



 CC-BY 4.0
CC-BY 4.0
1. Introduction
The introduction of fluorine atoms into biomolecules is now a well-established strategy in drug development [1, 2, 3, 4]. In particular, the incorporation of fluorinated amino acids and pseudopeptidic units into peptides is an outstanding tool for the modulation of their biophysical properties such as local hydrophobicity, membrane permeability [5] and metabolic stability [6]. Moreover, fluorine atoms can serve as highly sensitive probes for 19F NMR spectroscopy [7, 8]. The development of highly efficient syntheses of enantiopure fluorinated amino acids and pseudopeptide units is an essential prerequisite for their use in peptide chemistry [9, 10, 11, 12] and we present herein our main contributions in this area. Applications of the enantiopure fluorinated chiral auxiliary Fox will also be reviewed.
2. Synthesis of chiral fluorinated amino compounds
2.1. Amines, amino alcohols, diamines
Chiral amines and β-amino alcohols are very interesting compounds for biological use and for the design of chiral ligands or auxiliaries. This is why their asymmetric synthesis is a major topic of organic synthesis. Considering the interesting properties provided by fluorinated groups within biologically active molecules, our laboratory was interested in the synthesis of trifluoro- or difluoromethylated amines and amino alcohols in enantiopure form. One of the main synthetic strategies to produce non-racemic chiral amines and amino alcohols is the asymmetric Strecker reaction followed by the reduction of the so formed nitrile group. In 2010, our group described two efficient routes for the synthesis of both enantiomers of trifluoroalaninol in enantiopure form [13]. Starting from (R)-phenylglycinol derived fluorinated oxazolidines or imines, the first route relies on a Strecker type reaction whereas the more straightforward second one uses ethyl trifluoropyruvate as fluorinated starting material. This second more direct route involves, as a key step, the reduction of chiral oxazolidines or imines derived from ethyl trifluoropyruvate. The convenient separation of the two diastereomers of the common intermediate amino diol makes it possible to obtain trifluoroalaninols in enantiopure form in good yields. These two routes use common and cheap reactants and can be scaled up to gram quantities (Scheme 1).
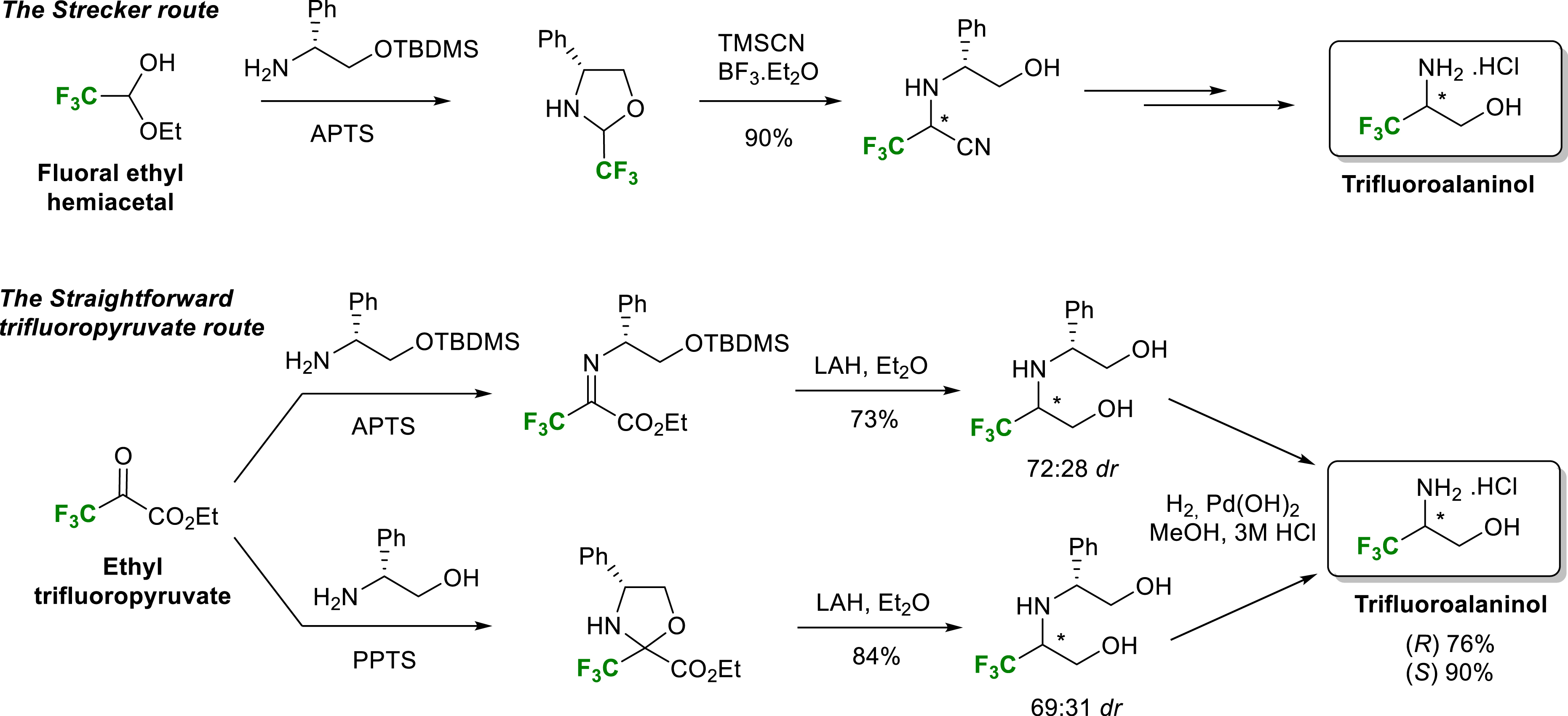
Synthesis of (R)- and (S)-trifluoroalaninol.
In order to provide simple and highly stereoselective access to β-alkyl β-trifluoromethyl β-amino alcohols, we decided to study addition reactions of organometallic species to a chiral 2-hydroxymethyl fluorinated oxazolidines [14]. These latter were obtained by chemoselective NaBH4 reduction of oxazolidine derived from trifluoropyruvate in very good yield as a 71:29 diastereomeric mixture. Each diastereomer could be easily isolated by chromatography on silica gel or by precipitation of the main diastereomer in pentane. The major diastereomer of reduced oxazolidine was treated with an excess of methyllithium at −78 °C in THF to give the amino diol (R) in 93% yield and full diastereoselectivity. We showed that the addition of methyllithium to the minor diastereoisomer gave the same amino diol (R) in 80% yield and 90% diastereoselectivity.
We therefore considered that the same transition state should be involved in the addition reaction of methyllithium to the two diastereomers of reduced oxazolidines. This result was confirmed by the fact that the addition of methyllithium to a diastereomeric mixture of oxazolidines also gave the unique diastereomer (R) with excellent diastereoselectivity. More generally, the addition of organometallic species to fluorinated oxazolidines made it possible to obtain, after cleavage of the phenylglycinol chain, quaternarized β-amino alcohols in the β position with good yields and excellent diastereoselectivity (Scheme 2).
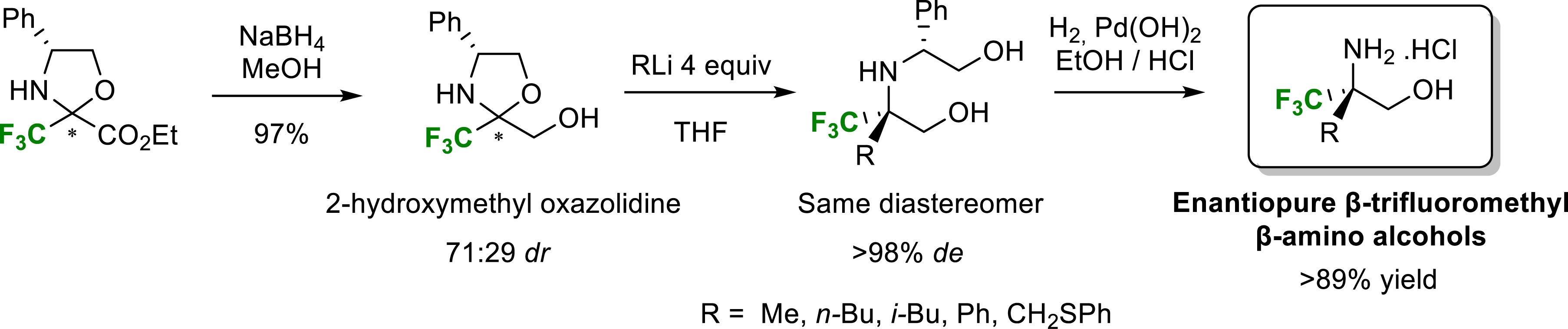
Synthesis of β-trifluoromethyl β-amino alcohols.
Our group has also developed a straightforward synthetic route for the synthesis of both enantiomers of various enantiopure diamines and amino alcohols from trifluoromethyl α-amino nitriles as key intermediates [15]. The Strecker-type reaction with TMSCN required Lewis acid activation of fluorinated imines or oxazolidines, and the reaction was promoted very efficiently under mild conditions with a catalytic amount of Yb(OTf)3. Despite the moderate diastereoselectivity of the Strecker-type reaction, the high efficiency of the chromatographic separation of each α-amino nitrile diastereomer assisted by the (R)-phenylglycinol side chain allowed the very convenient synthesis of enantiopure compounds (Scheme 3).
