



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Corynebacteriales belong to Actinobacteria and include numerus bacteria responsible of human diseases such as leprosy (Mycobacterium leprae), diphtheria (Corynebacterium diphtheriae) and tuberculosis (Mycobacterium tuberculosis). Nowadays, the latter is still responsible for more than 10 million infections per year [1]. In contrast, some Corynebacteriales are non-pathogenic, such as Corynebacterium glutamicum which is involved in the industrial production of glutamate [2]. All bacteria of this group possess an atypical cell envelope exceptionally resistant against external stress and antibiotics, this resistance being attributed to the unique organization of their cell envelope. In addition to the plasma membrane, this envelope is composed of a cross-linked peptidoglycan layer (PG) bounded to an arabinogalactan (AG) complex itself connected to long fatty acids, called mycolic acids (MA) through an ester linkage. The esterified arabinogalatan constitutes the inner part of the outer membrane, the so called mycomembrane (Figure 1).
Schematic representation of the cell envelope of Corynebacteriales. (MA: mycolic acid; TDM: trehalose dimycolate; TMM: trehalose monomycolate.)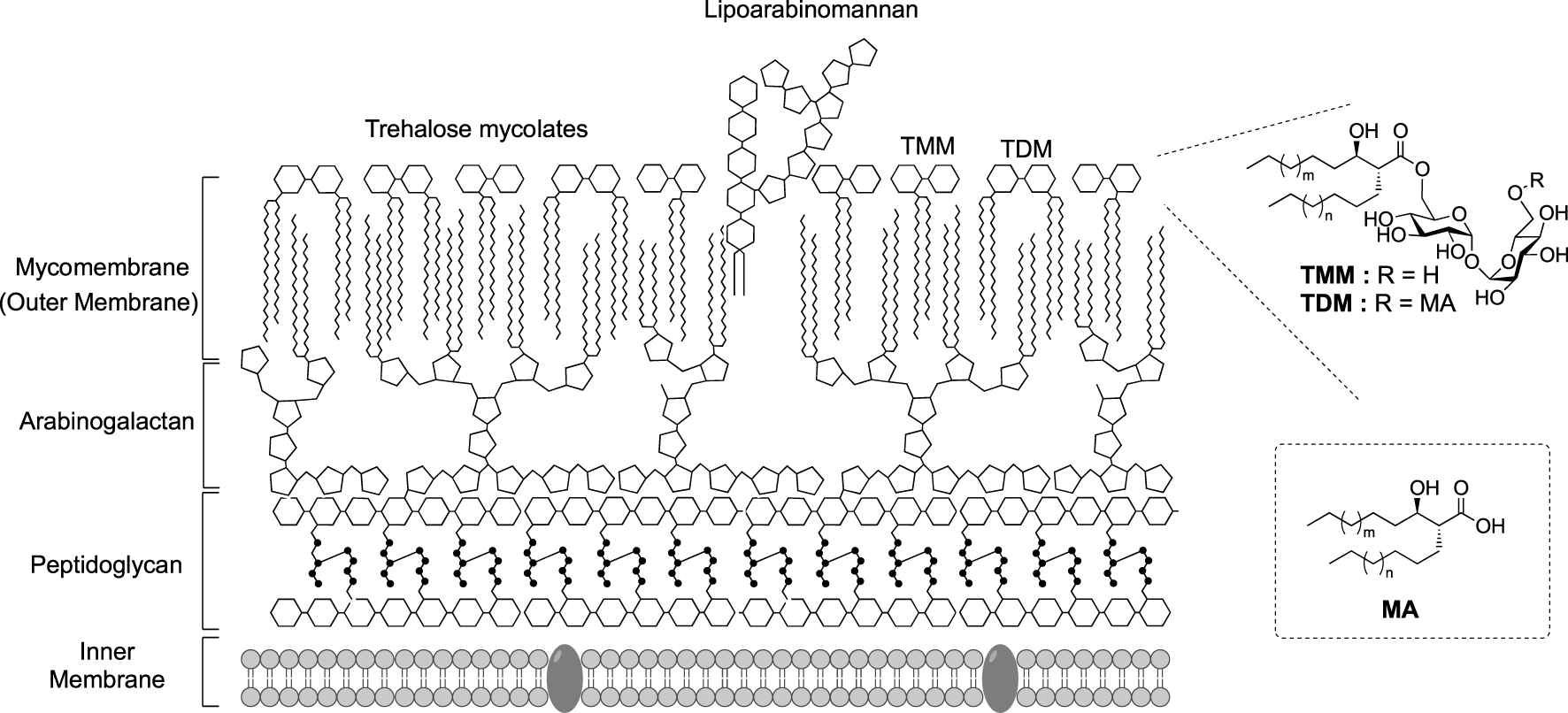
Mycolic acids (MA) are very long chain (C30–C90) α-branched and β-hydroxylated fatty acids, with an anti-relationship between the β-OH and the α-ramification. Beyond the mycoloylated AG (AGM), the outer leaflet of the mycomembrane is composed of non-covalently attached glycolipids esterified with mycolic acids. These glycolipids are mainly trehalose monomycolate (TMM) and trehalose dimycolate (TDM). After being biosynthesized in the cytoplasm, mycolic acids are esterified to trehalose by the polyketide synthase Pks13, giving rise to TMM, and then transferred across the inner membrane by a transporter of the MmpL3 family. TMM is then processed by enzymes called mycoloyltransferases, and the mycolate part of TMM is transferred to different acceptors. Four mycoloyltransferases have been identified in M. tuberculosis (Ag85), six in M. smegmatis (Fbp) and six in C. glutamicum (Myt) [3]. TMM plays the role of a mycolate donor for several mycolate acceptors and the mycolate part can be transferred to (i) another TMM to form trehalose dimycolate (TDM); (ii) arabinogalactan; and (iii) channel-forming porins in some cases [4, 5, 6] (Figure 2).
Schematic representation of the mycoloylation of various acceptors in the mycomembrane and labeling localization depending on the pathways used (green or red stars). (AG: arabinogalactan; AGM: mycoloylated arabinogalactan; Ag85: antigen 85; CmpL: corynebacterial membrane proteins large; MmpL: mycobacterial membrane proteins large; Myt: mycoloyltransferase; Pks: polyketide synthase; TMM: trehalose monomycolate; Tre: trehalose.)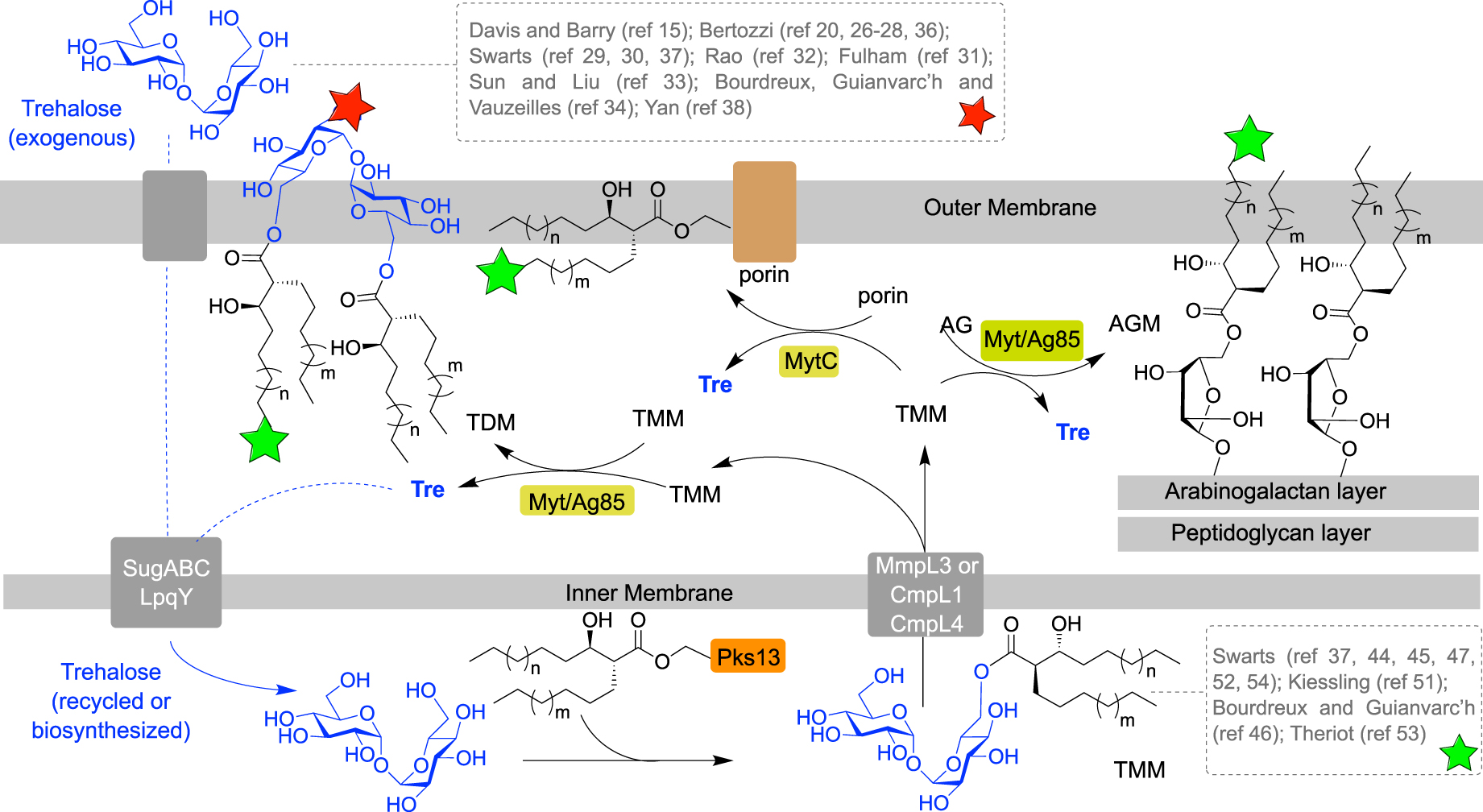
Due to its importance for mycobacteria, the mycomembrane biogenesis is highly studied. Over the last decades, several chemical reporters have been developed to investigate biological processes in bacteria [7, 8] and in this context, mycoloyltransferases have been widely targeted by trehalose- and TMM-based chemical reporters. Chemical labeling of the mycomembrane involves incorporation of chemical tools in the cell envelope and processing of these metabolic analogues by enzymes. The bacteria labeling can be performed in one or two steps, depending on the analogues used. It can be a detectable analogue of a mycomembrane metabolite, for example a mycomembrane precursor analogue conjugated with a fluorescent moiety. In such approach, the labeling is performed in one step allowing a direct mycomembrane imaging, but precautions should be taken in the design of such compounds since a bulky fluorophore could disrupt enzymatic processes. In another approach, the metabolic precursor analogues can be conjugated to a small bioorthogonal group, allowing the introduction of a detectable moiety in a distinct step using a bioorthogonal reaction with a fluorescent probe. Since the size of the bioorthogonal groups is small compared to fluorophores, this approach, called the bioorthogonal chemical reporter strategy, is supposed to be less disruptive for enzymes. From a metabolic point of view, the synthetic trehalose-based derivatives (Figure 2, dashed gray box at top) can be incorporated through the trehalose transporter LpqY-SugABC and then coupled to a mycolic acid and transferred into the mycomembrane, allowing specific labeling of the upper part of the mycomembrane, i.e., the TDM layer (Figure 2, red star). Alternatively, the TMM-based derivatives (Figure 2, dashed gray box at bottom), can be either directly inserted into the mycomembrane or processed by mycoloyltransferases, thus labeling both the inner and the outer part of the mycomembrane, AGM and TDM layers, respectively (Figure 2, green stars).
Trehalose is a non-reducing and C2 symmetrical disaccharide composed of two glucose units with an 1,1,α,α-linkage. It can be found in fungi, algae, insects, bacteria, and some invertebrates but not in mammalian. Trehalose has been extensively studied for its properties against stresses such as, for example, heat, oxidative stresses, or desiccation [9, 10]. Due to its symmetry, and taking into account that many trehalose-containing glycoconjugates are unsymmetrical, the most important challenges in the synthesis of trehalose derivatives are its desymmetrization and its selective modification among its eight hydroxyl groups. Such transformations are clearly exposed in recent reviews and most of the syntheses rely on two different approaches [11, 12, 13, 14]. The first approach relies on the formation of the trehalose core by an α,α-selective glycosylation, using selectively protected glucose derivatives. The second approach is based on the use of native trehalose as starting material and the selective post-modification of the eight hydroxyl groups. Both approaches are very challenging and were used for the selective synthesis of trehalose-based probes. In this review we will focus on the synthesis of trehalose- and TMM-based probes already used in bacteria for the study of the mycomembrane. We classified the probes in two categories: (i) the trehalose-based probes in which the detectable moiety or the bioorthogonal small functional group is directly linked to the trehalose core; and (ii) the TMM-based probes and analogues in which the detectable moiety or the bioorthogonal small functional group is attached at the end of the lipidic chain thus mimicking TMM. All the synthesized trehalose- and TMM-based probes, that have been successfully used so far for metabolic labeling experiments with diverse application, are presented in Figures 3 and 4 respectively.
