

1 Introduction
Since heterogeneous catalysts are easily separated and reused, many efforts have been dedicated to the development of heterogeneous catalysts [1–5]. MCM-41 (Mobil Composition of Matter No. 41) is one of the materials most commonly used as supports for the heterogenization of homogeneous catalysts [1,6–10]. Heterogenization can be achieved by the entrapment or grafting of the active catalysts on the surfaces or inside the pores of a solid support, such as silica, alumina, or ceria. Grafting can be achieved by covalent binding [11–18], electrostatic interactions [19,20] or by hydrogen bonding with the support [11,12,21–23]. Since magnetic field provides a convenient and low-cost method for the separation of solid particles of a suspension, using modified MCM-41s with Fe3O4 as ideal appropriate supporting materials, it has attracted much attention [5,7,24–27]. Magnetically recoverable materials have been used in a wide range of catalytic reactions, including oxidations, hydrogenations, photocatalysis, and CC bond formation, as well as in novel applications in asymmetric synthesis, hydration, Knoevenagel condensations, and CO2 cyclo-addition reactions [28–30]. On the other hand, dioxidomolybdenum(VI) complexes are of interest as models for molybdoenzymes [31–33] and used as catalysts for the epoxidation of olefins [34–40].
The objective of this work is to immobilize a molybdenum Schiff base complex onto magnetite nanoparticles with the hydrogen bonding method to obtain a heterogeneous molybdenum magnetic nanocatalyst for the epoxidation of olefins. The advantages of this system are the facile and fast recovery of the solid catalyst at the end of reaction as well as its good activity and selectivity for epoxide formation.
2 Experimental
2.1 Instruments and reagents
Infrared spectra were recorded on KBr pellets using a Unicam Matson 1000 FT-IR. A Bruker FT-NMR 500 (500 MHz) spectrometer was utilized to obtain the NMR spectra. A Varian AA240 atomic absorption spectrometer was used for molybdenum determination. The high-power ultrasonic cleaning unit Bandelin Super Sonorex RK-100H with ultrasonic peak output 320 W and HF power 80 Weff has been used. The melting points were measured on an Electro thermal 9100 apparatus. X-ray powder diffraction (XRD) was carried out on a Philips X’Pert diffractometer using the Cu Kα radiation. Scanning electron microscopy (SEM) images were obtained on a Hitachi S-1460 field emission scanning electron microscope using an ACC voltage of 15 kV. Transmission electron microscopy (TEM) image were obtained with a Zeiss EM-900 transmission electron microscopy device using an ACC voltage of 80 kV. The oxidation products were analyzed with a gas chromatograph (Shimadzu, GC-14B) equipped with a SAB-5 capillary column (phenyl methyl siloxane 30 m × 320 mm × 0.25 mm) and a flame ionization detector.
2,2′-Dimethylpropylenediamine, 5-bromo-2-hydroxybenzaldehyde, molybdenyl acetylacetonate and hydrogen peroxide (solution 27% in water) were used as received from commercial suppliers. The solvents were dried and distilled by standard methods before use. Other chemicals were purchased from Merck or Fluka chemical companies.
2.2 Preparation of the Schiff base ligand (H2L)
The Schiff base ligand was prepared by the reported methods [41,42].
2.3 Preparation of molybdenum(VI) complex
The molybdenum complex (Fig. 1) was prepared as follows: the Schiff base ligand, H2L (0.468 g, 1 mmol) was dissolved in 20 mL of ethanol. An ethanolic solution of molybdenyl acetylacetonate (0.326 g, 1 mmol) was added and the reaction mixture was refluxed for 1 h. The light orange solution was concentrated to yield orange powders. The products were washed with warm ethanol. Mo(O)2L: Yield: 83%, 0.493 g, D.p.: > 295, Selected FT-IR data, υ (cm−1): 2922 (CH), 1614 (CN), 1520 (CC), 826 and 913 (Mo–O). 1H NMR (δ): 0.75, 1.17 (s, 6H, NCH2C(CH3)2CH2N), 1.18 (s, 2H, NCH), 3.38–4.63 (4H, NCH2C(CH3)2CH2N), 6.54–7.60 (m, 6H, ArH), 8.04–8.12 (s, 2H, NCH).
