

{kind=link}
{kind=link}
{kind=link}
1 Introduction
Benzisoxazoles are aromatic ring heterocycles containing one oxygen atom adjacent to one nitrogen atom in a five-membered ring fused with a benzo moiety. There are two types of benzisoxazoles, namely 1,2-benzisoxazoles (indoxazene) and 2,1-benzisoxazoles (2,1-benzoisoxazoles or anthranil). The first one is more widely used due to its high therapeutic interest [1]. The second is more useful in the preparation of biologically active compounds. Indeed, several important heterocycles such as quinolones, acridines, or quinazolines can be prepared directly from 2,1-benzisoxazoles or via aminoarylketones easily obtained by treatment with iron in acetic acid [2].
Since the first described synthesis of 2,1-benzisoxazoles by reductive cyclization of ortho-nitrobenzaldehydes by Friedlander, in 1882, which wrongly assigned the resulting product as an anthranil lactam [3], several other methods have been described (see Supplementary information) and recently reviewed [2,4].
To the best of our knowledge, 2,1-benzisoxazoles containing an exo double bond at 3-position have been scarcely described [5]. In this context, in the reinvestigation of the synthesis of indoles from Baylis–Hillman adducts [5a], three new 2,1-benzisoxazoles possessing an exo double bond were obtained as secondary products. In fact, the reduction of ortho-nitro adducts of nitrobenzaldeyde using tin(II) chloride in refluxing 1,4-dioxane originated these 2,1-benzisoxazoles in 26% yield [5b]. Later, the intramolecular FeBr2-catalyzed NO bond formation from an aryl azide possessing an ortho-isopropylcarbonyl group affording the corresponding 1H-benzisoxazole in 72% yield after SiO2 purification was also described [5c].
2 Results and discussion
2.1 Synthesis and structural characterization
As a part of our ongoing research on the development of new barbiturate[4,5-b]quinolones 7 as potential xanthine oxidase (XO) inhibitors and antitumor agents, we herein report a new route to prepare 3-substituted-2,1-benzisoxazoles 8 from 5-(2-nitrobenzylidene)barbiturates 3 (Scheme 1).
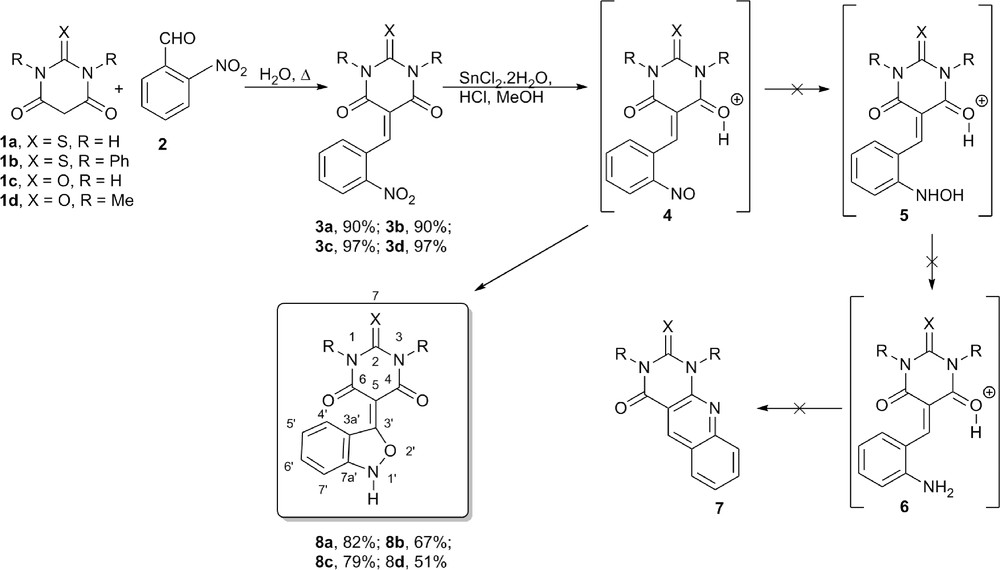
Attempt to prepare (thio)barbiturate[4,5-b]quinolines 7 with the formation of 2,1-benzisoxazoles 8.
