

1 Introduction
Metal-organic frameworks (MOFs), also called porous coordination polymers (PCPs) [1,2], are known as promising sorbent-materials with record-holding surface areas (> 6000 m2/g) [3,4], exceeding the values for zeolites, active carbons and silica gels [5,6]. High uniformity of pore-sizes in MOFs makes them promising materials [7] for hydrogen or methane storage [8–12], effective small molecule, e.g. CO2, separation [8–10,13–15], drug delivery [16–18], size-exclusion catalysis [19], as hosts for metal colloids or nanoparticles [20] and sensing [21,22]. Broad possibilities for functionalization of the organic linkers allows for fine tuning of ensuing MOFs’ properties. Effective achievement of this goal is, however, impossible without a good correlation of the functionalization with the sorptive properties of these promising materials. Prediction and exact quantification of the functional group influence on the sorptive properties of MOFs is important in the context of their further improvement.
Advances in reticular chemistry of carboxylate MOFs [23] allow one to reproduce some structural types, especially of pcu topology [24] with high predictability, tolerating functionalization, provided that it is sterically acceptable. The linker 4,4′-biphenyldicarboxylate (BPDC, Scheme 1) is an often used scaffold for functionalization, either on the ortho- or meta-position to the carboxylate group. Meta-functionalization does not normally affect the SBU-related properties, such as metal-enhanced physisorption or solvolytic stability as long as no sterical shielding of the metal atoms occur. Instead, meta-functionalization imparts implanarity to the biphenyl core, thus enabling the possibility of axial chirality, provided that the relative rotation of the phenyl moieties is restricted.
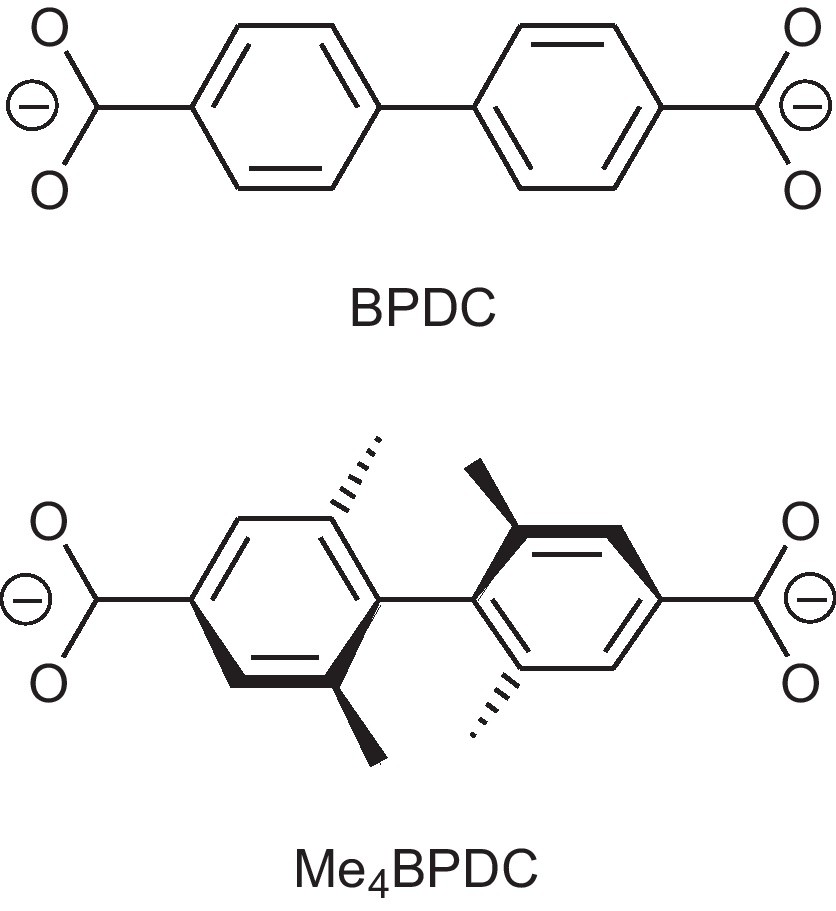
4,4′-Biphenyldicarboxylate (BPDC) and 2,2′,6,6′-tetramethyl-4,4′-biphenyl-dicarboxylate (Me4BPDC).
The linker 2,2′,6,6′-tetramethyl-4,4′-biphenyldicarboxylic acid (H2Me4BPDC, 1) is a representative of perfectly restricted non-planar biphenyl with maximal meta-substitution. Introduction of a single methyl group in the organic building linker increases per se the surface area of the resultant isotypic MOF, but normally decrease slightly the specific surface area (m2/g) as the methyl group introduces also some weight [25]. On the other hand, multiple functionalization by methyl groups, which has only a minor electronic effect, increases strongly the lipophilicity and introduces kinetic obstacles for diffusion of small molecules, which could increase selectivity towards sorption and separation. Multiple functionalization is thought to be additive but at some point, the general liphopilicity could reach a threshold which could influence the adsorption efficiency of such gases as methane and significantly improve the capabilities towards small molecule separation when lipophilic molecules are involved. The linker 1 bears the maximum number of methyl groups (four) which do not interfer with the immediate environment of the structure-defining metal secondary building units (SBUs).
In the present contribution, we report the synthesis and sorptive properties of the [Zn4O(Me4BPDC)3] and [Cu2(Me4BPDC)2] MOFs and discuss the influence of fourfold methyl-functionalization. The related compound [Zn4O(Me4BPDC)3] · x(diethylformamide) has been prepared in the context of nonlinear properties in coordination copolymers of randomly mixed ligands [26].
2 Results and discussion
2.1 Synthesis
The linker H2Me4BPDC, 1 is obtained in a one step modification of the bimesityl molecule (Fig. 1) by selective oxidation of the two sterically non-hindered methyl groups of the bimesityl molecule with dioxygen in the presence of trivalent cobalt as a catalyst which is formed from a Co(II) source (Fig. 1) [27,28].
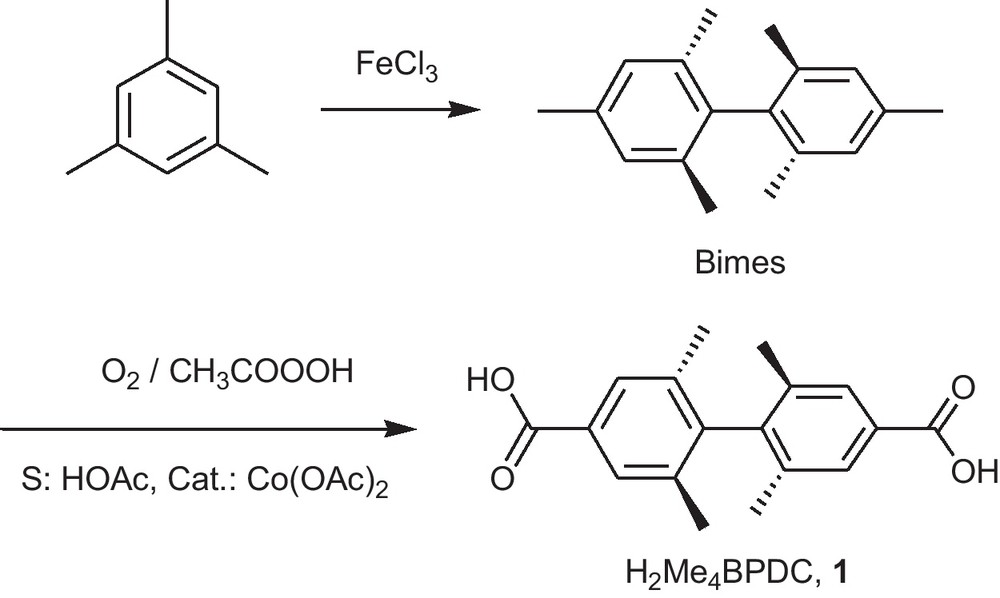
Synthesis of 2,2′,6,6′-tetramethyl-4,4′-biphenyldicarboxylic acid, H2Me4BPDC, 1.