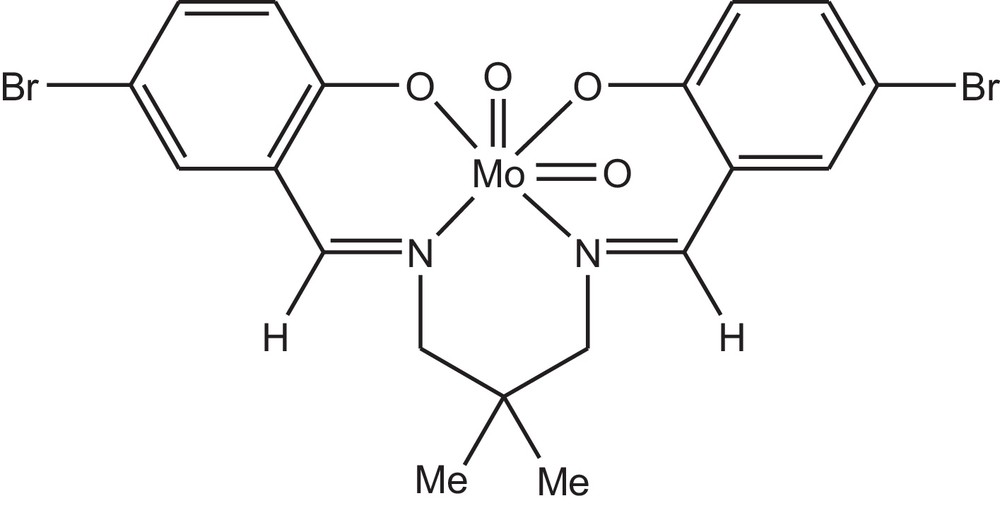
General structure of the used dioxomolybdenum complex.
2.4 Preparation of the (α-Fe2O3)–MCM-41–Mo(O)2L
Fe3O4 magnetic nanoparticles were prepared by the chemical co-precipitation method reported in the literature [10,43]. Briefly, a solution with molar composition of 3.2 FeCl3:1.6 FeCl2:1 CTABr:39 NH4OH:2300 H2O was used for the preparation of Fe3O4 nanoparticles at room temperature under N2 atmosphere. The magnetic MCM-41 was prepared by adding 20 mL of the Fe3O4 colloid after sonication to a 1 L solution with molar composition 292 NH4OH:1 CTABr:2773 H2O under vigorous mixing and sonication. Then, sodium silicate (16 mL) was added, and the mixture was allowed to react at room temperature for 24 h under consecutive mixed conditions. The magnetic MCM-41 was filtered and washed. The surfactant template was then removed from the synthesized material by calcination at 450 °C for 4 h and so (Fe3O4)–MCM-41 was converted to (α-Fe2O3)–MCM-41[10]. (α-Fe2O3)–MCM-41 (3 g) was suspended in 60 mL of chloroform with sonication. To this mixture, an excess amount of Mo(O)2L was added, and the resulting mixture was stirred for 12 h with a mild magnetic agitator at room temperature, then filtered to obtain a brick-red precipitate, which was washed with chloroform as a Mo(O)2L solvent, and finally dried in air. The preparation of (α-Fe2O3)–MCM-41–Mo(O)2L is shown in Fig. 2.
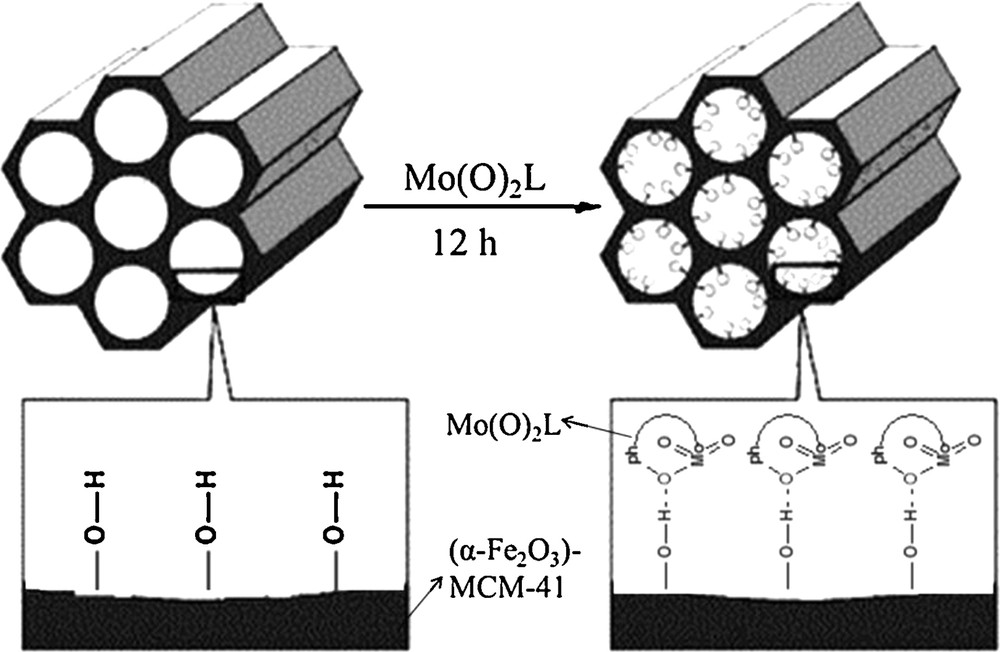
Schematic preparation of (α-Fe2O3)–MCM-41–Mo(O)2.