The starting materials thiobarbituric acid (1a), barbituric acid (1c), N,N-dimethylbarbituric acid (1d), and ortho-nitrobenzaldehyde were purchased from sigma aldrich, whereas N,N-diphenylthiobarbituric acid (1b) was prepared as described [6]. (2-Nitrobenzylidene)barbiturates 3a–d were prepared in a simple and mild way based on the method described by Deb and Bhuyan [7], using water as the solvent, in 90–97% yield. Surprisingly after refluxing for 2 h the methanolic acid suspension containing the respective 5-(2-nitrobenzylidene)barbiturate 3 and tin chloride dihydrate, the 5-benzo[c]isoxazolbarbiturates 8 were easily obtained by a direct filtration of the solid formed, in moderate to good yields (51–82%), instead of the expected barbiturate[4,5-b]quinolones 7 (Scheme 1).
Apart from precursors, all 5-benzo[c]isoxazolbarbiturates 8a–d prepared have not hitherto been described. Consequently, their full spectroscopic characterization was performed, including 1H and 13C NMR shift assignments, which were established with the aid of distortionless enhancement by polarization transfer 135, heteronuclear simple quantum coherence, and heteronuclear multiple bond correlation experiments. In the 1H NMR spectrum of 5-benzo[c]isoxazolbarbiturates 8a,c a characteristic signal at 11.31 or 10.69 ppm can be found, corresponding to the proton bond to the nitrogen of the pyrimidine scaffold suggesting the symmetry of this ring. In addition, four signals observed for all 8a–d in the range of 7.90–6.71 ppm indicated an ortho-substituted benzene ring. In the 8a–d 13C NMR spectrum, the signals at 178.6–174.5, 162.7–150.8, and 85.4–82.5 ppm were attributed to carbons directly attached to a sulfur atom, to an oxygen atom, and to a methylenic carbon, respectively. Within these, the signal at 162.7–159.6 ppm was associated with two carbonyls, confirming the symmetry of this pyrimidine scaffold. The 13C NMR spectrum of the benzisoxazoles 8a–d presented two signals at 167.4–163.6 and 156.4–155.9 ppm related to 3′ and 7a′ isoxazole ring carbon atoms. The remaining aromatic carbon atoms can be found between 141.8 and 112.9 ppm. In addition, attenuated total reflectance infrared (ATR-IR) spectroscopy and high-resolution mass spectrometry (HRMS) were also performed. In conclusion, all the novel compounds showed spectral and analytical data fully consistent with the assigned structures. In addition, single crystal X-ray data clearly confirmed the structure of 8d (Fig. 1 and Supplementary information).
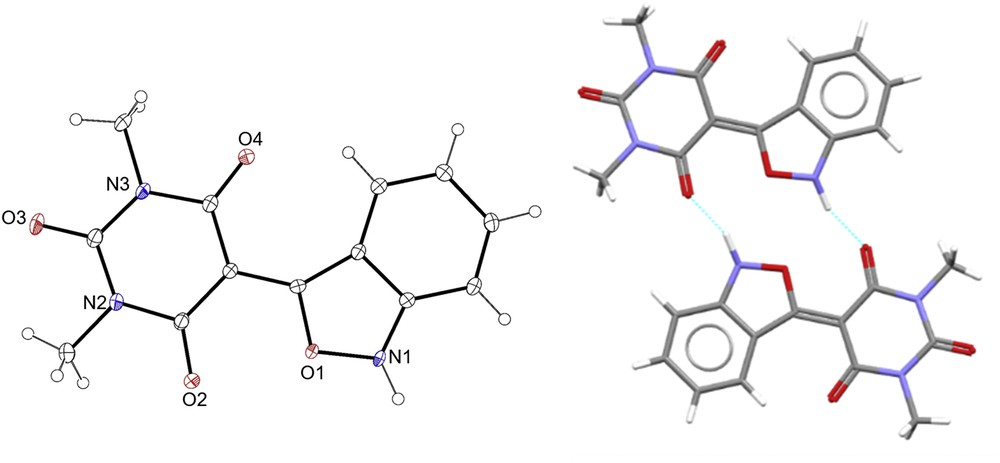
Molecular structure of 5-benzo[c]isoxazolbarbiturate 8d. (Left) ORTEP-3 plot (30% thermal ellipsoids). (Right) Packing showing N1H1⋯O2 hydrogen bonds (dashed light blue lines).