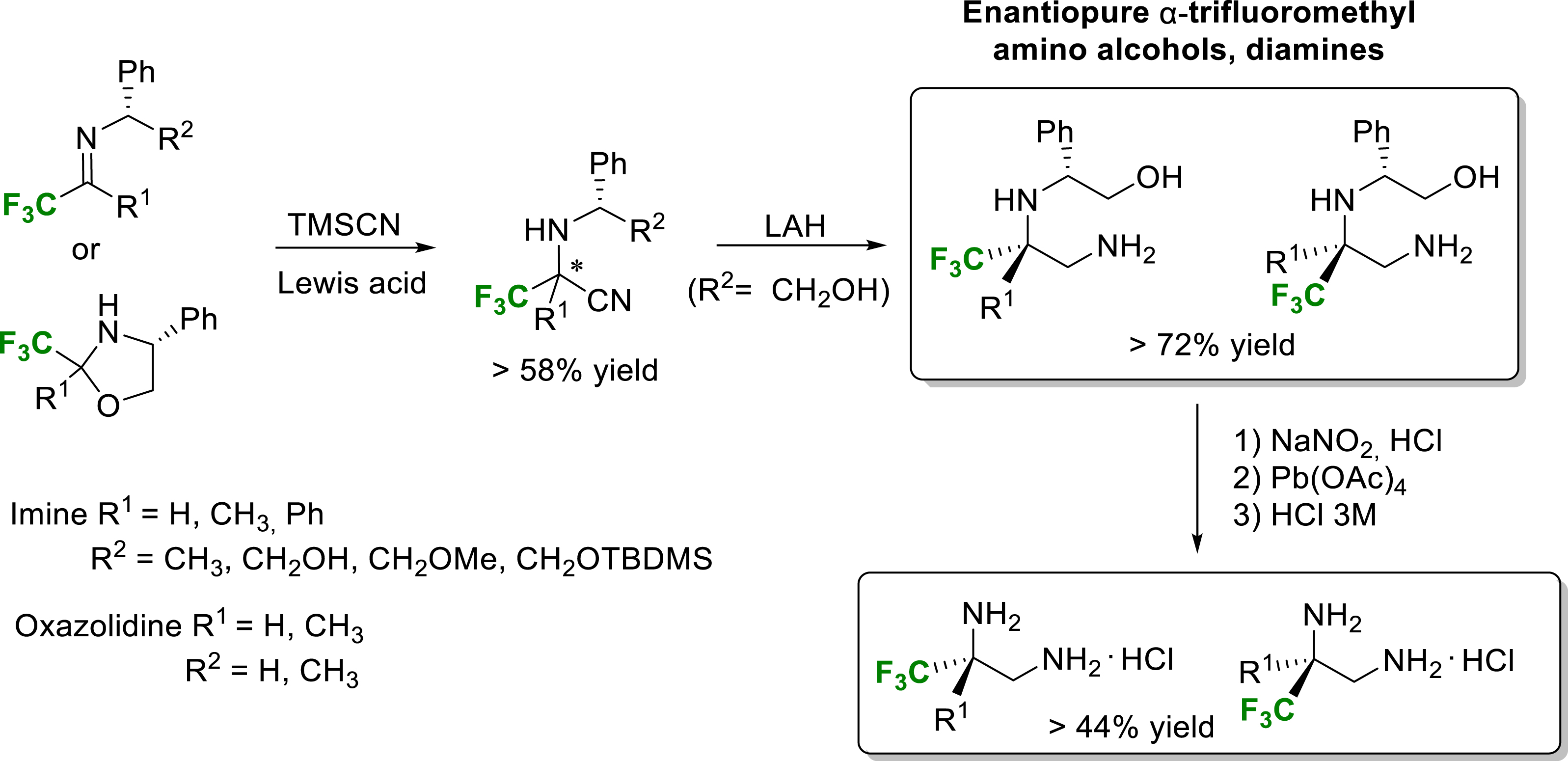
Synthesis of enantiopure α-trifluoromethyl diamines and amino alcohols.
In 2005 we proposed a highly stereoselective method for the synthesis of α-trifluoromethyl amines [16]. Indeed, we have shown that organolithium and Grignard reagents add efficiently with very high stereoselectivity (>98% de) to a trifluoroacetaldehyde hydrazone derived from (R)-N-benzylphenylglycinol. The addition proceeds on the Re face of the chelated hydroxyhydrazone providing quantitatively the (R)-α-trifluoromethylated amine after hydrogenolysis (Scheme 4).
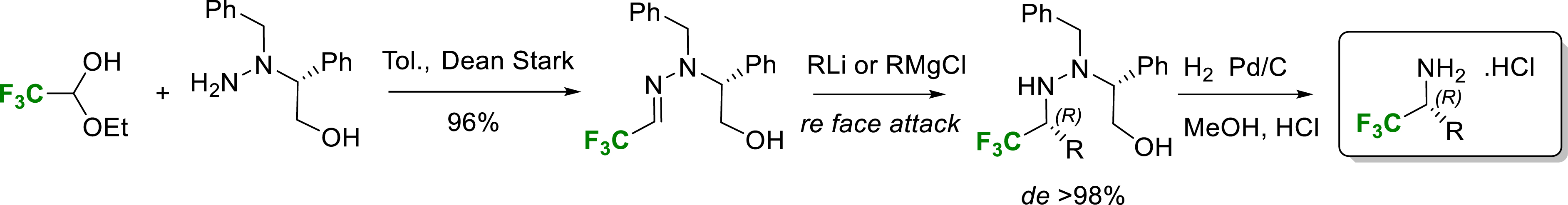
Synthesis of enantiopure α-trifluoromethylamines.
In 2008, our group reported a synthesis of enantiopure α-difluoromethylamines [17]. The silylated difluoroenols formed in a first step can behave as electrophilic reagents with amino alcohols, allowing the formation of oxazolidines. The use of (R)-phenylglycinol therefore made it possible to prepare 2-difluoromethyloxazolidines which proved to be good precursors of enantiopure α-difluoromethylamines by lithium aluminium hydride (LAH) reduction and oxidative cleavage of the intermediate amino alcohols (Scheme 5). The key 2-difluoromethyloxazolidine intermediates are prepared in high yield from acylsilanes, treated successively with trifluoromethyltrimethylsilane and the amine reagent.
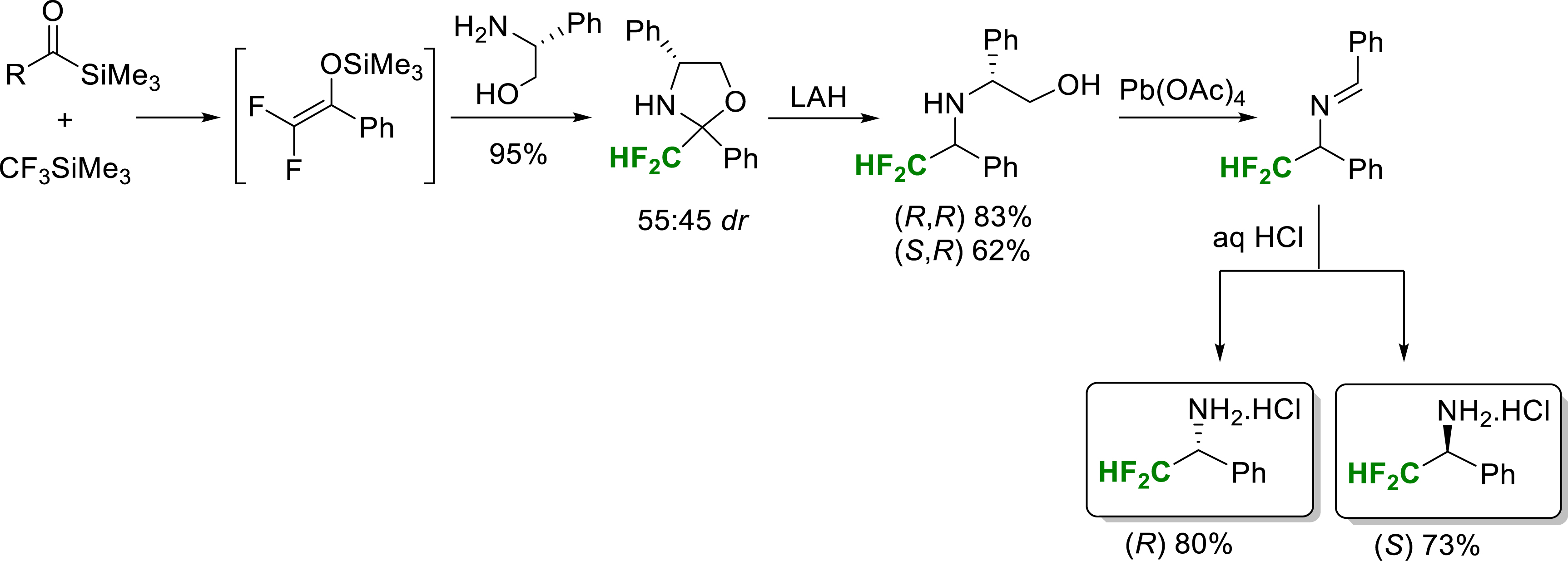
Synthesis of enantiopure α-difluoromethylamines.
