

1 Introduction
Developing solvents with less impact on the environment is currently a great concern. This aspect is identified as one of the 12 principles of green chemistry as defined by P.T. Anastas and J.C. Warner [1]. Although numerous studies are dedicated to this pursuit, the number of “green” solvents available today on the market is limited, and the search for alternative solvents remains a major challenge, notably in domains such as fine chemistry or catalysis. Recently, catalysis by transition metals in aqueous medium has been the object of increasing interest [2,3] because reactions in aqueous media are considerably safer, non-toxic, environmentally friendly and inexpensive. Since then, water has been introduced into numerous reactions of organic or organometallic synthesis. However, the lack of solubility of reagents and/or catalysts limits its use in organic synthesis. The use of transfer agents or surfactants, which are key compounds to the organized molecular systems can partially overcome these difficulties, while improving the catalytic activity [4]. Concerning hydrogenation processes, for example, the Rh-catalyzed hydrogenation of unsaturated substrates was one of the first widely described studies [5–7].
For several years, we have been concerned with the synthesis of surfactants from renewable resources [8–10]. We have already obtained pentosides with an octadienyl chain from the Pd-catalyzed telomerization of butadiene with pentoses [8] and prepared xylosides containing only one terminal double bond [9a-e] by classical glycosylation. These xylosides were then transformed into bolaamphiphilic surfactants by metathesis reactions [10]. Within this framework, we will describe how biobased alkylpentosides can be easily synthesized from d-xylose and their resulting interfacial properties. Molecular modeling was performed on four xylosides to understand the forces involved in the stability of the compounds.
2 Experimental
2.1 Materials and methods
2.1.1 Synthesis
2.1.1.1 General procedure for the glycosylation
In a three-necked flask equipped with a reflux condensor, d-xylose (1 equiv) was dissolved under argon atmosphere in anhydrous THF. The alcohol (2 equiv) was then added and the resulting mixture was stirred at 80 °C. The PTSA (0.6 equiv) was then added in three portions (0.2 equiv each hour). After 48 or 72 h of reaction, the mixture was neutralized by adding a 0.5 M methanolic solution of MeONa, and the products were purified by flash chromatography (eluting mixture CH2Cl2/MeOH 9:1).
Hexyl-α,β-d-xylopyranoside (1)
This compound was obtained as a yellow paste following the general procedure previously described with d-xylose (4 g, 0.026 mol) in 150 mL of THF, hexanol (6.46 mL, 2 equiv) and PTSA (3.08 g, 0.6 equiv). Yield: 37%, α/β: 3/7, Rf: 0.54 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3378, 2929, 2865, 1463, 1377, 1272, 1242, 1152, 1115, 1045, 726. 1H NMR (CD3OD, 250 MHz): δ (ppm) = 0.91 (t, 6H, J = 5 Hz, H6′α, H6′β), 1.33 (m, 12H, H5′α,H5′β, H4′α, H4′β, H3′α, H3′β), 1.60–1.63 (m, 4H, H2′α, H2′β), 3.18–3.81(m, 14H, H1′α, H1′β, H5α, H5β, H2α, H2β, H4α, H4β, H3α, H3β), 4.18 (d, 1H, J = 7.5 Hz, H1β), 4.69 (d, 1H, J = 3.5 Hz, H1α), 4.87 (s, 6H, 6OH). 13C NMR (CD3OD, 62.9 MHz): δ (ppm) = 14.9 (C6′), 24.1 (C5′α), 25.3 (C5′β), 27.2 (C4′α), 27.5 (C4′β), 31.0 (C3′α), 31.2 (C3′β), 33.2 (C2′α), 33.3 (C2′β), 63.4 (C5α), 67.4 (C5β), 69.7 (C1′α), 71.3 (C1′β), 71.7 (C4α), 72.0 (C4β), 74.0 (C2α), 75.4 (C2β), 75.6 (C3α), 78.3 (C3β), 100.8 (C1α), 105.5 (C1β). Anal calcd for C11H22O5: C, 56.39; H, 9.46. Found: C, 56.11; H, 9.46.