On the basis of the results mentioned previously and described in Scheme 1, a possible reaction mechanism for the formation of 8 is proposed considering an intramolecular attack of a nucleophilic intermediate in the reduction of the nitro group, specifically a nitroso [8] compound (Scheme 2). At first, the nitrosoarene 4 formed by the partial reduction of the nitro group behaves as a nucleophile capable to attack the C-5 of the (thio)barbiturate in a typical Michael addition reaction. Moreover, this cyclization path is activated by carbonyl acid catalysis. Several mechanisms proposed for the 2,1-benzisoxazole cyclization also involved radical anion nitroso or nitroso as nucleophilic (methods D, E G, Scheme S1) [2] or electrophilic (method F, Scheme S1) [2b] species. Besides, considering the local reactivity descriptors applied on the nitroso group of the nitrosobenzene, it has been shown that both N and O nitroso atomic centers can be suitable nucleophilic reactive sites in aqueous solutions [9]. Because tin chloride is a rather effective reducing agent [10], the unlikely partial reduction of the nitroarene to nitrosoarene can be explained by the intramolecular trapping of this unstable species by the reaction with the β-keto unsaturated system. Finally, elimination of a proton and the conversion of the enol form to the corresponding ketone leads to the formation of 2,1-benzisoxazole 8. Therefore, a novel system and associated reactivity different from all of those previously described for the preparation of 2,1-benzisoxazoles are reported in the present work. In addition, the simplicity and mildness of the reaction, the easy access to the starting materials, and the straightforward workup and good yields of the products obtained clearly are interesting advantages of this new method.
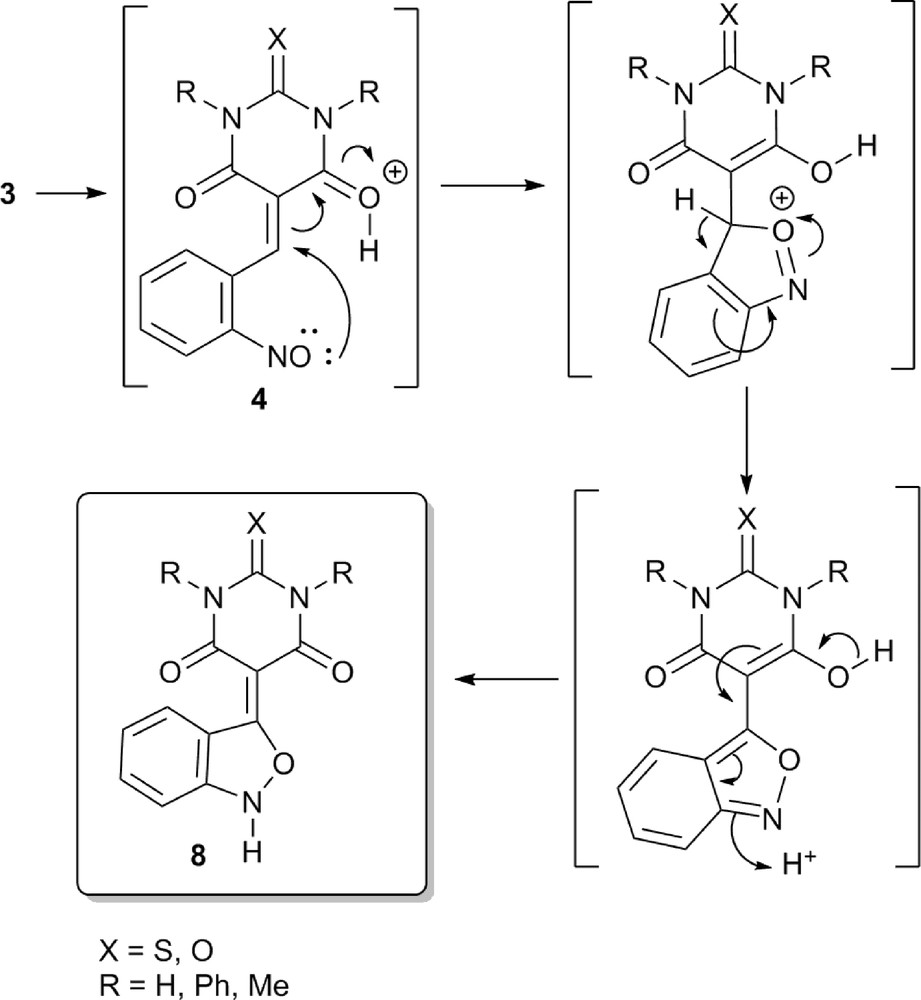
Proposed mechanism for the formation of 2,1-benzisoxazoles.