Structures of trehalose-based probes. (Az: azide; CDG: cephalosporinase-dependent green; Cz: carbazole; DMN: 4-(N,N-Dimethylamino)-1,8-naphthalimide; 2-FD: 2-deoxy-2-fluoro; FI: fluorescein; FITC: fluorescein-isothiocyanate; HC: hydroxychromone; NFC: nitrofuranyl calanolide; RMR: far-red molecular rotor; TMR: tetramethylrhodamine; Tre: trehalose.)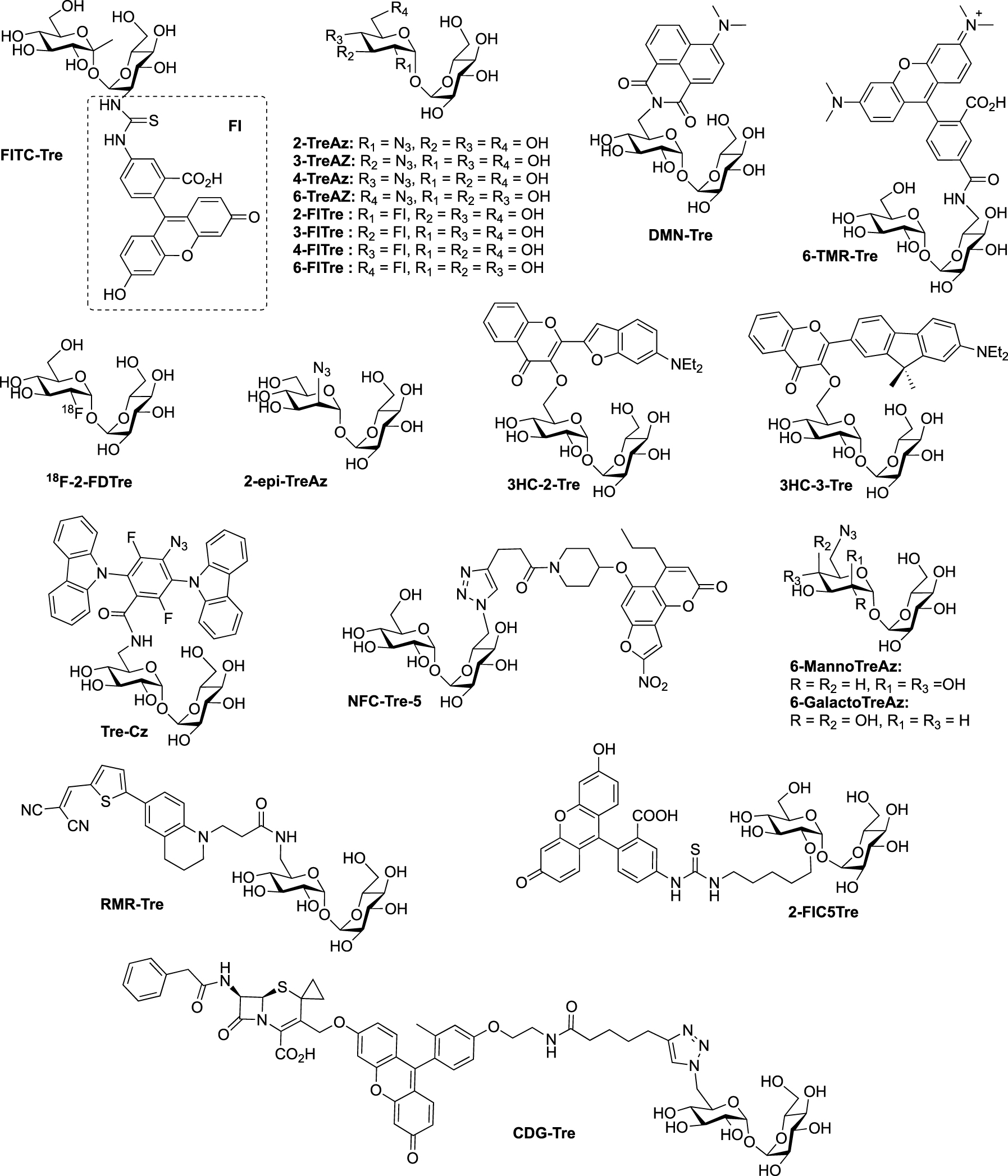
Structures of TMM-bases probes. (Alk: alkyne; Az: azide; DBF: dibromofluorescein; FRET: fluorescence resonance energy transfer; FITC: fluorescein-isothiocyanate; QTF: quencher-trehalose-fluorophore; RMR: far-red molecular rotor; TCO: trans-cyclooctene; TDM: trehalose dimycolate; TMM: trehalose monomycolate.)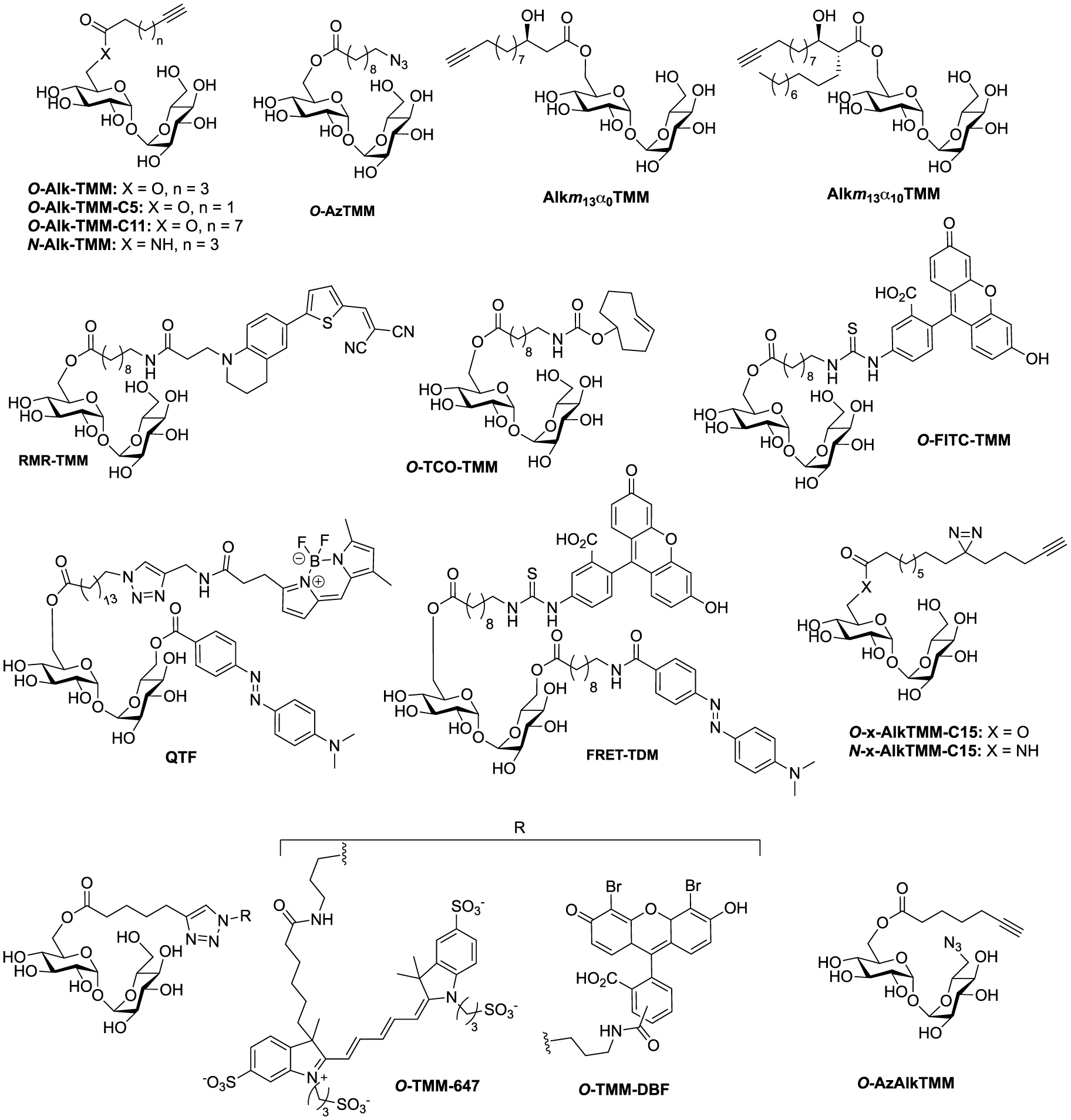
Synthesis of FITC-Tre [15].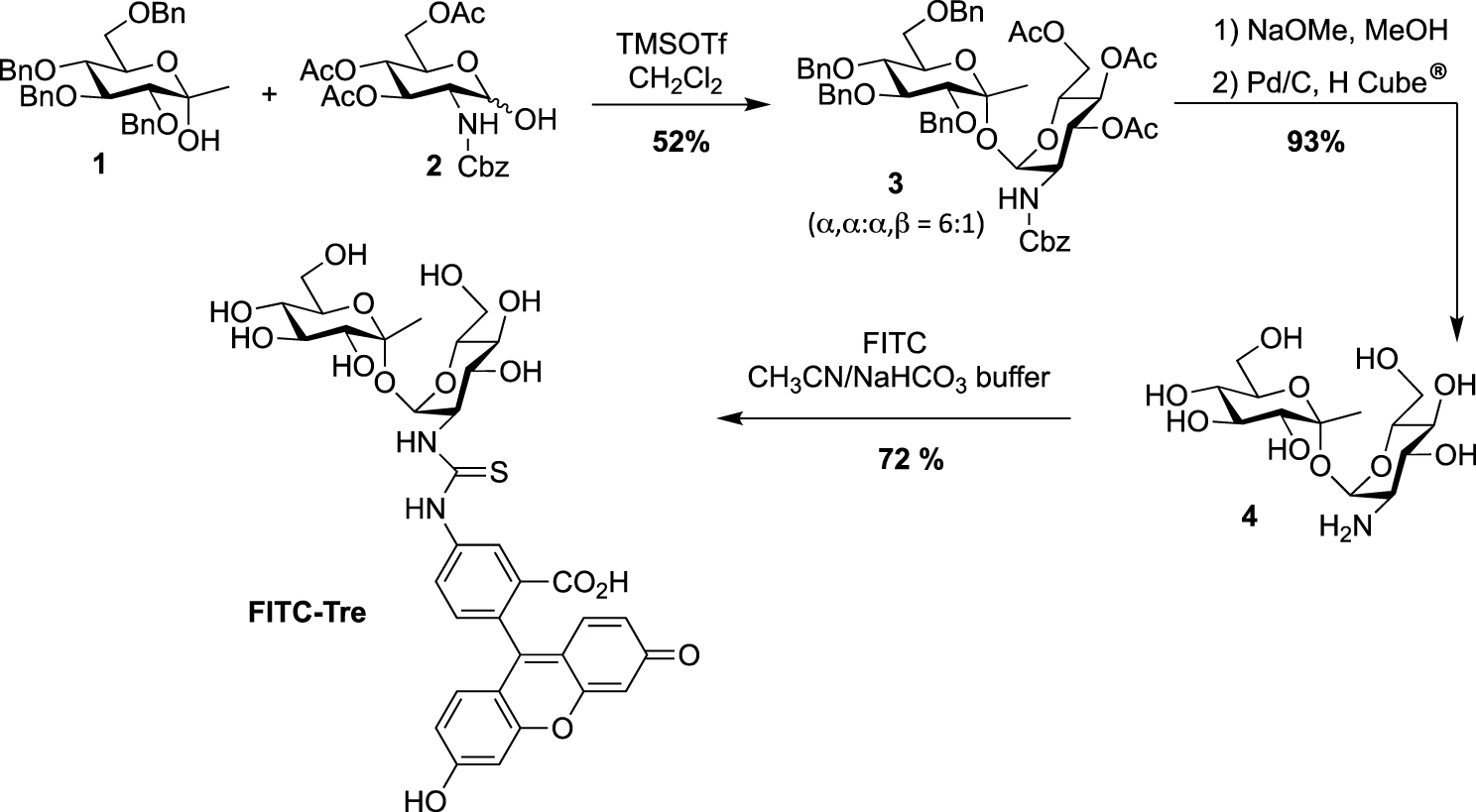
2. Synthesis of trehalose-based probes
The synthesis of trehalose-based probes can be either straightforward or challenging, depending on the hydroxyl group of trehalose to be functionalized (i.e., primary or secondary alcohol). The first synthesis of a trehalose-based probe used in a cellular context was reported by Davis, Barry and co-workers with the fluorescein-containing trehalose FITC-Tre [15], bearing an extra methyl group at the anomeric position of one of the glucose units. One of the challenging steps of the preparation of FITC-Tre was the α,α-selective glycosylation. In this study the authors used the α-selective approach developed by Ikegami [16], starting from the ketoside 1 as glycosyl donor and the glucosamine derivative 2 as acceptor and governed by the extra methyl group of compound 1. The glycosylation provided in one step the derivative 3 in 52% yield, functionalized with an equatorial amino group. Such glycosylations, in order to construct the trehalose core, are very challenging, and other studies were reported (see for example [17, 18, 19]). It should be noted that the synthesis of amino-trehaloses starting from native trehalose is very challenging and usually takes many synthetic steps. Thus, even if the preparation of donor and acceptor 1 and 2 could be time consuming, this approach furnished in one glycosylation step the expected derivative 3. After methanolysis of the acetate groups and hydrogenolysis of the benzyl carbamate, free derivative 4 was converted in FITC-Tre in high yield by treatment with fluorescein-isothiocyanate (FITC) (Scheme 1). Interestingly, this study revealed a strong Ag85 plasticity for trehalose processing and FITC-Tre was the first trehalose analogue used in metabolic labeling experiments on growing Mtb producing fluorescent bacteria.
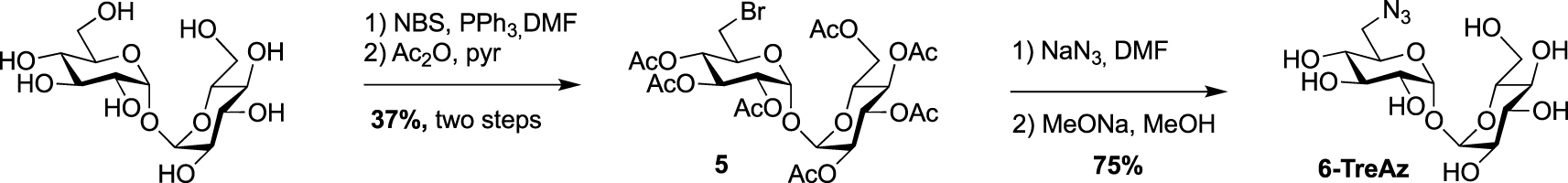
In 2012, Bertozzi published a mycomembrane labeling study using four azido-trehalose analogues referred to as 2-, 3-, 4- and 6-TreAz [20]. The latter1 was prepared according to a synthesis reported by Hanessian [21]. The synthesis started with the mono-O-bromination of commercially available α-α-d-trehalose in the presence of N-bromosuccinimide (NBS) and triphenylphosphine. Due to the C2 symmetry of trehalose, mono-O-bromination at C6 or C6′ gave a same and unique derivative. Desymmetrized derivative 5 was obtained after per-O-acetylation of the crude product, a step that facilitated its purification. 5 was then engaged in a nucleophilic substitution reaction in the presence of sodium azide followed by de-O-acetylation to obtain 6-TreAz (Scheme 2).