Octyl-α,β-d-xylopyranoside (2)
This compound was obtained as a yellow paste following the general procedure previously described with d-xylose (4 g, 0.026 mol) in 150 mL of THF, octanol (8.50 mL, 2 equiv) and PTSA (3.08 g, 0.6 equiv). Yield: 31%, α/β: 4/6, Rf: 0.67 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3381, 2926, 2857, 1463, 1378, 1271, 1242, 1151, 1115, 1044, 722. 1H NMR (CD3OD, 250 MHz): δ (ppm) = 0.87 (t, 6H, J = 7.5 Hz, H8′α, H8′β), 1.31 (m, 20H, H7′α, H7′β, H6′α, H6′β, H5′α, H5′β, H4′α, H4′β, H3′α, H3′β,), 1.58–1.63 (m, 4H, H2′α, H2′β), 3.18–3.82 (m, 14H, H1′α, H1′β, H5α, H5β, H2α, H2β, H4α, H4β, H3α, H3β), 4.18 (d, 1H, J = 7.5 Hz, H1β), 4.71 (d, 1H, J = 3.75 Hz, H1α), 4.87 (s, 6H, 6 OH). 13C NMR (CD3OD, 62.9 MHz): δ (ppm) = 14.9 (C8′), 24.2 (C7′), 27.5 (C6′), 27.8 (C5′), 30.9 (C4′), 31.2 (C3′), 33.5 (C2′), 63.4 (C5α), 67.3 (C5β), 69.7 (C1′α), 71.3 (C1′β), 71.6 (C4α), 71.9 (C4β), 74.0 (C2α), 75.3 (C2β), 75.6 (C3α), 78.3 (C3β), 100.8 (C1α), 105.5 (C1β). Anal calcd for C13H26O5, ½ H2O: C, 57.54; H, 10.03. Found: C, 57.71; H, 10.
Decyl-α,β-d-xylopyranoside (3)
This compound was obtained as a white paste following the general procedure previously described with d-xylose (5 g, 0.033 mol) in 188 mL of THF, decanol (13.18 mL, 2 equiv) and PTSA (3.762 g, 0.6 equiv). Yield: 25%, α/β: 4/6, Rf: 0.66 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3362, 2920, 2853, 1466, 1376, 1246, 1144, 1114, 1045, 720. 1H NMR (CD3OD, 250 MHz): δ (ppm) = 0.88 (t, 6H, J = 5 Hz, H10′α, H10′β), 1.25 (m, 28H, H9′α, H9′β, H8′α, H8′β, H7′α, H7′β, H6′α, H6′β, H5′α, H5′β, H4′α, H4′β, H3′α, H3′β,), 1.57–1.59 (m, 4H, H2′α, H2′β), 3.14–3.49 (m, 14H, H1′α, H1′β, H5α, H5β, H2α, H2β, H4α, H4β, H3α, H3β), 4.14 (d, 1H, J = 7.5 Hz, H1β), 4.64 (d, 1H, J = 3.75 Hz, H1α), 4.81(s, 6H, 6OH). 13C NMR (CD3OD, 62.9 MHz): δ (ppm) = 14.9 (C10′), 24.2 (C9′), 27.5 (C8′), 27.8 (C7′), 30.9, 31.0, 31.1, 31.2 (C6′α, C6′β, C5′α, C5′β, C4′α, C4′β, C3′α, C3′β,), 33.5 (C2′), 63.4 (C5α), 67.3 (C5β), 69.7 (C1′α), 71,3 (C1′β), 71.6 (C4α), 71,9 (C4β), 74.0 (C2α), 75,3 (C2β), 75.6 (C3α), 78.3 (C3β), 100.8 (C1α), 105.5 (C1β). Anal calcd for C15H30O5, ¼ H2O: C, 61.09; H, 10.42. Found: C, 61.26; H, 10.44.
Dodecyl-α,β-d-xylopyranoside (4)
This compound was obtained as a white powder following the general procedure previously described with d-xylose (8 g, 0.053 mol) in 350 mL of THF, dodecanol (23.66 mL, 2 equiv) and PTSA (6.02 g, 0.6 equiv). Yield: 40%, α/β: 7/3, Rf: 0.62 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3382, 2919, 2852, 1470, 1376, 1246, 1144, 1114, 1044, 719. 1H NMR (CD3OD, 250 MHz): δ (ppm) = 0.88 (t, 6H, J = 7.5 Hz, H12′α, H12′β), 1.27 (m, 36H, H11′, H10′ H9’, H8′, H7′, H6′, H5′, H4′, H3′), 1.57–1.62 (m, 4H, H2′α, H2′β), 3.13–3.78 (m, 14H, H1′α, H1′β, H5α, H5β, H2α, H2β, H4α, H4β, H3α, H3β), 4.16 (d, 1H, J = 7,5 Hz, H1β), 4.67 (d, 1H, J = 3,75 Hz, H1α), 4.87 (s, 6H, 6OH). 13C NMR (CD3OD, 62.9 MHz): 14.3 (C12′), 23.6 (C11′), 27.2 (C10′), 27.9 (C9′), 30.3, 30.4, 30.5, 30.6 (C8′, C7′, C6′, C5′, C4′,C3′), 32.9 (C2′), 62.9 (C5α), 66.8 (C5β), 69.1 (C1′α), 70.7 (C1′β), 71.0 (C4α), 71.4 (C4β), 73.5 (C2α), 74.8 (C2β), 75.0 (C3α), 77.7 (C3β), 100.2 (C1α), 104.9 (C1β). Anal calcd for C17H33O5: C, 64.12; H, 10.76. Found: C, 64.11; H, 10.87.
