (14.9), 117 [M–Br–H]+ (14.8), 102 (8.8), 91 (7.3), 90 (8.8), 89 [M–NCH3–Br]+ (12.6), 77 (11.1), 76 [M–CHNCH3–Br]+ (11.9), 75 (12.0), 63 (8.0), 62 (5.0), 59 (5.6), 51 (6.9), 50 (12.4), 42 [M–C6H4Br]+ (25.9); 13C NMR (CDCl3, 50 MHz): 48.1, 124.8, 129.3 (2 C), 131.8 (2 C), 135.1, 161.2; 1H NMR (CDCl3, 200 MHz): 3.50 (d, J = 1.6 Hz, 3H, NCH3), 7.50-7.60 (m, 4H, ArH), 8.22 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C8H8BrN: C 48.51, H 4.07, Br 40.34, N 7.07%; found: C 48.47, H 4.05, N 7.10%; RI (HP-5MS) 1316.
IR and 1H NMR spectroscopic data are in accordance with [66].
4.6.11 N-(2-methylbenzylidene)methanamine (2l)
Colorless oil; FTIR (neat): 2934, 2877, 2839, 2771, 1644 (C = N), 1601, 1483, 1454, 1400, 1368, 1286, 1001, 960, 753, 716 cm−1; EIMS m/z 133 [M]+ (38.5), 132 [M–H]+ (51.2), 118 [M–CH3]+ (100.0), 117 [M–CH3–H]+ (33.0), 105 (4.9), 104 [M–NCH3]+ (3.4), 103 [M–NH2CH2]+ (5.3), 91 [M–CHNCH3]+ (26.2), 90 (6.3), 89 [M–NCH3–CH3]+ (9.8), 77 (7.0), 65 (11.7), 63 (7.3), 51 (6.1), 42 [M–C7H7]+ (10.3); 13C NMR (CDCl3, 50 MHz): 19.2, 48.7, 126.2, 127.1, 130.0, 130.7, 134.2, 137.7, 161.0; 1H NMR (CDCl3, 200 MHz): 2.50 (s, 3H, CH3), 3.53 (d, J = 1.6 Hz, 3H, NCH3), 7.14-7.30 (m, 3H, ArH), 7.83 (dd, J = 7.2, 1.8 Hz, 1H, H6), 8.59 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H11N: C 81.16, H 8.32, N 10.52%; found: C 81.12, H 8.30, N 10.57%; RI (HP-5MS) 1153.
1H NMR spectroscopic data are in accordance with [54].
4.6.12 N-(3-methylbenzylidene)methanamine (2m)
Colorless oil; FTIR (neat): 2945, 2842, 2763, 1650 (C = N), 1603, 1586, 1480, 1455, 1400, 1362, 1282, 1249, 1158, 999, 955, 781, 692 cm−1; EIMS m/z 133 [M]+ (46.2), 132 [M–H]+ (100.0), 118 [M–CH3]+ (21.3), 117 [M–CH3–H]+ (7.5), 116 (5.0), 105 (10.1), 103 [M–NH2CH2] (3.2), 92 (5.3), 91 [M–CHNCH3]+ (26.6), 90 (3.5), 89 [M–NCH3–CH3]+ (6.4), 77 (5.2), 65 (9.4), 63 (5.5), 51 (4.3), 42 [M–C7H7]+ (11.3); 13C NMR (CDCl3, 50 MHz): 21.2, 48.1, 125.4, 128.1, 128.4, 131.3, 135.2, 138.3, 162.6; 1H NMR (CDCl3, 200 MHz): 2.38 (s, 3H, CH3), 3.51 (d, J = 1.6 Hz, 3H, NCH3), 7.24 (bd, J = 7.6 Hz, 1H, H4), 7.30 (t, J = 7.6 Hz, 1H, H5), 7.46 (bd, J = 7.6 Hz, 1H, H6), 7.56 (bs, 1H, H2), 8.25 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H11N: C 81.16, H 8.32, N 10.52%; found: C 81.15, H 8.34, N 10.54%; RI (HP-5MS) 1150.
