

1 Introduction
Crystalline porous hybrid solids known as Metal-Organic Frameworks (MOFs) are the most recent class of porous solids [1–5]. These materials are constructed of inorganic subunits (almost all the plausible di- tri- and tetravalent metals present in the periodic table) which are linked together through organic polycomplexing moieties (carboxylates, azolates, imidazolates, phosphonates, catecholates…). This offers an unprecedented structural and chemical diversity leading to thousands of architectures reported so far and many more to be discovered. Besides their promise for industrial application in diverse areas [6,7], this class of materials attracts tremendous curiosity from a fundamental point of view arising from the complexity and the flexibility of their architectures [1–5, 8, 9]. This makes this family of solids an ideal platform for the development and the validation of advanced experimental and computational tools.
The breathing behavior of some MOFs is one of the most fascinating structural behaviors encountered in the field of porous solids [8–10]. The fundamental understanding of the origin of such a peculiarity aroused great interest which led to the deployment of an arsenal of in situ characterization tools and multi-scale modeling techniques [9–18]. The complexity of their architectures also calls for a continuous interplay between experimental and modeling approaches integrating advanced characterization and computational tools fully intertwined during all stages of the structure determination [19–25]. Modeling tools involving energy minimization techniques at the force field (interatomic potential) and/or electronic (quantum) levels have been deployed in tandem with experimentation including X-ray diffraction and Nuclear Magnetic Resonance measurements to solve the crystal structure of several porous MOFs [19–28]. In particular, such a computational assisted structure determination strategy is currently indispensable for the structure resolution of complex (large unit cell and low symmetry) and/or poorly crystallized porous MOF solids [29], which is hardly achieved by the conventional ab initio direct methods applied to powder diffraction. In complement to high-throughput synthesis, there is nowadays growing interest in the development of large-scale computational toolboxes integrating efficient search algorithms and optimization methods, to boost the discovery of novel porous MOF structures and probe their adsorption/separation properties [30–37]. A huge collection of hypothetical MOF structures has been predicted; however only rarely synthesized so far.
The aim of this article is to emphasize how modeling is a valuable tool not only to aid the structure determination of complex architectures and to predict novel structures sometimes with targeted chemical and topological features for further applications, but also to capture and understand the complex structural behaviors of these MOFs upon diverse stimuli as for instance their spectacular breathing behavior triggered by the adsorption of guest molecules. The continuous interplay between experimental and computational tools in these three areas is highlighted through recent achievements made by our group in tandem with the experimentalists at the Institut Lavoisier Versailles (ILV). The readers can refer to recent review journals [30] and books [38] for an exhaustive review of the computational approaches applied to model MOFs.
2 Structure solution for MOFs
The relatively poor crystallinity of certain MOFs sometimes combined with large unit cells or low symmetries is still a hurdle on the way to obtain the structure solution of these hybrid solids solely from the indexation of the powder X-ray diffraction patterns using the conventional ab initio methods [39–41] as implemented in the most popular crystallography software. This called for the development of molecular simulation tools intertwined with experimental information in order to boost the resolution of the crystal structure of many MOFs. The Institut Lavoisier Versailles (ILV) is the pioneer in this field, particularly with the structure elucidation of the complex mesoporous MIL-100 [42] and MIL-101 [43] solids using a computational assisted structure determination approach. Following the concept of Secondary Building Blocks [20] and the chemical intuition raised from the analysis of the synthetic conditions, a series of hypothetical zeotype architectures built up from the assembly of inorganic trimers and benzene-1,3,5-tricarboxylate (MIL-100) /1,4-dicarboxylate (MIL-101) linkers were first computationally generated and their associated X-ray diffraction patterns were calculated. A direct comparison between the experimental and simulated diffraction diagrams allowed the selection of the most probable structure candidate which was used for a further Rietveld refinement on the experimental powder diffraction data. The resolution of these two giant pore MOFs with huge unit cell volumes (380,000 Å3 and 702,000 Å3 for MIL-100 and MIL-101, respectively) comparable to some proteins is the first spectacular success of the interplay between experimental and computational approaches.