Octadecyl-α,β-d-xylopyranoside (5)
This compound was obtained as a yellow powder following the general procedure previously described with d-xylose (8 g, 0.053 mol) in 350 mL of THF, octadecanol (28.67 mL, 2 equiv) and PTSA (10.02 g, 0.6 equiv). Yield: 23%, α/β: 7/3, Rf: 0.57 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3380, 2918, 2851, 1470, 1376, 1248, 1144, 1114, 1044, 719. 1H NMR (DMSO-d6, 250 MHz): δ (ppm) = 0.99 (m, 6H, H18′α, H18′β), 1.37(m, 60H, H17′, H16′, H15′, H14′, H13′, H12′, H11′, H10′ H9′, H8′, H7′, H6′, H5′, H4′, H3′), 1.63–1.65 (m, 4H, H2′α, H2′β), 3.00–3.82 (m, 14H, H1′α, H1′β, H5α, H5β, H2α, H2β, H4α, H4β, H3α, H3β), 4.18 (d, 1H, J = 7,5 Hz, H1β), 4.54 (d, 1H, J = 3.75 Hz, H1α), 4.94 (s, 6H, 6OH). 13C NMR (DMSO-d6, 62.9 MHz): 14.3 (C18′), 23.6 (C17′), 25.9 (C16′), 27.9 (C15′), 30. 3, 30.4, 30.5, 30.6 (C14′, C13′, C12′, C11′C10′, C9′, C8′, C7′, C6′, C5′, C4′, C3′), 31,7 (C2′), 62.3 (C5α), 66.0 (C5β), 67.5 (C1′α), 68.9 (C1′β), 69.9 (C4α), 70.4 (C4β), 72.3 (C2α), 73.6 (C2β), 73.7 (C3α), 77.0 (C3β), 99.3 (C1α), 104.0 (C1β). Anal calcd for C23H46O5: C, 68.61; H, 11.52. Found: C, 68.62; H, 11.70.
2.1.1.2 General procedure for the acetylation of pentosides
The previously synthesized pentosides (1–5) were acetylated in the presence of acetic anhydride (12 equiv) and sodium acetate (1 equiv) for 24 to 72 h at 50 °C under stirring. The reaction was followed by TLC. Diethylether (50 mL) was added to the resulting mixture and three washings (3 × 50 mL) with a Na2CO3 saturated solution were carried out until a neutral pH was reached. The organic phase was then dried with MgSO4, filtered and concentrated under reduced pressure. The residue was then purified by flash chromatography (silica gel, eluting mixture petroleum ether/AcOEt 8:2).
Dodecyl -2,3,4-tri-O-acetyl-β-d-xylopyranoside (4β′)
This compound was obtained as a white powder following the general procedure previously described with dodecyl-α,β-d-xylopyranoside (6.57 g, 0.02 mol) in 26.4 mL of acetic anhydride (12 equiv) and AcONa (2.584 g, 1 equiv) (reaction time: 24 h). Yield: 24%, Rf: 0.64 (petroleum ether/AcOEt, 8:2).
1H NMR (CDCl3, 250 MHz): δ (ppm) = 0.82 (t, 3H, J = 2.5 Hz, H12′), 1.19 (m, 18H, H3′, H4′, H5′, H6′, H7′ H8′, H9′, H10′, H11′), 1.49 (m, 2H, H2′), 1.97–1.99 (s, 9H, 3 CH3CO), 3.29–3.43 (m, 2H, H1′), 3.74 (dt, 1H, J = 5 Hz, J = 2.5 Hz, H5a), 4.06 (dd, 1H, J = 5 Hz, J = 2.5 Hz, H5eq), 4.40 (d, 1H, J = 7.5 Hz, H1β), 4.84–4.87 (m, 2H, H2, H4), 5.09 (t, 1H, J = 8.6 Hz, H3). 13C NMR (CDCl3, 62.9 MHz): 14.5 (C12′), 21.2 (3 C, CH3CO), 23.1 (C11′), 26.3 (C10′), 30.1, 30.0 29.8 (C3′, C4′, C5‘, C6′, C7′, C8′, C9′), 32.3 (C2′), 62.4 (C5), 69.4 (C1′), 70.2 (C4), 71.3 (C2), 71.9 (C3), 101.1 (C1β), 168.1, 168.0, 167.3 (3CH3CO). Anal calcd for C23H40O8: C, 62.14; H, 9.07. Found: C, 62.21; H, 9.12.