2.2 Biological evaluation
Following our initial aim of study and considering the growing interest on (thio)barbiturates by medicinal chemists because of their increasingly wide range of biological activities [11], in addition to their most important and classical use as sedative hypnotics [12], these new benzisoxazole–barbiturate systems were also screened as XO inhibitors, radical 2,2-diphenyl-1-picrylhydrazyl (DPPH) scavengers, and cytotoxic compounds (Table 1).
XO inhibitory, antioxidant, and antiproliferative effects on MCF-7 cells of 5-benzo[c]isoxazolbarbiturates 8a–d.a
| Compound | XO inhibition (IC50, μM) | DPPH scavenging (IC50, μM) | Relative MCF-7 cell proliferation |
| 8a | 12.65 | 24.62 (70.91) | 72.79 |
| 8b | 15.50 | 57.28 (46.99) | 58.06 |
| 8c | 52.26 (22.10) | 5.39 | 43.48 |
| 8d | 29.89 | −0.64 | 64.82 |
| Allopurinol | 80.41 (3.17) | n.d.b | n.d.b |
| Trolox | n.d.b | 70.35 (35.93) | n.d.b |
| 5-FU | n.d.b | n.d.b | 11.88 |
a The screening was effected at a concentration of 30 μM, and the results are presented as % of XO inhibition, % of DPPH reduction, and % of the negative control in cell proliferation, respectively. For the most relevant compounds, dose–response curves to determine the half-medium inhibitory concentrations (IC50) were then performed and presented within parentheses.
b n.d., Not determined.
From the results of Table 1, it is important to stress the moderate XO inhibition of the isoxazolbarbiturate 8c (IC50 = 22.10 μM). In addition, the thiobarbiturates 8a and 8b have some DPPH radical scavenger effects, especially for compound 8b. Moreover, a marked reduction in MCF-7 cell proliferation was observed with the benzisoxazole 8c. Therefore, these structures can be considered in further studies concerning the development of antioxidants and/or XO inhibitors.
3 Conclusions
A new, efficient, simple, and mild method to prepare bioactive 5-benzo[c]isoxazolbarbiturates from 5-(2-nitrobenzylidene)barbiturates has been developed and for the first time described. Single crystal X-ray data unequivocally confirmed the presence of this ring system, whose formation can involve a nitroso intermediate in the reduction of the nitroaromatic group. In addition, the isoxazolbarbiturate 8c has potential to be a starting point in the development of new and improved XO inhibitors.
4 Materials and methods
Reagents and solvents were purchased from standard sources and were of analytical grade. Melting points were measured in open capillary tubes in a Büchi B-540 apparatus and were uncorrected. ATR-IR spectra were collected using a Thermo-scientific Nicolet iS10: smart iTR, equipped with a diamond ATR crystal using OMNIC 8.2 software. For ATR data acquisition the sample was placed onto the crystal and the spectrum was recorded, using an air spectrum as a reference in absorbance calculations. The sample spectra were collected at room temperature in the 4000–600 cm−1 range by averaging 32 scans with a spectral resolution of 4 cm−1. NMR spectra were acquired at room temperature using a Brüker Avance III 400 MHz spectrometer (1H NMR at 400.13 MHz and 13C NMR at 100.62 MHz) and were processed with the software MestReNova 11.0.3 (trial). Deuterated dimethyl sulfoxide (DMSO-d6) and chloroform (CDCl3) were used as solvents and as an internal standard. The 13C NMR chemical shifts were attributed based on distortionless enhancement by polarization transfer 135, heteronuclear multiple bond correlation, and heteronuclear simple quantum coherence experiments. Electrospray ionization (ESI) HRMS was performed by the microanalysis service using a QSTAR XL instrument (Salamanca, Spain).