2.2. Amino acids
2.2.1. Fluoroalkylamino acids
2.2.1.1. Synthesis of acyclic α-fluoroalkyl non proteogenic amino acids
Over the past 20 years, our group has devoted a great deal of effort to developing a general methodology for accessing non-proteogenic fluoroalkyl amino acids. These studies have been carried out for the synthesis of non-proteinogenic α- or γ-trifluoromethylated or α-difluoromethylated amino acids, whether cyclic or linear. In most cases, the Strecker-type reaction and the use of a chiral inducer such as the commercially available phenylglycinol have been used for several reasons:
- The Strecker-type reaction provides the nitrile function precursor to the carboxylic function of the expected amino acids.
- Phenylglycinol enables the expected enantiopure amino acids to be synthesized.
- It provides the nitrogen atom of the expected amino acid.
- The phenylethanol group on the nitrogen atom can be easily removed by acid hydrolysis or hydrogenolysis at the end of synthesis, at the same time as hydrolysis of the nitrile function to carboxylic acid.
- The source of fluorinated groups is commercially available and therefore easily accessible.
In 2006, an allylation reaction on trifluoromethylated oxazolidine obtained by condensation of trifluoropyruvate with (R)-phenylglycinol enabled the construction of amino acid side chains and the synthesis of enantiopure α-Tfm-α-allylglycine and α-Tfm-norvaline thanks to possible separation of diastereomers during the morpholinone formation step [18]. The pure major diastereomer was then converted to the enantiopure (S)-α-Tfm-norvaline via two hydrogenolysis steps (Scheme 6).
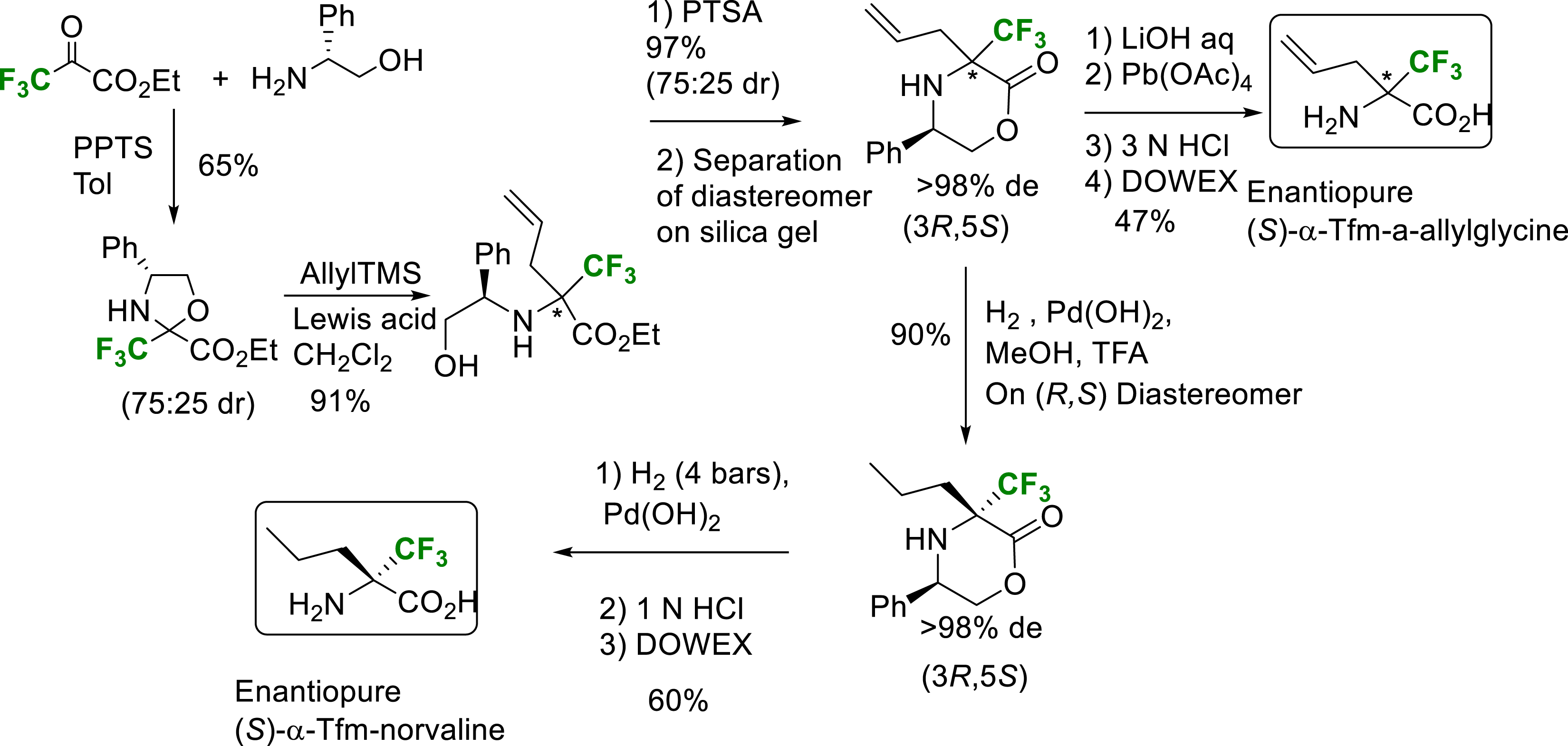
Synthesis of enantiopure (S) or (R)-α-Tfm-allylglycine and (S)-α-Tfm-norvaline.
In 2011, the Strecker-type strategy was implemented for the synthesis of enantiopure (S)- and (R)-α-Tfm-serine and α-Tfm-aspartic acid in good yields (Scheme 7) [19]. The ethyl trifluoropyruvate based oxazolidine was reduced and submitted to the Strecker-type reaction to provide the corresponding hydroxymethyl amino nitriles. After separation of each diastereomer and acidic hydrolysis, enantiopure (S)- and (R)-α-Tfm-serine were obtained in good yields. A similar strategy starting from the oxazolidine derived from ethyl 4,4,4-trifluoroacetoacetate provided enantiopure (S)- and (R)-α-Tfm-aspartic acid in good yields (Scheme 7) [19].
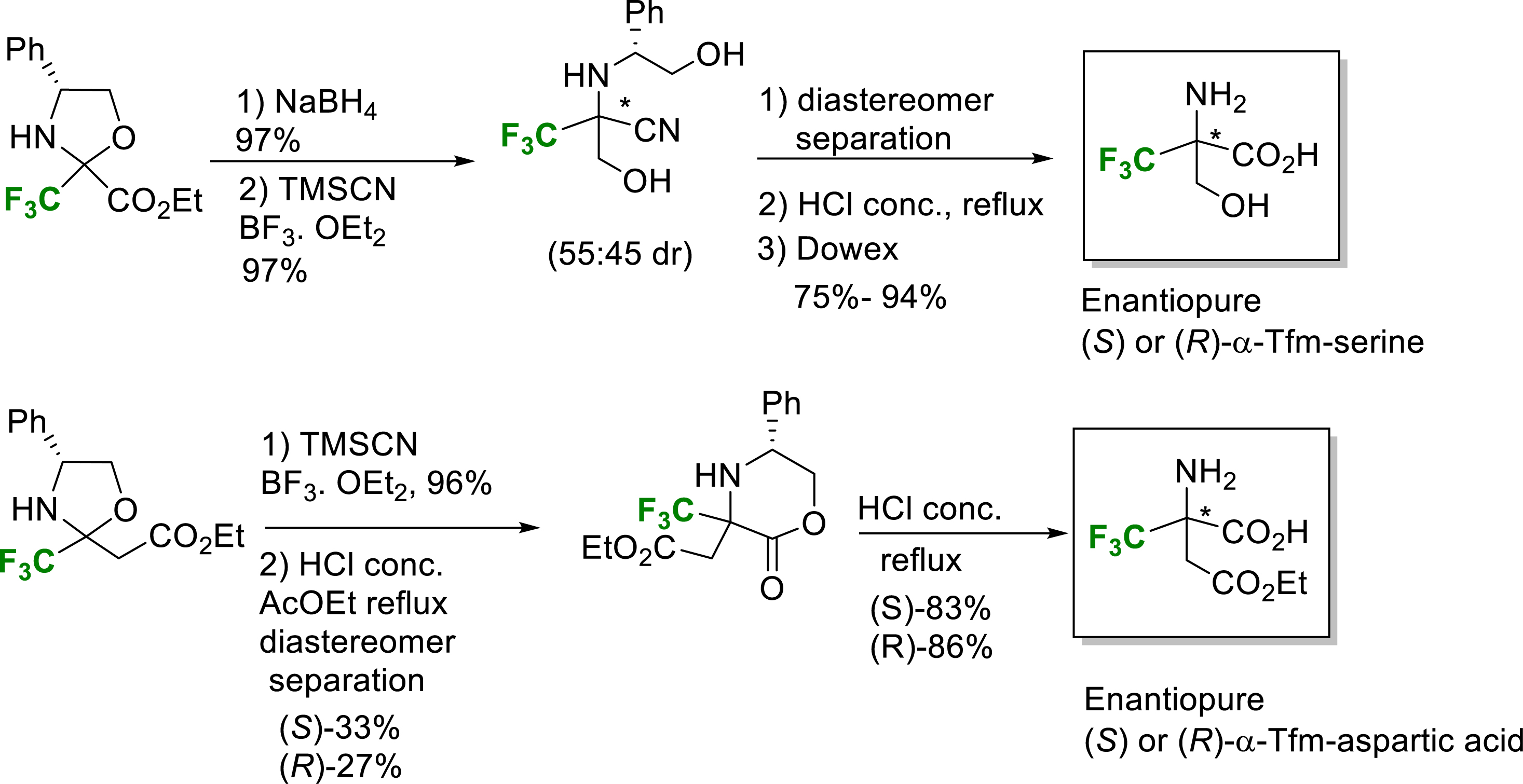
Synthesis of enantiopure (S) or (R)-α-Tfm-aspartic acid and (S) or (R)-α-Tfm-serine.