Synthesis of 2-TreAz [20].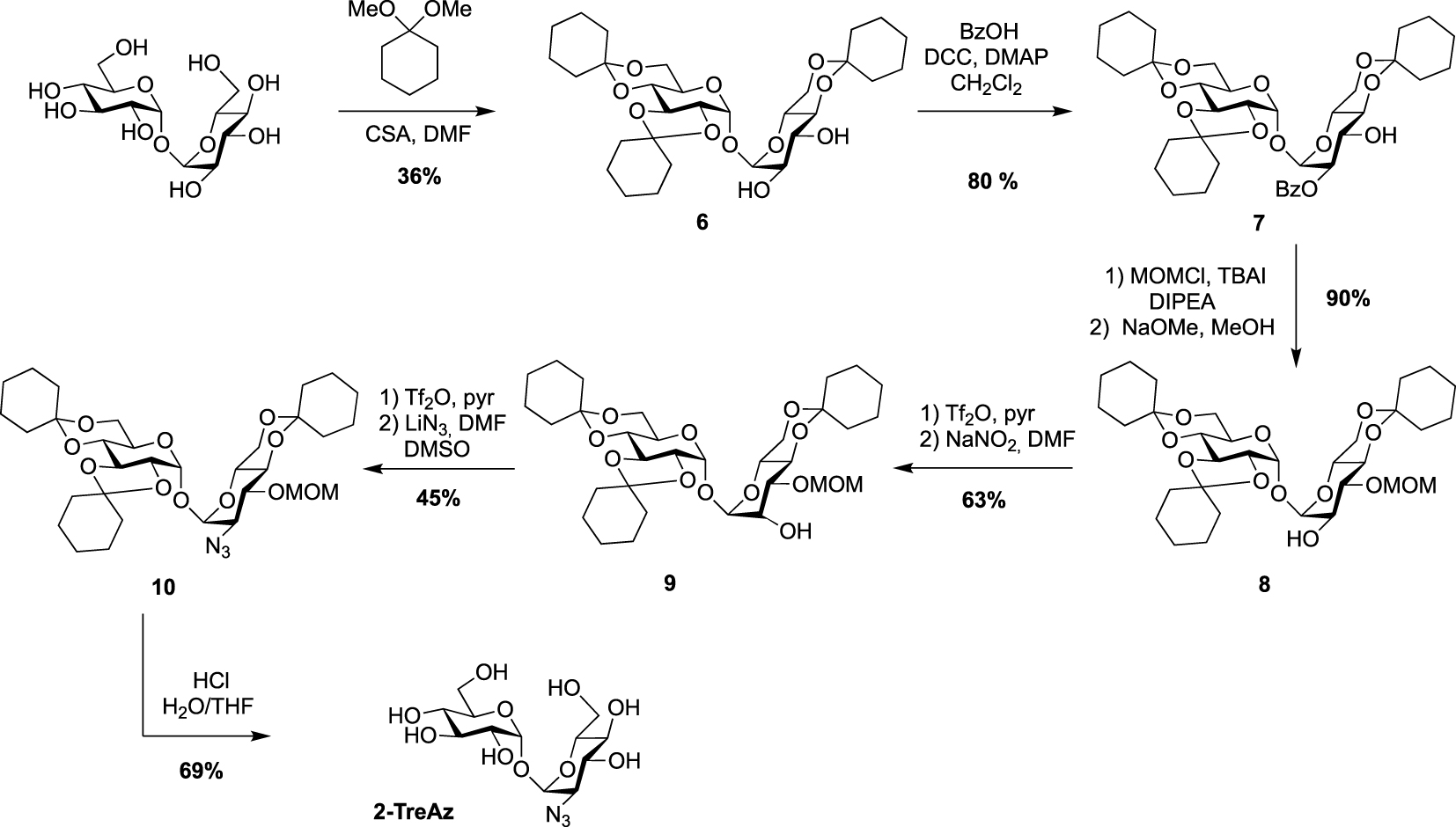
The preparation of the azido-trehaloses functionalized through their secondary alcohol is more challenging because it needs robust methods enabling selective protection to differentiate the eight hydroxyl groups. Furthermore, the introduced azido group should be positioned in equatorial position. The synthesis of 2- and 3-TreAz started from the same desymmetrized intermediate 6 obtained according to a procedure reported by Wallace and Minnikin [22]. From this common intermediate different protection/deprotection steps were carried out to selectively isolate the 2- or 3-hydroxyl groups. In order to get 2-TreAz, compound 6 was first protected on its C2 position via a selective esterification on the more reactive OH-2, leading to key compound 7 in good yield. The OH-3 was then protected using methoxymethyl chloride (MOMCl) and derivative 8 was obtained after de-O-benzoylation. This sequence resulted in the isolation of the 2-hydroxyl group, which was engaged on a double inversion process using the Lattrell–Dax nitrite-mediated inversion [23, 24]. After activation of the hydroxyl group of 8 using triflic anhydride, compound 9 was obtained after treatment in the presence of NaNO2. This sequence allowed the authors to isolate a protected trehalose with an axial hydroxyl on C2 position. Compound 9 was then reacted with triflic anhydride followed by lithium azide, and the azido derivative 10 was obtained in 45% yield. The difficulty of this sequence could be the separation of the target compound which can be obtained as a mixture with a side product resulting from an elimination reaction. Finally, acidic hydrolysis afforded 2-TreAz with 69% yield (Scheme 3).
For the preparation of 3-TreAz, Swern oxidation of key intermediate 7 led to 11 in excellent yield, and then derivative 12, with an axial OH group at C3 was obtained through the selective reduction of the ketone function of 11. Then, activation of the OH group of 12 using triflic anhydride followed by nucleophilic substitution of the triflate intermediate by lithium azide gave compound 13 in good yield. Finally, de-O-benzoylation and acidic hydrolysis led to expected 3-TreAz (Scheme 4).
Synthesis of 3-TreAz [20].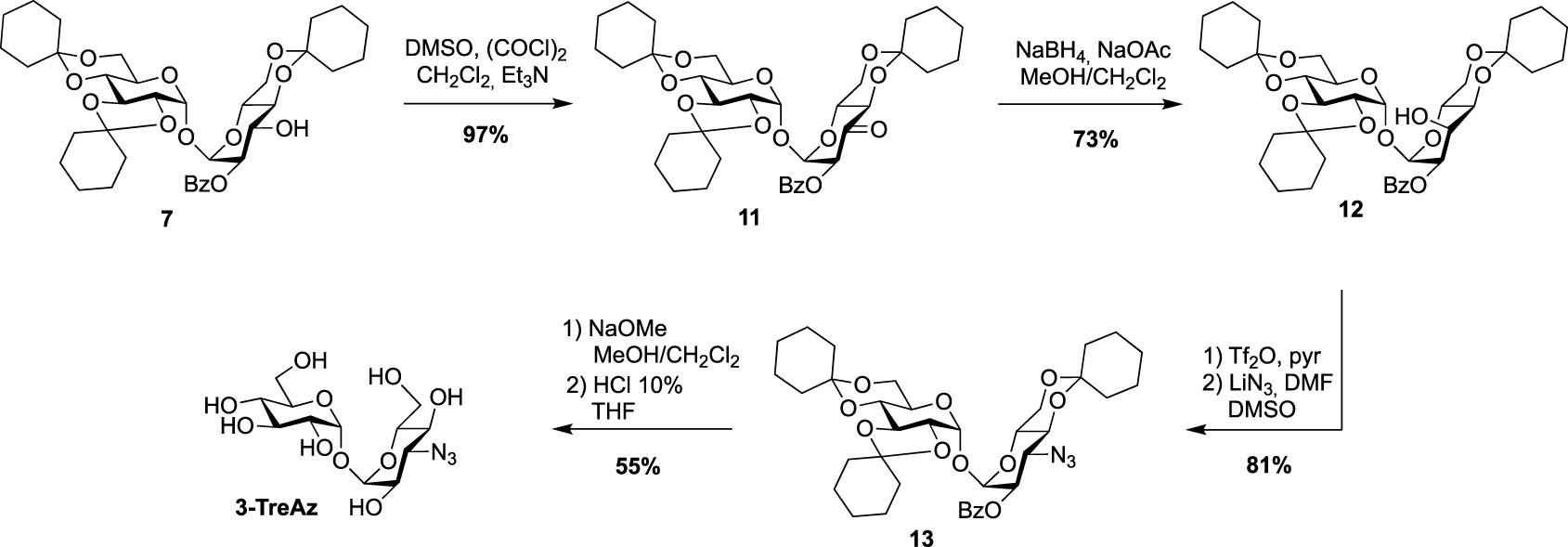
Synthesis of 4-TreAz [20].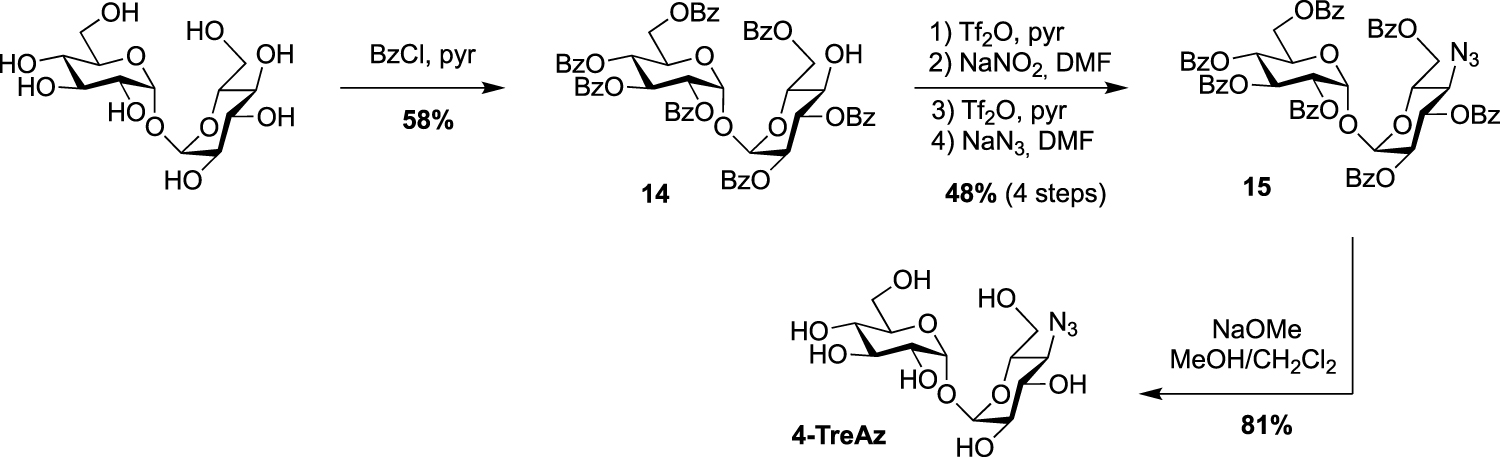
In the same study, the azido analogue, 4-TreAz was prepared using the hepta-O-benzoate derivative 14 described by Nashed and coworkers in 1993 [25]. Indeed, treatment of trehalose by eight equivalents of benzoyl chloride led to the desymmetrized trehalose 14 in good yield. It should be noted that, in these conditions, two other symmetrical derivatives were also obtained, i.e., the per-O-benzoylated trehalose and the 2,2′,3,3′,6,6′-hexa-O-benzoylated trehalose. The azide group was then incorporated through a double inversion using the same approach applied for 2-TreAz with the Lattrell–Dax nitrite-mediated inversion. Finally, 4-TreAz was obtained after de-O-benzoylation (Scheme 5).
All the four azido-trehalose analogues were used in metabolic labeling experiments in M. smegmatis strains using a two-step approach through the bioorthogonal chemical reporter strategy. The derivatives 2- and 6-TreAz were found to be the most efficient. Bertozzi next used these analogues as key precursors for the synthesis of new fluorescent trehaloses. Indeed, TreAz derivatives were subjected to hydrogenolysis to give the corresponding amino-trehaloses. All the four obtained amino-trehaloses were then reacted with fluorescein isothiocyanate (FITC) to afford 2-, 3-, 4- and 6-FITre (Scheme 6) [26]. These fluorescein-modified trehalose probes were more efficient in metabolic labeling experiments on Mycobacteria compared to the first reported FITC-Tre. The same approach was performed to obtain the tetramethylrhodamine-trehalose 6-TMR-Tre for super-resolution microscopy experiments [27]. Finally, in order to avoid the wash steps usually performed when using fluorescent probes, the solvatochromic dye DMN-Tre [28] was prepared starting from 6-amino-trehalose (Scheme 6). Indeed, solvatochromic tools show high shifts of fluorescence with solvent polarity modifications and are thus efficient for mycomembrane imaging without washing steps.
In 2014, the Swarts’s group published a new route for the synthesis of azido trehalose derivatives using a chemoenzymatic approach starting from several azido-glucoses and uridine diphosphate-glucose (UDP-glucose) in presence of trehalose synthase TreT (Scheme 7) [29]. Under these conditions, access to 3- and 6-TreAz were improved in comparison to the firstly reported chemical syntheses. Unfortunately, this enzymatic approach failed for the 2- and 4-modified trehaloses. For the latter, the chemical synthesis remains still more efficient. Additionally, the same team also developed an access to the radioprobe 18F-2-FDTre using the same methodology [30].