Dodecyl-2,3,4-tri-O-acetyl-α-d-xylopyranoside (4α′)
This compound was obtained as a colorless oil following the general procedure previously described with dodecyl-α,β-d-xylopyranoside (6.57 g, 0.02 mol) in 26.4 mL of acetic anhydride (12 equiv) and AcONa (2.584 g, 1 equiv) (reaction time: 24 h). Yield: 58%, Rf: 0.87 (petroleum ether/AcOEt, 8:2).
1H NMR (CDCl3, 250 MHz): δ (ppm) = 0.79 (t, 3H, J = 7.5 Hz, H12′), 1.18 (m, 18H, H3′, H4′, H5′, H6′, H7′ H8′, H9′, H10′, H11′), 1.48–1.51 (m, 2H, H2′), 1.94–1.96 (s, 9H, 3 CH3CO), 3.28–3.32 (m, 1H, H1’a), 3.53–3.66 (m, 2H, H1’b, H5a), 4.01–4.06 (m, 1H, H5eq), 4.70(dd, 1H, J = 2.5 Hz, H2), 4.87–4.80 (m, 2H, H1α, H4), 5.38 (t, 1H, J = 10 Hz, H3). 13C NMR (CDCl3, 62.9 MHz): 13.4 (C12′), 22.6 (3 CH3CO), 23.0 (C11′), 26.2 (C10′), 29.3, 29.5, 29.6 (C9′, C8′, C7‘, C6′, C5′, C4′, C3′), 31.9 (C2′), 61.9 (C5), 68.1 (C1′), 70.3 (C4), 72.3 (C2), 73.9 (C3), 99.1 (C1α), 170.6, 170.4, 170.6 (3 CH3CO). Anal calcd for C23H40O8: C, 62.14; H, 9.07. Found: C, 62.14; H, 9.23.
Octadecyl-2,3,4-tri-O-acetyl-β-d-xylopyranoside (5β′)
This compound was obtained as a white powder following the general procedure previously described with octadodecyl-α,β-d-xylopyranoside (3.102 g, 7,57 mmol) in 17 mL of acetic anhydride (12 equiv) and AcONa (1.029 g, 1 equiv) (reaction time: 24 h). Yield: 15%, Rf: 0.57 (petroleum ether/AcOEt, 8:2).
IR (KBr): 2918, 2850, 1756, 1470, 1375, 1226, 1144, 1079, 723. 1H NMR (CDCl3, 250 MHz): δ (ppm) = 0.81(t, 3H, J = 7.5 Hz, H18′), 1.18 (m, 30H, H3′, H4′, H5′, H6′, H7′ H8′, H9′, H10′, H11′, H12′ H13′ H14′ H15′, H16′, H17′), 1.49 (m, 2H, H2′), 1.97–2.03 (s, 9H, 3CH3CO), 3.29–3.40 (m, 2H, H1′), 3.73–3.76 (m, 1H, H5a), 4.05 (dd, 1H, J4–5eq = J5a-5eq = 5 Hz, H5eq), 4.40 (d, 1H, J = 7.5 Hz, H1β), 4.84–4.89 (m, 2H, H2, H4), 5.09 (t, 1H, J = 10 Hz, H3). 13C NMR (CDCl3, 62.9 MHz): 13.4 (C18′), 20.0 (3 CH3CO), 21.9 (C17‘), 25.2 (C16‘), 30.1, 30.0 29.9 (C15′, C14′, C13‘, C12′, C11′, C10′, C9′, C8′, C7′, C6′, C5′, C4′, C3′), 31.2 (C2′), 61.3 (C5), 68.2 (C1′), 68.9 (C4), 70.1 (C2), 70.8 (C3), 99.9 (C1β), 167.9, 169.1, 169.4 (3 CH3CO). Anal calcd for C29H52O8: C, 65.88; H, 9.91. Found: C, 65.19; H, 9.88.
