

1 Introduction
Coordination polymers have recently attracted much research attention due to their potential application in areas of synthetic chemistry, materials science and catalysis [1]. Coordination polymers are solid phase catalysts and as such, can easily be separated from the reaction mixtures and reused without any further treatment. We have recently been interested in the study of coordination polymers and potential catalytic activity of these compounds. The research has demonstrated that the silver (I)-based coordination polymer [Ag (μ-bpfb)(N3)]n is able to catalyze the oxidation of amines into hydroxylamines using urea hydrogen peroxide [2]. Studies on tungsten coordination polymers are rare and very little work has been reported on the catalytic activity of these compounds in oxidation reactions. A perusal of the related literature demonstrated that a number of methods have been developed for the oxidative coupling of the primary amines and oxidation of the secondary amines into imines using metal-organic frameworks (MOFs) and coordination polymers as the solid phase catalysts [3,4]. However, no precedent has yet been reported for the oxidative syntheses of nitrones and oximes from amines using coordination polymers as the catalyst, apart from the work reported by Abrantes et al. [5].
In this study, the investigation illustrated the efficient catalytic oxidation of secondary amines to nitrones using hydrogen peroxide as the primary oxidant in the presence of the organometallic coordination polymer [(nBu3Sn)2WO4]. Furthermore, this investigation examined the catalytic activity of this compound in the oxidation of the primary amines. Hydrogen peroxide was used as the stoichiometric oxidant because, it is non-toxic and water is produced as the only by-product; as a result, it is considered as environmentally safe.
There are a large number of related articles published on the synthesis of azomethine N-oxides, which are commonly known as nitrones. Nitrones are important because, they are relatively stable as compared to analogous azomethine imines [6] and are key synthetic intermediates. Nitrones are the building blocks in the synthesis of various bioactive compounds. α-Aryl-N-alkyl nitrones, have been recognized as potential agents for treating strokes, for example tert-butylbenzyl nitrone (PBN) acts as a radical spin trapping agent and exhibits antioxidant and neuroprotective activity against oxidative damage [7]. Several methods are available for the synthesis of nitrones. Selective catalytic oxidation of secondary amines has long been recognized as an atom efficient method for producing nitrones; which occurs with a variety of oxidizing agents and catalysts [8–10].
Organotin–oxotungstate coordination polymer [(nBu3Sn)2WO4], has been prepared by Abrantes group [11] and applied as an efficient catalyst for the epoxidation of olefins by hydrogen peroxide, selective oxidation of aniline derivatives to nitroso compounds [12], and for the oxyfunctionalization of monoterpenes [13]. In this present work, the investigation was focused on further examination of the catalytic potential of this compound in the oxidation of primary and secondary amines using aqueous hydrogen peroxide.
2 Results and discussion
The organometallic coordination polymer [(nBu3Sn)2WO4] was prepared according to a reported procedure [11], as shown on Scheme 1. It was synthesized from commercially available sodium tungstate and tri (n-butyl) tin chloride in 92% yield. The FT-IR spectra and the XRD diffraction patterns of the resulting white powder were the same as those reported in other studies and these observations confirm synthesis of [(nBu3Sn)2WO4] [11,12].
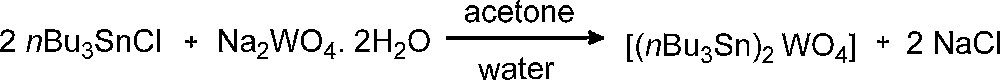
Preparation of [(nBu3Sn)2WO4] coordination polymer.
To determine the optimal conditions for the oxidation of amines, various loadings of coordination polymer [(nBu3Sn)2WO4] were initially tested as a catalyst or catalyst precursor for the oxidation of dibenzylamine at room temperature, using hydrogen peroxide (30 wt%, aqueous solution) as the stoichiometric oxidant with methanol as the solvent (Table 1). The absence of catalyst resulted in a non-selective oxidation of dibenzylamine (Table 1, entry 1). The optimum condition for the selective oxidation of 1a into the corresponding nitrone, 1b required the use of at least 4 mol% of the coordination polymer, [(nBu3Sn)2WO4] (Table 1, entry 3). The oxidation of dibenzylamine in the presence of 4 mol% [(nBu3Sn)2WO4], using other oxidants like tert-butylhydroperoxide (TBHP) and sodium hypochlorite (NaOCl) was unsuccessful (Table 1, entry 5–6), therefore hydrogen peroxide was selected as the optimal efficient oxidant.