Inspired by this advance, our group contributed in collaboration with ILV to solve the structure of a series of complex isoreticular MOFs including (i) the extra-large extended and functionalized mesoporous iron carboxylate solids of cubic symmetry with the MIL-100 and MIL-101 topology [44]; the typical example is the MIL-100(BTB) built up from the 1,3,5-tris(4-carboxyphenyl)benzene linker showing an extremely large unit cell volume up to 2 million Å3, and (ii) the carboxylate interwoven structured microporous based-MIL-142 series and their functionalized derivatives of rhombohedra symmetry and associated with intermediate unit cell volumes, the 4,4′-azobenzenedicarboxylate MIL-142E solid showing the largest volume (95,547 Å3) [45]. Most of these MOFs are of relatively poor crystallinity which rendered the resolution of their structures hardly feasible simply from X-ray diffraction data. A structureless pattern refinement of the experimental diffraction diagram was however possible, and the cell parameters and the space group symmetry of each phase were thus determined. A computational ligand replacement strategy was used and consisted of gradually substituting the benzene-1,3,5-tricarboxylate (btc) and the benzene-1,4-dicarboxylate (bdc) linkers of the parent MIL-100/MIL-101 and MIL-142(A) structures respectively with ligands of a longer length. Force field based-energy minimization using potential parameters selected from generic force fields was subsequently performed to optimize the geometry of these structures at fixed cell parameters experimentally obtained for each solid. The so-obtained structure models were further used for Rietveld refinement on the experimental powder diffraction data in order to deliver a final solution of the structures (Fig. 1).
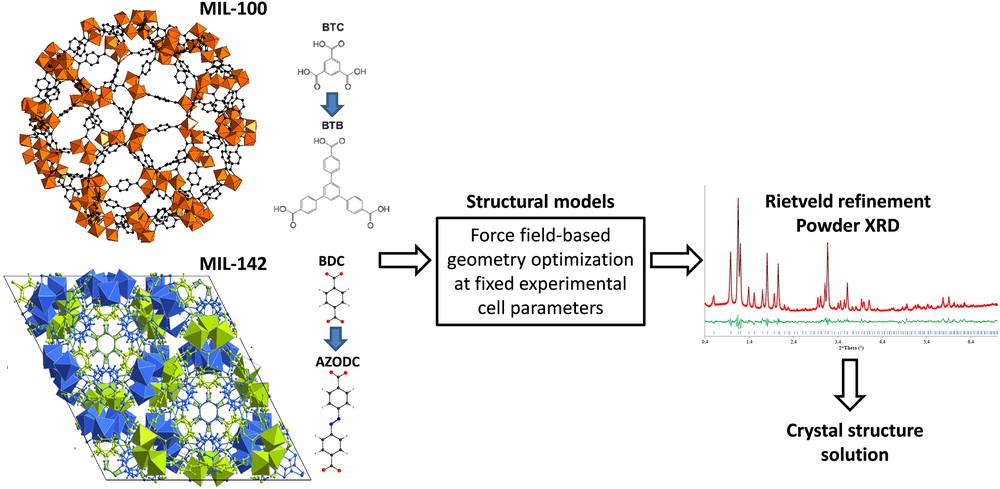
Illustration of the ligand replacement strategy applied to the MIL-100 [44] and MIL-142 [45] series for solving the crystal structure of MIL-100(BTB) and MIL-142D.
A variant of this strategy can be applied when MOFs of moderate/small unit cell sizes are considered. Typically, the series of porous zirconium di-carboxylate solids, denoted MIL-140A to MIL-140D [46], of general chemical formula [ZrO(O2C-R-CO2)] where R = C6H4 (MIL-140A), C10H6 (B), C12H8 (C) and C12N2H6Cl2 (D) with unit cell volumes ranging from 2000 to 4700 Å3, fall into this category of MOF solids. In this case, the cell size is not anymore a limiting step to run calculations at the electronic level. The crystal structure of MIL-140A which was solved by direct methods, exhibits triangular channels delimited by zirconium oxide chains along the channel axis connected to six other chains through the dicarboxylate linkers (see Fig. 2). The structure models of MIL-140B to MIL-140D, built up using a ligand replacement strategy applied to the resolved crystal structure of the parent MIL-140A, were thus energy minimized using DFT calculations including a full relaxation of atomic positions and cell parameters without fixing any constraint in terms of symmetry. The resulting optimized structures showed unit cell parameters and space groups in very good agreement with the experimental data issued from the structureless pattern refinement and indeed they were proposed as valuable structure solutions for the novel synthesized solids. It can be also noticed that simple and fast computational methods based on Monte Carlo integration techniques exist in order to estimate the pore size, the accessible surface area for a probe molecule and the pore volume from the crystal structure [47–50]. These can be used as additional characterization tools for judging the consistency of the proposed structure models by a direct comparison between the corresponding simulated and experimental data. This was done for the full validation of the MIL-140 series.