The synthesis (Fig. 1) is of general interest, as chiral polymesitylene species, like quatermesityl [29], could be converted to para-functionalized chiral di- (or poly-) acids for optically active coordination polymers which are further aims of our research [30–33]. Hence, the process of oxidation, initially based on a patent [25], was subjected to an independent optimization study, the results of which were found to be very close to the only published paper with the synthesis of H2Me4BPDC [28]. The achieved yields (50–60%) are significantly larger than in the patent and become practical for preparation of more expensive and advanced quatermesityl ligands noted above. Selective oxidation of the sterically least hindered methyl groups on an aromatic core by active oxygen in the presence of a metal catalyst have good prospects, with the Mn(III)/O3 combination being also worth mentioning [34].
The syntheses of the MOFs [Zn4O(Me4BPDC)3] · 9 DMF (2 · 9 DMF) and [Cu2(Me4BPDC)2] · 9 DMF (3 · 9 DMF) were carried out through the classical low temperature solvothermal synthesis in dimethylformamide, DMF, similar to the conditions described for the synthesis of IRMOFs [35]. Large cubic crystals for both compounds with dimensions up to 1 mm were grown at 70–80 °C. Higher temperatures (up to 110 °C) are possible for the synthesis of 2 · 9 DMF. In the case of 3 · 9 DMF, large deep-blue cubic crystals could only be grown at 70 °C. At higher temperatures, Cu2O starts to form after two days of heating.
2.2 Structural characterization
The structure of [Zn4O(Me4BPDC)3] · 9 DMF (2 · 9 DMF) represents a doubly interpenetrated [36] network of the well-known IRMOF-n series structure, based on the {Zn4O(O2CR)6} secondary building unit (Fig. 2). The 90° twist of the ligand is equivalent to a proper symmetry transformation of the SBU and, thus, the self-assembly results in the same pcu-a (alternatively cab) [37] primitive cubic network as in the case of a planar terephthalic acid homologue.
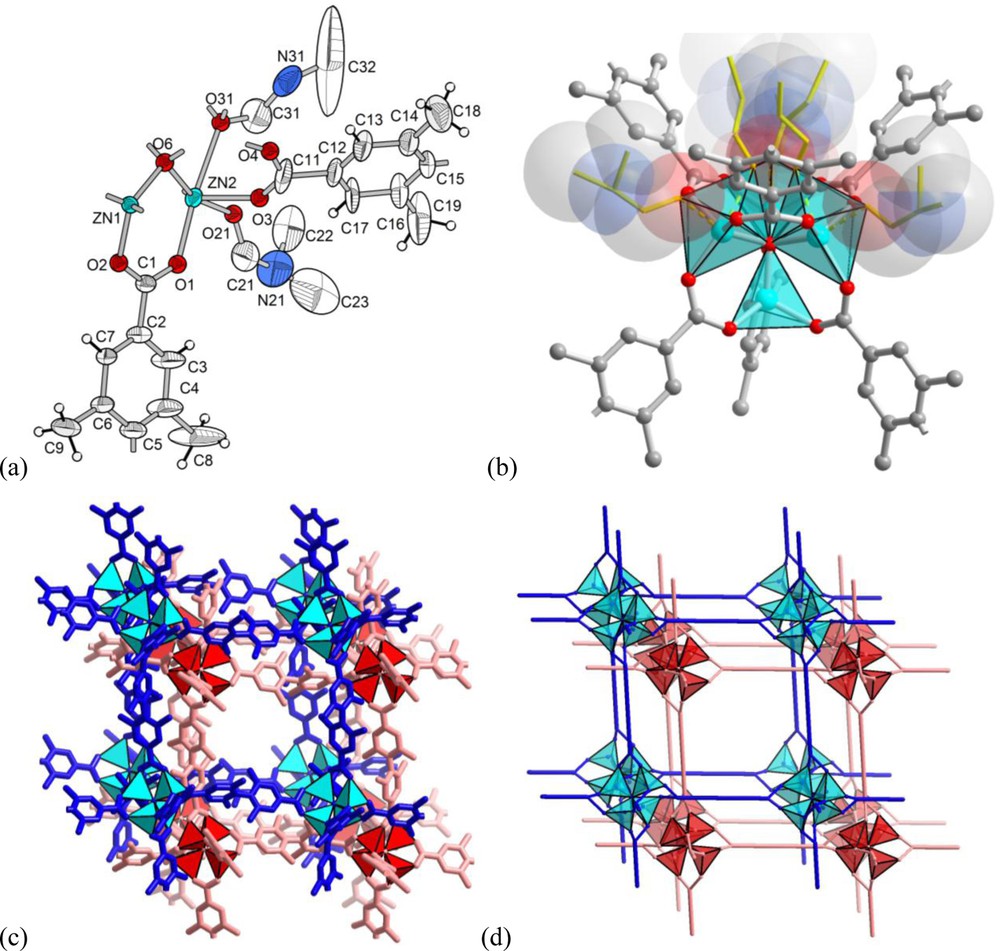
Crystal structure of [Zn4O(Me4BPDC)3] · 9 DMF (2 · 9 DMF): (a) ORTEP style representation (50% thermal ellipsoids). Atoms O31-C31-N31-C32 are part of the DMF molecule, which is delocalized over three positions by the crystallographic three-fold symmetry; (b) {Zn4O(O2CR)6} SBU with the weakly coordinated DMF molecules in transparent space-filling and wire mode. One of these DMF molecules, pointing upward, is delocalized over three equivalent positions by symmetry; (c) and (d) section of the doubly interpenetrated pcu-a (cab) network in stick mode (c) and schematic representation (d).
The quality of the structural experiment was sufficient to locate the DMF molecules that are weakly coordinated to the Zn atoms of the SBU and typically are not refined. Three of the four Zn atoms coordinate one DMF molecule each (in one case a single solvent molecule is delocalized over three positions generated by the three-fold crystallographic symmetry axis), which renders them close to a five-coordinated 4+1 capped tetrahedron configuration (Fig. 2b). The fourth Zn atom (Zn1 in Fig. 2a) does not coordinate any solvent molecules as the surrounding space is filled by atoms of the second interpenetrating framework.
The structure of [Cu2(Me4BPDC)2] · 9 DMF (3 · 9 DMF) is based on the well-known {Cu2(O2CR)4} paddle-wheel unit found in molecular form in the prototypal copper acetate (Fig. 3a,b) [38]. Two DMF molecules weakly coordinate to the axial positions of the paddle-wheel's copper atoms (Cu-O 2.10–2.15 Å, for three crystallographically different Cu atoms) bringing the copper coordination sphere to square-pyramidal (not counting the Cu ⋯ Cu contact of 2.621(2) and 2.625(2) Å) (Fig. 3a,c). The {Cu2(O2CR)4} paddle-wheel units are linked by a dicarboxylate bridge with a ∼90° twist (Fig. 3c), which predetermines formation of the three-dimensional nbo net (Fig. 3d,e).