Syntheses of 2-, 3-, 4-, 6-FITre [26]; 6-TMR-Tre [27] and DMN-Tre [28]. (TAMRA-NHS: 5-Carboxy-tetramethylrhodamine N-succinimidyl ester.)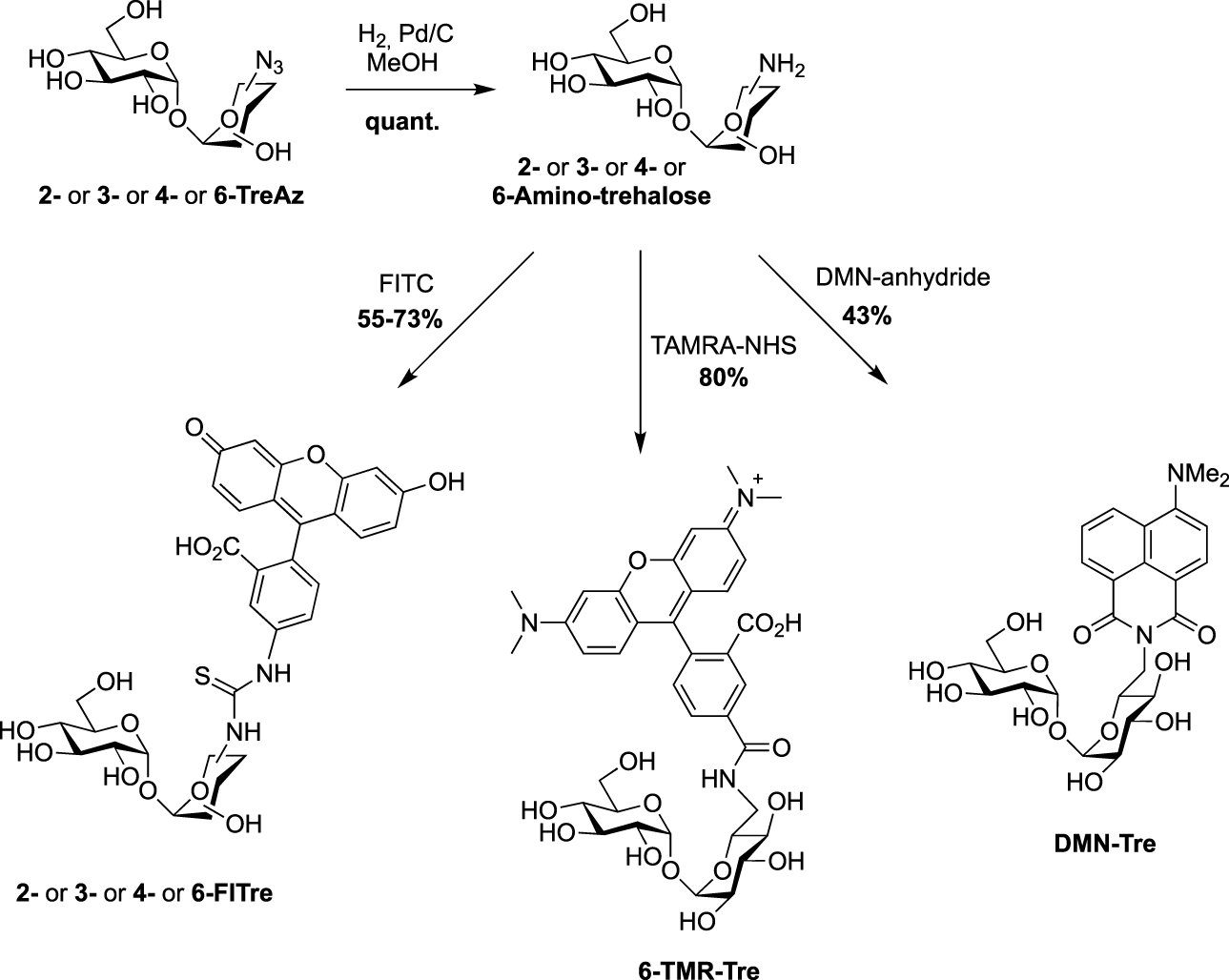
Chemoenzymatic approaches for the synthesis of 3- and 6-TreAz [29] and 18F-2-FDTre [30].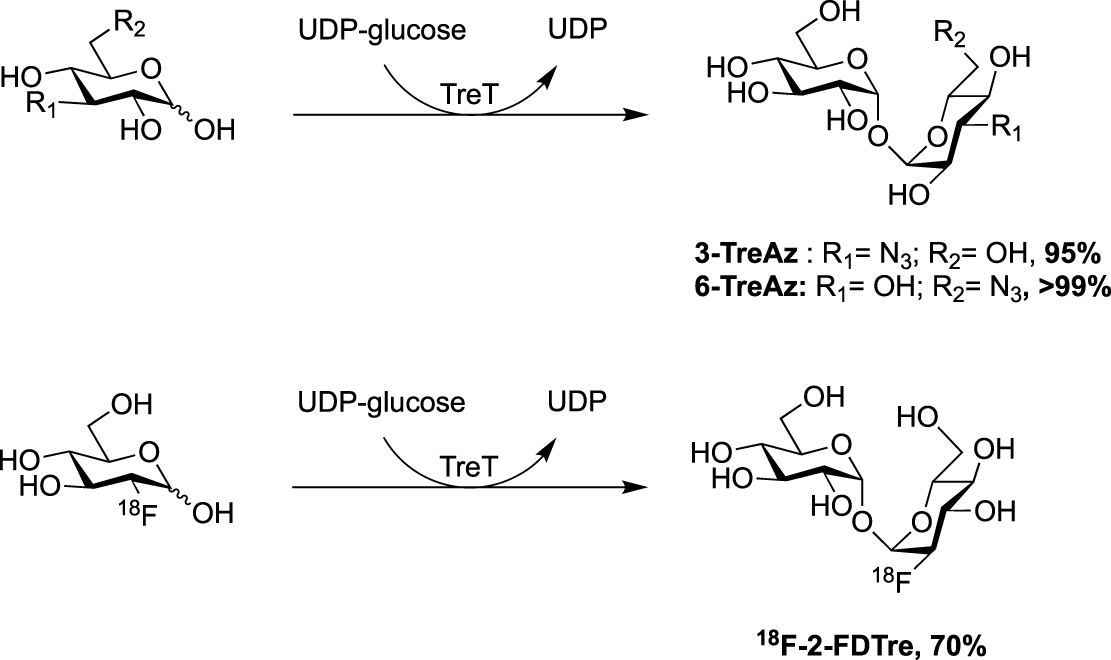
In 2020, the Fulham group employed this enzymatic approach for the synthesis of 6-azido-modified derivatives with the use of 6-azido-mannose and 6-azido-galactose in place of 6-azido-glucose [31]. This resulted in the obtention of the non-symmetrical trehalose analogues 6-MannoTreAz and 6-GalactoTreAz (Scheme 8). These bioorthogonal trehalose analogues were used in M. smegmatis labeling experiments, highlighting a good substrate tolerance of mycoloyltransferases.
Chemoenzymatic synthesis of 6-MannoTreAz and 6-GalactoTreAz [31].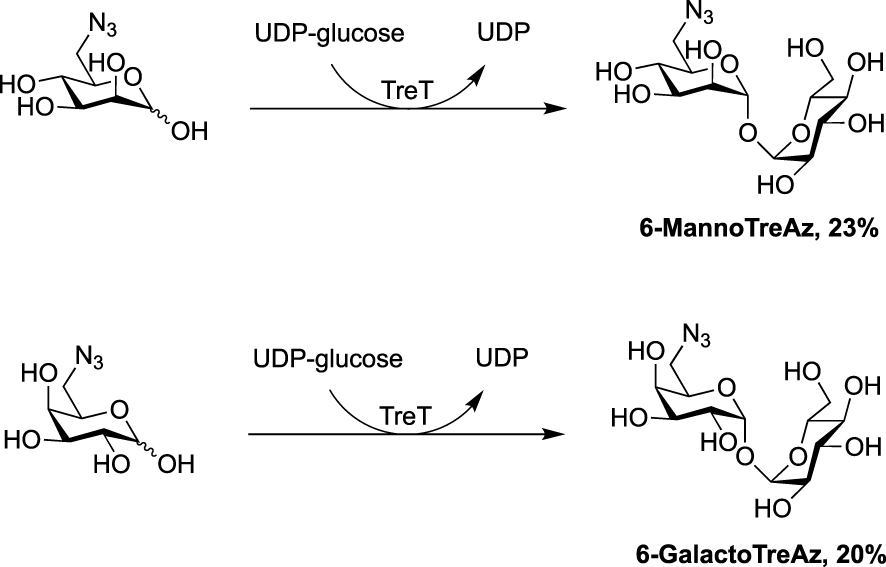
Synthesis of CDG-Tre probe targeting two mycobacterial enzymes [32].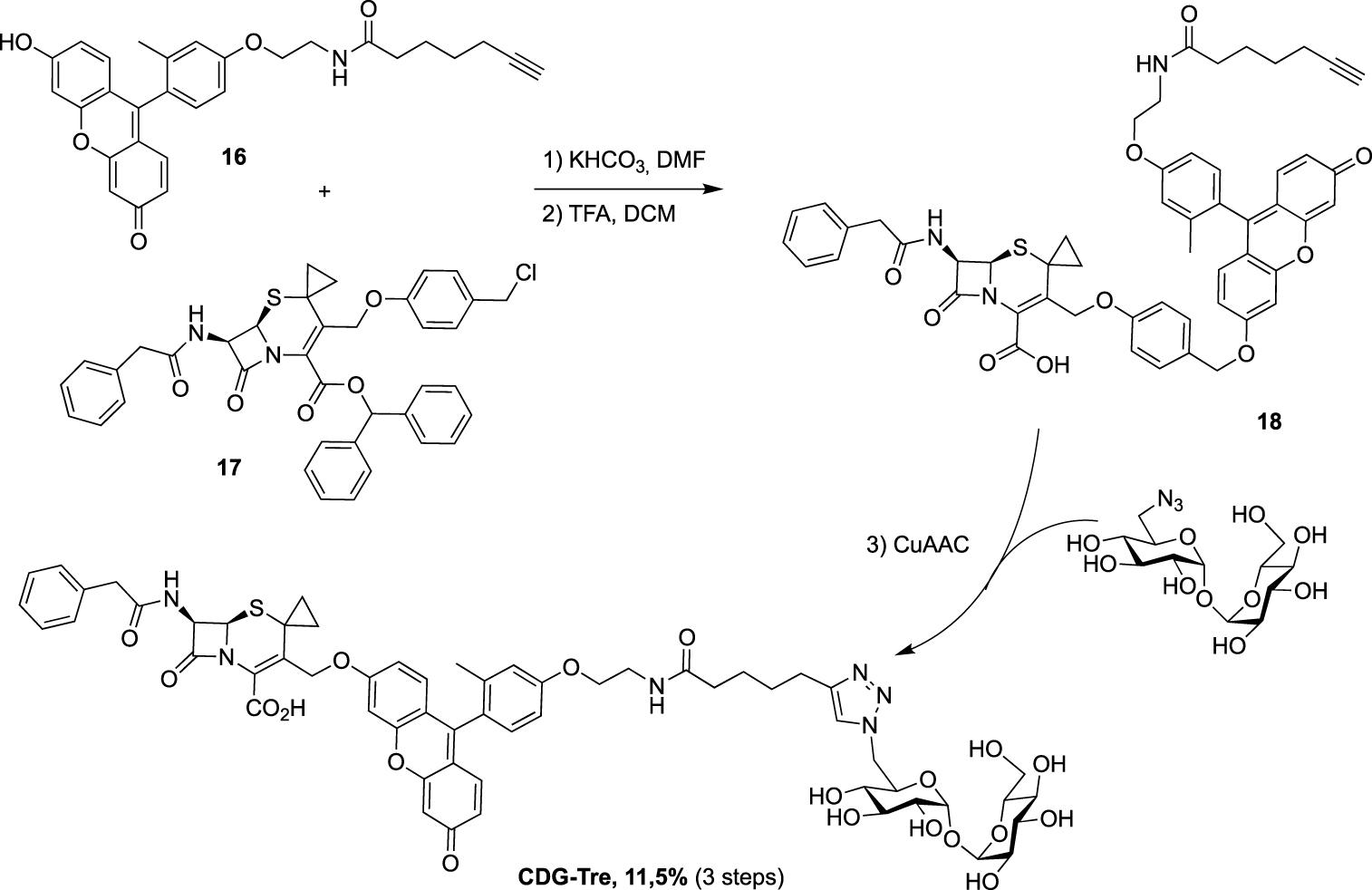
Still in 2020, the 6-TreAz derivative was used, by Rao group, as a precursor for the preparation of the new fluorogenic probe CDG-Tre activated by the β-lactamase BlaC expressed in M. tuberculosis [32]. In this approach, the fluorophore is quenched by the cephalosporin residue and the fluorescence is recovered after processing by the β-lactamase BlaC. The resulting fluorescent trehalose can then be processed by mycoloyltransferases and thus label the mycomembrane. Freshly prepared fluorescein derivative 16 and cephalosporin scaffold 17 were engaged in nucleophilic substitution followed by treatment under acidic conditions, leading to 18. The crude was then directly coupled without purification to 6-TreAz via copper-catalyzed azide-alkyne cycloaddition (CuAAC) (Scheme 9).
Later, Sun and Liu used 6-TreAz as precursor for the synthesis of the additional fluorescent trehalose-based probe NFC-Tre-5 [33]. This probe featured an off-on fluorescent group activated by the nitroreductase Rv2466 allowing the specific labeling of single cells. NFC-Tre-5 was obtained after CuAAC reaction between 6-TreAz and an alkyne-modified nitrofuranyl calanolide scaffold (Scheme 10).
Synthesis of NFC-Tre-5 [33].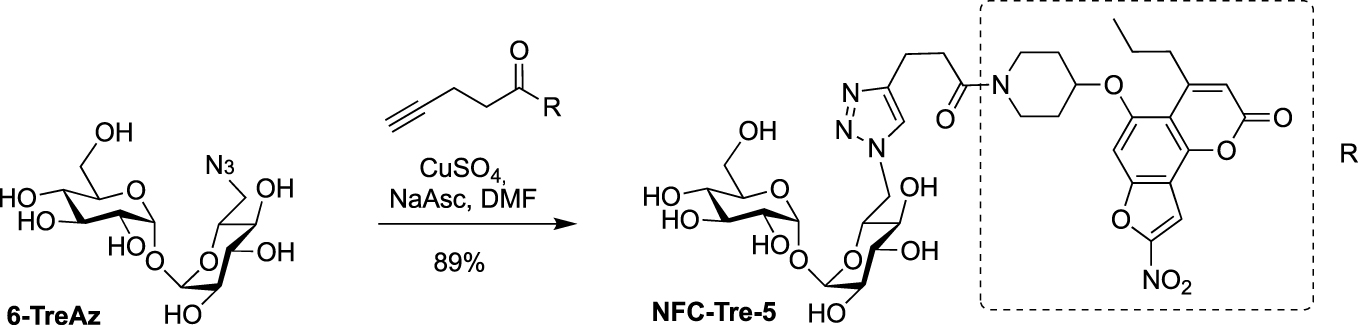
In 2022, Guianvarc’h, Vauzeilles and Bourdreux reported the synthesis of two new trehalose-based probes modified at C2 for the metabolic labeling of the mycomembrane [34] i.e., a native trehalose bearing a short ether linker functionalized with a fluorescein, 2-FIC5Tre, and 2-epi-TreAz, an epimer at C2 of the known 2-TreAz. In the course of this study, they also proposed a new route for the synthesis of 2-TreAz. The key step of these syntheses was a tandem one-pot protection and desymmetrization of trehalose mediated by FeCl3⋅6H2O. In this study, the authors revisited a first published protocol [35] in order to get a protected trehalose with only a free OH group at C2 position. Briefly, α,α-d-trehalose was firstly per-O-silylated in pyridine and the resulting derivative 19 was engaged, in a tandem protection protocol mediated by FeCl3⋅6H2O in the presence of benzaldehyde and triethylsilane (Scheme 11). Under these conditions the desymmetrized and protected trehalose 20 was obtained in 40% yield. This key compound was used as starting material for the synthesis of the three derivatives 2-epi-TreAz, 2-TreAz and 2-FIC5Tre. The tri-O-benzylated trehalose 20 was therefore converted into triflate 21 and then immediately engaged in a nucleophilic substitution reaction to introduce the azido moiety in presence of tetra-n-butylammonium azide (TBAN3). This sequence allowed to rapidly obtain an axial azido moiety at C2 of trehalose. Challenging deprotections were then carried out using anhydrous iron(iii) chloride in the presence of acetic anhydride followed by de-O-acetylation providing 2-epi-TreAz in good yield (Scheme 12). The known 2-TreAz was also synthesized during this study by performing the same double inversion protocol as the one used for its first synthesis by the group of Bertozzi. Triflate 21 was indeed treated with sodium nitrite and the resulting compound 23 with an axial OH group was then converted into 24 after an activation-nucleophilic substitution sequence using triflate anhydride in pyridine followed by a TBAN3 treatment. Then, the same deprotection protocols were performed leading to 2-TreAz. Finally, the authors also prepared a fluorescent probe starting from 20. A small linker bearing an azido moiety was first introduced by alkylation of the hydroxyl group and the resulting compound 25 was subjected to hydrogenolysis. Then, the amine function of 26 was coupled to the fluorophore using a treatment with FITC leading to 2-FIC5Tre. The three probes were successfully used in mycomembrane labeling experiments on C. glutamicum. 2-TreAz and 2-epi-TreAz appeared to have similar efficacy again indicating a good substrate tolerance of mycoloyltransferases.