4.6.13 N-(4-methylbenzylidene)methanamine (2n)
Colorless oil; FTIR (neat): 2942, 2883, 2839, 2774, 1650 (C = N), 1607, 1510, 1451, 1400, 1306, 1172, 1004, 961, 810, 758 cm−1; EIMS m/z 133 [M]+ (53.2), 132 [M–H]+ (100.0), 118 [M–CH3]+ (10.8), 117 [M–CH3–H]+ (6.1), 116 (6.1), 105 (10.8), 103 [M–NH2CH2]+ (5.5), 92 (4.9), 91 [M–CHNCH3]+ (26.0), 89 [M–NCH3–CH3]+ (6.2), 77 (5.3), 65 (9.3), 63 (5.2), 51 (4.6), 42 [M–C7H7]+ (10.3); 13C NMR (CDCl3, 50 MHz): 21.4, 48.1, 127.8 (2 C), 129.3 (2 C), 133.7, 140.7, 162.4; 1H NMR (CDCl3, 200 MHz): 2.37 (s, 3H, CH3), 3.50 (d, J = 1.6 Hz, 3H, NCH3), 7.21 (m, 2H, H3,5 (AA’)), 7.59 (m, 2H, H2,6 (BB’)), 8.24 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H11N: C 81.16, H 8.32, N 10.52%; found: C 81.18, H 8.30, N 10.49%; RI (HP-5MS) 1155.
1H NMR and IR spectroscopic data are in accordance with literature (1H NMR [51,66]; IR [66]).
4.6.14 N-(2-methoxybenzylidene)methanamine (2o)
Colorless oil; FTIR (neat): 3002, 2938, 2883, 2837, 2770, 1642 (C = N), 1598, 1581, 1487, 1464, 1437, 1367, 1285, 1245 (C–O–C, asym.), 1160, 1109, 1025 (C–O–C, sym.), 999, 958, 857, 785, 752, 634 cm−1; EIMS m/z 149 [M]+ (10.1), 148 [M–H]+ (49.0), 134 [M–CH3]+ (26.9), 133 [M–H–CH3]+ (22.6), 120 [M–NCH3]+ (29.5), 119 [M–H2NCH2]+ (100.0), 118 [M–OCH3]+ (75.2), 105 (15.0), 91 (49.2), 77 (21.3), 65 (10.3), 51 (13.3), 42 [M–C6H4OCH3]+ (21.4); 13C NMR (CDCl3, 50 MHz): 48.3 (C8), 55.3 (OCH3), 110.8 (C3), 120.6 (C5), 124.5 (C1), 126.9 (C6), 131.5 (C4), 158.3 (C7), 158.4 (C2); 1H NMR (CDCl3, 200 MHz): 3.50 (d, J = 1.6 Hz, 3H, NCH3), 3.83 (s, 3H, OCH3), 6.88 (d, J = 8.4 Hz, 1H, H3), 6.95 (t, J = 7.6 Hz, 1H, H5), 7.35 (ddd, J = 8.4, 7.6, 1.8 Hz, 1H, H4), 7.89 (dd, J = 7.6, 1.8 Hz, 1H, H6), 8.70 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H11NO: C 72.46, H 7.43, N 9.39, O 10.72%; found: C 72.39, H 7.46, N 9.48, O 10.67%; RI (HP-5MS) 1298.
1H NMR spectroscopic data are in accordance with [54].
4.6.15 N-(3-methoxybenzylidene)methanamine (2p)
Colorless oil; FTIR (neat): 3005, 2943, 2836, 2769, 1650 (C = N), 1582, 1486, 1454, 1432, 1400, 1367, 1317, 1262, 1194, 1167, 1154, 1122, 1038, 1000, 952, 868, 781, 756, 688 cm−1; EIMS m/z 149 [M]+ (51.4), 148 [M–H]+ (100.0), 134 [M–CH3]+ (5.7), 133 [M–H–CH3]+ (15.2), 119 [M–NH2CH2]+ (7.8), 118 [M–OCH3]+ (17.1), 117 [M–CH3OH]+ (4.6), 106 (6.3), 105 (8.3), 91 (5.2), 78 (8.2), 77 (11.3), 65 (5.5), 63 (6.3), 51 (5.6), 42 [M–C6H4OCH3]+ (13.8); 13C NMR (CDCl3, 50 MHz): 48.0, 55.2, 111.1, 117.3, 121.2, 129.4, 137.6, 159.8, 162.3; 1H NMR (CDCl3, 200 MHz): 3.50 (d, J = 1.6 Hz, 3H, NCH3), 3.82 (s, 3H, OCH3), 6.96 (ddd, J = 8.0, 2.6, 1.3 Hz, 1H, ArH), 7.18-7.34 (m, 3H, ArH), 8.23 (q, J = 1.6 Hz, 1H, N = CH); Calcd for C9H11NO: C 72.46, H 7.43, N 9.39, O 10.72%; found: C 72.42, H 7.44, N 9.43, 10.71%; RI (HP-5MS) 1291.