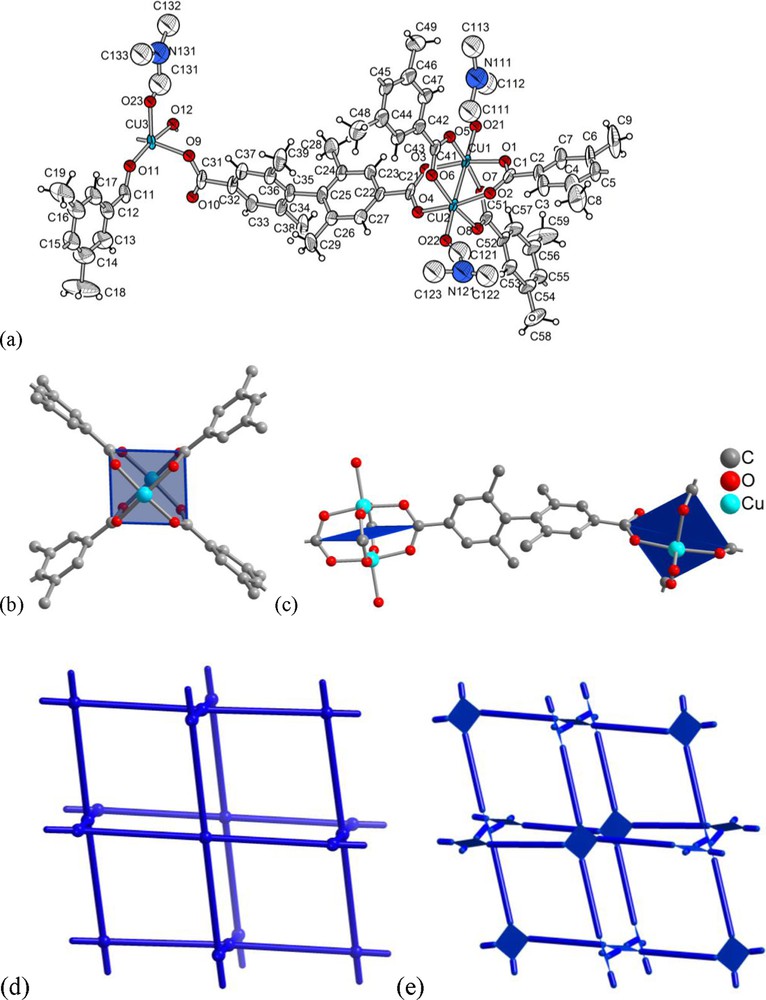
(a) Asymmetric unit of [Cu2(Me4BPDC)2] · 9 DMF, 3 · 9 DMF: ORTEP style representation (50% thermal ellipsoids); (b) {Cu2(O2CR)4} paddle-wheel structure in 3 · 9 DMF withouth the axially coordinated DMF molecules; (c) two {Cu2(O2CR)4} paddle-wheel units linked by 2,2′,6,6′-tetramethyl-4,4′-biphenyldicarboxylate to illustrate the twist of the square-planar SBUs. The single O-atom in the apical position on Cu belongs to a DMF molecule (cf. a); (d) nbo net and (e) augmented nbo-a net where the point vertices of the parent nbo net are replaced by the square paddle-wheel vertex figures [24].
In a planar ligand, such as the unsubstituted 4,4′-biphenyldicarboxylate (BPDC) ligand, a two-dimensional (4,4) net would form. This is the case in 2D-[Zn(BPDC)(DMSO)] where 4,4′-biphenyldicarboxylates as equatorial linkers connect the zinc paddle-wheel units [39]. According to the Cambridge Structure Database (CSD, version 5.33, November 2011), there are 296 coordination compounds and salts based on the 4,4′-biphenyldicarboxylate or its derivatives. Two hundred and sixty-eight of these are polymeric and 225 contain transition metals (see the summary in Table S1, Supplementary material). Many of the early Cu “proto-MOFs” (MOF 102-109) are layered structures based on (4,4)-net topologies [40]. These cases represent the most typical flat configuration of a mono- or a polyphenyl-ligand bi-functionalized with carboxylic groups. However, the energy of conjugation is though quite low and even a minor sterical influence could move the molecular parts out of plane. It was very typical for early works that the co-planarity of the carboxylate groups is lost by substituents on the aromatic core, as small as a chloride atom. This then leads to other topologies, including the nbo one (interplanar angle between carboxylates is ∼90°). Bromine-substitution on benzene-1,4-dicarboxlyate produces a 90° twist of the carboxylate groups and gives an nbo net as in [Cu(O2C-C6H3Br-CO2)] (MOF-101) [41]. Moving-out from planarity by means of steric repulsion of the 2,2′ substituents in the biphenyl moiety is an equivalent possibility, which is, however, synthetically not always easy to realize. The interest towards such ligands is now on the rise, as non-planarity is viewed as a prerequisite for the formation of true 3D frameworks while, at the same time, the limited “axial” degree of freedom allows for “self-adjustment” of most symmetric structures with a chance for dynamic porosity. An additional factor of interest is the use of the substituents as functional groups or anchors for postsynthetic modification.
The voids in a single nbo-a net are large enough to allow for double interpenetration of the nets in 3 · 9 DMF (Fig. 4). While the doubly interpenetrated nbo-a net may already appear like a dense-packed structure there still exist void space, notably open channels with about 10 Å diameter along the ac diagonal (Fig. 4).
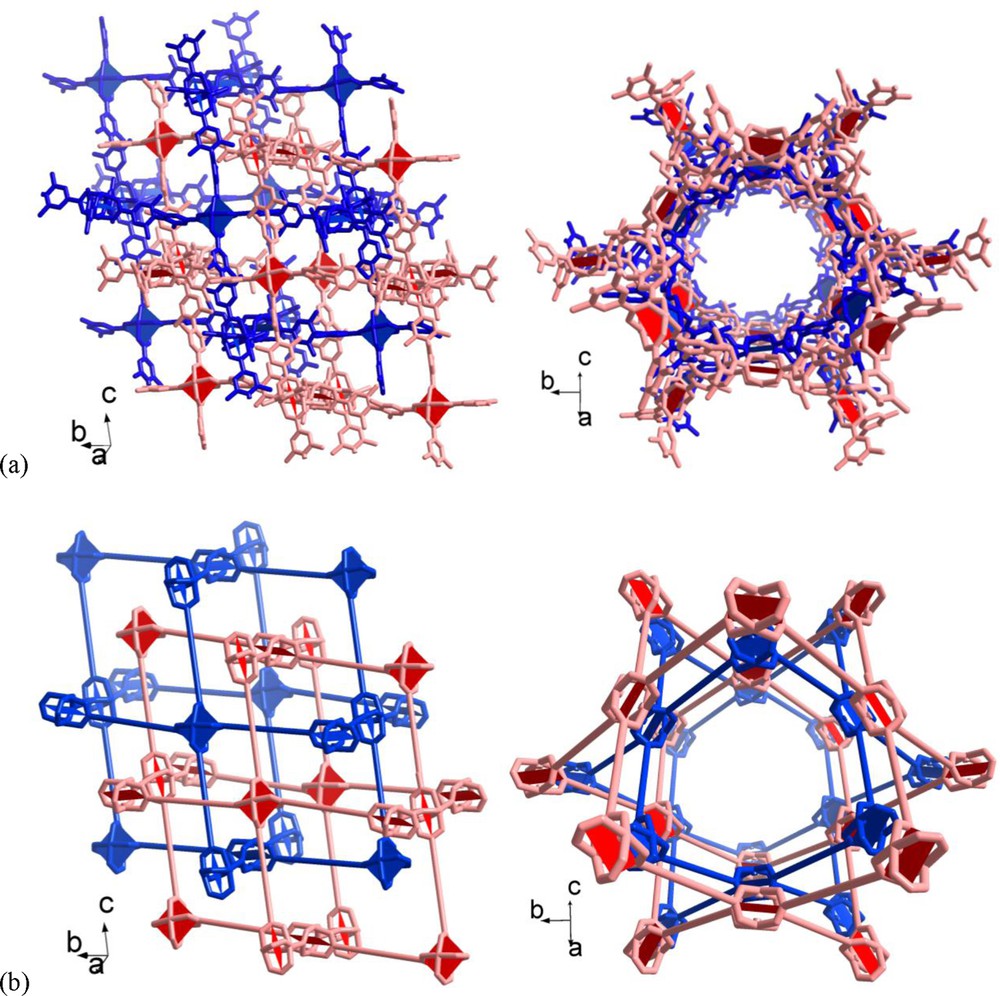
Section of the doubly interpenetrated nbo-a nets in 3 · 9 DMF (a) in wire mode and (b) in schematic representation with the ligands replaced by topological vertex figure connections. There are two viewing directions in (a) and (b) to illustrate the channel formation along the ac diagonal.
The formation of the nbo net by purposeful design through twisting of the biphenyl module is not new and was convincingly demonstrated by Yaghi in a recent paper, mostly devoted to the subject [42], but the number of observed cases is modest (Table S1). The difference for Me4BPDC is only in stricter pre-orientation of both halves of the molecules conditioned by higher sterical repulsion of four methyl groups compared to the typical bis-meta-substituted predecessors.