Octadecyl-2,3,4-tri-O-acetyl-α-d-xylopyranoside (5α′)
This compound was obtained as a white powder following the general procedure previously described with octadodecyl-α,β-d-xylopyranoside (3.102 g, 7,57 mmol) in 17 mL of acetic anhydride (12 equiv) and AcONa (1.029 g, 1 equiv) (reaction time: 24 h). Yield: 50%, Rf: 0.67 (petroleum ether/AcOEt, 8:2).
IR (KBr): 2920, 2851, 1739, 1470, 1376, 1234, 1141, 1045, 721. 1H NMR (CDCl3, 250 MHz): δ (ppm) = 0.82 (t, 3H, J = 5 Hz, H18′), 1.19 (m, 30H, H3′, H4′, H5′, H6′, H7′ H8′, H9′, H10′, H11′, H12′ H13′ H14′ H15′, H16′, H17′), 1.49–1.52 (m, 2H, H2′), 1.96–1.99 (s, 9H, 3CH3CO), 3.29–3.33 (m, 1H, H1’a), 3.55–3.73 (m, 2H, H5a, H1’b), 4.01–4.06 (m, 1H, H5eq), 4.72 (dd, 1H, J = 2.5 Hz, H2), 4.91–4.93 (m, 2H, H1α, H4), 5.41 (t, 1H, J = 10 Hz, H3). 13C NMR (CDCl3, 62.9 MHz): 14.5 (C18′), 21.09 (3 CH3CO), 21.1 (C17‘), 23.0 (C16‘), 29.3, 29.5 29.6 (C15′, C14′, C13‘, C12′, C11′, C10′, C9′, C8,’ C7′, C6′, C5′, C4′, C3′), 32.33 (C2′), 58.6 (C5), 68.9 (C1′), 69.9 (C4), 70.1 (C2), 71.6 (C3), 95.9 (C1α), 170.3, 170.4, 170.6 (3 CH3CO). Anal calcd for C29H52O8: C, 65.88; H, 9.91. Found: C, 64.32; H, 10.05.
2.1.1.3 General procedure for the deacetylation of acetylated pure anomeric pentosides
Each of the previously synthesized pentosides was deacetylated in the presence of MeONa in MeOH. In a Schlenk tube under argon atmosphere, the peracetylated pentoside was dissolved in a mixture of CH2Cl2/MeOH (1/1); the sodium methylate (6 equiv, 2 equiv per OH group) was then added. The evolution of the reaction was followed by TLC until completion; an ion-exchange resin was added to neutralize the excess of sodium methylate. The resulting mixture was then filtered and the solvent was evaporated under reduced pressure.
Dodecyl-β-d-xylopyranoside (4β)
This compound was obtained as a white powder following the general procedure previously described with dodecyl-2,3,4-tri-O-acetyl-β-d-xylopyranoside (2.044 g, 4.93 mmol) in 30 mL of CH2Cl2/MeOH (1/1) and MeONa (1.599 g, 6 equiv) (reaction time: 24 h). Yield: 96%, Rf: 0.62 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3358, 2919, 2852, 1456, 1378, 1142, 1114, 1045, 722. 1H NMR (CD3OD, 250 MHz): δ (ppm) = 0.89 (t, 3H, J = 5 Hz, H12′), 1.28 (m, 18H, H11′, H10′ H9′, H8′, H7′, H6′, H5′, H4′, H3′), 1.61 (quint, 2H, J = 10 Hz, H2′), 3.14–3.17 (m, 3H, H2, H5a, H3), 3.45–3.53 (m, 2H, H4, H1a), 3.77–3.86 (m, 2H, H1b, H5eq), 4.17 (d, 1H, J = 7, 5 Hz, H1β), 4.87 (s, 3H, 3 OH). 13C NMR (CD3OD, 62.9 MHz): 15.5 (C12′), 24.8 (C11′), 28.1 (C10′), 31.5, 31.6, 31.78, 31.9, 33.1 (C9′, C8′, C7′, C6′, C5′, C4′,C3′), 34.1 (C2′), 67.9 (C5), 71.9 (C1′), 72.3 (C4), 75.9 (C2), 78.9 (C3), 106.1 (C1β). Anal calcd for C17H34O5: C, 64.12; H, 10.76. Found: C, 64.37; H, 10.98.