Illustration of the ligand replacement strategy applied to the MIL-140 series [46].
Alternatively, the combination of high resolution solid state NMR spectroscopy and diffraction and molecular simulations, labeled as SMARTER [51–53], is highly valuable to obtain the structure solution of MOFs. We recently followed this approach in collaboration with ILV to validate the crystal structures of a series of functionalized zirconium carboxylate UiO-66(Zr) (UiO stands for University of Oslo) [54] MOFs which were initially proposed by DFT geometry optimization. These materials are built up from the inorganic building blocks of Zr6-octahedra [Zr6O4(OH)4] bound to twelve terephthalate (BDC-X with X = –H, –NH2, –Br, and –2OH) ligands, leading to a 3D architecture with the coexistence of octahedral and tetrahedral cages connected through triangular windows (see the inset in Fig. 3). It is well known that the 1H and 13C isotropic chemical shifts obtained by first principles calculations strongly depend on the local geometry of the structure. Indeed, the very good agreement obtained between the experimental and simulated NMR parameters for the MOFs investigated in this study (Fig. 3) was considered as a strong support for the validation of the predicted structures [26].
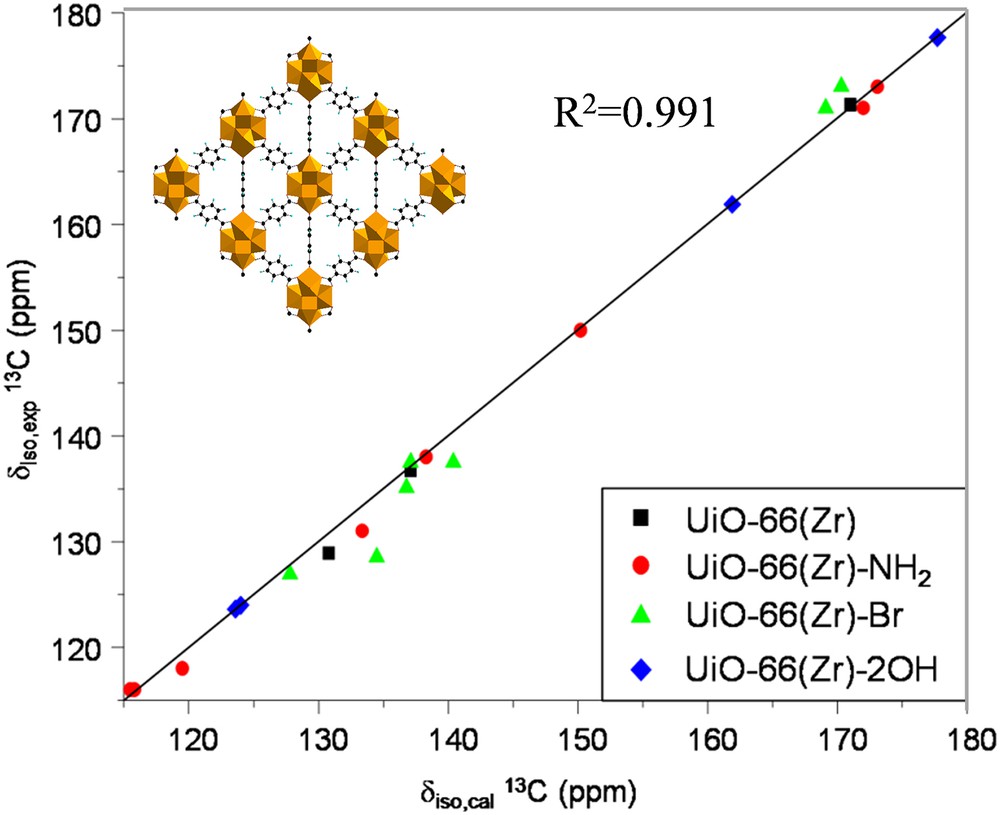
Comparison between the first-principles calculated (δiso,cal) and experimental (δiso,exp) 13C isotropic chemical shifts for the UiO-66(Zr) structure with the organic linkers grafted by different functions. Inset: 3D-crystal structure of UiO-66(Zr) [Ref. 26].