Enantiopure α-Tfm-homoserine has also been obtained as a ring-opening product during the synthesis of 2-trifluoromethyl-2-carboxyazetidine (Scheme 8) [20].
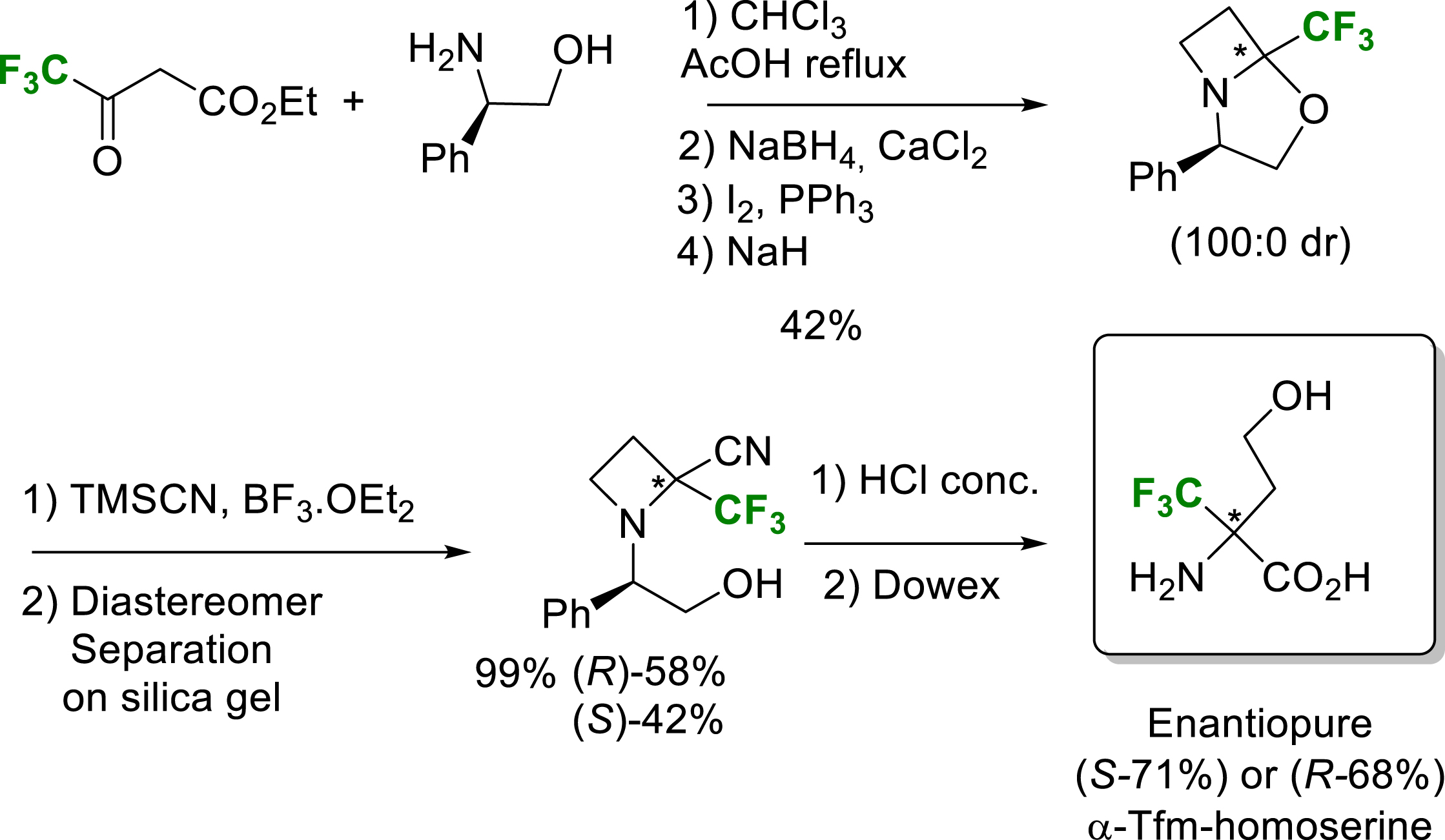
Synthesis of enantiopure α-Tfm-homoserine.
The laboratory was also interested in the synthesis of difluorinated amino acids. This work was initiated in 2008, with the synthesis of enantiopure (S) or (R)-α-difluomethylalanine from the condensation of acylsilanes and CF3TMS, via the in-situ formation of silyl ethers of difluoroenol. Condensation of this difluoroenol silyl ether with the (R)-phenylglycinol enabled the preparation of diastereomerically pure 2-difluoromethyl oxazolidines. A Strecker type reaction followed by an oxidation led to chiral cyanodifluoroimines. Acid hydrolysis gave access to the expected enantiopure (S) or (R)-α-difluoromethylalanine (Scheme 9) [17].
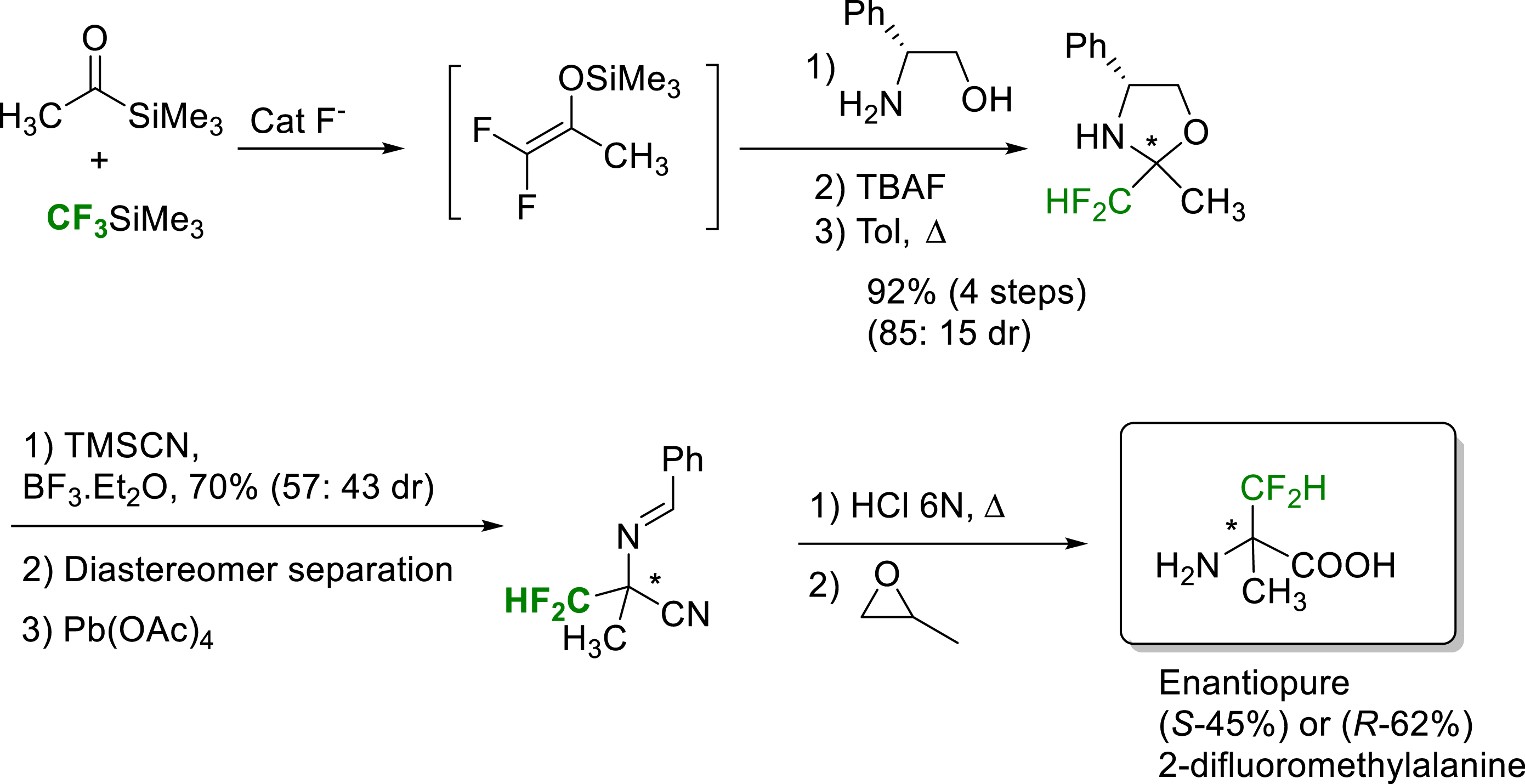
Synthesis of enantiopure (S) or (R)-α-difluoromethylalanine.
In 2023, we reported a synthesis of enantioenriched (>90% ee) N-Fmoc-β,β-difluoroalanine employing the Strecker’s strategy from a chiral difluorinated aldimine. The Strecker reaction produced two diastereomeric amino nitriles which were separated. Subsequent selective hydrolysis reactions yielded the final two enantiomers of difluoroalanine in good overall yield with an enantiomeric excess superior to 90% (Scheme 10) [21].