Dodecyl-α-d-xylopyranoside (4α)
This compound was obtained as a white solid following the general procedure previously described with dodecyl-2,3,4-tri-O-acetyl-α-d-xylopyranoside (4.979 g, 12.02 mmol) in 30 mL of CH2Cl2/MeOH (1/1) and MeONa (3.896 g, 6 equiv) (reaction time: 24 h). Yield: 78%, Rf: 0.60 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3391, 2918, 2852, 1472, 1372, 1142, 1111, 1043, 718. 1H NMR (CD3OD, 250 MHz): δ (ppm) = 0.85 (t, 3H, J = 7.5 Hz H12′), 1.20 (m, 18H, H11′, H10′ H9,, H8′, H7′, H6′, H5′, H4′, H3′), 1.51–1.54 (m, 2H, H2′), 3.20–3.71 (m, 7H, H1′, H5a, H5eq, H4, H2, H3), 4.61 (d, 1H, J = 2. 5 Hz, H1α), 4.97 (s, 3H, 3 OH). 13C NMR (CD3OD, 62.9 MHz): 13.4 (C12′), 22.6 (C11′), 23.1 (C10′), 26.2, 29.3, 29.5, 29.6 (C9′, C8′, C7′, C6′, C5′, C4′,C3′), 31.9 (C2′), 61.9 (C5), 68.1 (C1′), 70.3 (C4), 72.3 (C2), 73.9 (C3), 99.1 (C1α). Anal calcd for C17H34O5: C, 64.12; H, 10.76. Found: C, 64.50; H, 10.99.
Octadecyl-β-d-xylopyranoside (5β)
This compound was obtained as a white solid following the general procedure previously described with octadecyl-2,3,4-tri-O-acetyl-β-d-xylopyranoside (0.622 g, 1.17 mmol) in 18 mL of CH2Cl2/MeOH (1/1) and MeONa (0.381 g, 6 equiv) (reaction time: 24 h). Yield: 83%, Rf: 0.56 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3344, 2918, 2850, 1464, 1377, 1181, 1116, 1047, 723. 1H NMR (DMSO-d6, 250 MHz): δ (ppm) = 0.84 (t, 3H, J = 7, 5 Hz, H18′), 1.22 (m, 30H, H17′, H16′, H15′, H14′, H13′, H12′, H11′, H10′ H9′ H8′, H7′, H6′, H5′, H4′, H3′), 1.47 (m, 2H, H2′), 2.95–3.18 (m, 3H, H2, H5a, H3), 3.30–3.60 (m, 2H, H4, H1a), 3.62–3.68 (m, 2H, H1b, H5eq), 4.04 (d, 1H, J = 7, 5 Hz, H1β), 4.94 (s, 3H, 3 OH). 13C NMR (DMSO-d6, 62.9 MHz): 13.3 (C18′), 21.4 (C17′), 24.9 (C16′), 28.0, 28.2, 28.4, 28.7 (C15′, C14′, C13′, C12′, C11′, C10′, C9′, C8′, C7′, C6′, C5′, C4′,C3′), 30.6 (C2′), 65.01 (C5), 67.9 (C1′), 68.9 (C4), 72.6 (C2), 75.9 (C3), 102.9 (C1β). Anal calcd for C23H46O5: C, 68.61; H, 11.52. Found: C, 68.82; H, 11.77.
Octadecyl-α-d-xylopyranoside (5α)
This compound was obtained as a white solid following the general procedure previously described with octadecyl-2,3,4-tri-O-acetyl-α-d-xylopyranoside (1.956 g, 3.73 mmol) in 18 mL of CH2Cl2/MeOH (1/1) and MeONa (1.198 g, 6 equiv) (reaction time: 24 h). Yield: 44%, Rf: 0.59 (CH2Cl2/MeOH, 9:1).
IR (KBr): 3388, 2917, 2851, 1473, 1377, 1144, 1112, 1043, 717. 1H NMR (DMSO-d6, 250 MHz): δ (ppm) = 0.80 (m, 3H, H18′), 1.21 (m, 30H, H17′, H16′, H15′, H14′, H13′, H12′, H11′, H10′ H9′, H8′, H7′, H6′, H5′, H4′, H3′), 1.51–1.54 (m, 2H, H2′), 3.20–3.71 (m, 7H, H1′, H5a, H5eq, H4, H2,H3), 4.61 (d, 1H, J = 2. 5 Hz, H1α), 4.97 (s, 3H, 3 OH). 13C NMR (DMSO-d6, 62.9 MHz): 14.3 (C18′), 22.5 (C17′), 26.1 (C16′), 29.1, 29.3, 29.5, 29.6 (C15′, C14′, C13′, C12′, C11′, C10′, C9′, C8′, C7′, C6′, C5′, C4′,C3′), 31.7 (C2′), 62.3 (C5), 67.5 (C1′), 70.4 (C4), 72.3 (C2), 73.7 (C3), 99.4 (C1α). Anal calcd for C23H46O5: C, 68.61; H, 11.52. Found: C, 69.10; H, 11.68.