With this in mind, our very recent studies integrated molecular simulation, high resolution solid state NMR spectroscopy and X-ray diffraction approaches [29] to solve the structure of the fumarate A520 MOF, commercialized by BASF for methane storage in automotive applications [55,56]. Here, the cell parameters, the space group and the positions of the Aluminum, Carbon and Oxygen atoms of the hydrated phase were first obtained from a Rietveld refinement on the experimental powder diffraction data collected for a sample with a relatively good crystallinity. Starting with this information, DFT calculations allowing a full relaxation of the atomic positions and cell parameters were performed to deliver a plausible structure model which was subsequently considered for Rietveld refinement. A520 was revealed to be isostructural to MIL-53, the fumaric acid linker substituting the benzene-1,4-dicarboxylate, with the lozenge-shaped 1D pores (Fig. 4). The crystal structure was finally validated by a good agreement between the first-principles calculated and experimental NMR parameters for Aluminum, including the quadrupolar coupling constant CQ and the asymmetry parameter ηQ. In addition to the validation of the architectures, this permanent experimental-modeling interplay was also able to elucidate the positions of the water molecules confined into the pores of A520; the DFT-predicted distribution is very similar to the one obtained through successive difference Fourier maps.
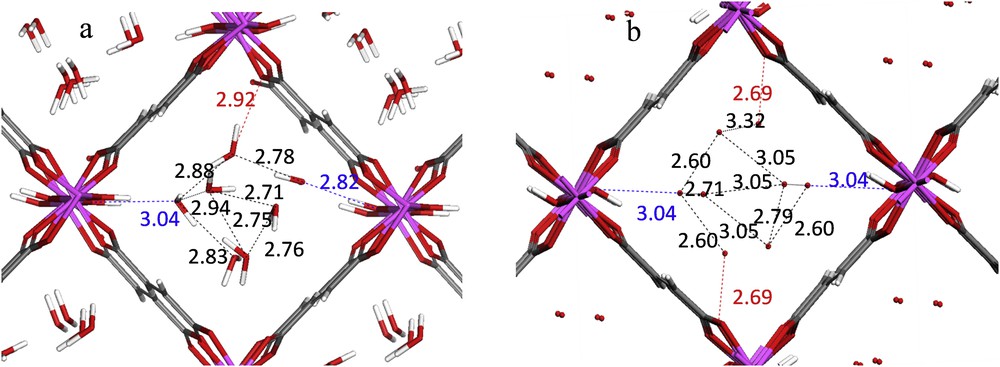
Distribution of the water molecules in the hydrated form of the A-520 MOF: a. DFT optimization and b. Powder XRD data [Ref. 29]. The Al, oxygen, carbon and hydrogen atoms are in purple, red, black and white, respectively. The interatomic distances between water/OH groups (blue), water/O of the carboxylate (red) and water/water (black) are reported in Å.