2.3 Thermogravimetric analysis
The approximate solvent contents in 2 · 9 DMF and 3 · 9 DMF were found by thermogravimetric analysis (TGA) in combination with elemental analysis. In the case of [Zn4O(Me4BPDC)3] · 9 DMF, the TGA curve is very well defined and gives 9.05 solvent molecules per formula unit (Fig. S2). This value can be correlated with the solvent accessible volume given by the “calc solv” or “calc void” procedure of PLATON [43] for 2. At 55.8% or 16071 Å3 solvent accessible volume per unit cell volume of 28785 Å3 in 2, 10.5 DMF molecules/per formula unit (Z) of 2 (with Z = 12 and VDMF = 127 Å3) could be housed with the same density as the bulk liquid phase.
This comparison helped to determine the solvent content in the case of [Cu2(Me4BPDC)2] · 9 DMF, where the somewhat uncertain elemental analysis predicted eight solvent molecules and no plateau was reached in the TGA because at temperatures exceeding 250 °C combustion catalyzed by copper and/or decarboxylation effects took place. With 66.9% or 16189 Å3 solvent accessible volume per unit cell volume of 24207 Å3 in 3, 15.9 DMF molecules/per formula unit of Z (with Z = 8) could be housed in the voids. In 3, a formula unit contains three Cu atoms or 1.5 {Cu2} paddle-wheel SBUs which led us, in analogy with 2, ascribe nine solvent molecules per {Cu2} unit, two of which are weakly coordinated.
2.4 Gas sorption studies
The activation of 2 · 9 DMF by soaking in MeOH with regular refreshing of the solvent proved to be incomplete, as witnessed by IR spectroscopy (the CO stretching band of DMF at ∼1650 cm−1 was used for monitoring; Table S2 and Figs. S4 to S7 in Supplementary material). 3 · 9 DMF could also be activated only partially; THF proved to be a better exchange solvent than MeOH for this purpose, as the last was detrimental to the porosity of the ensuing material. The best result was achieved for 3I (Table 1), where one molecule of DMF is still residing in the pores according to the results of the elemental analysis.
Results of N2 sorption measurements on samples 2 and 3 and data for comparison.
| Degassing, solvent exchange conditions for activation | SBET m2/g | SLangmuir m2/g | p/p0 range | Total pore volume exp., cm3/g at p/p0 | Total pore volume (DFT), cm3/g | Ref. | |
| 2a | 170°C, as synth. | 1735 | 1911 | 0.008–0.08 | 0.723 at 0.97 | 0.603 | This work |
| 2Ib | 70°C, MeOH exch. | 1653 | 1808 | 0.008–0.08 | 0.692 at 0.95 | 0.616 | This work |
| 2IIb | 120°C, MeOH exch. | 1599 | 1749 | 0.008–0.08 | 0.672 at 0.95 | 0.597 | This work |
| 2IIIb | 170°C, MeOH exch. | 1626 | 1802 | 0.01–0.09 | 0.694 at 0.96 | 0.616 | This work |
| MOF-5 (IRMOF-1) | 3362 | 2900 | 1.04 | [44,45] | |||
| IRMOF-9 | 1904 | 2613 | 0.9 | [46] | |||
| 3a | 170°C, as synth. | –c | – | – | – | – | This work |
| 3Ia | 70°C, THF exch. | 1041 | 1199 | 0.02–0.1 | 0.431 at 0.95 | 0.367 | This work |
| 3IIa | 120°C, THF exch. | 871 | 1000 | 0.02–0.1 | 0.364 at 0.95 | 0.302 | This work |
| MOF-601d | 980 | < 0.1 | 0.47 | [42] | |||
| MOF-602d | 910 | < 0.1 | 0.53 | [42] | |||
| MOF-603d | 430 | < 0.1 | 0.20 | [42] |
a Micromeritics ASAP 2020 automatic gas sorption analyzer. DFT calculations (“N2 DFT slit pore” model) with the ASAP 2020 v3.05 software.
b Quantachrome iQ MP sorption analyzer. DFT calculations with ASiQWin 1.2 software employing the “N2 at 77 K on carbon, slit pore, NLDFT equilibrium” model.
c Non-porous material.
d [Cu2L2] with L = 2,2′-R2-4,4′-biphenyldicarboxylate, R = CN (MOF-601), Me (MOF-602), I (MOF-603).
Direct degassing of freshly synthesized samples of zinc MOF 2 at 170 °C in high vacuum surprisingly was more efficient then the activation by solvent exchange yielding a material with a BET surface area of 1735 m2/g (p/p0 = 0.008–0.08, Table 1). Still, the copper MOF 3 not only collapses completely at 170 °C, but the compound activated through solvent exchange could be reliably degassed only until about 70 °C yielding a material with a BET surface area of 1041 m2/g (p/p0 = 0.02–0.1, Table 1).
The advantage of direct degassing over the typically reported activation involving solvent exchange is quite untypical for classical IRMOFs (hence, it is interesting to compare the results of different degassing modes for 2 and 3 as summarized in Table 1 and Fig. 5). When the solvent exchanged sample is subjected to degassing at 170 °C, the obtained material, 2III, still has lower surface area, which could be explained by slight deterioration of the material during the lengthy processing and probably short contacts with air, associated with it.
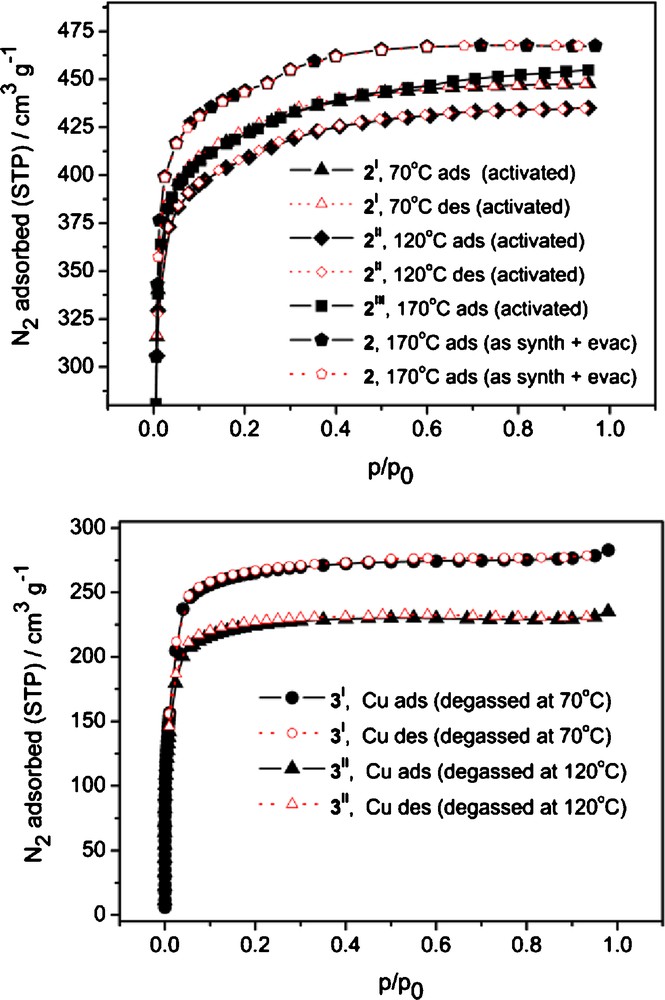
Top: N2 sorption isotherms for 2 (170 °C as synth, marked with pentagons) and samples activated by solvent exchange and degassed at different temperatures (2I, 2II, 2III). Bottom: N2 sorption isotherms for 3I and 3II. See Table 1 for details.