2.1.2 Surface tension measurement
2.1.2.1 Wilhelmy's method
Surface tension was measured at a constant temperature (298 K) by means of an automatic tensiometer K100-KRUSS using the Wilhelmy plate method. A mother solution of surfactants (concentration 10−1 mol·L−1 in 20 mL of ultrapure water) was prepared and then diluted to obtain a range of surfactant solutions with concentrations from 10−2 to 10−5 mol·L−1.
2.1.2.2 Method of the hanging drop
The superficial tension of solutions of decreasing surfactant concentrations were measured using the IT concept Tracker drop tensiometer by carefully measuring the superficial tension of water between two sets of measurements to ensure the cleanliness of the syringe. These experiments were carried out at 298 K by analyzing the profile of a drop of a surfactant solution in a tank under a water-saturated atmosphere. All measures were taken when the superficial tension was stabilized.
3 Results and discussion
Glycosylation of d-xylose was performed with hexanol, octanol, decanol, dodecanol and octadecanol using p-toluenesulfonic acid (PTSA) as an acidic catalyst following the previously described methods (Scheme 1) [9b,11].

Synthesis of xylosides.
The yields were moderate; at high temperature under thermodynamic control, major pyranic forms were obtained. Concerning the ratio α/β, β anomers prevailed for C6–10 derivatives as described by Schmidt for the O-alkylation of protected sugars in the presence of a Li-base [12] (Table 1, entries 1–3). With the alcohols C12 and C18, the ratio α/β was reversed and the presence of a major α-xyloside can be explained by the high hydrophobic character of the medium (Table 1, entries 4 and 5).
Glycosylation of d-xylose with C6–18 alcohols.
| Entry | Alcohol | Xyloside | Time (h) | Ratio (α/β)c | Yield (%) |
| 1a | Hexanol (C6) | 1 | 48 | 3/7 | 37 |
| 2a | Octanol (C8) | 2 | 48 | 4/6 | 31 |
| 3a | Decanol (C10) | 3 | 48 | 4/6 | 25 |
| 4a | Dodecanol (C12) | 4 | 48 | 7/3 | 40 |
| 5a | Octadecanol (C18) | 5b | 72 | 6/4 | 23 |
a bd-Xylose (1 equiv), alcohol (2 equiv), PTSA (0.6 equiv) in a large excess of THF, 80 °C.
c Determined by 1H NMR (integration of anomeric protons).
Then, α and β anomers of 1–5 were obtained in pure forms by O-deacetylation of the corresponding acetylated α- and β-xylosides, previously prepared and separated by flash chromatography.
The surface tension characteristics of the xylosides pentosides in pure anomeric forms or in mixtures are reported in Table 2.
Surface tension characteristics of xylosides.
| Entry | Compound | CMC (mmol·L−1) | CMC (mg·L−1) | γ (mN·m−1) | Γ (mol·m−2) | A (Å2) |
| 1 | 4.91 | 1150 | 26 | 3.2 | 52.4 | |
| 2 | 3.63 [13] | 953 [13] | 27 [13] | – | – | |
| 3 | 4.40 [15] | 1616[15] | 30 [15] | 3.4 [15] | 43.7 [15] | |
| 4 | 1.04 [13] | 301 [13] | 27 [13] | – | – | |
| 5 | 0.21 [15] | 62 | 39 [15] | 4.1 [15] | 37.6 [15] | |
| 6 | 0.021 | 6.7 | 49 | 4.7 | 35.1 | |
| 7 | 0.013 [15] | 4.1 [15] | 47.6 [15] | 5.1 | 32.3 | |
| 8 | 3.7. 10−5 | 1.5 | 55 | 5.9 | 27.9 | |
| 9 | 4.9. 10−5 | 1.973 | 51 | 6.5 | 25.4 |
Surface tension, CMC and the area of compounds 2 and 3 have already been reported in the literature [13] and have shown their potential as powerful environmentally friendly surfactants complying with biorefinery guidelines [14] (Table 2, entries 2–5). These values were confirmed by the work of Matsumura et al. where the interfacial properties of dodecylpentosides 4 (especially the β form) were also described [15] (Table 2, entries 6 and 7). This last study demonstrated that alkyl β-d-xylobiosides have the best physicochemical properties as surface-active agents and that in addition β anomers exhibited a greater surface activity than the corresponding α anomers.