1H NMR spectroscopic data are in accordance with [54].
4.6.16 N-(4-methoxybenzylidene)methanamine (2q)
Colorless oil; FTIR (neat): 2937, 2884, 2837, 2770, 1650 (CN), 1604, 1578, 1509, 1457, 1307, 1245 (C–O–C, asym.), 1163, 1007, 1027 (C–O–C, sym.), 961, 829, 775 cm−1; EIMS m/z 149 [M]+ (62.7), 148 [M–H]+ (100.0), 134 [M–CH3]+ (4.2), 133 [M–H–CH3]+ (18.4), 121 (10.5), 118 [M–OCH3]+ (2.4), 117 [M–CH3OH]+ (2.6), 105 (6.8), 91 (5.4), 77 (9.4), 65 (5.6), 51 (5.2), 42 [M–C6H4OCH3]+ (6.7); 13C NMR (CDCl3, 50 MHz): 47.5, 54.8, 113.5 (2 C), 128.9, 129.0 (2 C), 161.1, 161.4; 1H NMR (CDCl3, 200 MHz): 3.48 (d, J = 1.6 Hz, 3H, NCH3), 3.83 (s, 3H, OCH3), 6.92 (m, 2H, H3,5 (AA’)), 7.65 (m, 2H, H2,6 (MM’)), 8.21 (q, J = 1.6 Hz, 1H, NCH); Calcd for C9H11NO: C 72.46, H 7.43, N 9.39, O 10.72%; found: C 72.48, H 7.39, N 9.44, 10.69%; RI (HP-5MS) 1321.
NMR spectroscopic data are in accordance with literature (1H NMR [51,54] and 13C NMR [69]).
4.6.17 N-(2-methoxy-5-nitrobenzylidene)methanamine (2r)
Colorless oil; FTIR (neat): 3088, 3010, 2951, 2905, 2843, 2772, 1643 (CN), 1610, 1584, 1509 (C–NO2, asym.), 1480, 1455, 1425, 1340 (C–NO2, sym.), 1263 (C–O–C, asym.), 1184, 1139, 1075 (C–O–C, sym.), 1013, 953, 822, 747, 634 cm−1; EIMS m/z 194 [M]+ (9.5), 193 [M–H]+ (68.3), 179 [M–CH3]+ (21.1), 165 [M–NCH3]+ (24.9), 164 [M–NH2CH2]+ (100.0), 163 [M–OCH3]+ (39.9), 148 [M–NO2]+ (9.7), 147 [M–H–NO2]+ (47.4), 133 [M–NH2CH2–OCH3]+ (23.5), 118 [M–NH2CH2–NO2]+ (42.7), 107 (3.0), 105 (9.4), 104 (11.6), 90 (6.9), 89 (7.5), 77 (13.5), 76 (10.6), 65 (3.9), 63 (13.1), 51 (7.4), 42 [M–C6H3OCH3NO2]+ (33.5); 13C NMR (CDCl3, 50 MHz): 48.4, 56.3, 110.9, 123.2, 125.5, 126.9, 141.8, 156.1, 162.6; 1H NMR (CDCl3, 200 MHz): 3.56 (d, J = 1.6 Hz, 3H, NCH3), 4.00 (s, 3H, OCH3), 7.00 (d, J = 9.2 Hz, 1H, H3), 8.26 (dd, J = 9.2, 2.9 Hz, 1H, H4), 8.65 (q, J = 1.6 Hz, 1H, NCH), 8.78 (d, J = 2.9 Hz, 1H, H6); Calcd for C9H10N2O3: C 55.67, H 5.19, N 14.43, O 24.72%; found: C 55.62, H 5.17, N 14.47, O 24.74%; RI (HP-5MS) 1793.