A high quality activation of 2 by direct degassing is also reflected by a pronounced bimodal pore size distribution associated with a small kink in the adsorption isotherm at p/p0 ca. 0.25 and with the pore size peak values according to the DFT calculations at 11.0 Å and 12.3 Å (Fig. S8). The calculated values are significantly overestimated compared to the crystallographic data, which indicates ∼10 × 10 Å2 van-der-Waals cross-section for projection apertures of a single, non-interpenetrated pcu net. This overestimation should be associated with discrepancies of the DFT model for MOF materials, but those differences affects the absolute value only and not so much the relative sizes of the pores. Experimental differentiation of so close pore widths reflects a remarkable quality of the material and also suggests that the linkers behave as relatively rigid units, with low mobility of the phenyl moieties because of steric repulsion of the four methyl groups. The X-ray powder diffractogram confirms the crystallinity of 2 after direct degassing at 170 °C (Fig. S10a). It should be noted that this degassing temperature is probably close to the feasible limit, as DTA data (Fig. S2a) indicates a thermal effect centered at 200 °C and starting at about 175 °C.
Direct degassing of the Cu-MOF 3 · 9 DMF at 170 °C yields a non-porous material (Table 1), while the THF exchanged sample could be degassed with partial retention of porosity, but the resulting sample is semi-amorphous (Fig. S10b). According to DTA (Fig. S2b), a thermal effect starting from 110 °C is discernible for the partially exchanged sample of 3, which is associated with start of the framework collapse. The assumption is confirmed by N2 measurements, which shows a deterioration of the BET surface area from 1041 to 871 m2/g when the sample is again degassed at 120 °C following the 70 °C degassing conditions in the initial experiment (Table 1).
The two materials with best N2 sorption properties, 2 and 3I were subjected to further adsorption studies using H2, CO2 and CH4 as adsorptives. The sorption of H2 reaches 1.26 wt% at 760 Torr for 2 and 1.02 wt% for 3I (Fig. 6). For MOF-5 (IRMOF-1), interpenetrated and non-interpenetrated [Zn4O(BPDC)3] (IRMOF-9 and -10), the gravimetric hydrogen storage capacity is 1.32 wt% [44,47] 1.17 wt% [9,43,48] and (simulated) 1.42 wt% [47], respectively, at 1 bar, 77 K. The closest analogs of the [Cu2(Me4BPDC)2] MOF 3 are MOF-601, MOF-602, MOF-603 of composition [Cu2L2] with L = 2,2′-R2-4,4′-biphenyldicarboxylate, R = CN, Me, I respectively. The topologies of MOF-601 to -603 are also doubly interpenetrated nbo-a nets. The reported H2 sorption values are 0.83 wt%, 0.90 wt% and 0.46 wt%, respectively [42]. The introduction of two additional groups in 3 thus increases the sorption capacity of H2 by approximately 14%, however, one should be careful in analyzing those numbers. As far as hydrogen does not bind specifically at 77 K to such organic functional groups as CN, Me or I, there is a strong indication that the MOF materials obtained in ref. [42] were also of low crystallinity, a fact which was not documented in the reference. Thus the differences should be attributed more to the stability of the frameworks during the activation/degassing processes. This assumption is supported by the reported loss of crystallinity during attempts to remove the guest molecules after solvent exchange in another analog ([Cu2L2], L = 2,2′-dihydroxy-4,4′-dimethyl-4,4′-biphenyldicarboxylate) [49]. Thus, despite the loss of crystallinity in 3I upon activation from 3 · 9 DMF, the sorption of hydrogen is sizeable and follows the typically higher sorption capabilities of copper MOFs compared to zinc MOFs at low pressures [44].
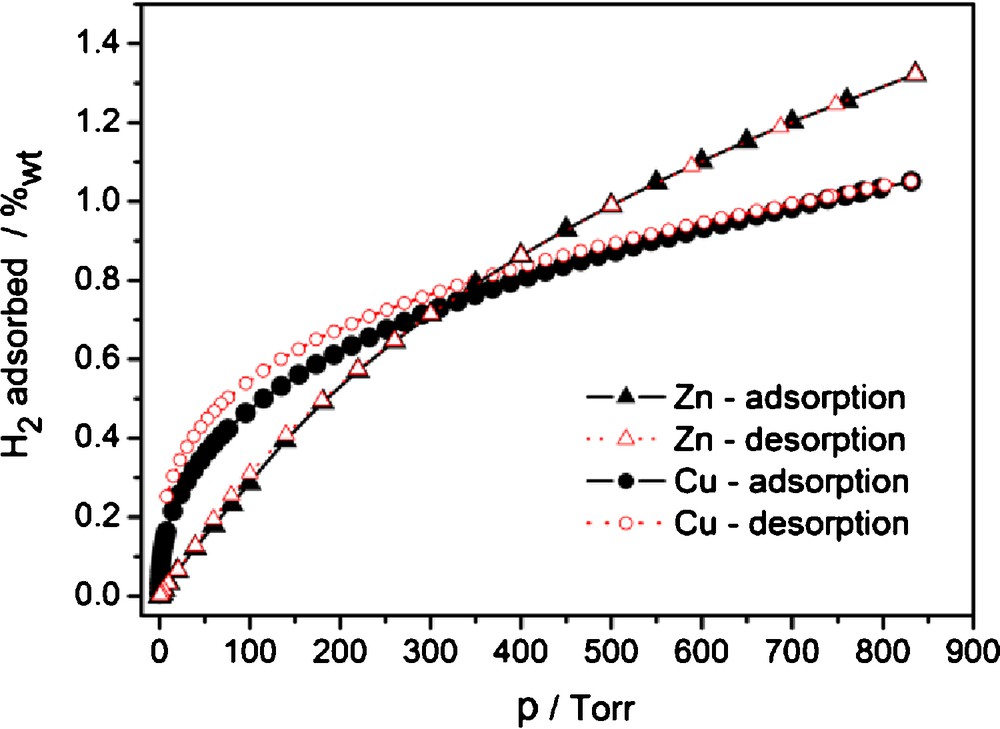
H2 sorption isotherms for 2 (Zn) and 3I (Cu) at 77 K.
Naked, coordinatively unsaturated copper ion sites physisorb hydrogen stronger than the analogous closed shell zinc complexes, which makes [Cu3(btc)2], HKUST-1, one of the best hydrogen adsorbents at low pressures [9,44,50] with an H2 uptake of 3.8 wt% vs 1.3 wt% for MOF-5 (IRMOF-1) at 77 K [51]. The tendency quickly reverses as the pressure grows and the amounts adsorbed are going to be more proportional to the surface area and at even higher pressures to the available free space [51]. Thus, the higher metal-site adsorption efficiency of 3I but higher surface area of 2 compared to 3I explains the more linear character of the sorption isotherm in the case of 2, with an initial lower adsorption which eventually surpasses the sorption capacity of 3 at pressures more than 300 Torr.
The sorption characteristics for CO2 are similar to the metal-site adsorption situation with H2. Pronounced metal-site specific binding of CO2 causes the initial adsorption to be much higher for 3I than for 2 (Fig. 7). The dominance (50 cm3/g at 0 °C, 31.5 cm3/g at 20 °C for 3I versus 30.8 cm3/g at 0 °C, 16.4 cm3/g at 20 °C for 2 at 760 Torr, Fig. 7) is evidently preserved also to higher pressures until probably a few bars. The reported CO2 sorption values for Zn-MOF-5 (IRMOF-1) are 51 cm3/g at 23 °C [52]; for Cu-MOF-601 to -603 they are 38, 24 and 24 cm3/g, respectively, at 0 °C, 1 bar [42]. The adsorption on the zinc complex 2 is characterized by almost exact linearity, which suggests minimal specific adsorbate-to-metal interaction. Low adsorption values of CO2 at these temperatures render the estimation of heats of adsorption (HoA, Fig. S10) very crude, but they could be used for illustrative comparison: HoA for the first ∼10% of adsorbed CO2 is 31 ± 2 kJ/mol for 3I versus 17 ± 3 kJ/mol for 2 (Fig. S10). The HoA is growing slightly, following a drop for 3I, with the quantities adsorbed, which could be partially explained by larger adsorbate-to-adsorbate interaction compared to the adsorbate-to-adsorbent interaction. MOF-5 (IRMOF-1) has an isosteric heat of adsorption for CO2 of about 34 kJ/mol [52].