4.1 Synthesis
4.1.1 1,3-Diphenylthiourea [13]
To a stirred solution of phenyl isothiocyanate (1 mmol; 135 mg) in dichloromethane (0.5 mL) at room temperature was added dropwise a solution of aniline (1 mmol; 93 mg) in dichloromethane (0.5 mL). The reaction was followed by thin-layer chromatography (dichloromethane) and completed in 3 h. The product so formed in suspension was filtered and washed with ethyl ether; 217 mg (95%); white crystals; mp 142–144 °C (lit. [14], 140–142 °C). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 9.79 (s, 2H), 7.48 (d, J = 7.4 Hz, 4H), 7.33 (t, J = 8.2 and 7.6 Hz, 4H), 7.12 (t, J = 7.4 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 179.6, 139.5, 128.5, 124.4, 123.7.
4.1.2 1,3-Diphenylthiobarbituric acid (1b) [6]
A stirred solution of 1,3-diphenylthiourea (1 mmol; 228 mg), malonic acid (1.3 mmol; 135 mg), and acetyl chloride (3 mmol; 235 mg; 214 μL) was heated at 60 °C for 30 min. The solid product so obtained was divided into finer pieces, filtered, washed with water, and recrystallized from acetic acid; 282 mg (95%); needle yellow crystals; mp 252–253 °C (lit. [6], 258–259 °C). 1H NMR (400 MHz, CDCl3) δ (ppm) 7.55 (m, 6H), 7.21 (d, J = 7.3 Hz, 4H), 4.10 (s, 2H); 13C NMR (101 MHz, CDCl3) δ (ppm) 181.6, 163.3, 138.8, 129.7, 129.2, 128.6, 41.2.
4.1.3 5-Benzylidenepyrimidines 3a–d [7]
A stirred mixture of (thio)barbituric acid 1a–d (1 mmol) and 2-nitrobenzaldehyde (2) (1 mmol) in water (5 mL) was refluxed for 2 h. The product so formed after cooling was filtered, washed with water, ethanol, and ethyl ether to afford the following 5-benzylidenepyrimidines.
5-(2-Nitrobenzylidene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3a), from thiobarbituric acid (1a); 250 mg (90%); pale yellow solid; mp 239–241 °C (lit. [15], 246–250 °C). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 12.56 (s, 1H), 12.33 (s, 1H), 8.63 (s, 1H), 8.24 (dd, J = 8.3 and 1.0 Hz, 1H), 7.80 (td, J = 7.6 and 1.1 Hz, 1H), 7.72–7.66 (m, 1H), 7.62 (dt, J = 7.7 and 1.2 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 179.1, 160.6, 159.2, 153.3, 146.3, 133.8, 131.6, 130.5, 130.4, 124.1, 120.6.
5-(2-Nitrobenzylidene)-1,3-diphenyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3b), from 1,3-diphenylthiobarbituric acid (1b); 387 mg (90%); pale orange solid; mp 235 °C decomposes (lit. [16], 232 °C). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.80 (s, 1H), 8.19 (d, J = 8.2 Hz, 1H), 7.71 (t, J = 7.5 Hz, 1H), 7.59 (t, J = 7.9 Hz, 2H), 7.45 (t, J = 7.6 Hz, 2H), 7.40–7.28 (m, 6H), 7.20 (d, J = 7.7 Hz, 2H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 181.4, 160.3, 158.8, 155.0, 146.2, 140.0, 139.7, 134.0, 131.8, 130.4, 130.2, 129.1, 128.9, 128.8, 128.7, 128.3, 128.1, 124.1, 121.3.
5-(2-Nitrobenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (3c), from barbituric acid (1c); 254 mg (97%); white solid; mp 274–275 °C (lit. [17], 274–276 °C). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 11.49 (s, 1H), 11.24 (s, 1H), 8.60 (s, 1H), 8.23 (d, J = 7.9 Hz, 1H), 7.79 (t, J = 7.4 Hz, 1H), 7.68 (t, J = 7.7 Hz, 1H), 7.57 (d, J = 7.7 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 162.4, 161.2, 152.5, 150.3, 146.3, 133.8, 131.7, 130.4, 130.2, 124.1, 120.5.