2.5 General heterogeneous green oxidation procedure
Catalytic experiments were carried out in a 10-mL glass flask fitted with a water condenser. In a typical procedure, 0.5 mmol cyclooctene, 0.015 g of (α-Fe2O3)–MCM-41–Mo(O)2L and 2 mL (20 mmol) of H2O2 were added in 3 mL of ethanol. The reaction mixture was refluxed for 8 h. The reaction products were monitored at periodic time intervals using gas chromatography (GC). The oxidation products were identified by comparison with authentic samples (retention times in GC).
3 Results and discussion
3.1 Characterization of the ligand and of the dioxomolybdenum(VI) complex
3.1.1 IR spectral studies
A practical list of IR spectral data is presented in Table 1. The comparison of the spectra of the complex with the ligand provides evidence for the coordination mode of the ligand in the dioxomolybdenum(VI) complex. A sharp band appearing at 1626 cm−1, due to υ(CN) (azomethine), shifts towards the lower wave numbers by 12 cm−1 and appears at 1614 cm−1. This observation indicates the involvement of the azomethine nitrogen in coordination with the molybdenum center [44]. The IR spectra of the Mo complex showed two characteristic strong bands in the regions 826 and 900 cm−1, which could be assigned to the symmetric and asymmetric stretching vibrations of cis-dioxo (MoO2), respectively [45–48].
IR spectral data of ligand and molybdenum complex.
| Compound | Selective IR bands (cm−1) | |
| MoO | CN | |
| H2L | – | 1626 |
| Mo(O)2L | 826, 900 | 1614 |
3.2 Characterization of (α-Fe2O3)–MCM-41–Mo(O)2L
3.2.1 Physicochemical characterization
Immobilization of the dioxomolybdenum complex on the surface of silica-coated magnetite nanoparticles is proposed to occur by hydrogen bond formation between the MoO and PhO groups of the complex as hydrogen bond acceptors, and the OH groups of the support as hydrogen bond donors. Also, the weak coordination of the OH groups to the metal center as a hard acid may be also involved. However, with respect to the negligible catalyst leaching, the formation of strong hydrogen bonds seems to be a more plausible explanation. A broad peak in the region 1050–1200 cm−1 and two peaks in 804 and 950 cm−1, respectively, are assigned to the stretching vibrations of SiOSi (mesoporous framework). The peak at 1627 cm−1 belongs to the FeO bond of α-Fe2O3. By entering the complex within the pores of α-Fe2O3–MCM-41, no change in the IR spectrum was observed. Peaks at 2921 cm−1 and 1458–1514 cm−1 can be related to the CH stretching vibrations and CC bonds, respectively (Fig. 3).
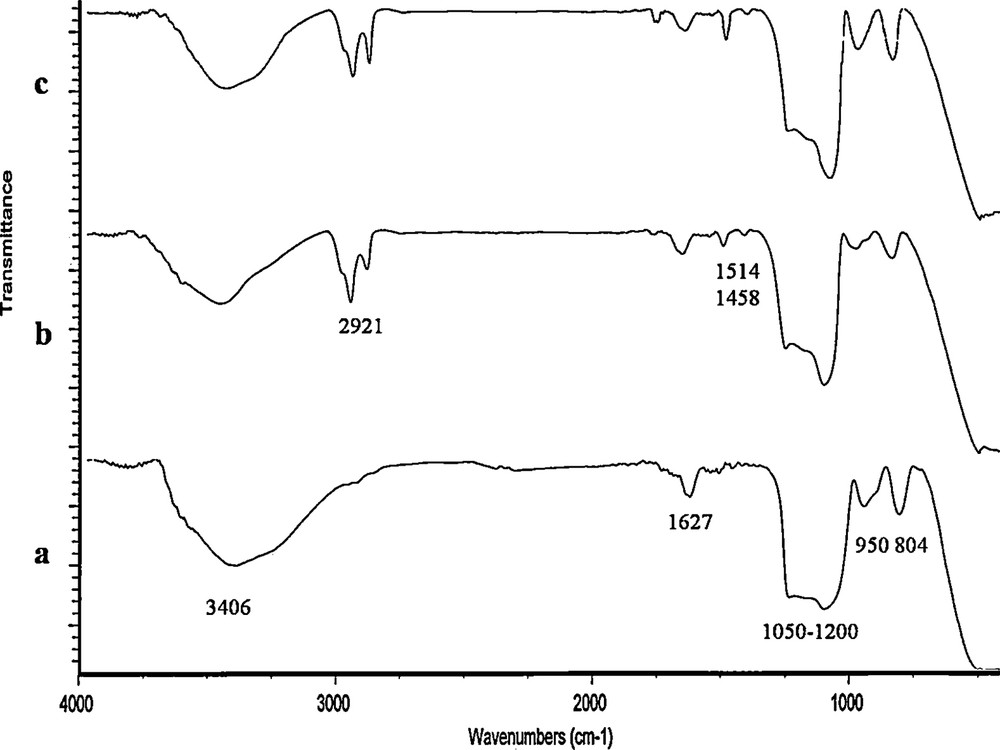
IR spectra of (a) (α-Fe2O3)–MCM-41, (b) (α-Fe2O3)–MCM-41–Mo(O)2L and (c) recovered (α-Fe2O3)–MCM-41–Mo(O)2L.