4.6.18 N-(3,5-dibromo-2-nitrobenzylidene)methanamine (2s)
White amorphous solid; FTIR (neat): 3065, 2943, 2877, 2828, 2761, 1649 (CN), 1572, 1553, 1454, 1410, 1377, 1353, 1219 (C–O–C, asym.), 1146, 1006, 988, 948, 864, 812, 708, 651 cm−1; EIMS m/z 309 [M81Br2]+ (2.0), 308 [M81Br2–H]+ (14.2), 307 [M81Br79Br]+ (4.8), 306 [M81Br79Br–H]+ (25.7), 305 [M79Br2]+ (2.2), 304 [M79Br2–H]+ (12.9), 294 [M81Br2–CH3]+ (11.4), 293 [M81Br2–H–CH3]+ (7.3), 292 [M81Br79Br–CH3]+ (23.8), 291 [M81Br79Br–H–CH3]+ (13.6), 290 [M79Br2–CH3]+ (14.3), 289 [M79Br2–H–CH3]+ (8.8), 279 [M81Br2–NH2CH2]+ (52.1), 278 [M81Br2–OCH3]+ (54.3), 277 [M81Br79Br–NH2CH2]+ (100.0), 276 [M81Br79Br–OCH3]+ (84.5), 275 [M79Br2–NH2CH2]+ (50.4), 274 [M79Br2–OCH3]+ (39.6), 267 [M81Br2–CHNCH3]+ (2.1), 265 [M81Br79Br–CHNCH3]+ (5.6), 263 [M79Br2–CHNCH3]+ (4.6), 198 (33.5), 196 (40.5), 118 (11.6), 117 (17.5), 75 (25.4), 74 (23.4), 63 (16.3), 62 (17.3), 42 [M–C6H2OCH3Br2]+ (75.5); 13C NMR (CDCl3, 50 MHz): 48.5, 62.4, 117.9, 118.4, 129.5, 132.3, 137.2, 155.8, 156.2; 1H NMR (CDCl3, 200 MHz): 3.57 (d, J = 1.6 Hz, 3H, NCH3), 3.85 (s, 3H, OCH3), 7.73 (d, J = 2.4 Hz, 1H, H6), 8.02 (d, J = 2.4 Hz, 1H, H4), 8.50 (q, J = 1.6 Hz, 1H, NCH); Calcd for C9H9Br2NO: C 35.21, H 2.96, N 4.56, O 5.21%; found: C 35.24, H 3.00, N 4.52%; RI (HP-5MS) 1722.
4.6.19 N-(4,5-dimethoxybenzylidene)methanamine (2t)
Colorless amorphous solid; FTIR (neat): 3002, 2936, 2837, 2771, 1646 (CN), 1585, 1509, 1452, 1418, 1263 (C–O–C, asym.), 1235, 1134, 1021 (C–O–C, sym.), 956, 867, 807, 749, 731 cm−1; EIMS m/z 179 [M]+ (100.0), 178 [M–H]+ (98.5), 164 [M–CH3]+ (24.4), 163 [M–H–CH3]+ (10.4), 148 [M–OCH3]+ (38.9), 133 [M–OCH3–CH3]+ (13.5), 120 [M–NCH3–CH3–CH3]+ (13.1), 119 [M–OCH3–NCH3]+ (9.1), 105 (5.1), 104 (5.7), 95 (9.7), 92 (7.6), 91 (4.7), 77 (13.9), 65 (8.6), 51 (8.1), 42 [M–C6H3(OCH3)2]+ (14.7); 13C NMR (CDCl3, 50 MHz): 47.8, 55.7 (2 C), 108.1, 110.2, 122.7, 129.3, 149.1, 151.0, 161.9; 1H NMR (CDCl3, 200 MHz): 3.49 (d, J = 1.4 Hz, 3H, NCH3), 3.91 (s, 3H, C3–OCH3)*, 3.93 (s, 3H, C3–OCH3)*, 6.87 (d, J = 8.2 Hz, 1H, H5), 7.12 (dd, J = 8.2, 1.9 Hz, 1H, H6), 7.41 (d, J = 1.9 Hz, 1H, H2), 8.19 (q, J = 1.4 Hz, 1H, N−CH); Calcd for C10H13NO2: C 67.02, H 7.31, N 7.82, O 17.85%; found: C 66.95, H 7.32, N 7.80, O 17.93%; RI (HP-5MS) 1533.
*Interchangeable protons.