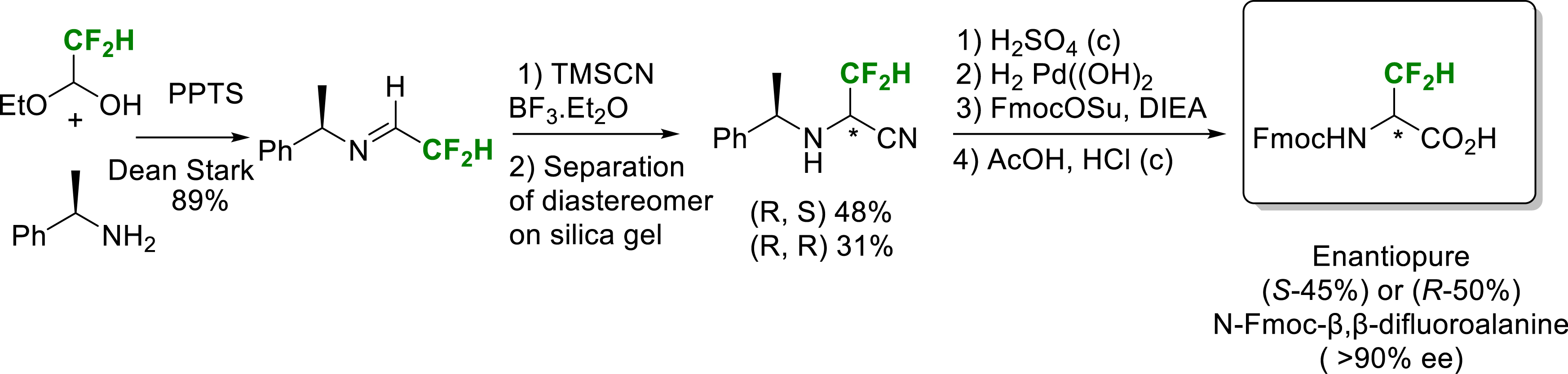
Synthesis of enantiopure (S) or (R)-N-Fmoc-β,β-difluoroalanine.
2.2.1.2. Synthesis of cyclic α-fluoroalkyl non proteogenic amino acids
Another area of research was the synthesis of more constrained cyclic α-trifluoromethylated amino acids. The synthesis routes were the same as those used for acyclic ones. The main strategy is based on the condensation of various commercially available trifluoromethylated ketoesters with enantiopure (R)-phenylglycinol. The corresponding chiral oxazolidines were obtained in good yields. Through various chemical transformations, monocyclic and bicyclic intermediates were synthesized. To introduce the nitrile group, precursor of the carboxy group, a Strecker-type reaction assisted by boron trifluoride Lewis acid on these chiral cyclic intermediates was chosen. While these Strecker type reactions proceeded in very high yields, only modest diastereoselectivities were probably obtained due to the passage through an iminium intermediate. Nevertheless, the diastereomeric mixture was resolved via silica gel separation to obtain enantiomerically pure batches of each amino nitrile. Once these aminonitriles synthesized, it was necessary to find the conditions for nitrile hydrolysis and protection of the amine function (Scheme 11).
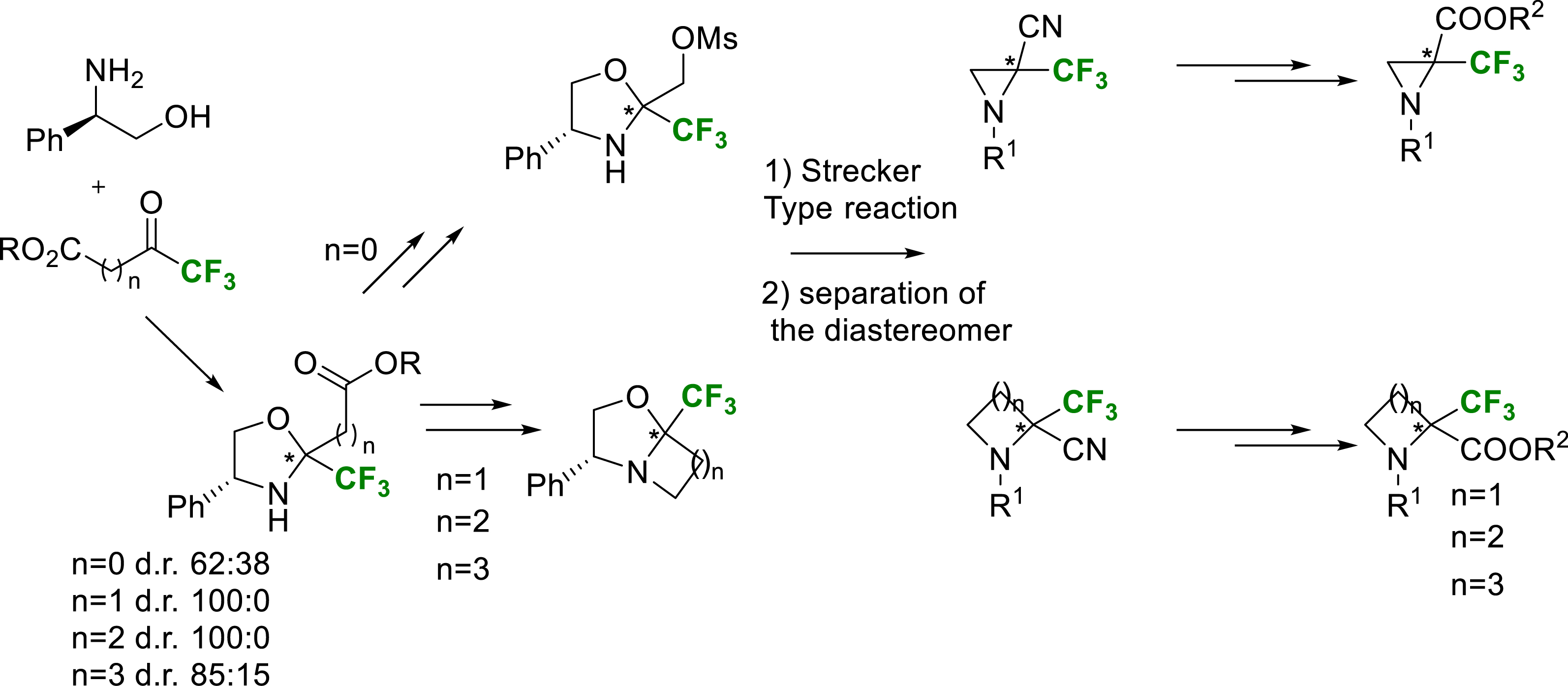
General pathway for the synthesis of enantiopure cyclic (R) or (S)-α-Tfm-N-protected amino acids.
In the case of aziridine, strong acid conditions led to the aziridine ring opening and formation of a complex mixture. Nevertheless, after mesylation of the primary alcohol function of oxazolidine followed by a Strecker-type reaction and separation of the diastereomer, the expected (R,S)-N-protected α-trifluoromethyl aziridine-2-carboxylic acid (α-TfmAzy) was obtained after basic treatment (Scheme 12) [22].
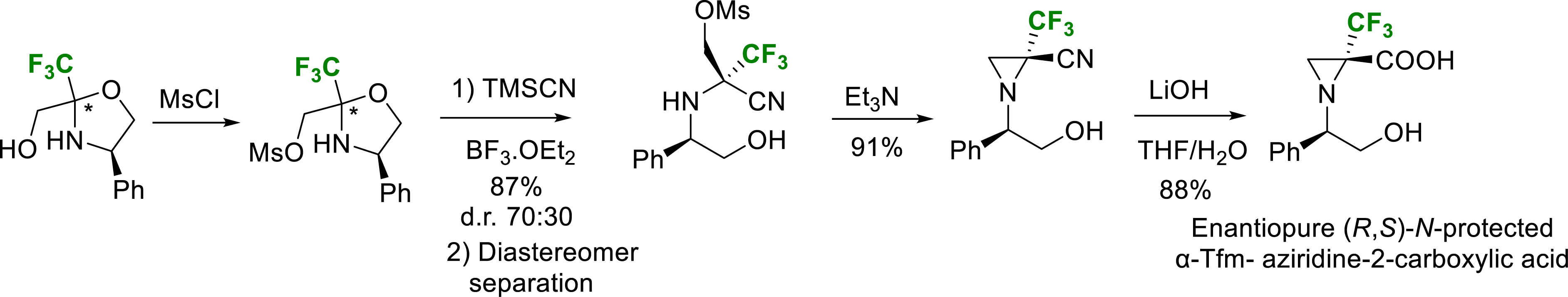
Synthesis of enantiopure N-protected-α-Tfm-aziridine 2-carboxylic acid.
Another strategy to access these enantiopure α-TfmAzy was via the intramolecular cyclization of enantiopure (S) or (R)-α-trifluoromethyl serine derivatives already synthesized in the laboratory (Scheme 13) [22].