FeCl3⋅6H2O mediated one-pot desymmetrization of trehalose [34, 35].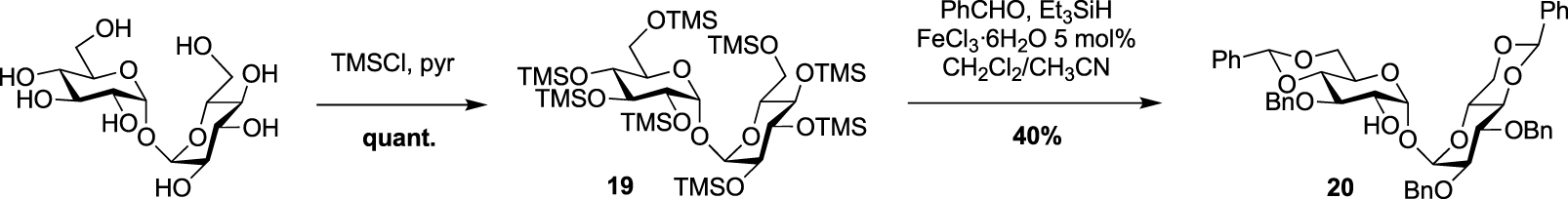
Syntheses of 2-epi-TreAz; 2-TreAz and 2-FIC5Tre [34].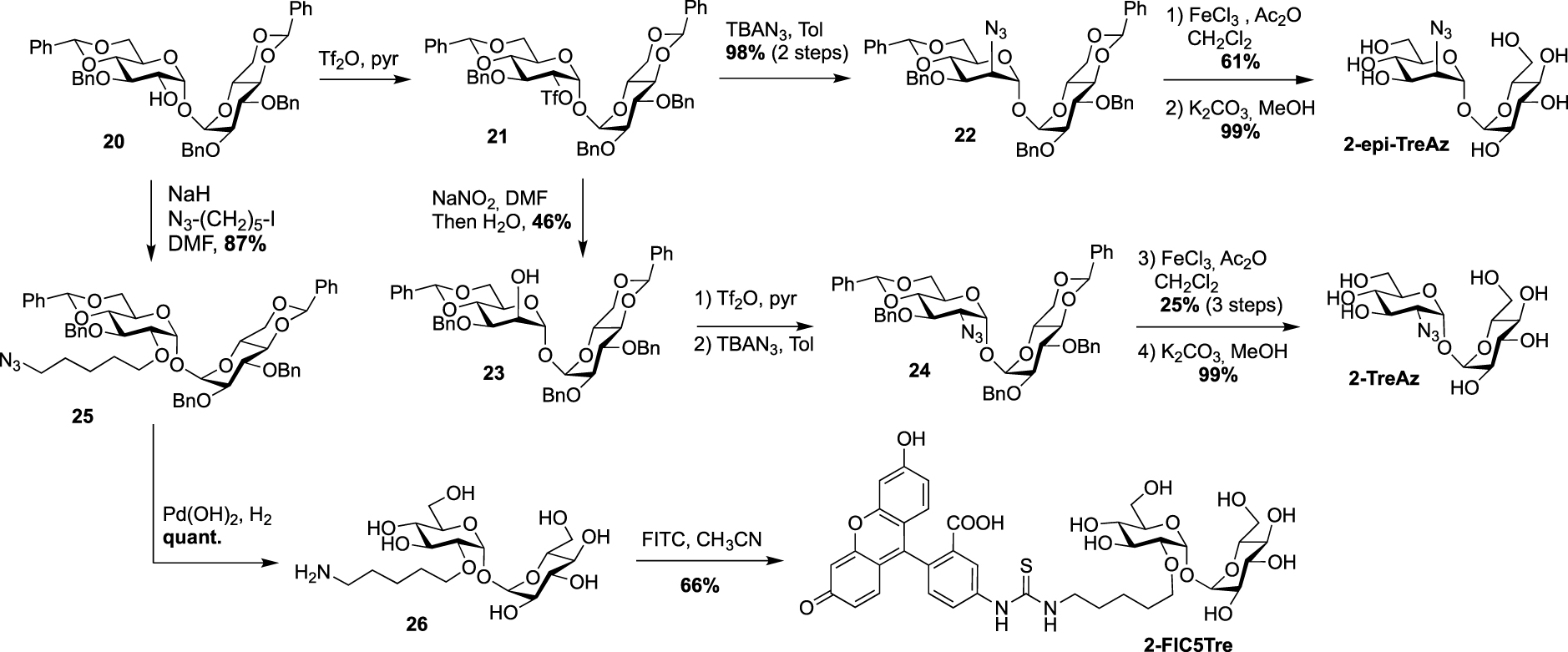
Recently, Bertozzi published the synthesis of new solvatochromic trehalose-based probes featuring 3-hydroxychromone (3HC) dyes, 3HC-3-Tre and 3HC-2-Tre, the first displaying a 10-fold increase in fluorescence intensity compared to the previous report on DMN-Tre [36]. These fluorescent probes are known to have high fluorescence quantum yields and they proved to efficiently label M. tuberculosis cells within 10 min of probe treatment. For these syntheses, the authors took advantage of the known desymmetrized and monobrominated derivative 5 previously described by Hanessian. Compound 5 was engaged in nucleophilic substitution with the two 3-hydroxychromones 27 and 28, providing mono-O-coupled derivatives 29 and 30. 3HC-3-Tre and 3HC-2-Tre were finally obtained after de-O-acetylation with sodium methoxide (Scheme 13).
Synthesis of 3HC-2-Tre and 3HC-3-Tre [36].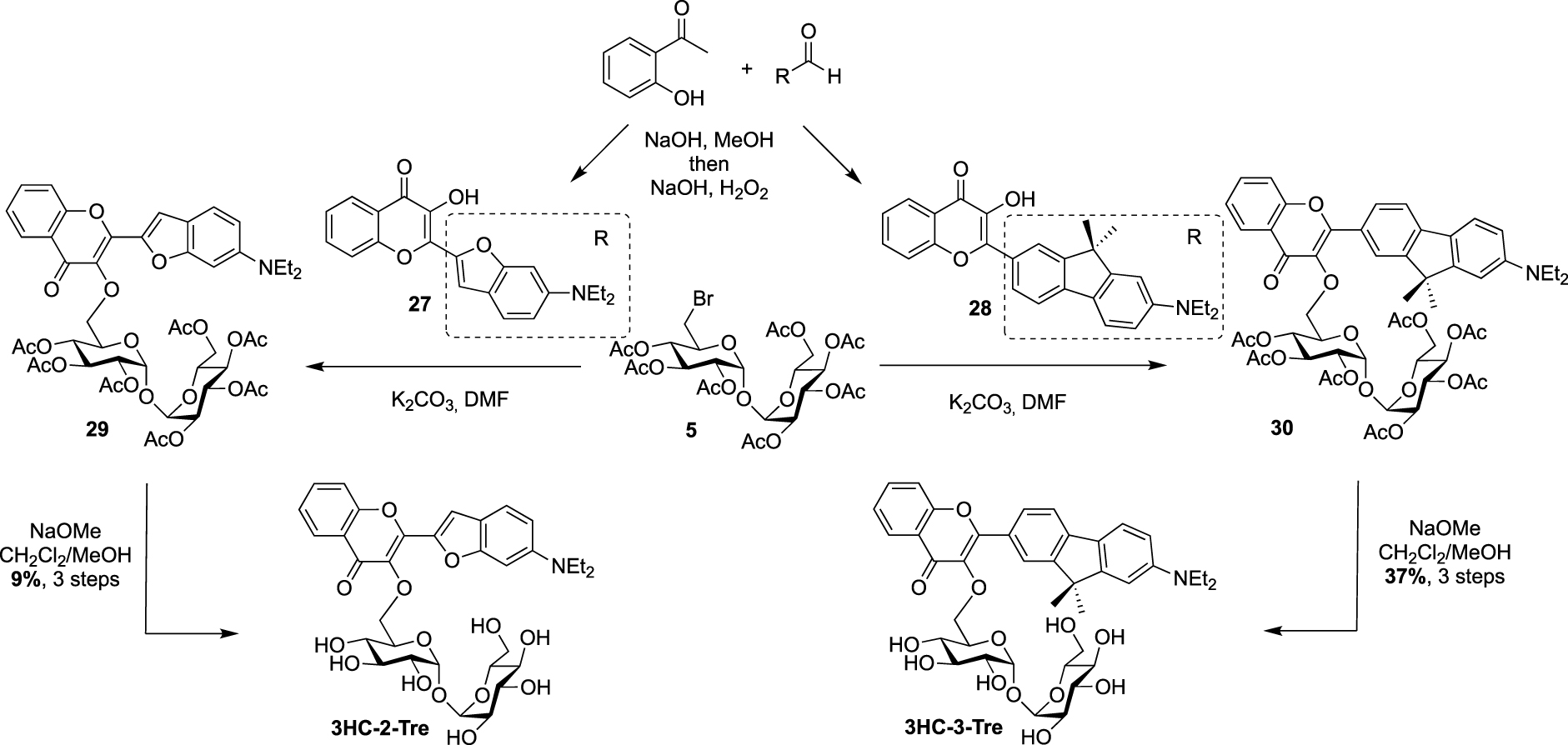
Very recently, Swarts’s lab added a new fluorogenic trehalose analogue to the trehalose-based probes collection [37]. This probe, RMR-Tre, featured a molecular rotor allowing turn-on far-red fluorescence sensor upon interaction with constraint environment such as the mycomembrane. Derivative 31, obtained after Knoevenagel condensation between the known aldehyde 32 and malonitrile, was coupled, using N,N,N′,N′-tetramethyl-O-(N-succinimidyl)uronium tetrafluoroborate (TSTU), to 6-amino trehalose 33 providing RMR-Tre in good yield (Scheme 14).
Synthesis of RMR-Tre [37].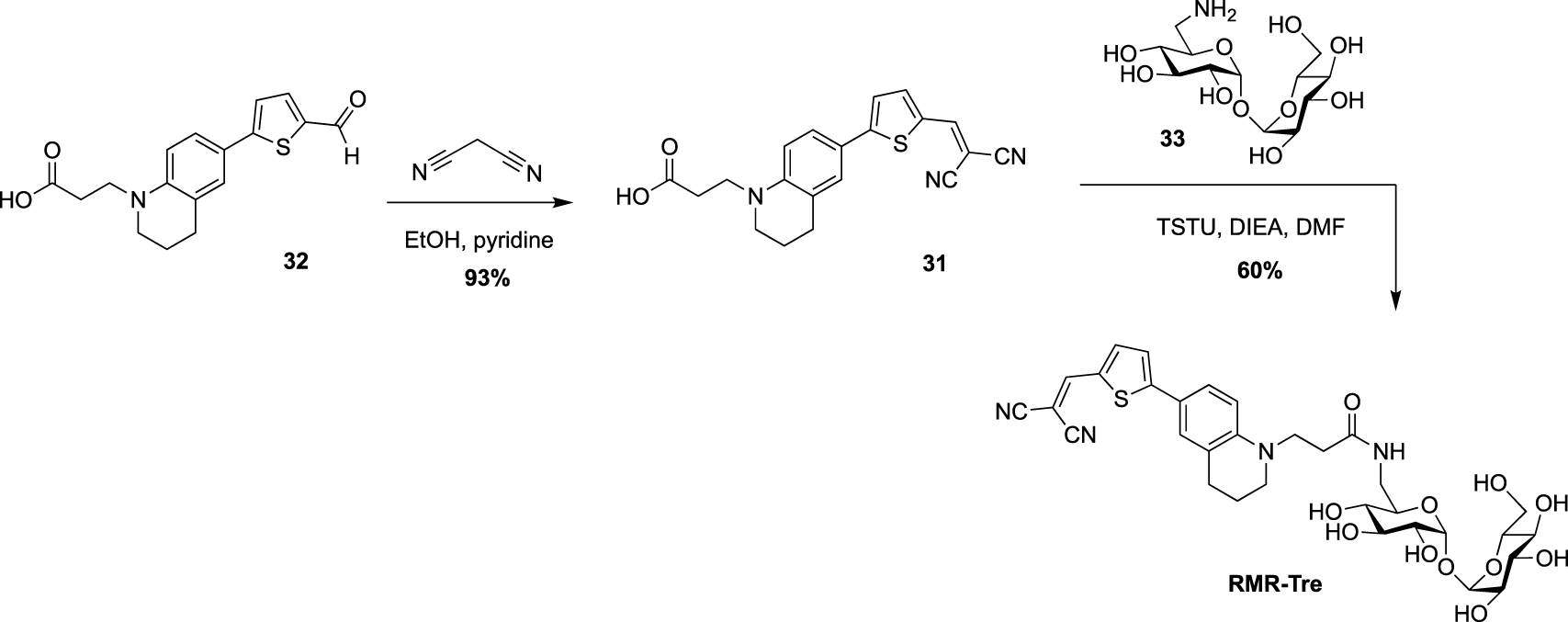
Finally, during the preparation of this review, Yan and coworkers reported the synthesis of a fluorescence turn-on probe Tre-Cz and its use for imaging mycobacteria after photoactivation of the aryl azide allowing the formation of the fluorescent product [38]. It was prepared starting from the known 6-amino-trehalose 33 and a N-hydroxysuccinimide(NHS)-functionalized carbazole derivative (Scheme 15).
Synthesis of Tre-Cz [38].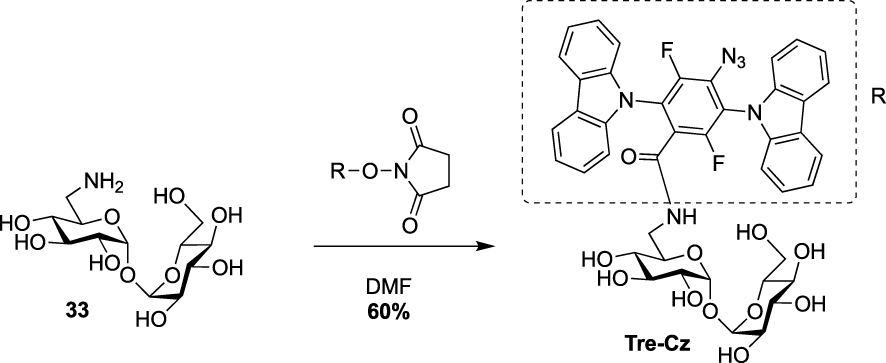
3. Synthesis of TMM-based probes
In parallel to trehalose-based probes, other useful chemical tools, i.e., acylated trehalose-based probes mimicking TMM, were synthesized to label the mycomembrane. In contrast to trehalose-based probes that are processed through the recycling trehalose pathway, TMM-based probes target mycoloyltransferases allowing the labeling of both the outer leaflet and the inner part of the mycomembrane (TDM and AGM layers, respectively).