4.6.20 N-(3,4,5-dimethoxybenzylidene)methanamine (2u)
Colorless amorphous solid; FTIR (neat): 2939, 2839, 1649 (CN), 1583, 1500, 1455, 1416, 1369, 1325, 1229 (C–O–C, asym.), 1121, 1001, 831, 713, 624 cm−1; EIMS m/z 209 [M]+ (100.0), 208 [M–H]+ (53.8), 194 [M–CH3]+ (29.0), 178 [M–OCH3]+ (24.3), 164 [M–NH2CH2–CH3]+ (8.2), 163 [M–OCH3–CH3]+ (8.0), 162 [M–CH3OH–CH3]+ (8.1), 151 (13.5), 136 [M–CHNCH3–OCH3]+ (11.4), 125 (17.2), 110 (10.0), 104 (2.8), 95 (9.1), 79 (6.5), 77 (5.0), 66 (5.9), 65 (5.4), 53 (5.5), 42 [M–C6H2(OCH3)3]+ (13.0); 13C NMR (CDCl3, 50 MHz): 47.9, 56.0 (2 C), 60.8, 104.7 (2 C), 131.7, 140.0, 153.3 (2 C), 161.9; 1H NMR (CDCl3, 200 MHz): 3.51 (d, J = 1.5 Hz, 3H, NCH3), 3.89 (s, 3H, C4–OCH3), 3.91 (s, 6H, C3–OCH3, C5–OCH3), 6.96 (s, 2H, H2,6), 8.18 (q, J = 1.5 Hz, 1H, NCH); Calcd for C11H15NO3: C 63.14, H 7.23, N 6.69, O 22.94%; found: C 63.17, H 7.22, N 6.74, O 22.87%; RI (HP-5MS) 1671.
IR and 1H NMR spectroscopic data are in accordance with [53].
4.6.21 N-(3-iodo-4,5-dimethoxybenzylidene)methanamine (2v)
Colorless oil; FTIR (neat): 3087, 2990, 2968, 2926, 2850, 1646 (CN), 1586, 1560, 1461, 1403, 1364, 1267 (C–O–C, asym.), 1228, 1136, 1164, 1042 (C–O–C, sym.), 1004, 946, 835, 798, 747, 641 cm−1; EIMS m/z 305 [M]+ (100.0), 304 [M–H]+ (39.0), 290 [M–CH3]+ (17.4), 274 [M–OCH3]+ (10.9), 259 [M–CH3–OCH3]+ (4.2), 246 [M–NCH3–CH3–CH3]+ (4.8), 178 [M–I]+ (4.0), 163 [M–I–CH3]+ (7.6), 162 [M–H–I–CH3]+ (8.4), 148 [M–I–NH2CH2]+ (11.5), 132 [M–I–CH3–OCH3]+ (12.9), 118 [M–I–NCH3–OCH3]+ (12.2), 90 (4.5), 89 (10.4), 77 (9.9), 65 (6.0), 63 (7.7), 51 (4.7), 50 (5.7), 42 [M–C6H2(OCH3)2I]+ (18.0); 13C NMR (CDCl3, 50 MHz): 48.0, 56.0, 60.5, 92.0, 110.2, 131.5, 134.7, 150.9, 152.8, 160.3; 1H NMR (CDCl3, 200 MHz): 3.51 (d, J = 1.6 Hz, 3H, NCH3), 3.86 (s, 3H, C5–OCH3)*, 3.90 (s, 3H, C4–OCH3) *, 7.36 (d, J = 1.8 Hz, 1H, H2), 7.57 (d, J = 1.8 Hz, 1H, H6), 8.13 (q, J = 1.6 Hz, 1H, NCH); Calcd for C10H12INO2: C 39.36, H 3.96, N 4.59, O 10.49%; found: C 39.42, H 3.94, N 4.56%; RI (HP-5MS) 1832.
*Interchangeable protons.