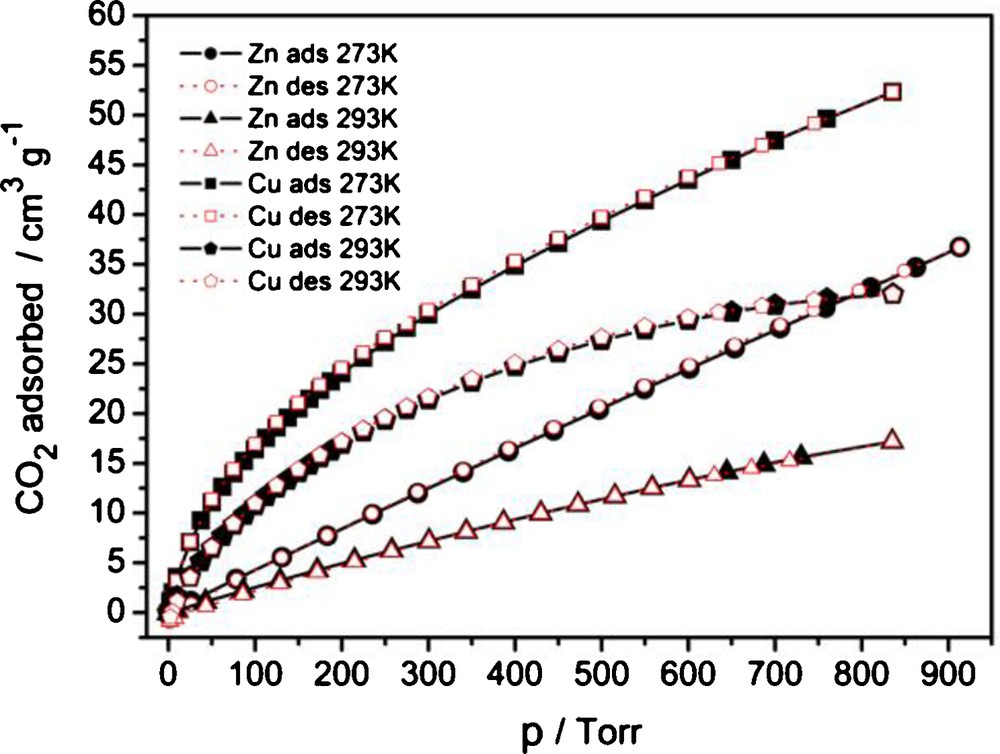
CO2 sorption isotherms for 2 (Zn) and 3I (Cu) at two different temperatures.
A simulated comparison of CO2/H2 adsorption of interpenetrated IRMOF-9 with non-interpenetrated IRMOF-10 showed that the interpenetrated one has a much higher CO2/H2 selectivity. This was attributed mainly to electrostatic interactions with the strength correlated to the pore size. The electrostatic interactions between CO2 and MOFs are dominant at low pressures and the electrostatic interactions of CO2–CO2 become evident and eventually dominant at high pressures [13,53].
Contrary to CO2, CH4 is not strongly adsorbed by metal sites, so the difference between 2 and 3I is quite small (12.9 cm3/g at 0 °C, 6.4 cm3/g at 20 °C for 2 and 11.4 cm3/g at 0 °C, 5.2 cm3/g at 20 °C for 3I at 760 Torr, Fig. 8). The adsorption is mainly caused by non-specific van der Waals interactions and is proportional to surface area.
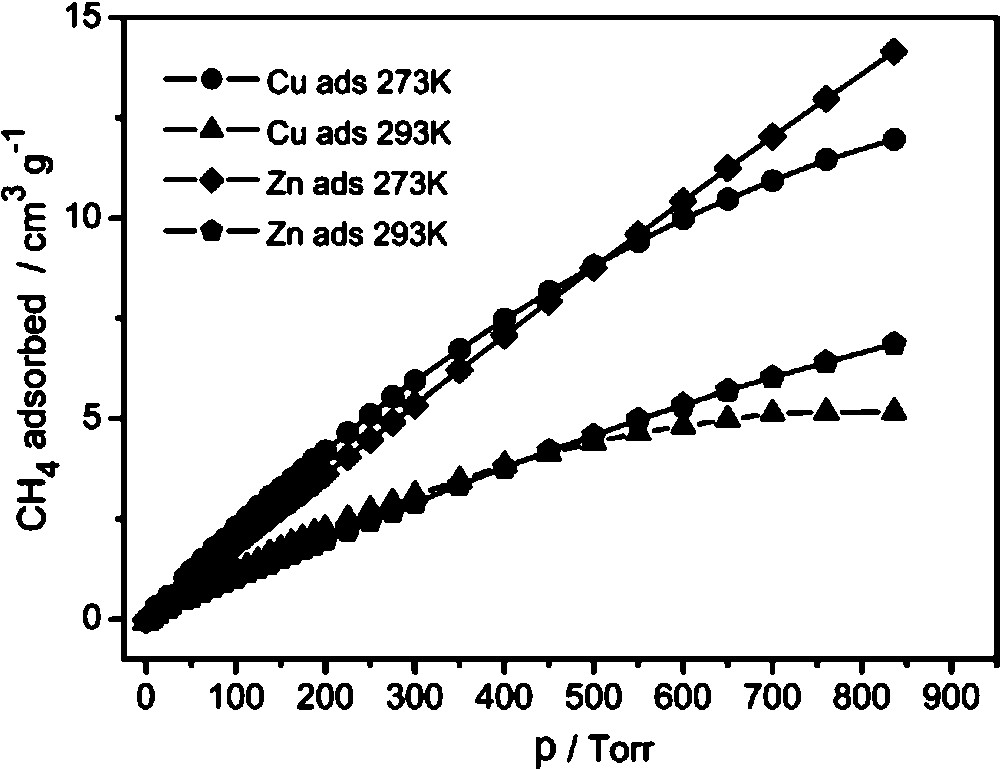
CH4 adsorption isotherms for 2 and 3I at two different temperatures.
Besides the gas sorption experiments, we were interested in the possibility of increased water stability of 2. Unfortunately, the surface area of a degassed sample of 2 deteriorates significantly within a few hours in air as well as 16 h soaking in water with subsequent cautious degassing to yield a completely non-adsorbing material. It could be concluded that for efficient hydrophobic protection of the SBU, the methyl group should be disposed in ortho-position to the carboxylate group, as it was shown recently [54]. No special experiments on water stability were performed on 3, as its quality deteriorates slowly even in anhydrous methanol.
3 Conclusions
Optimization of the selective oxidation of sterically non-hindered methyl groups demonstrated on the example of 2,2′,6,6′-tetramethyl-4,4′-biphenyldicarboxylic acid (H2Me4BPDC) that future work in this direction could make carboxylate ligands, including chiral ones, accessible by using polymetylated aromatics as precursors. The IRMOF [Zn4O(Me4BPDC)3] · 9 DMF and MOF [Cu2(Me4BPDC)2] · 9 DMF are constructed with a ligand bearing the maximum number of methyl groups meta to the carboxylate group on the biphenyl frame. DMF could be used instead of DEF which is mostly obligatory for high porous MOF-5 and IRMOF-9 or IRMOF-10 samples. [Zn4O(Me4BPDC)3] with pcu-a (cab) topology demonstrates good thermal stability up to 170 °C coupled with the possibility of direct degassing of the freshly synthesized sample without the necessity in tedious activation based on solvent exchange. For [Zn4O(Me4BPDC)3], the N2 BET surface area of 1735 m2/g for the obtained doubly interpenetrated modification is slightly lower than that of the BPDC analogue (1904 m2/g, IRMOF-9). The adsorptive properties towards H2, and CO2 are similar with [Zn4O(BDC)3], MOF-5 (IRMOF-1) and better than for [Zn4O(BPDC)3], IRMOF-9. Coupled with seemingly lower sensitivity towards atmospheric moisture, the more hydrophobic [Zn4O(Me4BPDC)3] may have a slight edge as a material for applications.