1,3-Dimethyl-5-(2-nitrobenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (3d), from 1,3-dimethylbarbituric acid (1d); 281 mg (97%); white solid; mp 158–159 °C (lit. [16], 159–161 °C). 1H NMR (400 MHz, DMSO-d6) δ (ppm) 8.71 (s, 1H), 8.26 (dd, J = 8.3 and 1.2 Hz, 1H), 7.80 (td, J = 7.6 and 1.3 Hz, 1H), 7.69 (t, J = 7.7 Hz, 1H), 7.53 (d, J = 7.8 Hz, 1H), 3.25 (s, 3H), 3.06 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 161.2, 160.0, 153.7, 151.1, 146.2, 133.9, 132.0, 130.1, 130.0, 124.1, 120.1, 28.4, 27.8.
4.1.4 3-Substituted-2,1-benzisoxazoles 8a–d
A stirred suspension of related 5-benzylidenepyrimidine 3a–d (1 mmol), tin chloride dihydrate (2 mmol), and concentrated hydrochloric acid (1.1 mL) was refluxed in methanol (35 mL) for 2 h. The obtained hot mixture was filtered. The solid was washed with ethanol and ethyl ether to afford the following 3-substituted-2,1-benzisoxazoles.
5-(Benzo[c]isoxazol-3(1H)-ylidene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (8a), from 5-(2-nitrobenzylidene)-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3a); 215 mg (82%); yellow solid; mp 207–208 °C decomposes. 1H NMR (400 MHz, DMSO-d6) δ 11.05 (s, 2H, 1- and 3-NH), 7.88 (d, J = 8.8 Hz, 1H, 4′-ArCH), 7.34 (d, J = 9.0 Hz, 1H, 7′-ArCH), 7.22 (dd, J = 9.0 and 6.3 Hz, 1H, 6′-ArCH), 6.73 (dd, J = 8.8 and 6.3 Hz, 1H, 5′-ArCH); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 174.5 (2-CS), 166.1 (3′-C), 161.0 (4 and 6-CO), 156.3 (7a′-ArC), 130.3 (6′-ArCH), 125.4 (4′-ArCH), 119.3 (5′-ArCH), 114.2 (3a′-C), 113.1 (7′-ArCH), 85.0 (5-C); IR νmax (cm−1) 3200–2200 (br), 3381, 2879, 2709, 1664, 1607, 1506, 1466, 1357, 1282, 1251, 1164, 1005, 939, 907, 785, 754, 714; HMRS (ESI-TOF) m/z: (M++H) calcd for C11H8N3O3S 262.0281, found 262.0281.
5-(Benzo[c]isoxazol-3(1H)-ylidene)-1,3-diphenyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (8b), from 5-(2-nitrobenzylidene)-1,3-diphenyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (3b); 278 mg (67%); yellow solid; mp 255–256 °C. 1H NMR (400 MHz, DMSO-d6) δ (ppm) 7.80 (d, J = 8.8 Hz, 1H, 4′-ArCH), 7.40 (t, J = 7.7 Hz, 4H, 3″- and 5″-ArCH), 7.35 (d, J = 8.9 Hz, 1H, 7′-ArCH), 7.29 (t, J = 7.4 Hz, 2H, 4″-ArCH), 7.24–7.19 (m, 5H, 2″-, 6″- and 6′-ArCH), 6.71 (dd, J = 8.9 and 6.2 Hz, 1H, 5′-ArCH); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 178.6 (2-CS), 166.3 (3′-C), 159.6 (4 and 6-CO), 156.1 (7a′-ArC), 141.8 (1″-ArC), 130.2 (6′-ArCH), 129.6 (2″- and 6″-ArCH), 128.4 (3″- and 5″-ArCH), 126.9 (4″-ArCH), 125.3 (4′-ArCH), 119.3 (5′-ArCH), 114.3 (3a′-ArC), 113.1 (7′-ArCH), 85.4 (5-C); IR νmax (cm−1) 3200–2200 (br), 3041, 2791, 2718, 1684, 1614, 1592, 1467, 1412, 1347, 1297, 1246, 1183, 955, 902, 764, 741, 684, 603; HMRS (ESI-TOF) m/z: (M++H) calcd for C23H16N3O3S 414.0907, found 414.0908.