3 Structure anticipation for MOFs
The majority of the MOFs reported so far were discovered by serendipity than true design [1]. The conventional trial-and-error experimental methods are tedious and time-consuming while the high-throughput experimental synthesis and combinatorial chemistry generally still cover only a small fraction of synthetic variables which prevents a full scan of the possible MOF structures. ILV was the pioneer for the computational prediction of hypothetical porous hybrid solids with the development of an extended version of the Automated Assembly of Structure Building Unit (AASBU) approach in the early 2000’s [20]. This method is based on a simulated annealing Monte Carlo algorithm to search for the lowest-energy arrangement of inorganic and organic building blocks with predefined linkages, initially introduced randomly in a simulation box with a given molar ratio. Thousands of plausible novel MOF structures were thus predicted. D.W. Lewis et al. further proposed the construction of a series of hypothetical Zeolitic Imidazolate frameworks (ZIFs) starting with various zeolite crystal structures and substituting the Si center and the Si-O-Si bridge by a Zn ion and the imidazole linker, respectively. These structures were geometry optimized using the Density Functional Theory and their stabilities were assessed through the determination of their total electronic energies [57]. Later, the database of hypothetical MOFs was considerably enriched by the development of novel computational tools which aimed to explore the recombination of building blocks simply driven by geometric and topological consideration. The Lego brick-like approach reported by Snurr et al. [30,31] was thus able to generate more than 130,000 hypothetical MOFs resulting from the assembly of building blocks derived from existing MOFs. This in silico discovery of novel MOFs was coupled with the development of large scale computational tools to assess rapidly their adsorption and separation performances with respect to societally relevant applications including natural gas purification and carbon dioxide capture [30,35–37]. Other groups constructed a large variety of hypothetical MOFs starting with nets available from the Reticular Chemistry Structure Resource (RSCR) and mapping onto these nets diverse building blocks [32–34]. However resulting from this, none of the MOFs discovered by such high-throughput virtual screening has been synthesized so far. Only two MOFs were really computationally designed in a predictive way taking into account the application requirements, later successfully synthesized and found to show properties in very good agreement with those simulated. The first one emanated from the group of Snurr, the NU-100 (NU stands for Northwestern University) [58] which was constructed by a ligand replacement strategy applied to existing NOTT-112 (NOTT stands for Nottingham University) [59]. The replacement of the existing 1,3,5-tris(3′,5′-dicarboxy[1,1′-biphenyl]-4-yl)benzene ligand by an hexatopic carboxylate linker was followed by force field-based energy minimization and this results in a MOF with a high BET area (∼6000 m2/g) combined with outstanding H2 and CO2 uptakes which were experimentally confirmed. The second computationally designed MOF comes from our recent exploration of this family of materials for the selective capture of CO2 from N2 [60]. The hypothetical UiO-66(Zr)-(CO2H)2 3D cage-type MOF (see Fig. 5a), we constructed by applying a ligand replacement strategy on the parent UiO-66(Zr) followed by DFT-geometry and cell optimizations, was predicted to show excellent separation performance for the mixture of interest. A synthesis effort was successfully deployed by ILV to prepare this novel material. The powder X-ray diffraction of the synthesized sample was revealed to coincide well with the one calculated from the predicted crystal model (see Fig. 5b). The experimental structure analysis led to unit cell parameters and space groups in very good agreement with those of the predicted structure (Table 1). The experimental BET area and pore volume were also in good accordance with the N2-accessible surface area and the free pore volume calculated using the geometric method (Table 1). Finally, the excellent agreement obtained between the NMR 13C isotropic chemical shifts and the first-principles calculated ones (Fig. 5c) allowed us to unambiguously validate the synthesis of the desired sample. Beyond the validation of the predicted structure, it was also confirmed that the separation performance of the synthesized sample is similar to that anticipated for the designed material.

UiO-66(Zr)-(CO2H)2: (a) Illustration of the tetrahedral and octahedral cages present in the 3D crystalline structure. The large green spheres represent the void regions inside the cages (Zr polyhedra; C, black; O, red, and H, cyan), (b) comparison between the powder X-ray diffraction pattern experimentally obtained (black) and calculated from the predicted structure (red), and (c) first-principles calculated (δiso,cal) and experimental (δiso,exp) 13C isotropic chemical shifts [60].