The nbo-a topology in the [Cu2(Me4BPDC)2] complex owes its formation to the 90° twist of the ligand's phenyl moieties due to steric repulsion of the methyl substituents. [Cu2(Me4BPDC)2] is mechanically much less stable than [Zn4O(Me4BPDC)3], obviously due to a lower connectivity of the square-planar {Cu2(O2CR)4} paddle-wheel unit compared to the octahedrally coordinated {Zn4O(O2CR)6} unit. The also doubly interpenetrated framework [Cu2(Me4BPDC)2] collapses easily and only a semi-amorphous sample (SBET = 1041 m2/g) was obtained. [Cu2(Me4BPDC)2] is stable at least until 70 °C provided some initial solvate molecules are still in the framework. The adsorptive properties towards N2, H2, and CO2 are better than for MOF-601, MOF-602, MOF-603 ([Cu2L2] with L = 2,2′-R2-4,4′-biphenyldicarboxylate, R = CN, Me, I, respectively).
Thus, methyl substituted analogs of established carboxylate ligands could be used for the synthesis of MOFs with properties similar or even better than that of the non-methylated parent ligand.
4 Experimental
4.1 Physical methods
The melting point measurement was performed in an open capillary using a Büchi-B450 apparatus. FT-IR spectra were recorded using a Bruker Tensor 37 system equipped with an ATR unit (Platinum ATR-QL, Diamond) in the 4000–550 cm−1 region with 2 cm−1 resolution. 1H and 13C spectra were recorded on Bruker Avance DRX-200 Bruker Avance DRX-500 instruments respectively. Thermogravimetric analysis (TGA) was performed on a Netzsch STA 449 C Jupiter instrument coupled with a Pfeiffer Thermostar GSD 300 mass spectrometer at 10 °C/min heating rate using corundum sample holders and air as carrier gas. Powder X-ray diffraction measurements on samples sealed in 0.5 mm “Mark” capillary tubes were performed on a Panalytical X’Pert Pro diffractometer in transmission mode using a filtered Cu-Kα radiation source at 40 kV/40 mA generator setting. The simulated PXRD patterns were generated using Mercury 2.3 [55], with 0.02° step and FWHM(2θ) = 0.1° scaled by an adapting exponential function aiming smoother intensity distribution for better visibility of the pattern's details at higher angles.
4.2 Syntheses
All the inorganic salts were of reagent grade and used without future purification. Dimethylformamide, DMF (VWR, ACS), MeOH (Acros, spectroscopic grade) and THF (VWR, ACS) were used as delivered.
4.2.1 2,2′,6,6′-tetramethyl-4,4′-biphenyldicarboxylic acid, H2Me4BPDC, 1
The procedure employed (Fig. 1) is a slight modification of a process described in reference [25]. The reaction was carried out in fume hood, accounting the danger associated with the high-flammability of the acetic acid vapors mixed with oxygen.
In a three necked 250 mL flask equipped with an efficient magnetic stirrer, a gas bubbler with a fritted glass exhaust, an internal thermometer, a condenser and a 10 mL dropping funnel, 60 mL of acetic acid and 4.5 g (18.1 mmol) of Co(OAc)2 · 4H2O were placed. To the partially dissolved slurry, 6 mL of peracetic acid (81.9 mmol) in a three-necked flask was added dropwise under intensive stirring during ∼10 min. An exothermic process took place which resulted in formation of a warm (45 °C) homogenous deep brownish-green solution of Co(III) compounds to which 3.00 g (12.6 mmol) of bimesityl [56] was added at once. The temperature of the mixture was raised to 100 °C using a heating bath and under continued efficient stirring a rotameter-controlled oxygen flow was passed through the reaction mixture for 5 h at 7.5 L/h then an additional 5 h at 3 L/h.
The obtained solution with some residue was poured in 400 mL of water. The formed precipitate was filtered-off after 3 h of staying and was washed multiple times with conc. HCl until the washings were practically colorless. The slightly greenish residue, after a thorough washing with water and drying is a mixture of H2Me4BPDC and 4-(2,4,6-trimethylphenyl)-3,5-dimethylphenylcarboxylic acid with a molar ratio of about 2.1:1.0 (Fig. S1 in Suppl. mater.; combined mass 2.21 g). The mixture was partitioned between 100 mL Et2O and 50 mL of 3% NaHCO3. The aqueous layer was acidified (pH = 2) by aq. HCl, the formed precipitate was filtered-off and dried at 100 °C, yielding 0.87 g of quite pure product in a form of a white powder. Repeated extraction of the ethereal solution by 50 ml of 3% NaHCO3 and analogous processing yields 0.45 g of slightly impure crop. Combined yield 1.32 g (35.2% versus ∼52% in the pre-work-up mixture). M.p. (in open capillary, 20 °C/min, starting from 375 °C) 386–395 °C with partial decomposition. 1H NMR (DMSO-d6, 200 MHz): δ = 2.12 (s, 12H, CH3); 7.80 (s, ArH, 4H). 13C NMR (DMSO-d6, 500 MHz): δ = 19.7 (CH3), 129.0 (CPhH), 130.1 (CPhCPh’), 135.5 (CPhCH3), 143.8 (CPhCOOH), 167.8 (COOH). FT-IR (neat), νmax/cm−1: 2981 (w), 2917 (w), 2861 (w), 2703 (w), 2604 (w), 2533 (w), 1684 (s), 1602 (w), 1571 (w), 1429 (m), 1304 (m), 1237 (m), 1130 (s), 1037 (vw), 1001 (w), 937 (w), 899 (w), 775 (m), 722 (m), 604 (vw), 566 (vw).
4.2.2 [Zn4O(Me4BPDC)3] · 9 DMF, 2 · 9 DMF and [Zn4O(Me4BPDC)3], 2
Amounts of 594 mg of [Zn(NO3)2] · 6H2O (2 mmol) and 152 mg of H2Me4BPDC (0.5 mmol) were dissolved in 10 mL of DMF inside a 25 mL vial sealed with a Teflon lined screw cap. The solution was heated for 2 days at 80 °C which resulted in deposition of large (up to ∼1 mm) cubic crystals after cooling. The supernatant solution was removed by suction under inert gas, rinsed a few times with MeOH and dried in vacuum at 10 Torr. Yield: 195 mg (64 %) of slightly yellow crystals. FT-IR (neat), νmax/cm−1: ∼3300 (w, vbr), 2919 (w), 2861 (w), 1666 (m), 1601 (s), 1559 (s), 1423 (s), 1395 (s), 1371 (s), 1266 (m), 1095 (w), 1003 (w), 907 (w), 789 (s), 760 (s), 663 (vw), 628 (vw). Elemental analysis, calcd for C81H111N9O22Zn4 C 53.33, H 6.13, N 6.91; found C 51.25, H 6.33, N 6.38%.
2 · 9 DMF was activated by soaking the crystals three times in approximately 10 mL of MeOH at ∼60 °C, changing the solvent after each two days and allowing only a very short contact of the wet crystals with air. The crystals were dried at 70 °C and 10−7 Torr vacuum (using a turbo-molecular pump) for 12 h to yield crystal-solvent depleted 2I. According to the N2 sorption properties it was found that the activated sample is almost identical to a directly dried sample of 2 · 9 DMF at 170 °C and 10–7 Torr. FT-IR (neat), νmax/cm−1: 2921 (vw), 2861 (vw), 1602 (s), 1560 (s), 1427 (s), 1399 (s), 1376 (s), 1268 (m), 1004 (w), 907 (w), 788 (s), 759 (s), 629 (vw). Elemental analysis, calcd for C54H48O13Zn4 C 55.60, H 4.15, N 0.0; found 54.91, H 4.46, N 0.0%.