5-(Benzo[c]isoxazol-3(1H)-ylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (8c), from 5-(2-nitrobenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (3c); 195 mg (79%); yellow solid; mp 266–268 °C decomposes. 1H NMR (400 MHz, DMSO-d6) δ 10.69 (s, 1H), 7.77 (dt, J = 8.8 and 1.7 Hz, 1H, 4′-ArCH), 7.46 (dt, J = 9.1 and 1.7 Hz, 1H, 7′-ArCH), 7.32 (ddd, J = 9.1, 6.4, and 0.8 Hz, 1H, 6′-ArCH), 6.89 (ddd, J = 8.8, 6.4, and 0.8 Hz, 1H, 5′-ArCH); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 163.6 (3′-C), 162.7 (4 and 6-CO), 156.4 (7a′-ArC), 150.8 (2-CO), 131.8 (6′-ArCH), 124.1 (4′-ArCH), 122.0 (5′-ArCH), 116.2 (3a′-C), 113.7 (7′-ArCH), 82.7 (5-C); IR νmax (cm−1) 3200–2200 (br), 3174, 2960, 2813, 1722, 1666, 1648, 1601, 1440, 1403, 1360, 1320, 1286, 1238, 1142, 906, 841, 766, 752; HMRS (ESI-TOF) m/z: (M++H) calcd for C11H8N3O4 246.0509, found 246.0511.
5-(Benzo[c]isoxazol-3(1H)-ylidene)-1,3-dimethylpyrimidine-2,4,6(1H,3H,5H)-trione (8d), from 1,3-dimethyl-5-(2-nitrobenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (3d); 140 mg (51%); yellow solid; yellow crystals by crystallization in dichloromethane/ethanol; mp 211–213 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.90 (dt, J = 8.8 and 1.1 Hz, 1H, 4′-ArCH), 7.37 (dt, J = 8.9 and 1.0 Hz, 1H, 7′-ArCH), 7.29 (ddd, J = 8.9, 6.2, and 1.1 Hz, 1H, 6′-ArCH), 6.80 (ddd, J = 8.8, 6.3, and 0.9 Hz, 1H, 5′-ArCH), 3.17 (s, 6H, 1- and 3-NCH3); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 167.4 (3′-C), 160.9 (4- and 6-CO), 155.9 (7a′-ArC), 152.3 (2-CO), 131.1 (6′-ArCH), 125.5 (4′-ArCH), 120.0 (5′-ArCH), 114.9 (3a′-C), 112.9 (7′-ArCH), 82.5 (5-C), 27.7 (1 and 3-NCH3); IR νmax (cm−1) 3200–2200 (br), 2922, 2852, 2715, 1707, 1644, 1604, 1445, 1353, 1230, 1175, 1142, 1068, 899, 760, 749, 683; HMRS (ESI-TOF) m/z: (M++H) calcd for C13H12N3O4 274.0822, found 274.0827.
4.2 X-ray analysis
Crystals suitable for single-crystal X-ray analysis were grown from a mixture of ethanol and dichloromethane. Selected crystals were covered with polyfluoroether oil (Fombling, Sigma Aldrich) and mounted on a nylon loop. The data were collected at 110(2) K using a Bruker D8 Venture diffractometer equipped with a Photon 100 detector and an Oxford Cryostem Cooler, using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). Table S1 (Supplementary information) contains crystallographic experimental data and structure refinement parameters. CCDC 1557900 contains the supplementary crystallographic data for this article. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/structures.
4.3 Biological evaluation
Preparation of sample solutions and experimental procedures are presented in Supplementary information.
Acknowledgments
This work is supported by FEDER funds through the POCI—COMPETE 2020—Operational Programme Competitiveness and Internationalisation in Axis I—Strengthening research, technological development and innovation (Project POCI-01-0145-FEDER-007491) and National Funds by FCT—Foundation for Science and Technology (Project UID/Multi/00709/2013). The SCXR determinations were performed at UCIBIO/FCT-NOVA, financed by national funds from FCT/MEC (UID/Multi/04378/2013), and cofinanced by the ERDF under the PT2020 Partnership Agreement (POCI-01-0145-FEDER-007728).