From a synthetic point of view, different challenges have to be addressed for the preparation of TMM-based probes. The first one is the trehalose selective esterification of the lipidic part mimicking the mycolate chain, which relies on the difference of reactivity between primary and secondary alcohols. The most important challenge is the preparation of the mycolic acid pattern which implies (i) the control of the absolute configuration of its two stereogenic centers and (ii) the introduction of a bioorthogonal or a detectable tag on one of the long fatty acid chains. Some enantioselective syntheses of mycolic acids have already been reported in the literature (for a few examples see [39, 40, 41]), but the main difficulty for the preparation of such TMM-based probes is to make them compatible with a bioorthogonal or detectable moiety. Several teams have been interested in the synthesis of TMM-based chemical reporters and most of them reported simplified structures by replacing the complex native mycolic pattern by a simple acyl chain including the tag at the end. These probes were prepared using the same approach for the esterification of the lipidic chain of interest with trehalose. It relies on the per-O-silylation of trehalose followed by a controlled deprotection of the two primary positions leading to the symmetrical diol 34 according to a procedure reported by Toubiana [42]. Then, the deprotected derivative 34 is monoesterified in the presence of a coupling agent and the resulting ester is fully deprotected under acidic conditions (Scheme 16) [43].
Approach for selective synthesis of 6-O-acylated trehaloses.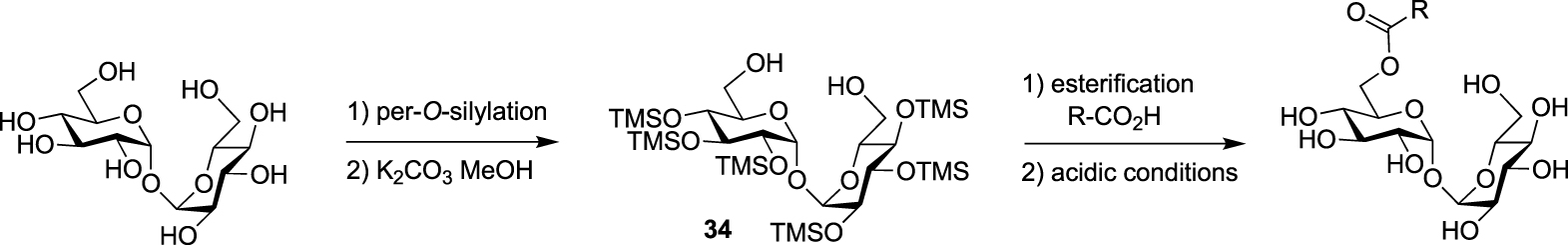
Synthesis of N- and O-AlkTMM [44].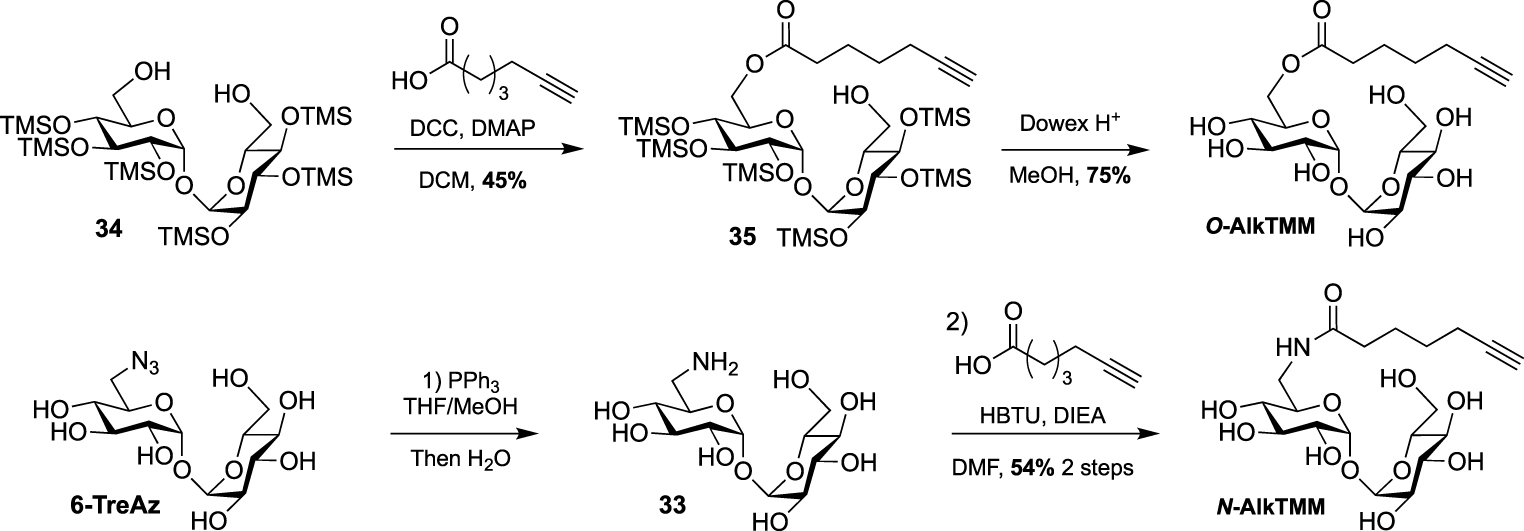
In 2016, Swarts took advantage of this approach and published the first bioorthogonal analogue related to TMM using commercially available 6-heptynoic acid as simplified mycolic acid mimic [44]. This analogue was obtained very efficiently by reaction between compound 34 and 6-heptynoic acid in the presence of dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP). The esterified intermediate 35 was then fully deprotected under acidic conditions leading to O-AlkTMM (Scheme 17). This simplified TMM analogue was successfully used for metabolic labeling experiments in M. smegmatis. During this study, 6-heptynoic acid was also used for the preparation of an amide-linked analogue. The known 6-TreAz was first converted to compound 33 using Staudinger reduction and the resulting amine was coupled to 6-heptynoic acid in the presence of O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) affording N-AlkTMM. The main difference between these two probes is the fact that after being processed by mycoloyltransferases, O-AlkTMM can label both the inner and the outer leaflets of the mycomembrane whereas N-AlkTMM can only label the outer part (i.e., the TDM layer).
A few years later, the same team extended the range of TMM-based probes by publishing a collection of new analogues differing by the chain length, the bioorthogonal group or by the introduction of a fluorophore [45]. On the basis of their previous work, the authors prepared the two other alkyne-TMM O-AlkTMM-C5 and O-AlkTMM-C11 with 5 and 11 carbon atoms on the lipidic moiety, respectively. Compound 34 was also esterified with 10-azido-decanoic acid providing O-AzTMM, an acylated trehalose with an azide as bioorthogonal group. This compound was finally converted to the corresponding amine allowing the coupling to a trans-cyclooctene N-hydroxysuccinimide ester (NHS-TCO) and to FITC, leading to O-TCO-TMM for tetrazine ligation and O-FITC-TMM respectively (Scheme 18). Very recently, the same lab expanded the far-red molecular rotor probe family by coupling compound 31 to intermediate 36 furnishing RMR-TMM (Scheme 18) [37].
Syntheses of O-AzTMM, O-TCO-TMM, O-FITC-TMM [45] and RMR-TMM [37].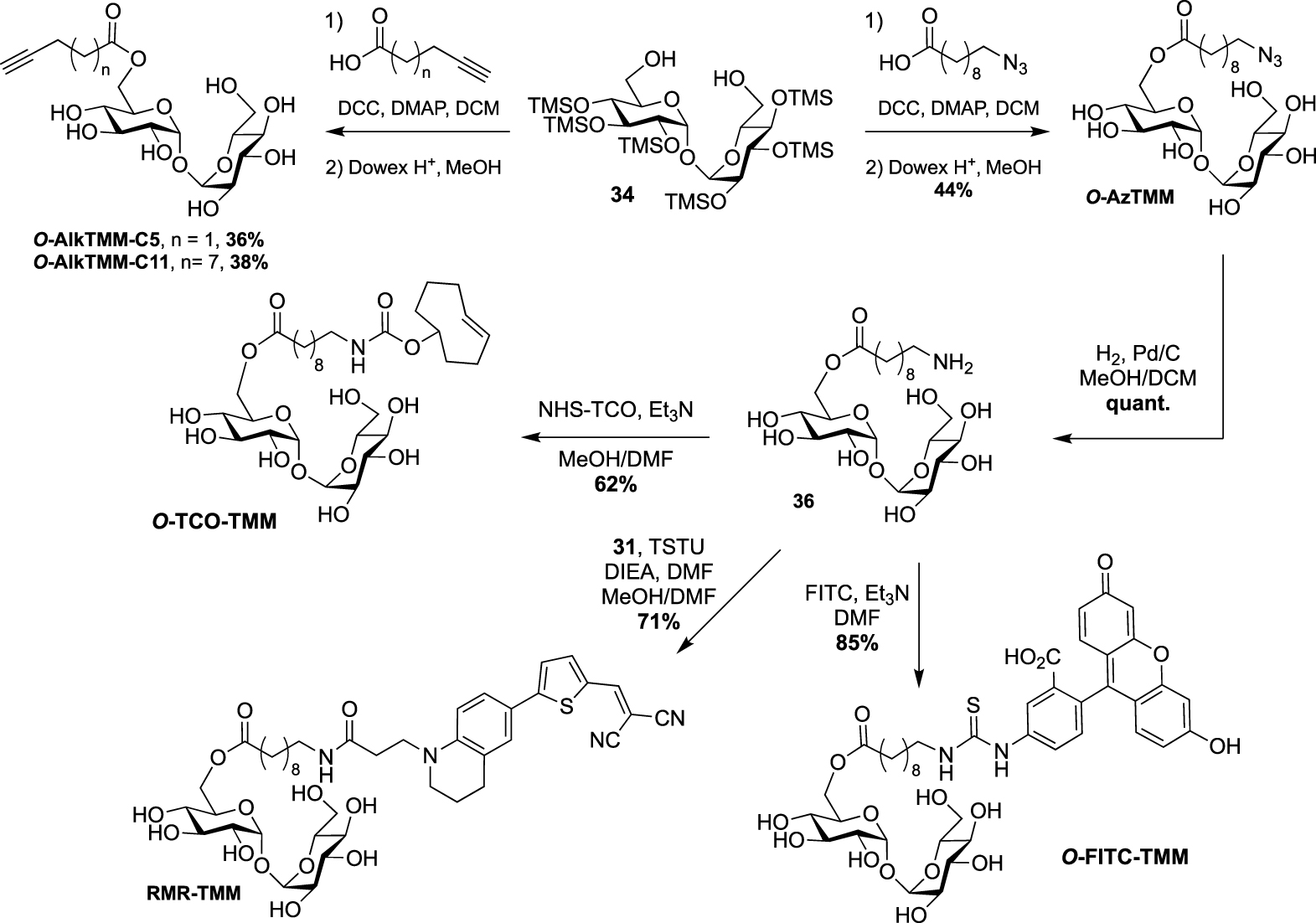
These studies have provided an impressive molecular diversity with different bioorthogonal or fluorescent moieties attached to the end of the ester-linked mono-acyl chain, but none of these included the native pattern of mycolic acids which represents an important synthetic challenge. In this way, Guianvarc’h and Bourdreux reported in 2019 the first access to trehalose monomycolate bioorthogonal probes closely mimicking the complex structure of native TMM [46]. Both Alkm13α0TMM and Alkm13α10TMM analogues featured an OH group in β position with an (R) stereochemistry (see Scheme 20). Moreover, Alkm13α10TMM also had the branched α-lipidic chain of mycolic acids, with an anti-relationship between the two α and β substituents. The β-ketoester 37, obtained after a two-carbon Masamune homologation of the corresponding alkyne-protected carboxylic acid, was engaged in an enantioselective reduction in the presence of Noyori’s catalyst (R)-BinapRuBr2. Under these conditions, the triisopropylsilyl protecting group prevented the undesired concomitant reduction of the alkyne. Thus, the expected β-hydroxyester 38, isolated in excellent yield and enantioselectivity, was then alkylated in the presence of 1-iododecane and hexamethylphosphoramide (HMPA) furnishing diastereoselectively 39 in a moderate yield. After n-tetrabutylammonium fluoride (TBAF) deprotection of the alkyne, the ester was saponified and the β-OH group was protected as triethylsilylether in anticipation of the future esterification with trehalose, providing carboxylic acid 42 (Scheme 19). During this study, the authors also prepared a bioorthogonal β-hydroxylated fatty acid lacking the α-side chain of mycolic acids. β-hydroxyester 38 was firstly desilylated, saponified and the β-OH group protected, leading to carboxylic acid 44.