4.6.22 N-(2-isopropoxybenzylidene)methanamine (2y)
Colorless oil; FTIR (neat): 2976, 2935, 2885, 2771, 1641 (CN), 1598, 1579, 1482, 1452, 1371, 1284, 1239 (C–O–C, asym.), 1159, 1114, 1000, 1043, 951, 867, 752 cm−1; EIMS m/z 177 [M]+ (2.9), 176 [M–H]+ (5.5), 162 [M–CH3]+ (43.6), 147 [M–NH2CH2]+ (16.1), 135 [M–C3H6]+ (14.3), 134 [M–C3H7]+ (100.0), 119 [M–C3H7–CH3]+ (27.0), 118 [M–OC3H7]+ (67.0), 107 (18.8), 91 (10.4), 77 (12.6), 65 (8.3), 51 (6.5), 42 [M–C6H4OCH3]+ (12.3); 13C NMR (CDCl3, 50 MHz): 22.1 (2 C), 48.5, 70.9, 113.9, 120.7, 125.8, 127.1, 131.5, 157.1, 158.8; 1H NMR (CDCl3, 200 MHz): 1.37 (d, J = 6.1 Hz, 6H, OCH(CH3)2), 3.52 (d, J = 1.6 Hz, 3H, NCH3), 4.60 (sep, J = 6.1 Hz, 1H, OCH(CH3)2), 6.87–7.05 (m, 2H, ArH), 7.34 (ddd, J = 8.2, 7.5, 1.8 Hz, 1H, H4), 7.90 (dd, J = 7.6, 1.8 Hz, 1H, H6), 8.71 (q, J = 1.6 Hz, 1H, NCH); Calcd for C11H15NO: C 74.54, H 8.53, N 7.90, O 9.03 %; found: C 74.60, H 8.52, N 7.86, O 9.02 %; RI (HP-5MS) 1371.
4.6.23 N-(2-(hexyloxy)benzylidene)methanamine (2z)
Colorless oil; FTIR (neat): 2930, 2857, 2770, 1642 (CN), 1599, 1581, 1487, 1454, 1365, 1296, 1286, 1243 (C–O–C, asym.), 1159, 1044, 1000, 958, 752 cm−1; EIMS m/z 219 [M]+ (2.2), 218 [M–H]+ (3.9), 204 [M–CH3]+ (6.1), 190 [M–C2H5]+ (4.2), 189 [M–NH2CH2]+ (4.7), 176 [M–C3H7]+ (3.0), 162 [M–C4H9]+ (9.6), 148 [M–C5H11]+ (100.0), 135 [M–C6H12]+ (18.5), 134 [M–C6H13]+ (41.7), 120 [M–CH3–C6H12]+ (24.1), 118 [M–OC6H13]+ (63.5), 107 (13.8), 91 (12.5), 77 (9.8), 65 (5.9), 51 (3.9), 42 [M–C6H4OCH3]+ (13.8); 13C NMR (CDCl3, 50 MHz): 14.0, 22.5, 25.7, 29.1, 31.5, 68.3, 48.5, 111.9, 120.5, 124.7, 126.8, 131.6, 158.1, 158.4; 1H NMR (CDCl3, 200 MHz): 0.92 (t, J = 6.6 Hz, 3H, H6’), 1.30–1.42 (m, 4H, H4’,5’), 1.43–1.53 (m, 2H, H3’), 1.82 (qui, J = 6.4 Hz, 2H, H2’), 4.00 (t, J = 6.4 Hz, 2H, H1’), 3.52 (d, J = 1.6 Hz, 3H, NCH3), 6.91 (d, 8.4 Hz, H3), 6.97 (t, 7.4 Hz, H5), 7.34 (ddd, J = 8.4, 7.4, 1.8 Hz, 1H, H4), 7.90 (dd, J = 7.7, 1.8 Hz, 1H, H6), 8.73 (q, J = 1.6 Hz, 1H, NCH); Calcd for C14H21NO: C 76.67, H 9.65, N 6.39, O 7.29%; found: C 76.74, H 9.70, N 6.37, O 7.19%; RI (HP-5MS) 1736.
4.6.24 2-((methylimino)methyl)phenol (2aa)
Colorless oil; FTIR (neat): 3054, 3007, 2954, 2887, 2854, 2750 br (O–H···N), 1634 (CN); 1582, 1495, 1450, 1417, 1401, 1368, 1277 (C–O), 1211, 1151, 1036, 1005, 965, 837, 752, 647 cm−1; EIMS m/z 135 [M]+ (100.0), 134 [M–H]+ (95.1), 120 [M–CH3]+ (59.2), 118 [M–OH]+ (17.5), 107 (17.6), 91 (8.3), 77 (14.5), 65 (11.8), 51 (10.4), 42 [M–C6H4OH]+ (14.8); 13C NMR (CDCl3, 50 MHz): 45.9, 116.9, 118.4, 118.8, 130.9, 131.9, 161.1, 166.1; 1H NMR (CDCl3, 200 MHz): 3.47 (d, J = 1.4 Hz, 3H, NCH3), 6.86 (td, J = 7.6, 1.1 Hz, 1H, H5), 6.95 (d, J = 8.2 Hz, 1H, H3), 7.23 (dd, J = 7.6, 1.6 Hz, 1H, H6), 7.29 (ddd, J = 8.2, 7.6, 1.6 Hz, 1H, H4), 8.33 (q, J = 1.4 Hz, 1H, NCH); Calcd for C8H9NO: C 71.09, H 6.71, N 10.36, O 11.84%; found: C 71.01, H 6.76, N 10.31, O 11.92%; RI (HP-5MS) 1230.