4 Structural behavior of MOFs upon stimuli: the complex case of breathing MOFs
Most of the MOFs with small or moderate pore sizes including some ZIFs [61,62], ELM-11 [63] and a series of MILs [64,65] show a certain degree of flexibility in the organic and/or organic sub-units (ligand flipping, pore gate opening, and small unit cell volume changes) The family of breathing MOFs is the most spectacular example of flexible porous solids [8–10]. This phenomenon that can be initiated by chemical, thermal or mechanical stimuli is associated with a structural transition between two states separated by energy barriers higher than the thermal vibration energy. MOFs such as DMOF-1 [66], DUT-8 [67], Co(BDP) [21], and some of the MIL-n series (MIL stands for Materials of the Institut Lavoisier), e.g. the MIL-53 [9,10, 68] and the MIL-88 [69–71] solids, are the most representative illustrations of this class of materials, showing a drastic expansion of their unit cell volumes up to 230 % upon diverse stimuli. Such a complex structural behavior is still scarcely explored computationally [13, 16,72–76]. Our contribution on the MIL-53 solids [74–76] and the one emanating from Miyahara et al. [13] on catenated jungle gym-type MOFs are pioneering studies in developing and applying force field-based molecular simulation tools to predict and understand at the atomic scale the breathing phenomenon in MOFs. One of the key challenges was to derive a specific force field with the appropriate mathematical function and set of potential parameters to treat accurately the intra-framework interactions of MIL-53(Cr), a 3D-MOF built-up from infinite chains of corner-sharing Cr3+O4(OH)2 octahedra, interconnected by terephthalate groups that form 1D diamond-shaped channels with pores of free diameter close to 8.5 Å (see the inset in Fig. 6). The force field parameters of the intra-framework energy terms (bond stretching, bond bending, torsion…) were determined using a semi-empirical approach defined as follows: an initial set of potential parameters was selected from generic force fields and a systematic refinement was conducted with the criteria to accurately reproduce the experimental vibrational properties of the MOF framework via an energy minimization procedure at 0 K. This derived force field was further combined to the one selected for representing CO2, and implemented in NσT Molecular Dynamics simulations to follow the structural transformations of the porous solid at 300 K along the whole adsorption process. This allows successful capture of the two-step structural switching from a large pore (orthorhombic symmetry, space group Imcm) to a narrow pore (monoclinic symmetry, space group C2/c) form, as induced by CO2 adsorption at ambient temperature which implies large atomic displacements up to 4.7 Å corresponding to a unit cell volume change of about 30 % (Fig. 6a). A full validation of these predictions with experimental data issued from in situ X-ray diffraction observations made this force field as the first one reported in the literature to capture such spectacular structural modifications in a porous MOF solid [74]. This force field was further revealed to also accurately capture the structural changes of MIL-53(Cr) under thermal and mechanical stimuli as experimentally evidenced [17]. It was thus established that MIL-53(Cr) undergoes a transition from the large pore to the narrow pore version within a range of temperatures (110 K, Fig. 6b) and applied pressures (55 MPa, Fig. 6c) in excellent agreement with the data issued from neutron diffraction and mercury porosimetry measurements, respectively.
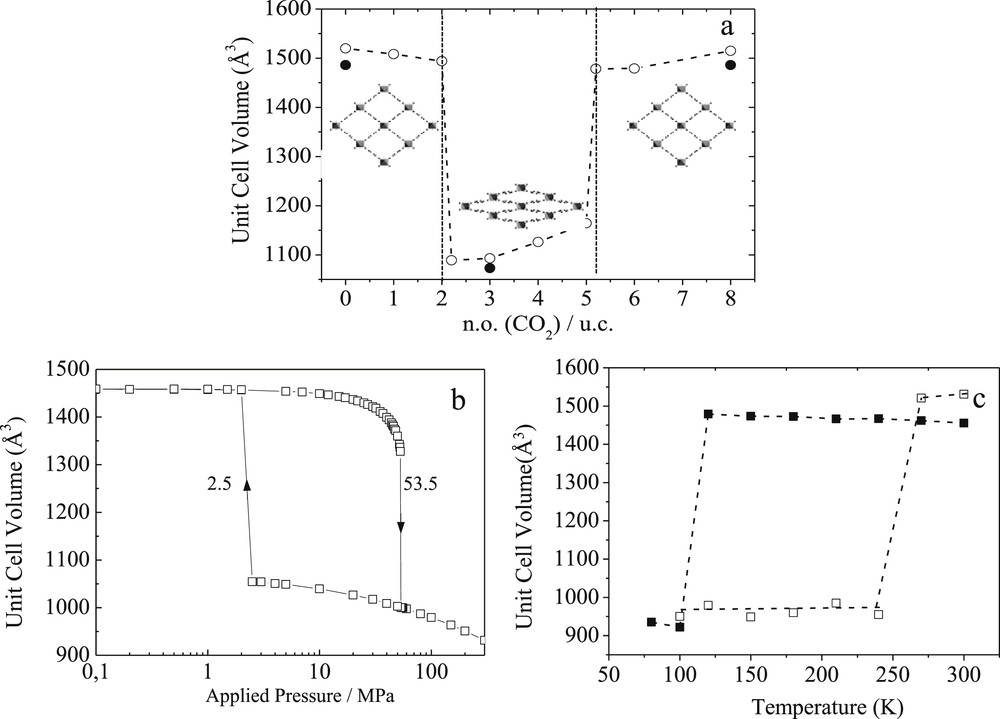
Evolution of the simulated unit cell volume for MIL-53(Cr) as a function of (a) the CO2 concentration inside the pore (solid symbols correspond to in situ X-ray diffraction data) at 303 K, (b) the applied external pressure at 303 K and (c) the temperature (cooling: solid symbols; heating: empty symbols) by Molecular Dynamics simulations. Inset of (a): illustration of the large pore and the narrow pore forms [Ref. 17 and 74].