4.2.3 [Cu2(Me4BPDC)2] · 9 DMF, 3 · 9 DMF and [Cu2(Me4BPDC)2], 3
Amounts of 384 mg of Cu(NO3)2 · 2.5H2O (1.5 mmol) and 90 mg (0.3 mmol) of H2Me4BPDC were dissolved in 10 mL of DMF inside a 25 mL vial sealed with a Teflon lined screw cap. The solution was heated for 2 days at 70 °C that resulted in the deposition of large deep-blue cubic crystals. The supernatant solution was removed by suction under inert gas, rinsed a few times with MeOH and dried in vacuum at 10 Torr. Yield: 135 mg (65%) of deep-blue crystals. Heating for more than two days or using higher temperature causes appearance and subsequent accumulation of copper(I) oxide, Cu2O. FT-IR (neat), νmax/cm−1: 3384 (w, vbr), 2920 (vw), 2857 (vw), 1649 (m), 1599 (m), 1559(m), 1394 (vs), 1422(s), 1371 (vs), 1308 (s), 1266 (m), 1104 (w), 1045 (m), 1002 (m), 904 (w), 788(s), 765 (s), 698 (m) 667 (m). Elemental analysis, calcd for C63H95Cu2N9O17 C 54.93, H 6.95N 8.59, found C 55.62, H 6.68, N 8.31(%).
3 · 9 DMF was activated by soaking the crystals three times in approximately 10 mL of THF at ∼60 °C, changing the solvent after each two days and allowing only a short contact of wet crystals with air. The crystals were dried at 70 °C and 10−7 Torr vacuum (using a turbo molecular pump) for 12 h to yield crystal-solvent depleted 3. Direct drying of 3 · 9 DMF is impossible, as 3 demonstrated instability at temperatures exceeding about 80 °C. Only for a short time of about 10 min, a heating up to 120 °C is tolerated according to the sorption measurements. FT-IR (neat), νmax/cm−1: 2918 (vvw), 2860 (vvw), 1602 (m), 1531(m), 1421 (s), 1391 (s), 1370 (s), 1266 (m), 1002 (m), 902 (w), 786(s), 762 (s). Elemental analysis, calcd (%) for C36H32Cu2O8 C 60.08, H 4.48, N 0.0 found: C 55.66, H 4.99, N 0.5.
4.3 Gas sorption measurements
Sorption isotherms were measured using a Micromeritics ASAP 2020 automatic gas sorption analyzer equipped with oil-free vacuum pumps (ultimate vacuum < 10−8 mbar) and valves, which guaranteed contamination free measurements.
For the sorption measurements 50 to 100 mg of freshly synthesized samples (see Section 4.2) were subjected to a solvent-exchange procedure and were processed immediately afterwards. After filtration and washing with dry MeOH (for 2 · 9 DMF) or THF (for 3 · 9 DMF), the samples were soaked in 5 ml of the same solvent for 2 days at about 60 °C in 6 ml screw cap vials with septum. The solvents were replaced by fresh one with a syringe and the soaking was repeated two more times. This way the original high-boiling DMF guest-solvent was exchanged to low-boiling MeOH for 2 and THF for 3, respectively. The solvent exchanged samples (in the form of their last soaking slurry) were transferred to nitrogen filled and pre-weighed sample tubes capped with a septum. The tube was immediately purged with nitrogen after the transfer and the solvent removed with a syringe and through drying in mild vacuum (∼ 10 Torr), which was followed by a control weighing. Then the sample was connected to the preparation port of the sorption analyzer and degassed under vacuum until the outgassing rate, i.e., the rate of pressure rise in the temporarily closed manifold with the connected sample tube, was less than 2 μTorr/min at the specified temperature (70–170 °C). After weighing, the sample tube was then transferred to the analysis port of the sorption analyzer. All used gases (H2, He, N2, CO2, CH4) were of ultra high purity (UHP, grade 5.0, 99.999%) and the STP volumes are given according to the NIST standards (293.15 K, 101.325 kPa). Helium gas was used for the determination of the cold and warm free space of the sample tubes. H2 and N2 sorption isotherms were measured at 77 K (liquid nitrogen bath), whereas CO2 and CH4 sorption isotherms were measured at 293 ± 1 K (passive thermostating) and 273.15 K (ice/deionized water bath). The heat of adsorption values and the DFT calculations (“N2 DFT slit pore” model) were done using the ASAP 2020 v3.05 software.
Some of the secondary measurements, specified in the description, were performed using a Quantachrome iQ MP automatic sorption analyzer. The DFT calculations were made using the native ASiQWin 1.2 software employing the “N2 at 77 K on carbon, slit pore, NLDFT equilibrium” model.
4.4 X-ray structure determination
X-ray quality crystals were selected in Kel-F-oil (Riedel deHaen) at ambient temperatures. All samples were cooled to 173(2) K during measurement. The data were collected on a Bruker-Nonius Apex X8 CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073). The structures were solved by direct methods (SHELXS-97) and refined by full-matrix least-squares on F2 using the SHELXL-97 program suite [57,58]. Semi-empirical absorption corrections were applied (SADABS) [59]. All non-hydrogen positions (except for disordered DMF atoms) were refined with anisotropic temperature factors. Hydrogen atoms of the main residue (except the coordinated DMF molecules) were included in the refinement at calculated positions using a riding model with the thermal displacement parameter being 1.2 and 1.5 of the bonded aromatic or aliphatic carbon respectively. The weakly coordinated DMF molecules were refined; partial occupancy factors were assigned and isotropic restraints were applied where necessary (C32 atom of 2 · 9 DMF with very large thermal displacement factor models two neighbouring atoms of disordered components). The relatively poor data quality obtained from crystals with high mosaicity at a relatively high temperature prevented the refinement of the heavily disordered non-coordinated DMF molecules in the voids of the structure and the data was corrected by removing the corresponding reflexes with the PLATON SQUEEZE routine [42]. Crystal data and details on the structure refinement are given in Table 2. Graphics were drawn with DIAMOND [60].
Crystal data and structure refinement for [Zn4O(Me4BPDC)3] · 9 DMF, 2 · 9 DMF and [Cu2(Me4BPDC)2] · 9 DMF, 3 · 9 DMF (SQUEEZE procedure applied).
| 2 · 9 DMF | 3 · 9 DMF | |
| Empirical formula | C63H69N3O16Zn4 | C63H69Cu3N3O15 |
| Mr/g/mol−1 | 1385.69 | 1298.83 |
| Crystal system | Trigonal | Monoclinic |
| Space group | R-3c | C2/c |
| a/Å | 22.7345(13) | 22.5641(9) |
| b/Å | 22.7345(13) | 37.1408(15) |
| c/Å | 64.308(7) | 30.0021(11) |
| β/° | 90 | 105.6860(10) |
| γ/° | 120 | 90 |
| V/Å3 | 28785(4) | 24207(2) |
| Z | 12 | 8 |
| Calc. density/g cm–3 | 0.959 | 0.713 |
| μ/mm–1 | 1.033 | 0.557 |
| F(000) | 8592 | 5400 |
| Crystal size/mm3 | 0.28 × 0.22 × 0.20 | 0.44 × 0.43 × 0.31 |
| θ range/° | 1.21 to 28.14 | 1.09 to 25.07 |
| Index ranges/h; k; l | ±30; ±30; ±84 | –25,26; –44,41; –32,35 |
| Reflections collected | 157947 | 81098 |
| Independent reflections | 7806 (Rint = 0.0735) | 21379 (Rint = 0.0584) |
| Completeness/% to θ/° | 99.3/28.14 | 99.6/25.07 |
| Data/restraints/parameters | 7806/13/299 | 21379/4/685 |
| Max./min. Δρ/eÅ–3a | 1.987/–1.675 | 2.004/–1.715 |
| R1/wR2 [I > 2σ(I)]b | 0.0874/0.2539 | 0.0808/0.2255 |
| R1/wR2 (all reflect.)b | 0.1094/0.2691 | 0.1120/0.2465 |
| Goodness-of-fit on F2c | 1.102 | 1.030 |
a Largest difference peak and hole.
b .
c .
Acknowledgement
Funding by the Federal German Ministry of Economics (BMWi) under grant 0327851A is gratefully acknowledged.