13C NMR and IR spectroscopic data are in accordance with literature (13C NMR [69]); IR [70,71]); 1H NMR spectroscopic data are in accordance with [70], except for the signal of H8.
4.6.25 2-methoxy-5-((methylimino)methyl)phenol (2ad)
Colorless oil; FTIR (neat): 3400 br (O–H), 2932, 2840, 1646 (CN), 1605, 1583, 1507, 1438, 1271 (C–O–C, asym.), 1166, 1126, 1020 (C–O–C, sym.), 958, 873, 804, 759 cm−1; EIMS m/z 165 [M]+ (75.8), 164 [M–H]+ (100.0), 149 [M–H–CH3]+ (32.3), 148 [M–OH]+ (7.1), 134 [M–OCH3]+ (7.6), 122 [M–H–CHNCH3]+ (4.5), 77 (4.7), 65 (6.5), 51 (6.9), 42 [M–C6H3OHOCH3]+ (17.4); 13C NMR (CDCl3, 50 MHz): 47.8, 55.9, 110.3, 113.4, 120.9, 129.8, 145.9, 148.8, 162.1; 1H NMR (CDCl3, 200 MHz): 3.47 (d, J = 1.4 Hz, 3H, NCH3), 3.91 (s, 3H, OCH3), 6.03 (bs, 1H, OH), 6.86 (d, J = 8.2 Hz, 1H, H5), 7.18 (dd, J = 8.2, 2.0 Hz, 1H, H6), 7.31 (d, J = 2.0 Hz, 1H, H2), 8.14 (q, J = 1.4 Hz, 1H, NCH); Calcd for C9H11NO2: C 65.44, H 6.71, N 8.48, O 19.37%; found: C 65.43, H 6.73, N 8.44, O 19.40%; RI (HP-5MS) 1515.
IR and 1H NMR spectroscopic data are in accordance with [72].
4.6.26 N-(quinolin-4-ylmethylene)methanamine (2ag)
Colorless oil; FTIR (neat): 3054, 2902, 2815, 2775, 1648 (CN), 1581, 1566, 1506, 1463, 1393, 1352, 1294, 1251, 1212, 1138, 1047, 999, 986, 948, 844, 754, 633 cm−1; EIMS m/z 170 [M]+ (90.6), 169 [M–H]+ (100.0), 168 (17.8), 155 [M–CH3]+ (30.6), 154 [M–CH3–H]+ (5.9), 129 (8.3), 128 [M–CHNCH3]+ (19.9), 127 (5.9), 115 (4.5), 102 (7.4), 101 (12.8), 85 (3.4), 75 (10.9), 74 (4.9), 63 (4.2), 51 (5.4), 42 [M–C9H6N]+ (12.7); 13C NMR (CDCl3, 50 MHz): 49.1, 120.4, 123.9, 125.5, 127.3, 129.2, 129.9, 138.8, 148.7, 150.0, 159.7; 1H NMR (CDCl3, 200 MHz): 3.65 (d, J = 1.7 Hz, 3H, NCH3), 7.58 (ddd, J = 8.4, 6.9, 1.1 Hz, 1H, ArH), 7.66 (d, J = 4.4 Hz, 3H, H3), 7.72 (ddd, J = 8.4, 6.9, 1.1 Hz, 1H, ArH), 8.15 (dd, J = 8.4, 1.1 Hz, 1H, ArH), 8.66 (dd, J = 8.4, 1.1 Hz, 1H, ArH), 8.85 (q, J = 1.6 Hz, 1H, NCH), 8.95 (d, J = 4.4 Hz, 3H, H2); Calcd for C11H10N2: C 77.62, H 5.92, N 16.46%; found: C 77.63, H 5.90, N 16.50%; RI 1622 (HP-5MS).
Acknowledgements
The authors acknowledge support from the Ministry of Education, Science and Technological development of the Republic of Serbia (Project 172061).