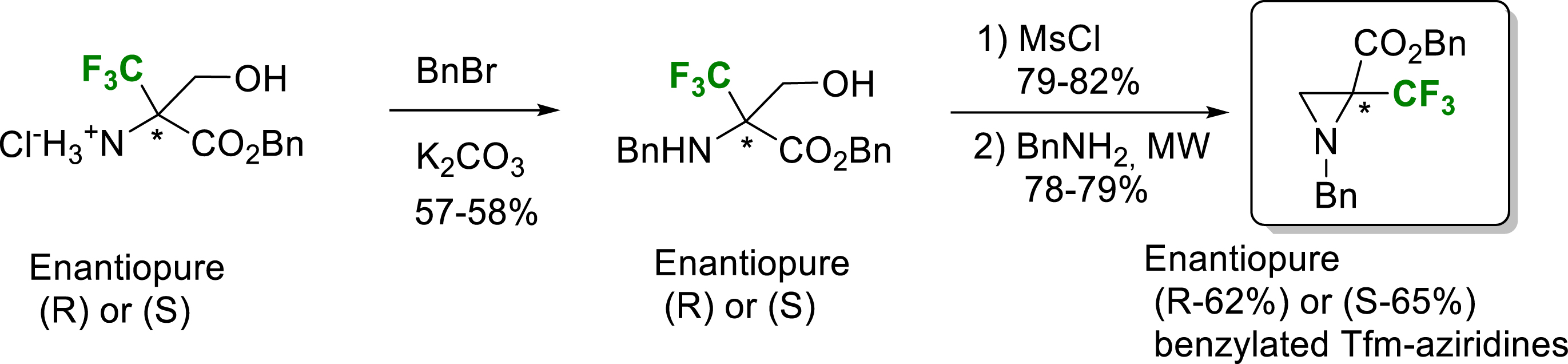
Synthesis of enantiopure N-Bn-α-Tfm-aziridine 2-benzyloxycarbonyl.
For α-trifluorometylated azetidine, the use of strong acidic conditions cleaved the azetidine ring to give α-trifluoromethylated homoserine (see Scheme 8). Milder conditions were found, and the synthesis via an intermediate bicyclic compound followed by hydrogenolysis gave access to the enantiomerically pure (2R)-2-trifluoromethyl-2-carboxyazetidine (Scheme 14) [20].
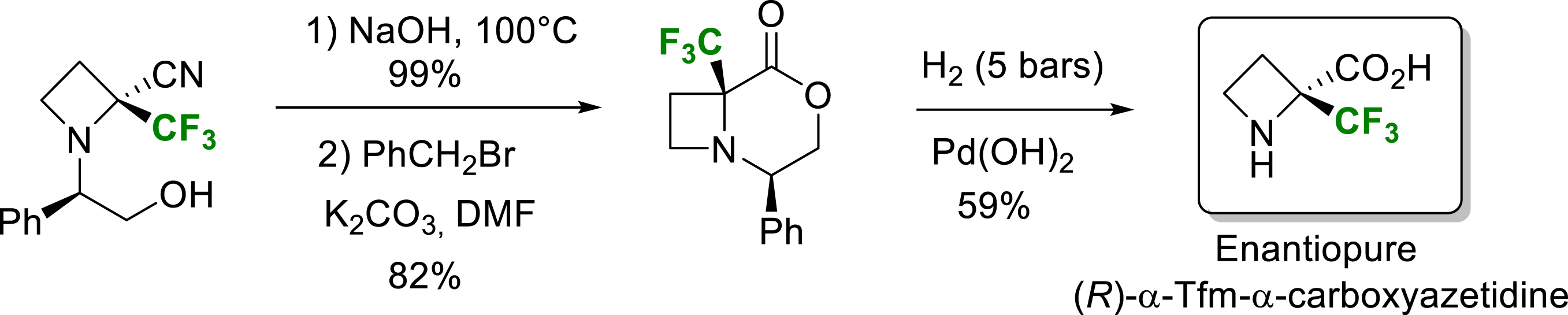
Synthesis of enantiopure (2R)-2-trifluoromethyl-2-carboxyazetidine.
The synthesis of constrained α-trifluoromethylated or 5-Tfm-proline has been a major research focus in the laboratory. Three synthetic routes have been developed. The first one was based on an allylation reaction on chiral 2-trifluoromethyloxazolidines derived from ethyl trifluoropyruvate to build up the amino acid side chain, the second one used a Strecker-type reaction to introduce the carboxy function, and finally the third one used Claisen-type reactions.
Starting from the Tfm-allylmorpholinone intermediate resulting from the pseudoproline allylation reaction, several synthetic routes are possible.
• Synthetic Route 1.
Hydroboration followed by oxidation led to the formation of the hydroxymorpholinone which then undergoes a cyclization reaction via mesylation. The bicycle formed then undergoes a hydrogenolysis reaction, giving access to the two enantiomers of α-Tfm-proline [18].
• Synthetic Route 2.
An iodocyclization reaction on Tfm-allylmorpholinone led to a bicyclic iodo compound, which via a hydrogenolysis reaction, also led to the formation of enantiopure α-Tfm-proline on a gram scale. The bicyclic iodo compound, by elimination of the iodine in a basic medium followed by a dihydroxylation reaction, gave access to the α-Tfm-dihydroxyproline [23].
• Synthetic Route 3.
A hydroboration reaction on CF3-allylmorpholinone followed by two oxidation reactions, saponification and deprotection of the amine function yields enantiopure α-Tfm-pyroglutamic acid (Scheme 15) [24].
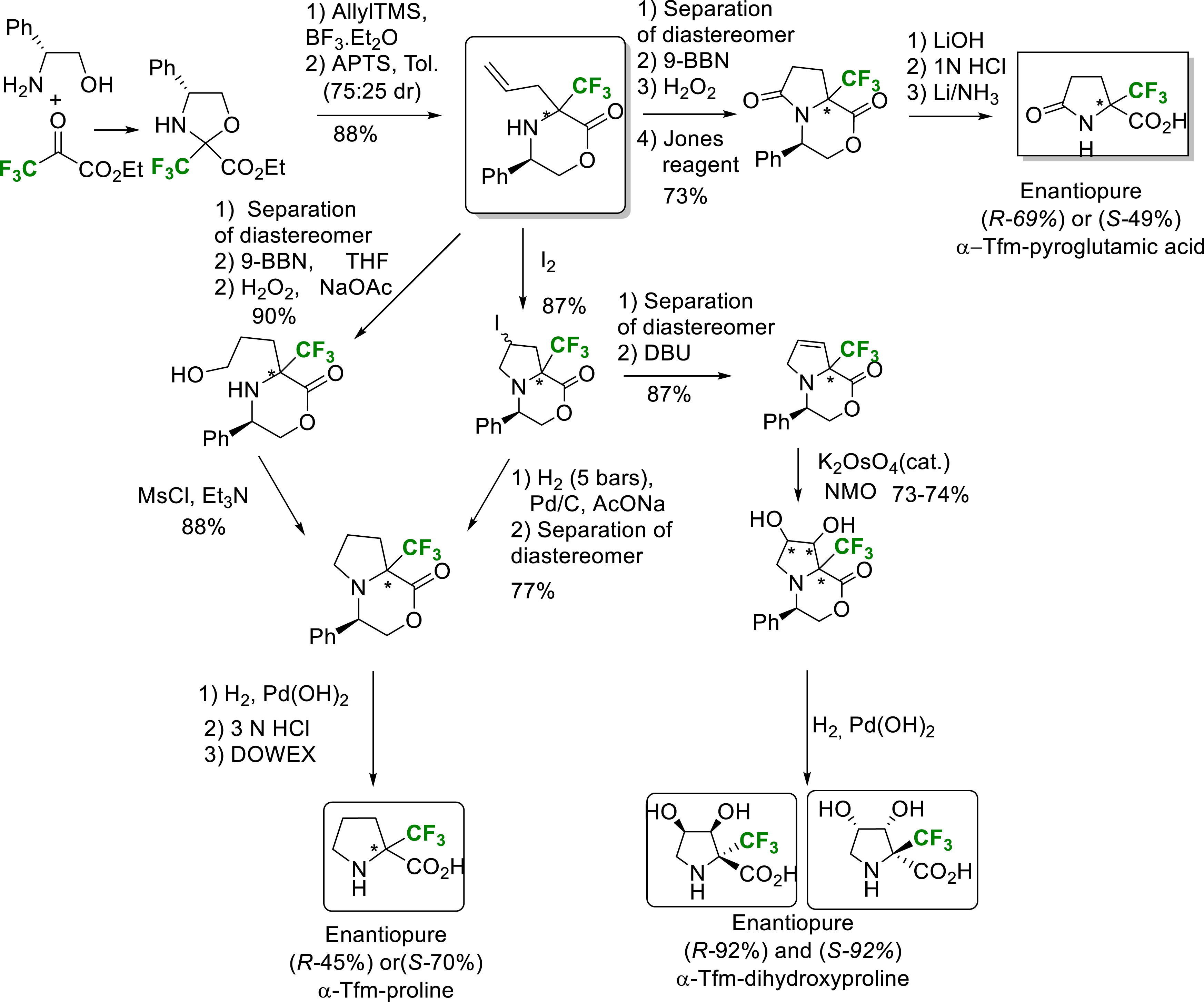
Synthesis of enantiopure α-Tfm-proline, α-Tfm-dihydroxyproline and α-Tfm-pyroglutamic acid.