Optimization of the reaction conditionsa.
| Entry | Oxidant | Catalyst (mol%) | Time (h) | Yield (%)b |
| 1 | H2O2 | No catalyst | 24 | Trace |
| 2 | H2O2 | 3 | 12 | 70 |
| 3 | H2O2 | 4 | 5 | 85 |
| 4 | H2O2 | 5 | 5 | 85 |
| 5 | TBHP | 4 | 12 | n.r. |
| 6 | NaOCl | 4 | 12 | n.r. |
a Reaction conditions: dibenzylamine (1 mmol), catalyst, oxidant (3–4 equiv), CH3OH (2 mL), r.t.
b Isolated yield of N-benzylidenebenzylamine N-oxide.
In the optimum conditions, oxidation of dibenzylamine by hydrogen peroxide proceeded in 5 hours at room temperature in the presence of 4 mol% of the catalyst ([(nBu3Sn)2WO4]), to give N-benzylidenebenzylamine N-oxide, in 85% yield (Table 1, entry 3). Turnover number of the catalyst was approximately 21 (Scheme 2).
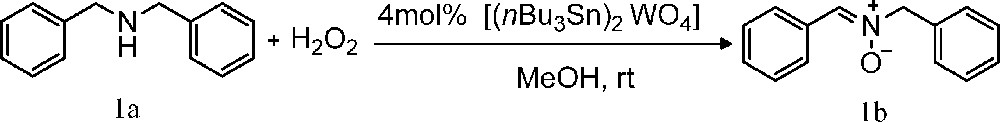
Oxidation of dibenzylamine using (30 wt% aqueous solution) promoted by [(nBu3Sn)2WO4].
The catalytic activity of coordination polymer ([(nBu3Sn)2WO4]), was evaluated in the oxidation of various secondary amines with aqueous hydrogen peroxide (30 wt%), in methanol, at room temperature. Results are presented in Table 2. Benzylic and symmetrical secondary amines such as dibenzylamine (1a), di (4-methylbenzyl)amine (2a), di (2-methoxylbenzyl)amine (3a) were readily converted into their corresponding nitrones, as anticipated (Table 2, entries 1–3).
Oxidation of amines in the presence of coordination polymer, [(nBu3Sn)2WO4] using hydrogen peroxide (30 wt%, aqueous solution) as the oxidanta.
| Entry | Amine | Product | Time (h) | Yield (%)b | TONc | Reference |
| 1 | 5 | 85 (82, 80, 76, 75)d | 21 | [26] | ||
| 2 | 2 | 78e | 19 | [27] | ||
| 3 | 2 | 80 | 20 | [27] | ||
| 4 | 4 | 90 | 22 | [8] | ||
| 5 | 3 | 80 | 20 | [8] | ||
| 6 | 4 | 89e | 22 | [8] | ||
| 7 | 5 | 85e | 21 | – | ||
| 8 | 8 | 85 | 21 | [15] | ||
| 9 | 3 | 60 | 15 | [9] | ||
| 10 | 10 | 73 | 18 | [15] | ||
| 11 | 2 | 78 | 19 | [21] | ||
| 12 | 0.5 | 56 | 14 | [24] | ||
| 13 | 0.5 | 52 | 13 | [21] |
a Reaction conditions: amine (1 mmol), catalyst (4 mol%), 30 wt% H2O2 (3–4 equiv), CH3OH (2 mL), r.t.
b Isolated yields.
c Turnover number.
d The reaction was accomplished with recycled catalyst.
e Small amount of benzaldoxime was formed according to GC, using an authentic sample as the calibrant.