The molybdenum content of the catalyst was determined by atomic absorption spectroscopy. Accordingly, the metal content of the catalysts was found to be ca. 0.0023 wt %. Scanning electron micrographs (SEM) were recorded to obtain the shape and diameter of the catalyst (Fig. 4a). The results show the spherical morphology of the catalyst; particles were well distributed and the average particle final size was 80–90 nm for (α-Fe2O3)–MCM-41–Mo(O)2L.

The scanning electron microscopy (a) and transmission electron microscopy (b) images of (α-Fe2O3)–MCM-41–Mo(O)2L.
Fig. 4b shows the transmission electron microscopy (TEM) images of (α-Fe2O3)–MCM-41–Mo(O)2L. The TEM micrograph of the prepared catalyst shows an ordered hexagonal pore system with embedded α-Fe2O3 nanoparticles. As it can be seen, the products are formed from particles with a mean diameter of 80–90 nm ((α-Fe2O3)–MCM-41–Mo(O)2L), which is in agreement with other results.
X-ray diffraction analysis was performed in order to investigate the effect of the incorporation of the molybdenum complex on the texture properties of the materials. In the XRD pattern of (α-Fe2O3)–MCM-41 (Fig. 5), a strong peak at 2θ = 2.75 and two weaker peaks at higher reflection angles of the hexagonal unit cell are observed. The XRD pattern of the supported molybdenum catalyst (α-Fe2O3)–MCM-41–Mo(O)2L is shown in Fig. 5. All the peaks are weaker than the former one, and the strongest peak shifted to a lower angle (2θ = 2.65), which is probably due to the incorporation of the molybdenum complex into (α-Fe2O3)–MCM-41.
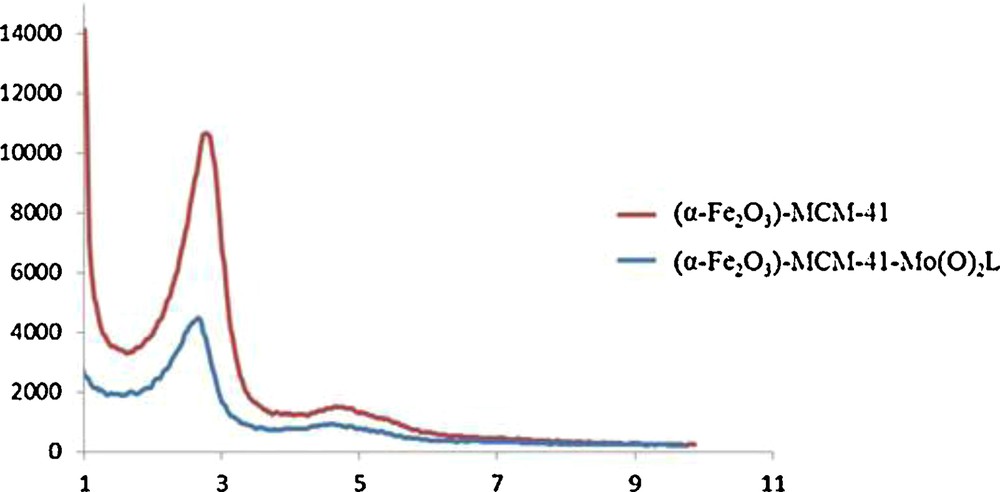
X-ray diffraction pattern of the prepared α-Fe2O3–MCM-41 (top) and α-Fe2O3–MCM-41–Mo(O)2L (bottom) in the 1.0° (2θ) to 10.0° (2θ) region.
3.3 Heterogeneous catalytic green epoxidation of olefins
The catalytic performance of (α-Fe2O3)–MCM-41–Mo(O)2L was investigated in the epoxidation of olefins with hydrogen peroxide (H2O2) and tert-butyl hydroperoxide (TBHP) as oxygen donors. A series of blank experiments (Table 2) revealed that the presence of both catalyst and oxidant is essential for an effective catalytic reaction. In the absence of the catalyst (entry 1 in Table 2) or oxidant (entry 2 in Table 2), the reactions did not proceed under reflux. Also, in the presence of (α-Fe2O3)–MCM-41 as a catalyst and TBHP as oxidant, no product was formed (entry 3 in Table 2).
Experimentsa on catalytic oxidation of cyclooctene at room temperature.
| No. | Catalyst | Oxidant | Conversionb (%) | Time (h) |
| 1 | None | H2O2 | 0 | 10 |
| 2 | (α-Fe2O3)–MCM-41–Mo(O)2L | None | 0 | 10 |
| 3 | (α-Fe2O3)–MCM-41 | TBHP | 0 | 10 |
| 4 | (α-Fe2O3)–MCM-41–Mo(O)2L | H2O2 | 100 | 8 |
| 5 | (α-Fe2O3)–MCM-41–Mo(O)2L | TBHP | 100 | 3 |
a Reaction conditions: catalyst (0.015 g), alkene (0.5 mmol), TBHP (2 mmol) or H2O2 (20 mmol) and solvent: ethanol (3 mL).
b GC yields based on the starting olefin.