For the synthesis of the enantiopure 5-Tfm-proline, an interesting constrained non-natural amino acid, our laboratory devised the following synthetic scheme (Scheme 16) [25]. The first step was the condensation of fluoral with (R)-phenylglycinol, yielding the CF3-oxazolidine of two inseparable diastereomers. Addition of an allyllithium reagent gave a mixture of the expected enol ether again in an inseparable mixture of diastereomers. Treatment under acidic conditions led to a fluorinated oxazolopyrrolidine also in a mixture of diastereomers. An enantiomerically pure oxazolopyrrolidine intermediate was obtained after efficient kinetic resolution by addition of 0.7 equivalent PhMgBr based on selective consumption of the undesirable minor diastereomer. A classical Strecker type reaction on this bicyclic intermediate followed by a separation of the diastereomer led by an acid hydrolysis step to a mixture of diastereomer. Basic conditions on a pure diastereomer led after hydrogenolysis the enantiopure (2S,5R)-5-trifluoromethylproline. The synthetic potential of this methodology was demonstrated by the scalable synthesis of the highly valuable enantiopure (2S,5R)-5-trifluoromethylproline (Scheme 16) [25].
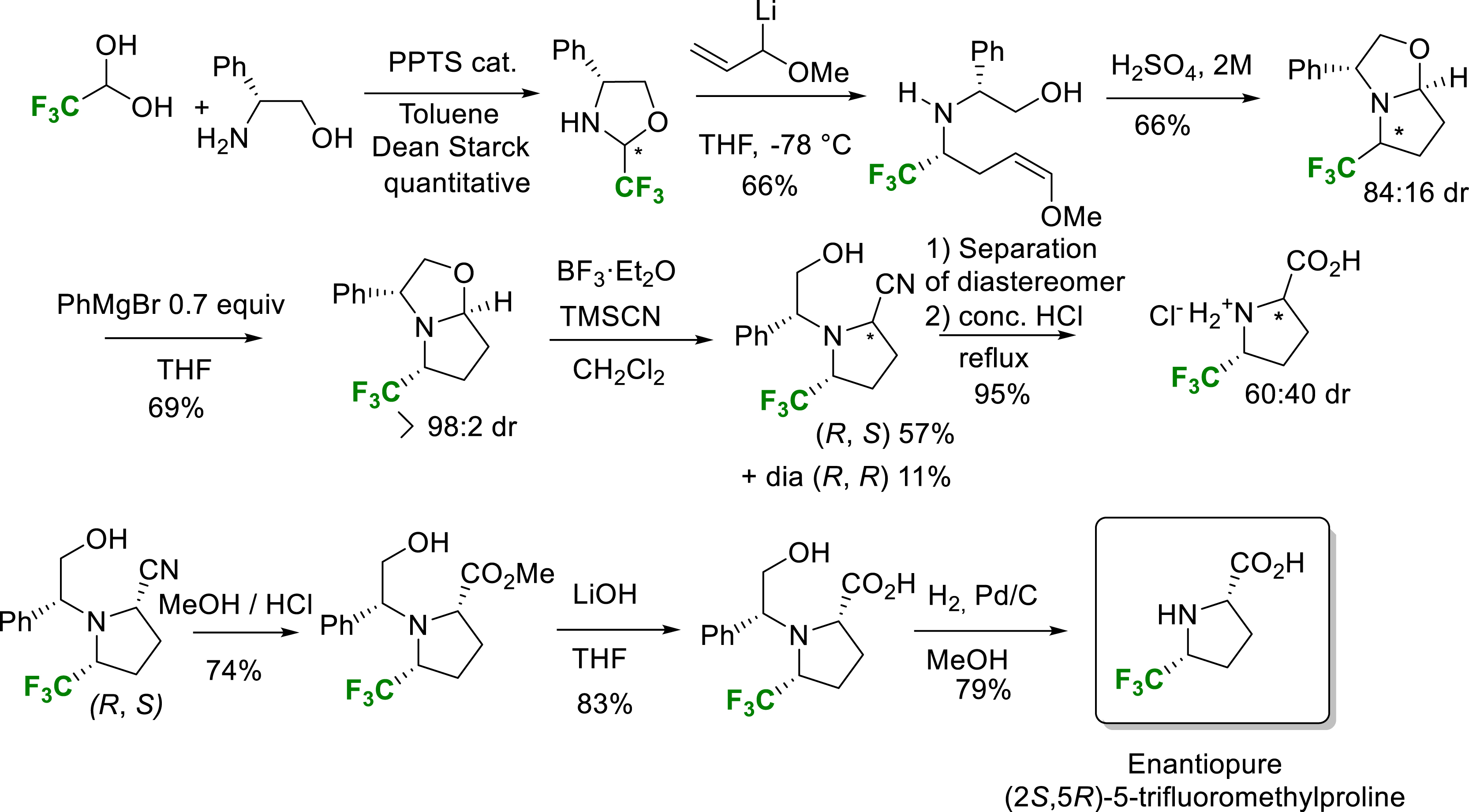
Synthesis of enantiopure (2S,5R)-5-trifluoromethylproline.
In 2021, we described a new straightforward synthesis of enantiopure 5-(R)-and 5-(S)-trifluoromethyl proline via a shorter multistep procedure avoiding tedious diastereomers separation (Scheme 17) [26]. The key steps are a Ruppert–Prakash reagent addition on the properly protected L-pyroglutamic acid followed by an elimination reaction and a diastereoselective reduction occurring on the less hindered face of the imine to provide the cis isomers. In order to obtain the trans isomer, the hydride reduction of the imine was investigated and the best results were obtained with NaBH3CN giving access to the trans benzyl ester of 5-CF3-proline after chromatographic separation on silica gel. Finally, an hydrogenolysis of the benzyl group provided trans 5-Tfm-proline (S) and the cis 5-Tfm-proline (R) (Scheme 17) [26].
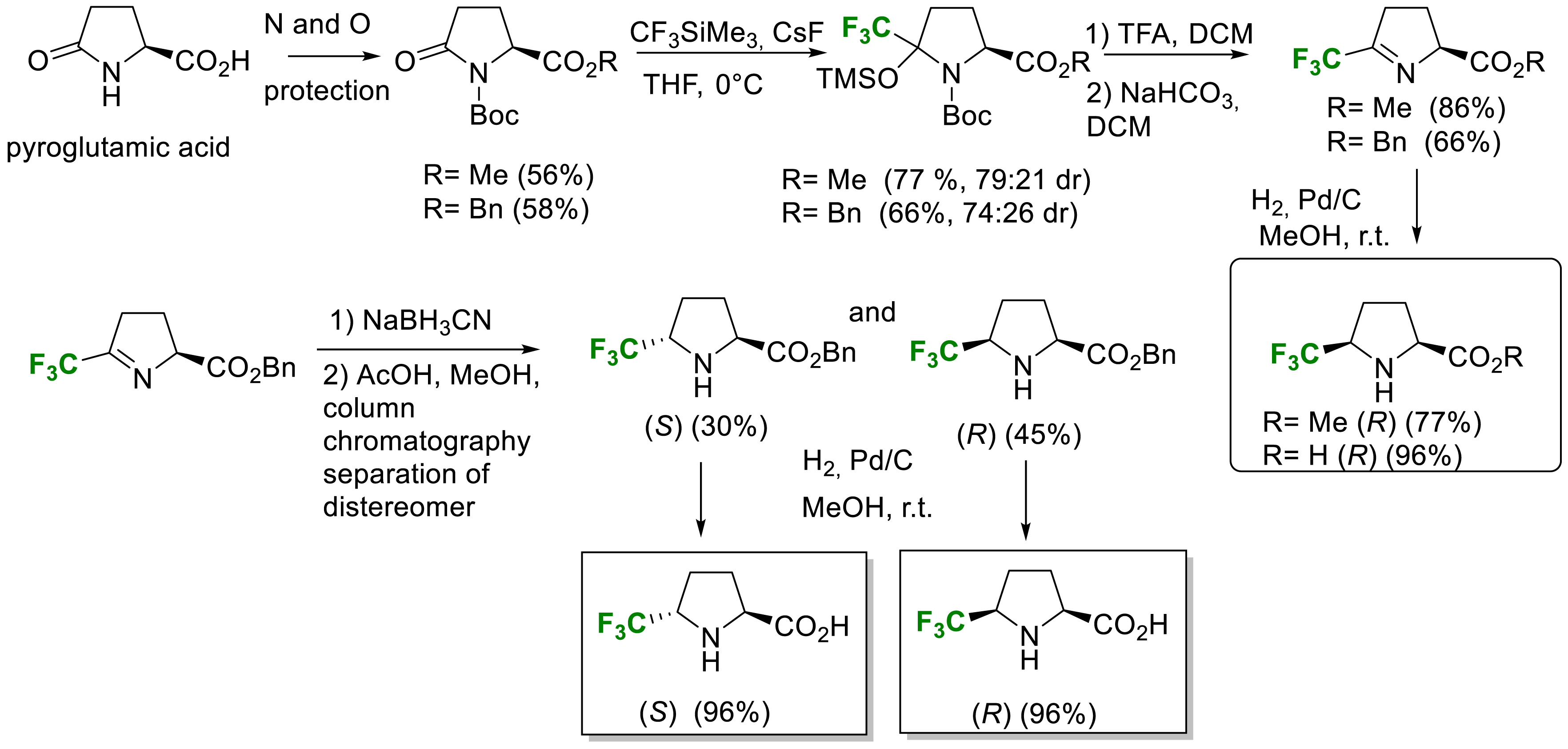
Synthesis of enantiopure 5-trifluoromethylproline.
We also reported, in 2021, a new simple and scalable synthesis of the two enantiomers of α-Tfm-proline [27]. In order to generalize to higher cyclic proline analogues, the synthesis of the two enantiomers of α-Tfm-pipecolic acid was also reported. Enantiopure α-Tfm-proline and α-Tfm-pipecolic acid were synthesized starting from commercially available diesters and ethyl trifluoroacetate via Claisen condensation (Scheme 18).