Synthesis of the first mycolic acid with an alkyne bioorthogonal moiety [46].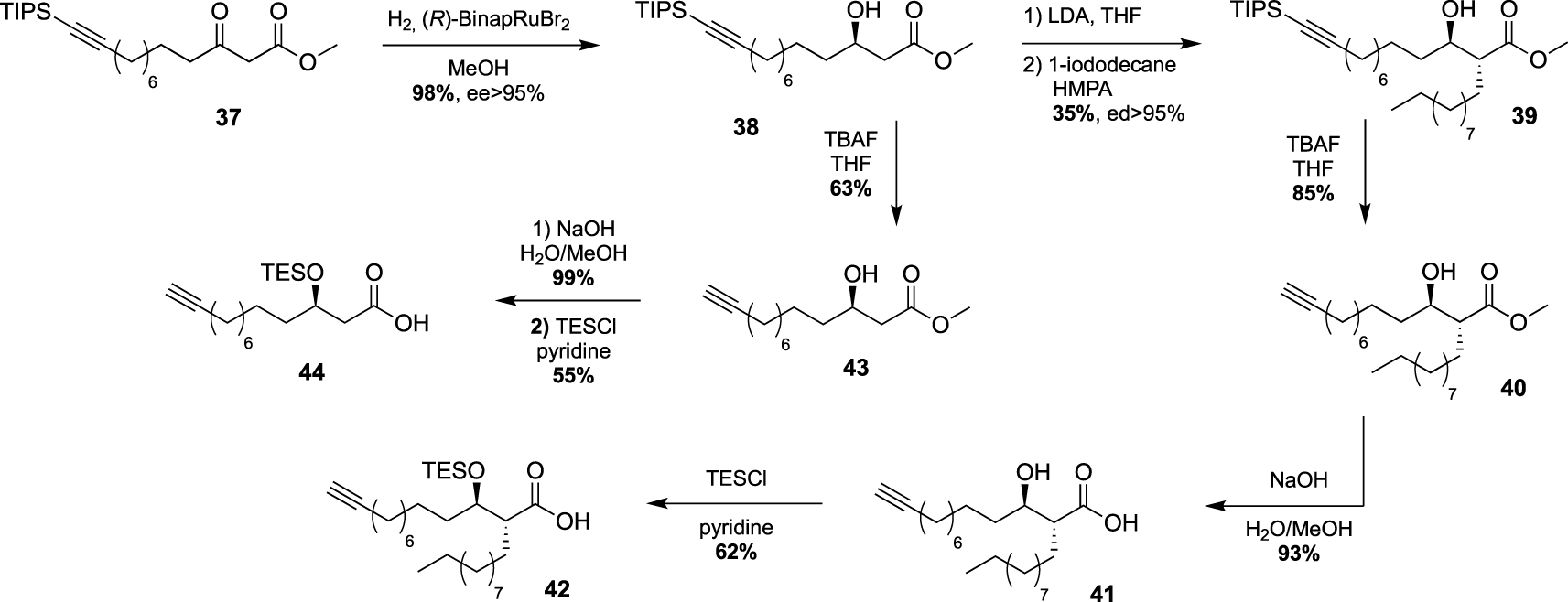
Synthesis of Alkm13α0TMM and Alkm13α10TMM [46].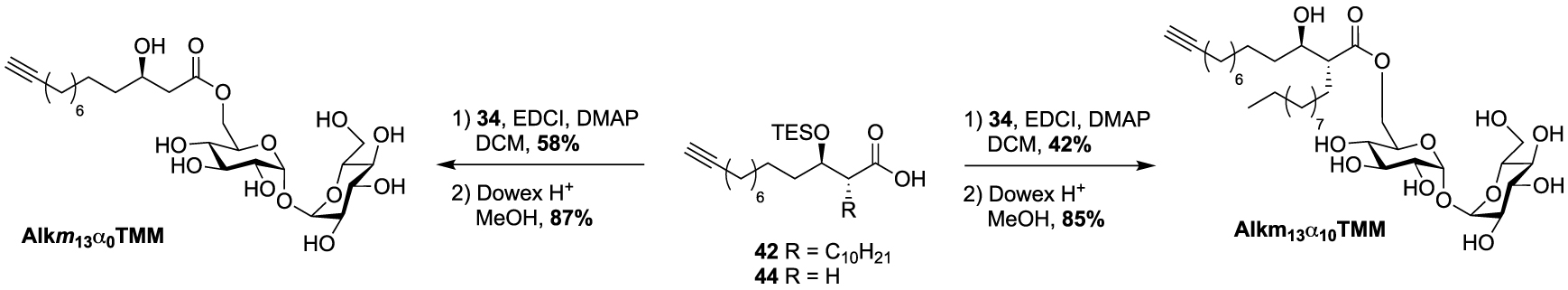
Synthesis of O-x-AlkTMM-C15 and N-x-AlkTMM-C15 [47].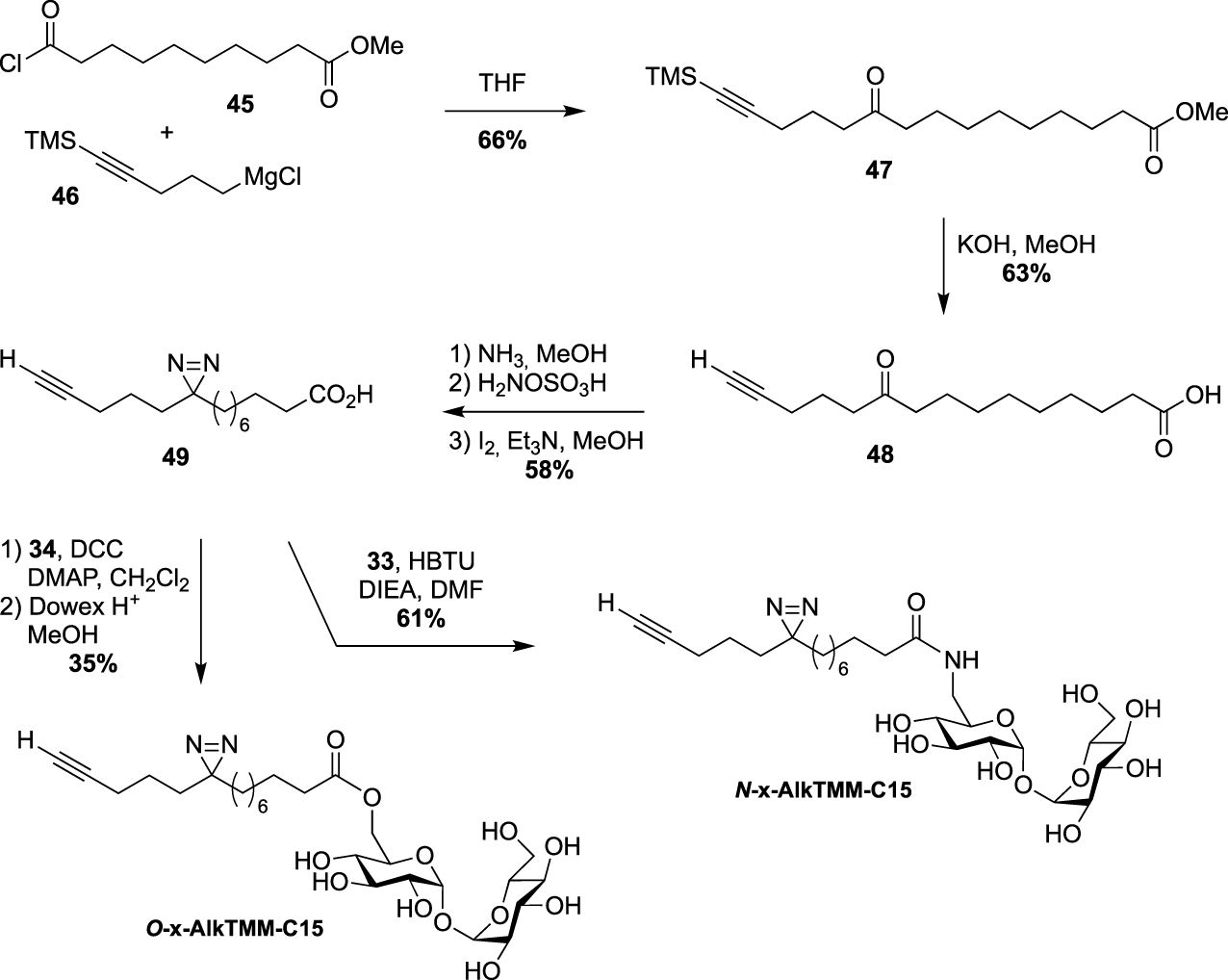
Finally, the two probes Alkm13α10TMM and Alkm13α0TMM were obtained using the same approach as previously described for the syntheses of the previous TMM analogues. Fatty acids 42 and 44 were esterified to trehalose 34 in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) and the resulting intermediates were fully deprotected under acidic conditions, providing the native TMM-based probes (Scheme 20). These two tools proved to be highly efficient for the labeling of the C. glutamicum mycomembrane even at low concentration. They are supposed to avoid the substantial acylhydrolase activity observed with unbranched lipid and to improve the native transfer activity of mycolate to its acceptors.
In 2020, Swarts proposed the TMM-based probes, O-x-AlkTMM-C15 and N-x-AlkTMM-C15, with an extra photoactivatable diazirine into the lipidic part in order to identify mycolate-protein interactions [47]. The synthesis of these tools relied on the preparation of the known bifunctional fatty acid 49 [48]. The latter was prepared by nucleophilic addition of the Grignard 46 to acyl chloride 45 and subsequent deprotections under alkaline conditions. The diazirine was then installed as previously reported [49] and coupled to trehalose derivatives 34 and 33 providing O-x-AlkTMM-C15 and N-x-AlkTMM-C15, respectively (Scheme 21).
A few years ago, Kiessling reported an ingenious strategy based on an observed acylhydrolase activity of mycoloyltransferases in the presence of TMM analogues with unbranched lipids [50]. She designed the fluorogenic QTF that is structurally closer to TDM rather than TMM analogues [51]. Indeed, one primary alcohol of trehalose is esterified to an acyl chain bearing a fluorescent BODIPY-based-probe, whereas the second primary alcohol is linked to a fluorescence quencher. When QTF is processed by mycoloyltransferases, the unbranched lipid bearing the fluorophore moiety can promote hydrolysis of the acyl-enzyme intermediate thus restoring the fluorescence. QTF was prepared by two successive selective esterifications with 4-dimethylaminoazobenzene-4-carboxylic acid (DABCYL-OH) and 15-azidopentadecanoic acid, respectively. Di-O-esterified compound 51 was next coupled to a fluorophore through CuAAC reaction leading, after deprotection, to QTF (Scheme 22).
Synthesis of QTF [51].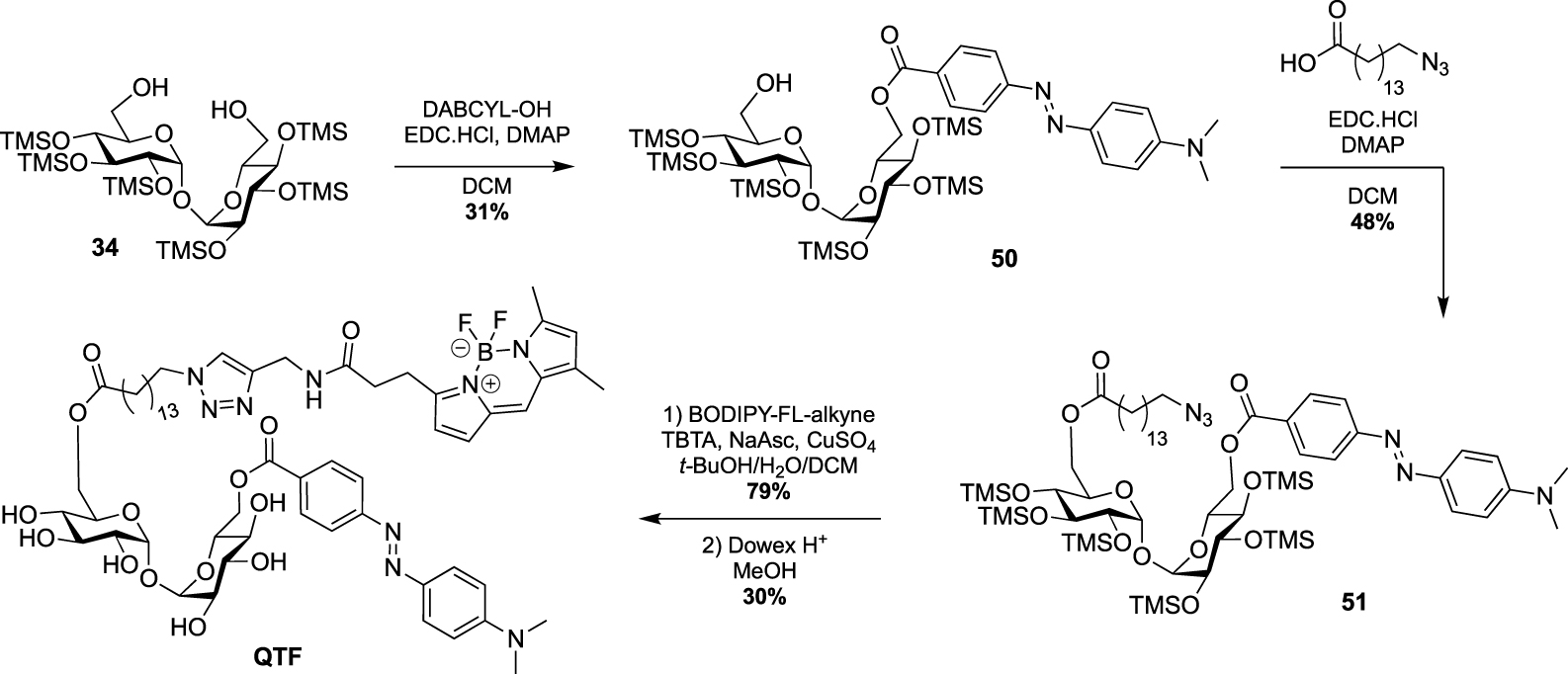
This strategy was next used by Swarts in a study targeting remodelling enzymes in mycobacteria, such as TDM hydrolase (Tdmh) [52]. The fluorogenic probe was obtained starting from 34 which was first fully esterified with 10-azidodecanoic acid in the presence of DCC and DMAP. The esterified derivative 52 was next converted to the diamine derivative 53 by reduction of the two azido groups. A fluorescein moiety was then introduced by treatment with FITC and the quencher of fluorescence was installed to provide the FRET-TDM probe (Scheme 23).