In order to find the suitable reaction conditions, the effect of various reaction parameters that may affect the conversion and selectivity of the reaction were studied. Solvent, catalyst concentration, the nature and the concentration of oxidant are the factors that have been evaluated. The influence of different solvents on the oxidation of cyclooctene was studied using nanoparticles of (α-Fe2O3)–MCM-41–Mo(O)2L as the catalysts; the results are presented in Fig. 6. Chloroform, acetonitrile, methanol and ethanol were used as solvents and the highest conversion (100% with H2O2) was obtained in ethanol. According to the data in Fig. 6, the conversions are comparable in methanol and acetonitrile. Aqueous hydrogen peroxide has a low solubility in dichloromethane, but is soluble in the other solvents. Therefore, solvents with higher dielectric constants, i.e. methanol, ethanol and acetonitrile are much better than dichloromethane. As it may be seen from Scheme 1, the presence of unoccupied coordination sites on the metal center of the catalyst is crucial for its catalytic performance [49]. On the other hand, methanol and acetonitrile are of smaller size compared to ethanol; their coordination to the metal center is expected to be more facile than with ethanol. Accordingly, the lower catalytic activity of the title catalyst observed in acetonitrile and methanol, compared to that in ethanol, seems to be due to the greater competition between the peroxide and the former for coordination sites on the metal center in comparison with the latter. Also, the higher conversions in ethanol relative to methanol are possibly due to the higher boiling point of the former.
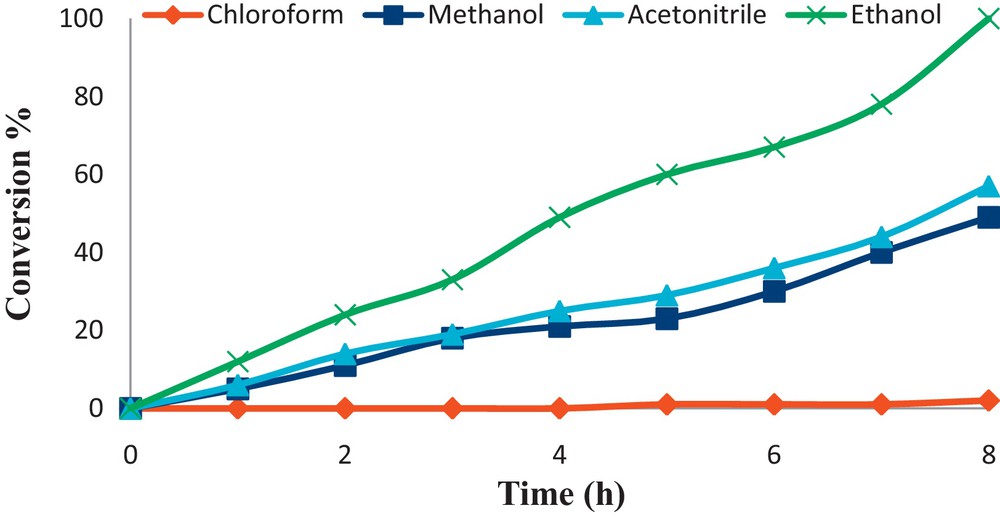
The effect of various solvents on the epoxidation of cyclooctene with H2O2 using (α-Fe2O3)–MCM-41–Mo(O)2L. Reaction conditions: cyclooctene (0.5 mmol), catalyst (0.015 g), solvent (3 mL), H2O2 (20 mmol); the reactions were run for 8 h under reflux.