The success of this computational approach motivated in the past five years numbers of our modeling studies on MIL-53 [77–90] but also from other groups on diverse MOFs and in particular the jungle-gym-type DMOF-1 MOF [66]. In this latter case, our force field was shown to be transferable leading to an accurate description of the breathing behavior of this MOF upon adsorption of benzene and alcohol. Ab initio and Density Functional Theory calculations on periodic solids or representative clusters were proposed along the last few years, as an alternative route to derive flexible force field parameters for MOFs [91–94]. In particular, the very recent work reported by Vanduyfhuys et al. provides a nice illustration of development and implementation of new methodologies and programs to facilitate the determination of flexible DFT-based force field parameters [95]. The emergence of high computing power and efficient software made also the exploration of guest and thermal assisted-structural transitions of diverse MIL-53(Al, Cr, Ga, and Sc) possible using DFT and ab initio Molecular Dynamics calculations [96–99]. Thermodynamic and analytical models have been also proposed to rationalize the dynamic behaviors of this family of materials [16, 72, 73].
Our contribution in this field also emphasized that beyond the derivation of accurate flexible force field parameters, the complex adsorption behavior of breathing MOFs requires the use of advanced computational tools. Typically, the standard Grand Canonical Monte Carlo (GCMC) approach failed to reproduce the step-wise shape of the CO2 adsorption isotherm experimentally obtained for MIL-53(Cr). A more elaborated and time consuming approach consisted of combining Molecular Dynamics (MD) and GCMC techniques in a hybrid osmotic Monte Carlo (HOMC) scheme [17]. In such a situation, the implementation of MD moves is crucial to allow an efficient sampling of the volume fluctuations for the MOF framework. This is particularly true when the volume variations reach spectacular values as it is the case for the MIL-53(Cr) solid upon CO2 adsorption. Combined with the flexible force field as derived above, this methodology offers a unique way to capture quantitatively (transition pressure and amount adsorbed) the unusual shape of the experimental CO2 adsorption isotherm in the whole range of pressures as shown in Fig. 7.