Synthesis of FRET-TDM [52].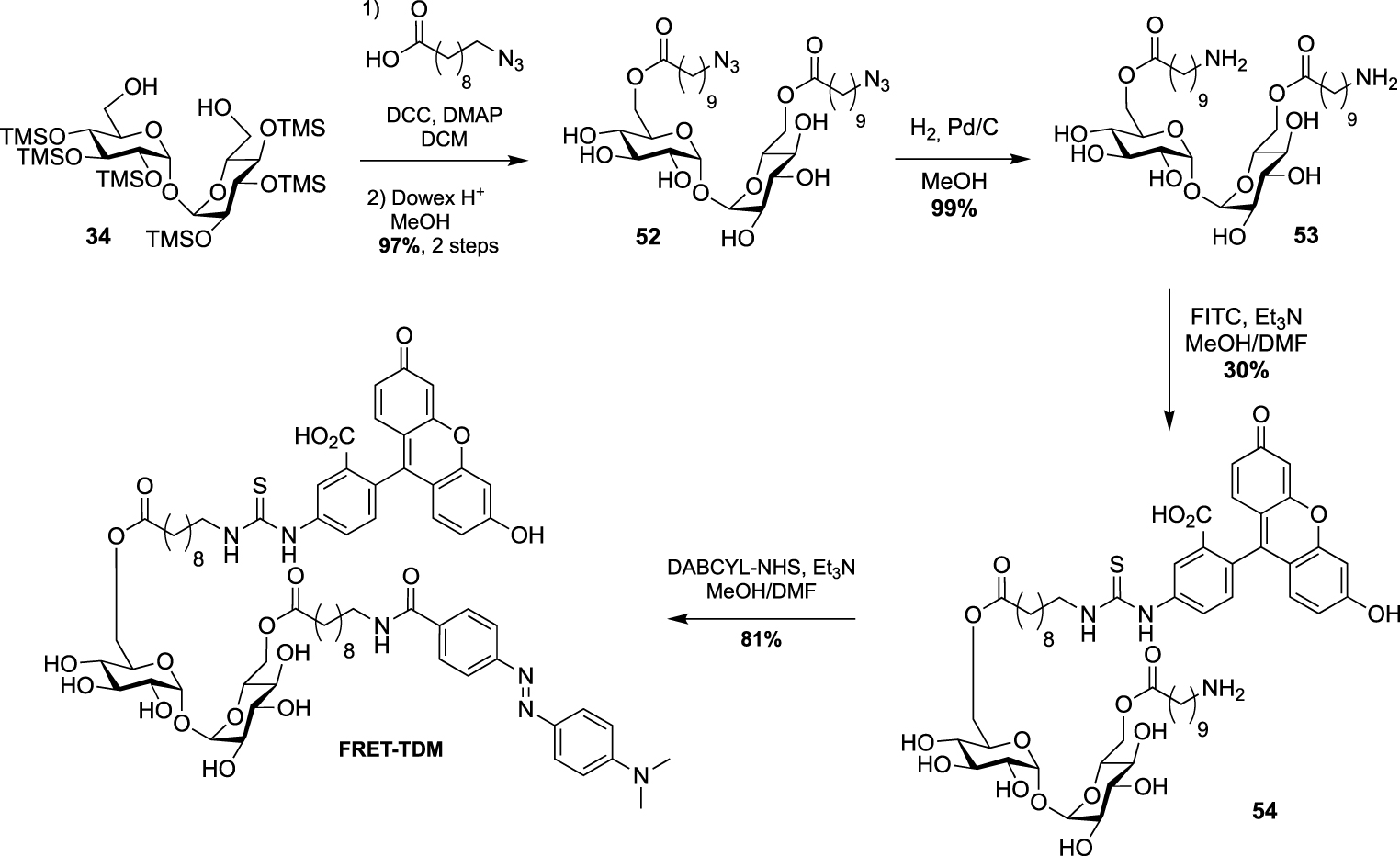
Synthesis of O-TMM-647, O-TMM-DBF [53].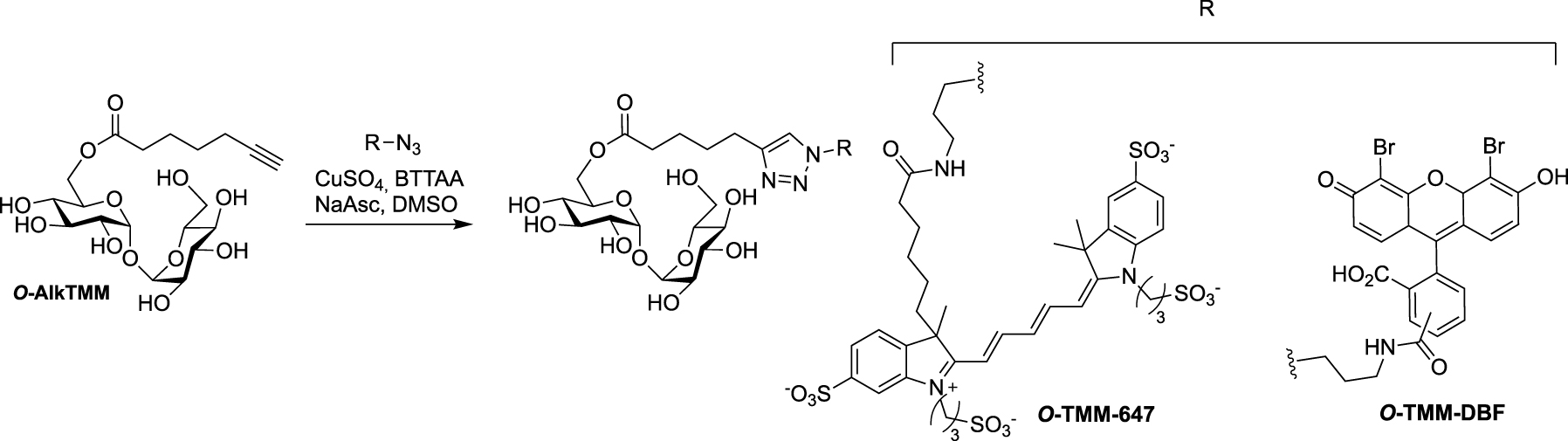
Following its description (see Scheme 17), O-AlkTMM was used as starting material for the preparation of the two additional fluorescent probes O-TMM-647 and O-TMM-DBF. Indeed, Theriot reported in 2019 the use of these two probes, synthesized by CuAAC reaction between O-AlkTMM and two different azide fluorophores, in a study on C. glutamicum (Scheme 24) [53].
Synthesis of O-AzAlkTMM [54].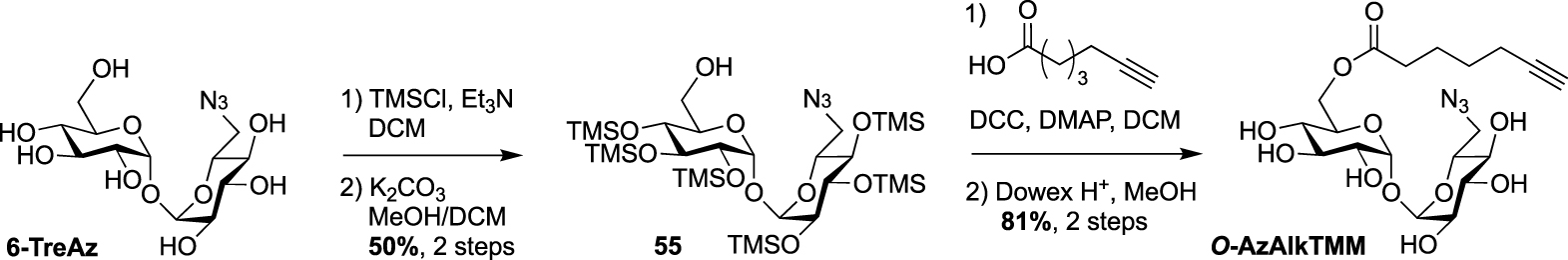
Finally, 6-TreAz and O-AlkTMM were merged giving rise to the bifunctional reporter O-AzAlkTMM [54]. It was synthesized using the approach used for most of the described TMM analogues as indicated in Scheme 16, starting from 6-TreAz. Indeed 6-TreAz was first per-O-silylated and then treated under mild basic conditions, leading to 55. The free alcohol of 55 was then esterified with 6-heptynoic acid and the resulting compound was fully deprotected to afford the expected bifunctional chemical reporter O-AzAlkTMM (Scheme 25) with an azide moiety on the trehalose core, and an alkyne on the lipidic chain mimicking the mycolic acid. These two bioorthogonal groups can react successively with two different fluorophores. Furthermore, the azide group can label only the TDM layer of the mycomembrane, whereas the alkyne moiety can label both the TDM and AGM layers. This bifunctional probe was used to study different metabolic processes by targeting mycoloyltransferase activities as well as trehalose recycling process.
Recapitulative table on the principal features and uses of trehalose-based probes described so far
| Classification | Probe name and referencea | Principal features |
|---|---|---|
| Fluorescein and tetramethylrhodamine conjugated trehalose | FITC-Tre [15] | ∙ First trehalose-based fluorescent probe with a C1 methyl group at the anomeric position ∙ Metabolic labeling experiments on growing Mtb and within infected macrophages ∙ Revealed a strong Ag85 plasticity |
| 2-,3-,4-,6-FITre [26] | ∙ First natural trehalose-based fluorescent probe ∙ More efficient in labeling than the FITC-Tre (Msmeg, Cglut) ∙ Applied to elucidate the mobility of labeled glycolipids in mycobacteria in several conditions |
|
| 2-FIC5Tre [34] | ∙ Fluorophore attached to trehalose through an ether-linker ∙ Enable efficient one step labeling of mycobacteria (Cglut) |
|
| 6-TMR-Tre [27] | ∙ Mycomembrane visualization with super-resolution microscopy ∙ Study of the mycomembrane and peptidoglycan dynamics using a dual metabolic labeling strategy (Msmeg, Mm, Cglut) |
|
| Azido-trehalose | 2-,3-,4-,6-TreAz [20, 29, 34] | ∙ First bioorthogonal trehalose for chemical reporter strategy ∙ Study of trehalose metabolism in live mycobacteria (Msmeg, Mtb, BCG) ∙ 2- and 6-TreAz more efficient |
| 6-MannoTreAz [31] 6-GalactoTreAz [31] |
∙ Good substrate tolerance toward mycoloyltransferases (Msmeg, BCG) |
|
| 2-epiTreAz [34] | ∙ Good substrate tolerance toward mycoloyltransferases ∙ Similar efficiency than 2-TreAz in labeling experiments (Cglut) |
|
| Trehalose conjugated to solvatochromic dye | DMN-Tre [28] | ∙ Sensitive to changes in the polarity of the medium ∙ 700-fold increase in fluorescence intensity in hydrophobic environment ∙ Does not require sample washing ∙ Strong potential to detect Mtb in patients’ sputum (Mtb, Msmeg, Mm) |
| 3HC-3-Tre [36] 3HC-2-Tre [36] |
∙ 10-fold increase in fluorescence intensity compared to DMN-Tre ∙ Efficiently label Mtb within 10 min of probe treatment (Msmeg, Mtb, Cglut) |
|
| RMR-Tre [37] | ∙ Rotor turn-on fluorophore, no-wash and fast detection of live cells ∙ Bright far-red emission and low-background fluorescence detection ∙ 100-fold enhancement in Mtb labeling compared to previous fluorogenic trehalose probes (Msmeg, Mtb) |
|
| Trehalose conjugated to off-on fluorescent dye activated by enzyme | CDG-Tre [32] | ∙ Release of the caged fluorophore conjugated to trehalose by the β-lactamase BlaC (Msmeg, BCG) ∙ low concentration for the labeling of phagocytosed live BCG within macrophages |
| NFC-Tre-5 [33] | ∙ Activated by the nitroreductase Rv2466 ∙ Specific labeling of single cells (Mmseg, Mtb, BCG) ∙ Strong potential for exploring the relationship between the host and pathogen |
|
| Trehalose conjugated to off-on fluorescent dye activated by light | Tre-Cz [38] | ∙ Photoactivation and detection using a handheld UV lamp (Msmeg, Mtb) ∙ Trehalose uptake pathway hijacked: intracellular incorporation through the LpqY-SugABC transporter |
| Trehalose functionalized with radioprobe | 18F-2-FDTre [30] | ∙ Positron emission tomography (PET) imaging to allow in vivo imaging of trehalose metabolism (Msmeg) |
aStructures of the probes are shown in Figure 3.
Recapitulative table on the principal features and uses of TMM-based probes described so far
| Classification | Probe name and referencea | Principal features |
|---|---|---|
| Bioorthogonal simplified analogs of TMM | O-AlkTMM [44] N-AlkTMM [44] |
∙ First bioorthogonal TMM analog with unbranched acyl chain, used in metabolic labeling experiments (Msmeg, Cglut) ∙ O-AlkTMM processed by mycoloyltransferases and enabling selective in situ detection of the mycomembrane components in living mycobacteria ∙ N-AlkTMM suitable to analyze the outer leaflet of the mycomembrane |
| O-AlkTMM-C5 [45] O-AlkTMM-C11 [45] O-Az-TMM [45] O-TCO-TMM [45] |
∙ Expansion of the set of TMM reporter analogs (Cglut, Msmeg) ∙ Rapid cell labeling (Msmeg) through tetrazine ligation |
|
| TMM mycolic acid-based bioorthogonal reporter | Alkm13α0TMM [46] Alkm13α10TMM [46] |
∙ First bioorthogonal TMM featuring the complex mycolic acid moiety ∙ Labeling of mycomembrane with a shorter labeling time and very low doses (Cglut) |
| Simplified analogs of TMM functionalized with a fluorescent dye | O-FITC-TMM [45] | ∙ First fluorescent TMM analog ∙ Enable one step labeling of live cells using TMM analog (Cglut, Msmeg) |
| RMR-TMM [37] | ∙ Far-red molecular rotor probe ∙ Enable no-wash and fast detection of live cells (Msmeg, Mtb) |
|
| O-TMM-647 [53] O-TMM-DBF [53] |
∙ Fluorescent TMM analog used to follow the assembly dynamics of the mycomembrane in live cells (Cglut, Msmeg) |
|
| Photoactivatable and bioorthogonal simplified analog of TMM | O-x-AlkTMMC15 [47] N-x-AlkTMMC15 [47] |
∙ First photoactivatable TMM analog enabling in vivo photo-cross-linking and click-chemistry-mediated analysis of mycolate-interacting proteins (Msmeg, Cglut) |
| Quencher-trehalose-fluorophore probe | QTF [51] | ∙ First FRET trehalose mycolate-based probe (Mtb, Msmeg, Cglut) ∙ Unbranched lipid bearing the fluorophore moiety to promote hydrolysis of the acyl-enzyme intermediate enabling real-time imaging in native cellular environment |
| FRET-TDM [52] | ∙ Unbranched TDM-based probe used to study remodeling enzymes such as Tdmh (TDM hydrolase) for in vitro and in vivo assays (Msmeg, Mtb) ∙ Probe also activated by hydrolases from other bacteria species |
|
| Bifunctional TMM chemical reporter | O-AzAlkTMM [54] | ∙ First bifunctional TMM-based probe ∙ Simultaneously marks mycomembrane biosynthesis by targeting mycoloyltransferases and subsequent trehalose recycling (Msmeg, Mtb) |
aStructures of the probes are shown in Figure 4.
4. Conclusion
In recent years, many smart probes have been designed and successfully used in metabolic labeling experiments to explore the mycomembrane, which is specific to mycobacteria and crucial for their survival. They have provided new insights into the mycomembrane biogenesis as well as potential new diagnostic tools for the detection of M. tuberculosis [7, 8]. A summary of the main features of these probes and their use is given in Tables 1 and 2 (see below). These trehalose-based probes, based on the selective trehalose functionalization, require important synthetic developments, especially in glycochemistry, in order to obtain more and more efficient tools, and we contributed to this exciting adventure. This review focused on the chemical challenges for the synthesis of the trehalose- and TMM-based probes described so far. Most of the reported trehalose-based analogues are modified at C6. Indeed, selective reaction on primary alcohols of trehalose is fast and convenient, and the C2-symmetry of trehalose makes the two primary positions equivalent. Modifications at C2, C3 or C4 are more challenging and require more chemical steps to retain an equatorial position on glucose unit. However, several studies have shown a great tolerance of mycoloyltransferases. Indeed, the synthesis of trehalose-based probes bearing the tag on axial position of the carbohydrate ring could make faster the access to C2, C3 or C4 modified trehalose-based probes, by suppressing double inversion protocols required to preserve the absolute configuration at C2, C3 or C4. The second major chemical reporters are mimics of TMM. In this case the same approach is used for most of the probes: a selective esterification on the same protected trehalose followed by acidic deprotection. The main difficulty for the preparation of such TMM-based probes is to synthesize the bioorthogonal functionalized mycolic acid part of this glycolipid. Most of the reported tools are simplified fatty acyl chain without ramification and β-OH group, thus making their syntheses fast and efficient. In a recent work, our group reported the first synthesis of a bioorthogonal TMM-based probes with the natural pattern of mycolic acid which was an important synthetic challenge. Interestingly, it proved to be very efficient in labeling C. glutamicum and such TMM-based probes may help to elucidate the mycomembrane biogenesis at the molecular level. Notably, they should help to decipher the enzymatic mechanism and specificity of mycoloyltransferases. Altogether, the molecular tools described here, in cooperation with biochemical approaches, could help to better understand the role of all mycoloyltransferases involved in the biogenesis of the mycomembrane, and may have a significant impact in the characterization of new targets to develop innovative therapeutic approaches.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgments
The authors thank the Ministère de l’enseignement supérieur et de la recherche for grants to EL and PR and for financial support. The authors also thank the Agence Nationale de la Recherche (PTMyco, grant NO. ANR-22-CE44-0005-03).
1 6-TreAz was also prepared in the study of Davis, Barry and coworkers, see reference [15].