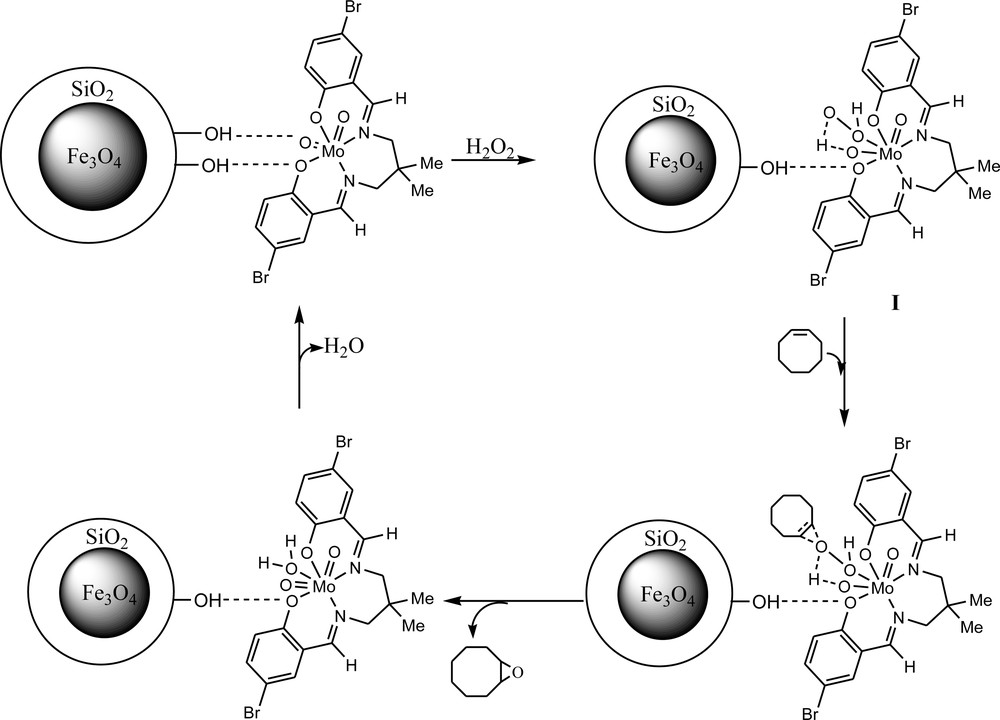
Proposed catalytic cycle.
In order to investigate the effect of the oxidizing agent in the oxidation reaction, TBHP and hydrogen peroxide were used. In the presence of TBHP, the reaction requires 3 h for competition, while the reaction was completed after 8 h with eco-friendly hydrogen peroxide.
Different catalyst concentrations have been used in the oxidation of cyclooctene (Fig. 7). It was observed that the oxidation of cyclooctene required 0.015 g of catalyst for completion. The highest conversion was obtained with 0.15 g of catalyst and further increments of catalyst hardly improved conversion. This may be due to the higher degradation of the oxidant at higher concentration of catalysts [49].
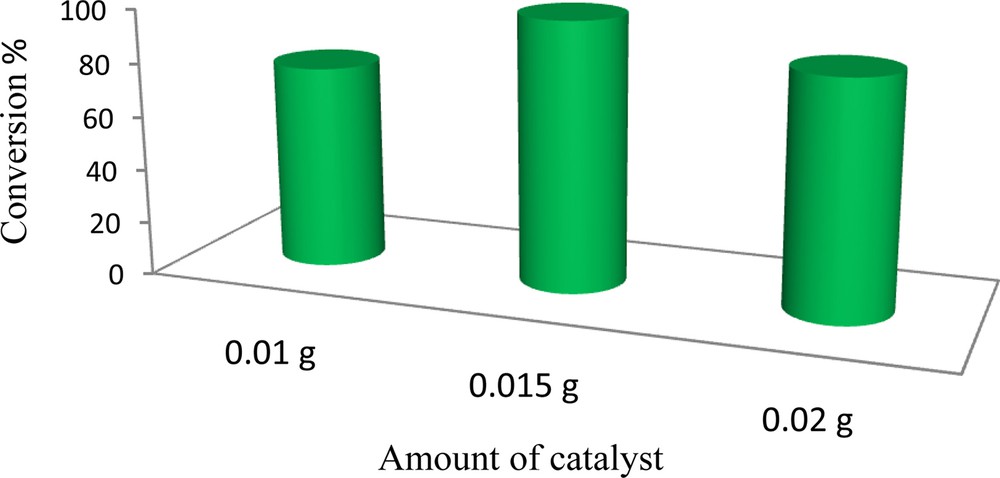
The influence of catalyst concentration on the oxidation of cyclooctene using H2O2 catalyzed by (α-Fe2O3)–MCM-41–Mo(O)2L. Reaction conditions: cyclooctene (0.5 mmol), solvent (3 mL), H2O2 (20 mmol); the reactions were run for 8 h under reflux.
Different oxidant (H2O2) concentrations and addition methods have been studied in the oxidation of cyclooctene (Fig. 8). It was observed that oxidation of cyclooctene required 20 mmol of H2O2 for completion. Gradual or immediate addition of H2O2 to the reaction mixture led to nearly similar results, though the latter gave relatively higher conversion. The result shows that there is little disproportionation of hydrogen peroxide in the used concentration.