Our results concerning xyloside 1 showed a good surface tension, but a high CMC due to the high solubility of this pentoside in water. On the contrary, for pentoside 5, surface tension is high for a very tiny solubility in water. The values of CMC, γ and A are in accordance with the reported results concerning the xylosides 2–4.
To complete this study, molecular modeling with Hyperchem® 4 software and the semi-empirical method PM 3 were used to determine the spatial conformation of xylosides (Fig. 1). The strength field MESSRS+ was used and the conformations were determined under vacuum.
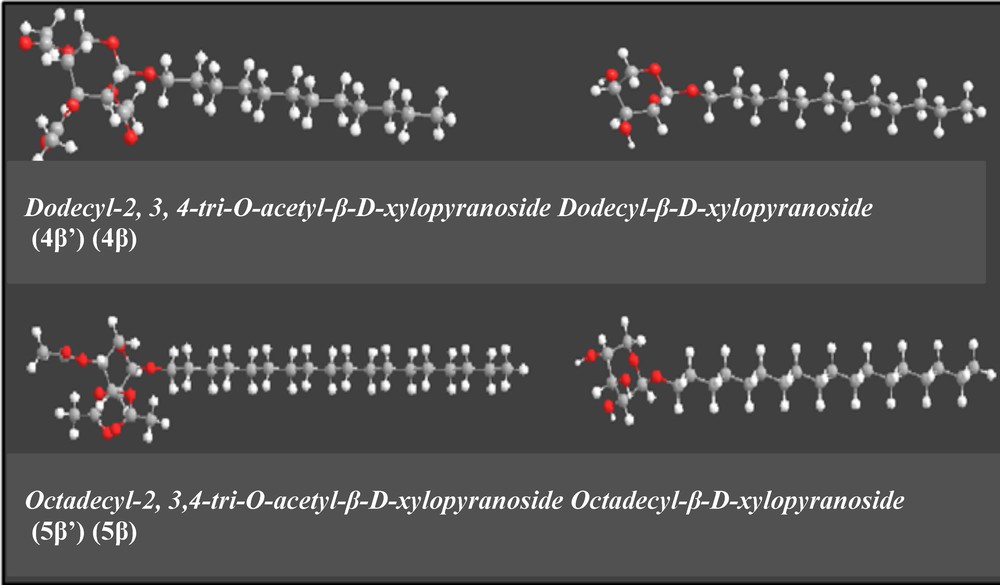
(Color online.) Molecular modeling for xylosides 4β, 4′β, 5β and 5′β.
According to the results (Table 3), the β xylosides are consistently more stable than the α ones. This observation is in agreement with the study of Gorin concerning the stability of methylglycopyranosides [16]. Increasing the length of the hydrophobic chain from 12 to 18 carbons leads to decreased energy and consequently to better stability due to the hydrophobic effect.
Molecular calculations for xylosides 4 and 5.
| d-Xylosides | Energy (kcal·mol−1) | ΔE (kcal·mol−1) |
| 4α | –276.66 | 1.840 |
| 4β | –278.50 | |
| 5α | –309.29 | 1.838 |
| 5β | –311.13 | |
| 4α′ | –386.81 | 3.378 |
| 4β′ | –390.19 | |
| 5α′ | –419.44 | 7.533 |
| 5β′ | –426.97 |
Furthermore, the compound 5β′ that presents the lowest value of energy is the most stable. We also noticed the formation of hydrogen bonds for β anomers 4β and 5β. These bonds are neither present for α anomers nor for acetylated compounds 4′ and 5′. As the acetylated compound 5β′ is the most stable, we can conclude that its stability is governed by the hydrophobic interactions.
4 Conclusion
d-Xylose derived surfactants were easily synthesized and fully characterized through classical analytical methods. Specific methods such as Wilhelmy's method or the hanging-drop method allowed their interfacial properties to be determined. The molecular modeling studies revealed that the β xylosides are consistently more stable than the α ones and that xylosides containing a long carbon chain are more stable. This stability can be attributed to the importance of hydrophobic interactions.
Acknowledgments
Financial support from Reims Champagne-Ardenne University, the CPER 2007–2013 framework (Pentoraffinerie program), and the FEDER is gratefully acknowledged.