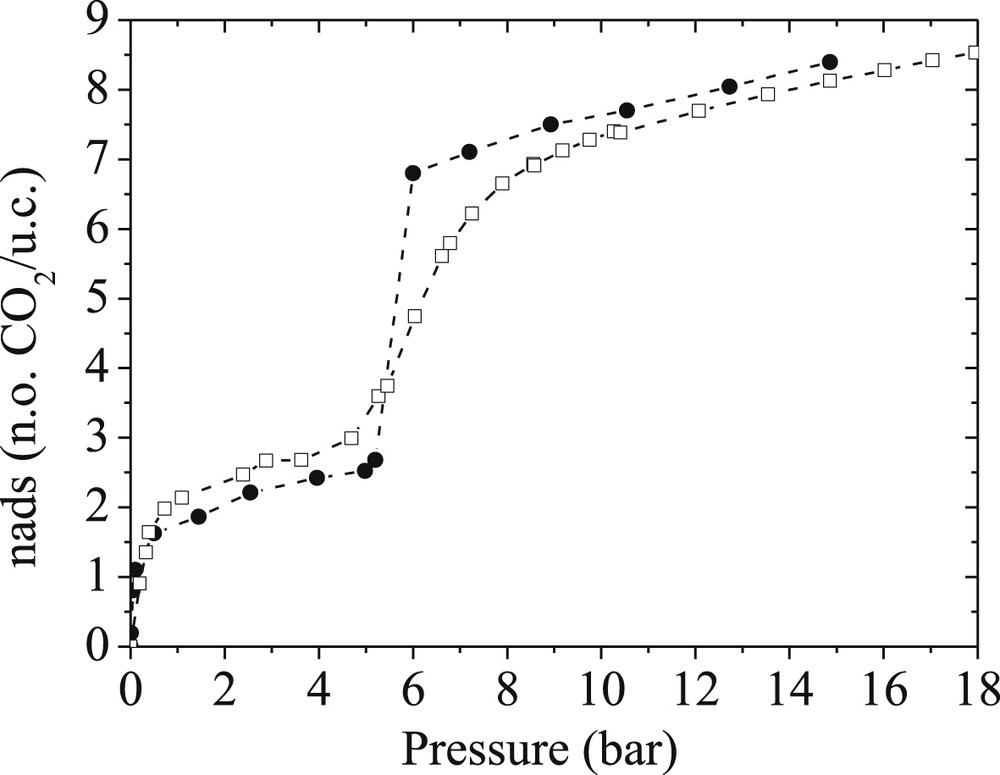
Experimental (empty squares) and Hybrid Osmotic Monte Carlo simulated (full circles) adsorption isotherms for CO2 in MIL-53(Cr) at 303 K [Ref. 17].
Finally, we were also able to capture the physics behind such a drastic structural change of the framework. It was demonstrated that once the external stimuli reaches a critical value, a soft mode appears and triggers the structural switching of the MIL-53(Cr) [75,76].
5 Conclusion
The illustrations presented in this article emphasize the crucial need to integrate advanced experimental and computational methods in order to not only solve the crystal structure of existing MOFs which would be not feasible without the implementation of such a dual multidisciplinary approach, but also to anticipate the design of a few crystalline porous solids that can be a posteriori synthesized and to characterize their structural behaviors upon diverse stimuli that can be at the origin of promising performances for certain applications, e.g. gas adsorption, mechanical energy storage, etc. The interplay between NMR, XRPD and molecular simulations has been already proved to be successful for the structure elucidation of some MOFs and this trend is expected to be reinforced by the incorporation of other experimental tools including Infra-red/Raman and Pair Distribution Function. A refinement of the computational tools for structure prediction is however required to really boost the discovery of novel MOFs. Besides the ability of the large-scale computational program to create huge libraries of hypothetical MOFs, there is a need to improve the simulation models for a real in silico design of materials with topological, chemical and electronic features specifically targeted for a given application that can be then prepared by the experimentalists. This challenge should be addressed in the near future with the development of advanced multi-scale approaches ranging from ab initio calculations to QSPR statistical tools again in strong interrelation with the experimental side. All MOFs show some flexibility degree of their frameworks as it is the case for the small pore (ligand flipping, pore gate opening, unit cell changes…) or breathing MOFs. There is a crucial need to devise an appropriate computational approach to account for this in order to accurately predict the performances of this family of materials for diverse applications. One of the great challenges will be to develop fast and efficient automated tools that are able to derive accurate and transferable flexible force fields.
Acknowledgments
G.M. thanks A. Ghoufi (IPR UMR 6251, Université de Rennes); F. Salles, N.A. Ramsahye, P.G. Yot (ICGM UMR 5253 CNRS UM ENSCM); Q. Ma, Q. Yang and C.L. Zhong (State Key Laboratory of Organic-Inorganic Composites, Beijing University of Chemical Technology); A. Vittadini (ISTM-CNR, Dipartimento di Scienze Chimiche and INSTM, UdR Padova, Universit degli Studi di Padova); V. Guillerm, H. Chevreau, F. Ragon, F. Nouar, E. Alvarez, C. Martineau, F. Taulelle, N. Guillou, T. Devic, P. Horcajada, C. Serre and G. Férey (Institut Lavoisier Versailles UMR 8180 CNRS Université Versailles–Saint-Quentin-en-Yvelines) for their contributions to this work and the Institut Universitaire de France for the support.