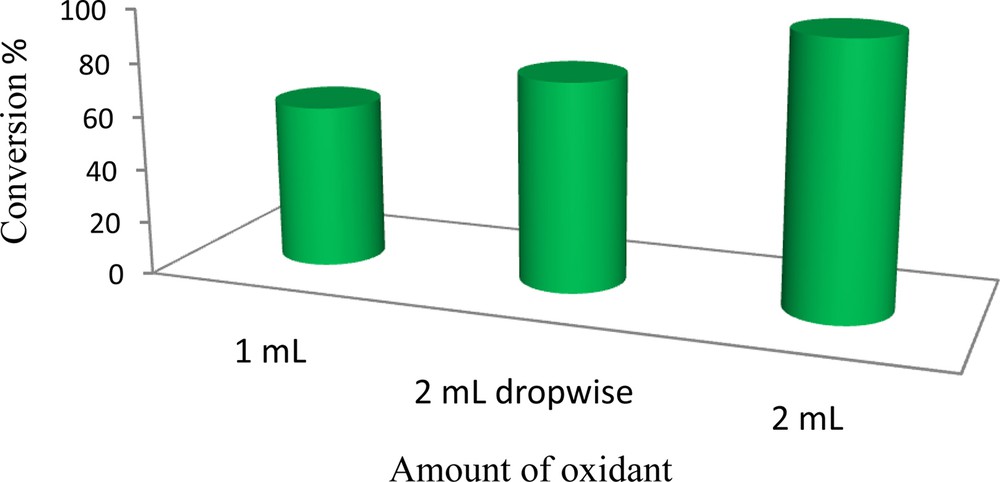
The influence of oxidant concentration on the oxidation of cyclooctene catalyzed by (α-Fe2O3)–MCM-41–Mo(O)2L. Reaction conditions: cyclooctene (0.5 mmol), solvent (3 mL), H2O2 (20 mmol); the reactions were run for 8 h under reflux.
In order to establish the general applicability of this catalytic system, under optimized conditions, the oxidation of different olefins was performed in the presence of a catalytic amount of (α-Fe2O3)–MCM-41–Mo(O)2L and the results are presented in Fig. 9. The oxidation of cyclooctene, cyclohexene, 1-methylcyclohexene, 1-octene and indene led to the corresponding epoxide as the sole product, while the oxidation of styrene led to the formation of benzaldehyde (Fig. 9, column 4), and that of substituted styrenes giving the substituted benzaldehydes (Fig. 9, columns 5–7); also, in the oxidation process of α-methyl styrene, acetophenone (Fig. 10, column 8) was detected as the sole product [50]. The production of benzaldehyde in the oxidation of styrene is due to the over-oxidation of styrene oxide with H2O2. The lower catalytic activity of α-methylstyrene with respect to styrene seems to be due to the steric hindrance of the methyl substituent of the former [51]. It should be noted that if electronic effects due to the methyl substituents were important, then, α-methylstyrene as a more electron-rich olefin would be expected to show higher reactivity relative to styrene. Epoxidation of the terminal alkene (oct-1-ene) proceeds in fair yield (73%).
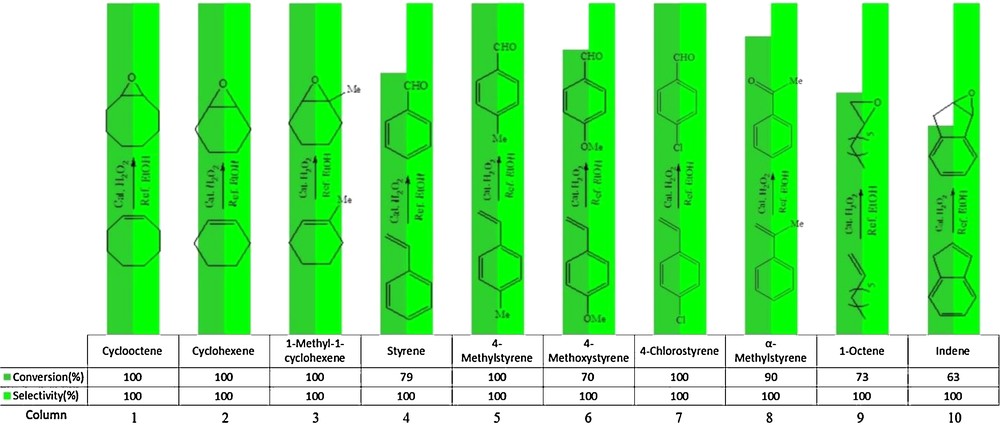
Epoxidation of olefins using H2O2 catalyzed by (α-Fe2O3)–MCM-41–Mo(O)2L. Reaction conditions: cyclooctene (0.5 mmol), solvent (3 mL), H2O2 (2 mL); the reactions were run for 8 h under reflux.