The oxidation processes of unsymmetrical benzylic amines such as N-tert-butylbenzylamine (4a), N-n-butylbenzylamine (5a), N-iso-propylbenzylamine (6a), and N-cyclohexylbenzylamine (7a) were tested. The nature of the alkyl group demonstrated no significant affect on the nitrone yield, however, those hindered amines reacted slightly slower than did the others (Table 2, entries 4–7). Tert-butylbenzylamine (4a) have no alpha protons on one side, therefore, the oxidation reaction gave only the expected nitrone, N-benzylidene-tert-butylamine N-oxide (PBN), which is a useful spin trapping reagent [14], in a 90% yield (entry 4). In the case of N-n-butylbenzylamine, N-iso-propylbenzylamine and N-cyclohexylbenzylamine(5a–7a), the major product deriving from the oxidation of the more favorable benzylic methylene and other regioisomeric nitrones were not seen (the ratio of isomers determined by 1H NMR analysis; Table 2, entries 5–7).
In a few cases, a small amount of benzaldoxime (4–6%) was isolated as the minor product according to gas chromatography (GC), using an authentic sample as the calibrant (Table 2, entries 2, 6, 7) [15]. The oxidation of N-phenylbnenzylamine, which has an aromatic ring directly linked to the nitrogen atom, was messy due to the competitive electron transfer from the amine to the oxidant [16].
The oxidation of symmetrical non-benzylic secondary amines such as dibutylamine (8a) and pyrrolidine (9a) were carried out and their corresponding nitrones were obtained as predicted (Table 2, entry 8–9).
Methyl prolinate was converted into the thermodynamically stable 2-substituted nitrone, regioselectively (entry 10).
The C-phenyl nitrone 1b (N-benzylidenebenzylamine N-oxide), was isolated as the Z-isomer, as usual [17–19]. In the case of dibutylamine, the formation of Z-isomer was also observed as the only product (referring to 1H NMR data compared to literature data) [5].
The oxidant activity of coordination polymer [(nBu3Sn)2WO4] may be attributed to the formation of peroxo complexes that have reportedly been isolated and characterized in several cases [20]. This oxidation process can follow the steps from amine to hydroxylamine and subsequently to nitrone.
This study additionally, investigated the oxidation of primary amines. It was determined using the same experimental procedure (Table 2). In the presence of 4 mol% of the catalyst, [(nBu3Sn)2WO4], the reaction of α-phenylethylamine, with 4 equiv of 30% aqueous H2O2 resulted in the selective formation of (E)-acetophenone oxime in 80% yield as the only product (Table 2, entry 11). In the case of other primary and benzylic amines such as benzylamine and 4-methylbenzylamine the corresponding oximes were isolated in moderate yields (Table 2, entries 12–13). The stereochemistries of oximes were confirmed by comparison on the 1H NMR spectrum with reported values [21]. Oximes are important intermediates for the synthesis of amides via a Beckmann rearrangement [22] and the selective catalytic oxidation of primary amines into oximes assumes great importance [23,24].
Less activated cyclohexylamine, a non-benzylic primary amine, was not converted to cyclohexanone oxime at all [25]. The oxidation of tert-butylamine was also performed and the process did not provide any products.
To complete this study, reusability of the catalyst was investigated in the oxidation of dibenzylamine. A set of experiments was carried out to examine the catalyst activity along five oxidation runs. The model reaction was carried out using 50 mg of the catalyst and the experiment was suitably scaled up. When the reaction was complete, the catalyst was filtered and washed with acetone several times. The remaining catalyst was dried under vacuum and reused in subsequent reactions. More than 98% of the catalyst could usually be recovered from each reaction. The catalyst could be reused for five cycles with no significant loss of its catalytic activity. The product yield was 85% in the first run after 5 hours. In second and third runs, the product yields decreased to 82% and 80%, respectively (Table 2, entry 1). The nature of the recovered catalyst was studied by FT-IR spectroscopy and XRD analysis and assignments were in good agreement with the reported values [12]. To verify the heterogeneity of the catalyst in the oxidation of the secondary amines, in an experiment, the catalyst was separated from the reaction mixture after 30 minutes of the reaction and the filtrate was then stirred at room temperature for a duration of 10 hours. The results demonstrated that no further selective conversion of dibenzylamine was observed in the absence of the catalyst.
3 Experimental
3.1 Preparation of [(nBu3Sn)2WO4] coordination polymer
The complex [(nBu3Sn)2WO4] was prepared as described previously [11]. Five mmol nBu3SnCl was dissolved in a mixture of water (3 mL) and acetone (13.5 mL). Then, a solution of 2.5 mmol Na2WO4. 2H2O in 5 mL distilled water was added slowly to the nBu3SnCl solution while stirring magnetically. A white precipitate formed immediately and stirring was continued for a further 5 min. The resultant solid was filtered, washed with water and acetone and then dried at 100 °C. FT–IR spectrum (KBr disc) was in agreement with results of preceding reports. IR (KBr, ν cm−1): 2960 (vs), 2925 (vs), 2859 (s), 1512 (w), 1458(s), 1417 (w), 1377 (m), 1342 (w), 1292 (w), 1250 (w), 1182 (w), 1155 (w), 1078 (m), 1049 (w), 1003 (w), 964 (w), 883 (m), 850 (m), 815 (vs), 744 (m), 670 (s), 669 (s), 613 (w), 513 (w).
3.2 General experimental procedure for the preparation of nitrones and oximes
Four mol% (33 mg) of organometallic coordination polymer [(nBu3Sn)2WO4], was added to a stirred solution of methanol (2 mL) and amine (1.0 mmol) and the resultant suspension was stirred for 2 min. To this, 3–4 mmol aqueous hydrogen peroxide (30 wt%) was added, after which the reaction mixture was stirred at r.t., until completion of the reaction as indicated by thin-layer chromatography (TLC). The catalyst was filtered and washed with acetone several times. Excess hydrogen peroxide was decomposed by adding small portions of sodium hydrogen sulphite. The solution was extracted with dichloromethane. The combined organic extracts were dried over anhydrous sodium sulfate. The mixture was filtered and the solvent was removed by a rotary evaporator and the crude residue was purified by preparative thin-layer chromatography using ethyl acetate and n-hexane (1:4) as eluents to give corresponding nitrone or oxime derivatives. All the products are known compounds (except 7b) and were characterized by comparison of their IR, 1H NMR and 13C NMR spectroscopic data and respective melting points with the reported values.
The spectroscopic data of nitrone 7b is reported since no literature data are available.
3.3 N-Benzylidenecyclohexylamine N-oxide (Table 2, 7b)
Pale yellow solid, 85% yield. Mp 69–70 °C; 1H NMR (CDCl3, 500 MHz) δ 8.16–8.19 (m, 2H, ArH ortho to CHN(O)CH2Ph), 7.36 (s, 1H, CHN(O)), 7.31–7.35 (m, 3H, ArH), 3.74–3.81 (m, 1H, CH), 1.60–2.03(m, 10H, CH2 cyclohexyl); 13C NMR (CDCl3, 100 MHz) δ 132.3, 132.2, 130.8, 130.7, 130.1, 127.6, 128.5, 75.7, 31.1, 25.1; FT-IR (KBr): IR (KBr): 3062 (m), 2931 (s), 2858 (s), 1571 (m), 1453 (s), 1345 (m), 1300 (m), 1227 (w), 1149 (s), 1022 (w), 937 (w), 901 (w), 805 (w), 751 (m), 693 (m) cm−1; MS m/z: 203.2 (M+).
4 Conclusion
In summary, this catalyst was able to catalyze mild and selective oxidation of the secondary and primary amines to nitrones and oximes, respectively, with a catalyst loadings of 4 mol%. This one-pot synthesis is a simple and selective process, which occurs under mild conditions and has high atom economy and releases water as the only by-product.
Acknowledgments
We thank the Tarbiat Modares University for financial support.