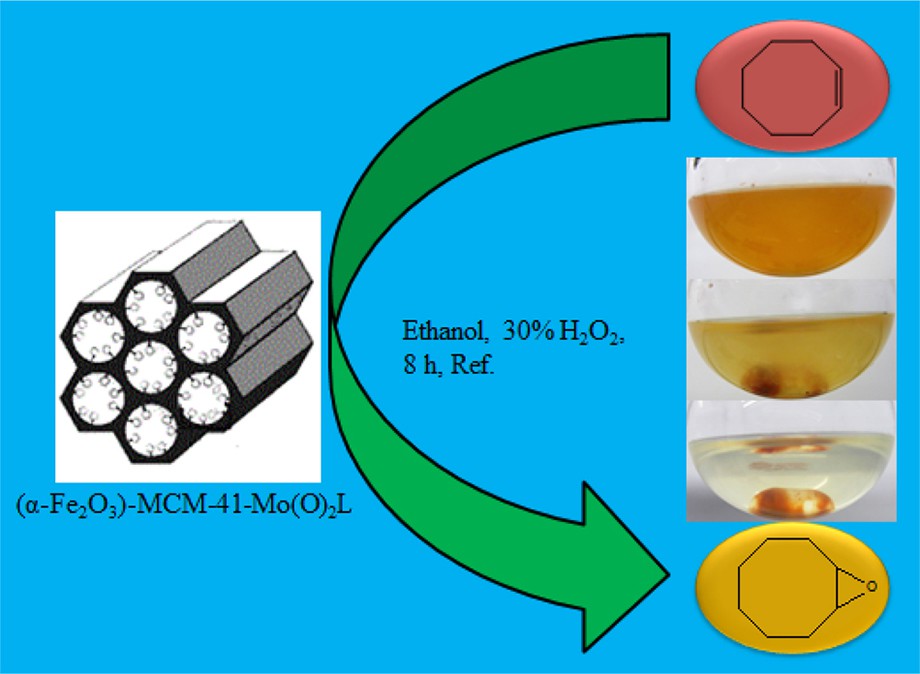
Overview of the reaction catalyzed by a magnetic nanocatalyst, i.e. attraction by a magnet.
In the proposed catalytic cycle, the oxidant will be activated by coordination to the molybdenum center and will form a hepta-coordinated molybdenum intermediate (I, Scheme 1) [52–56]. Then, olefin as a nucleophile will attack the oxygen atom of the coordinated hydrogen peroxide.
3.4 Catalyst separate, reuse and stability
One of the most attractive features of heterogeneous catalysts is their separation property. Catalyst recovery and reuse are the two most important features for many catalytic processes, and most heterogeneous systems require a filtration or centrifugation step and/or a tedious work up of the final reaction mixture to recover the catalyst. Magnetically supported catalysts can be recovered with an external magnet (Fig. 10). The homogeneous molybdenum complex cannot be recovered even once; in contrast, the heterogeneous catalysts can be magnetically separated and reused multiple times without significant loss of catalytic activity. The stability of the magnetic nanocatalyst was monitored using a multiple sequential epoxidation reaction. For each one of the repeated reactions, the catalyst was separated, washed with chloroform and methanol and dried before being used. The catalyst was consecutively reused six times (Fig. 11) without detectable catalyst leaching or significant loss of activity. The results indicated that the catalyst, after having been reused several times, showed no detectable leaching or change in its IR spectra.
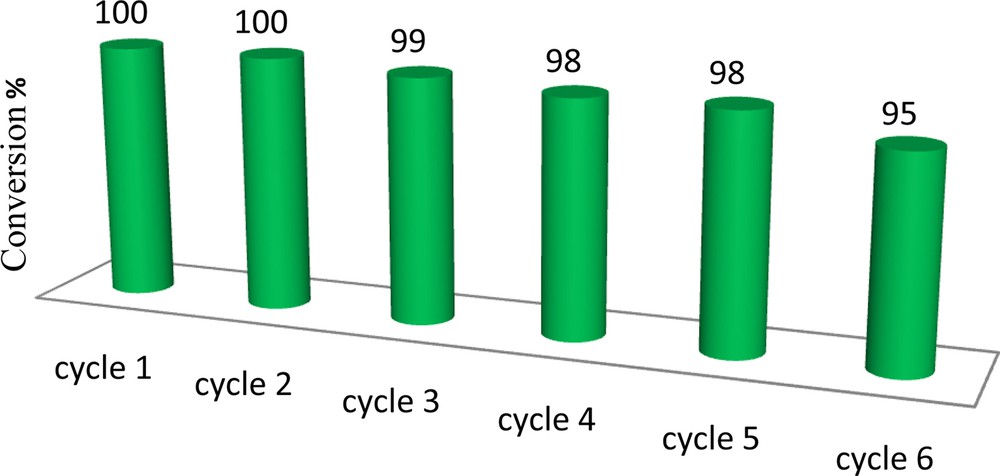
The results obtained in the oxidation of cyclooctene with H2O2 by (α-Fe2O3)–MCM-41–Mo(O)2L with a catalyst reused several times. Reaction conditions: cyclooctene (0.5 mmol), solvent (3 mL), H2O2 (20 mmol); the reactions were run for 8 h under reflux.
4 Conclusions
In summary, the dioxomolybdenum(VI) complex (Mo(O)2L) has been supported on the surface of silica-coated magnetite nanoparticles via O–H…O hydrogen bonds. This nanocatalyst was employed as a catalyst for the epoxidation of olefins with hydrogen peroxide. Excellent selectivity (100%) for epoxide formation was obtained in the case of cyclooctene, 1-octene, indene, and 1-methyl cyclohexene. The catalyst was reused six consecutive times without significant loss of catalytic activity or selectivity.
Acknowledgements
The financial support of this work by the K.N. Toosi University of Technology research council is acknowledged.