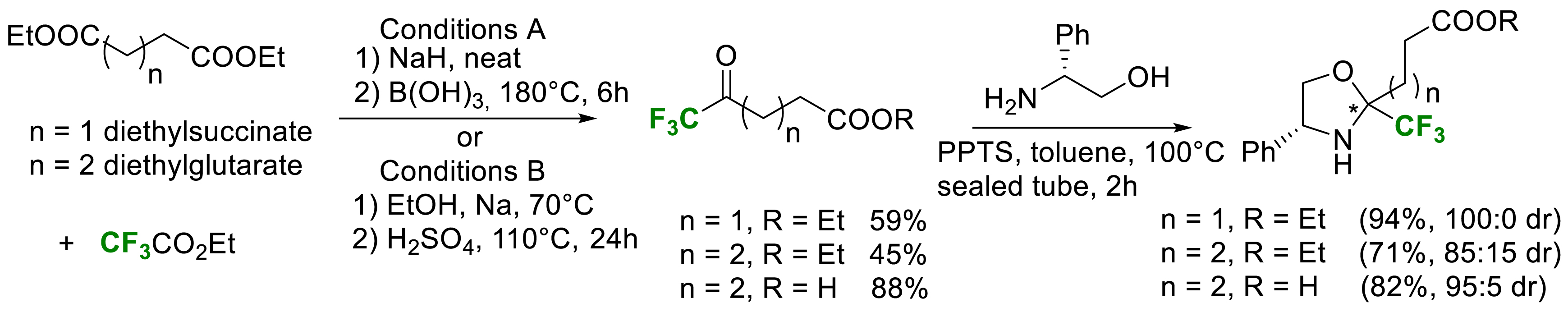
Synthesis of a common intermediate for α-Tfm-proline and α-Tfm-pipecolic acid.
At this stage, two different pathways were used for the proline and the pipecolic acid series. For the proline route, the oxazolidine was reduced with NaBH4/CaCl2 to give the alcohol. Its iodocyclization gave access to the Tfm-oxazolopyrrolidines. For the pipecolic route, a simple cyclization gave a lactam which was then reduced to give Tfm-oxazolopiperidines (Scheme 19).
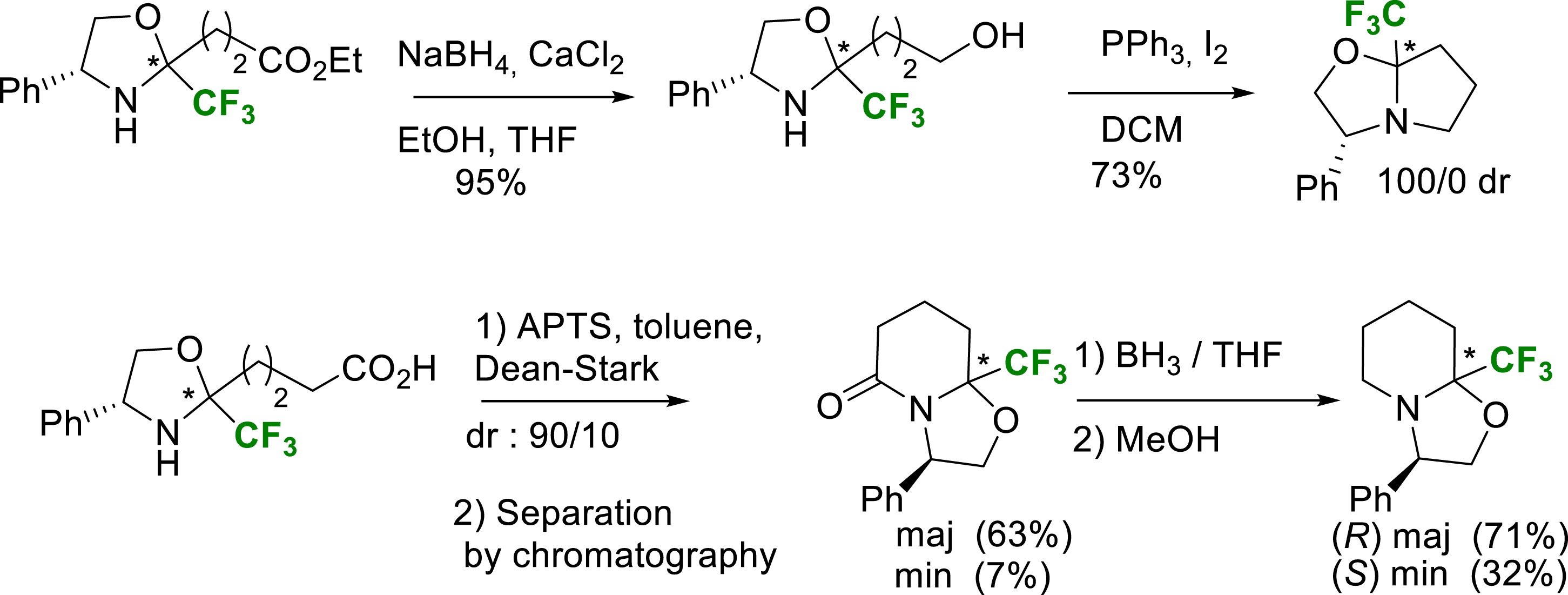
Synthesis of Tfm-oxazolopyrrolidines and Tfm-oxazolopiperidines intermediates.
A Strecker type reaction on chiral Tfm-oxazolo-pyrrolidine and piperidine intermediates yielded the corresponding nitriles, precursors of enantiopure (R) and (S) α-Tfm-proline and α-Tfm-pipecolic acid (Scheme 20) [27].
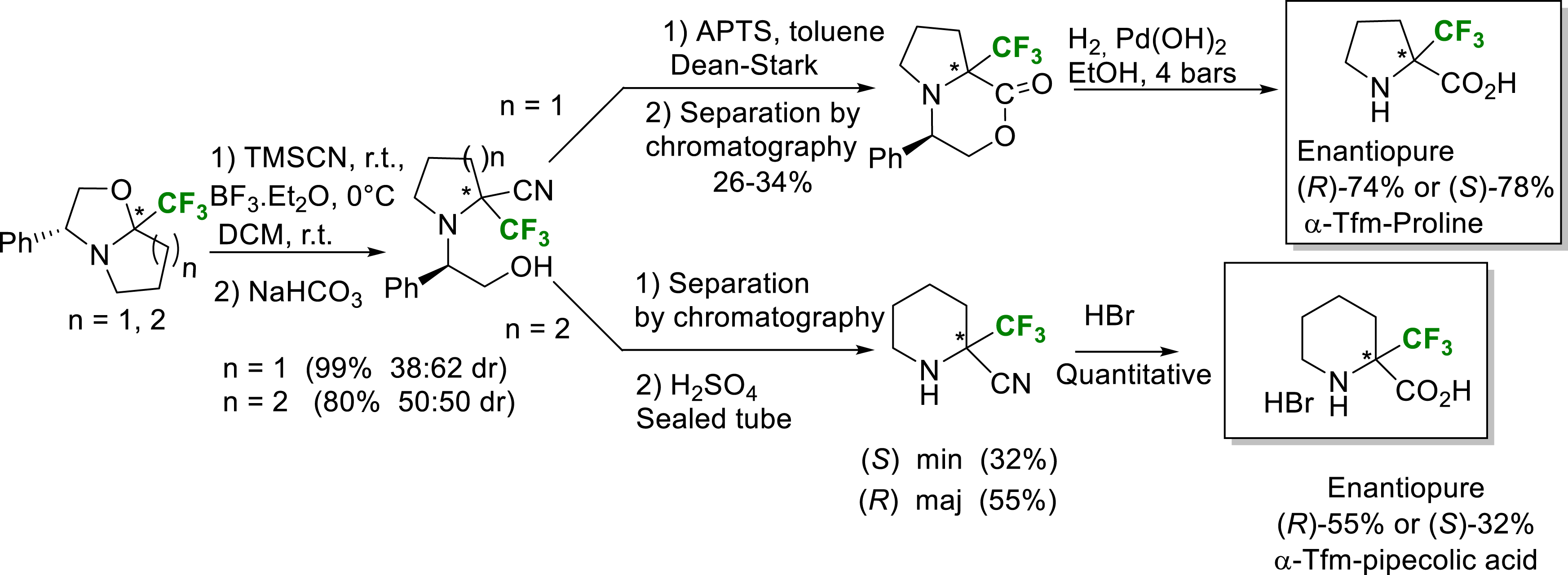
Synthesis of enantiopure (R) and (S) α-Tfm-proline and α-Tfm-pipecolic acid.
2.2.2. Pseudoprolines
Fluorinated oxazolidines are conveniently synthesized by condensation reaction of serine esters and hemiacetal trifluoroacetaldehyde or trifluoroacetone [28]. These oxazolidines are stabilized towards ring opening by the electron-attracting character of the trifluoromethyl group and can be considered as pseudoprolines (Ψpro) (Scheme 21). Each diastereomer can be isolated and they are not in equilibrium in neutral conditions. Despite the deactivation of the nitrogen atom, these oxazolidines can be efficiently N-acylated to give stable pseudoproline-type structures as cis/trans amide conformers. Conformational study of Tfm-pseudoprolines and cis/trans isomerization are the subject of a following part of this review. For future applications in peptide chemistry, pseudoprolines are hydrolytically stable authentic proline surrogates, compatible with the SPPS Fmoc/t-Bu strategy.
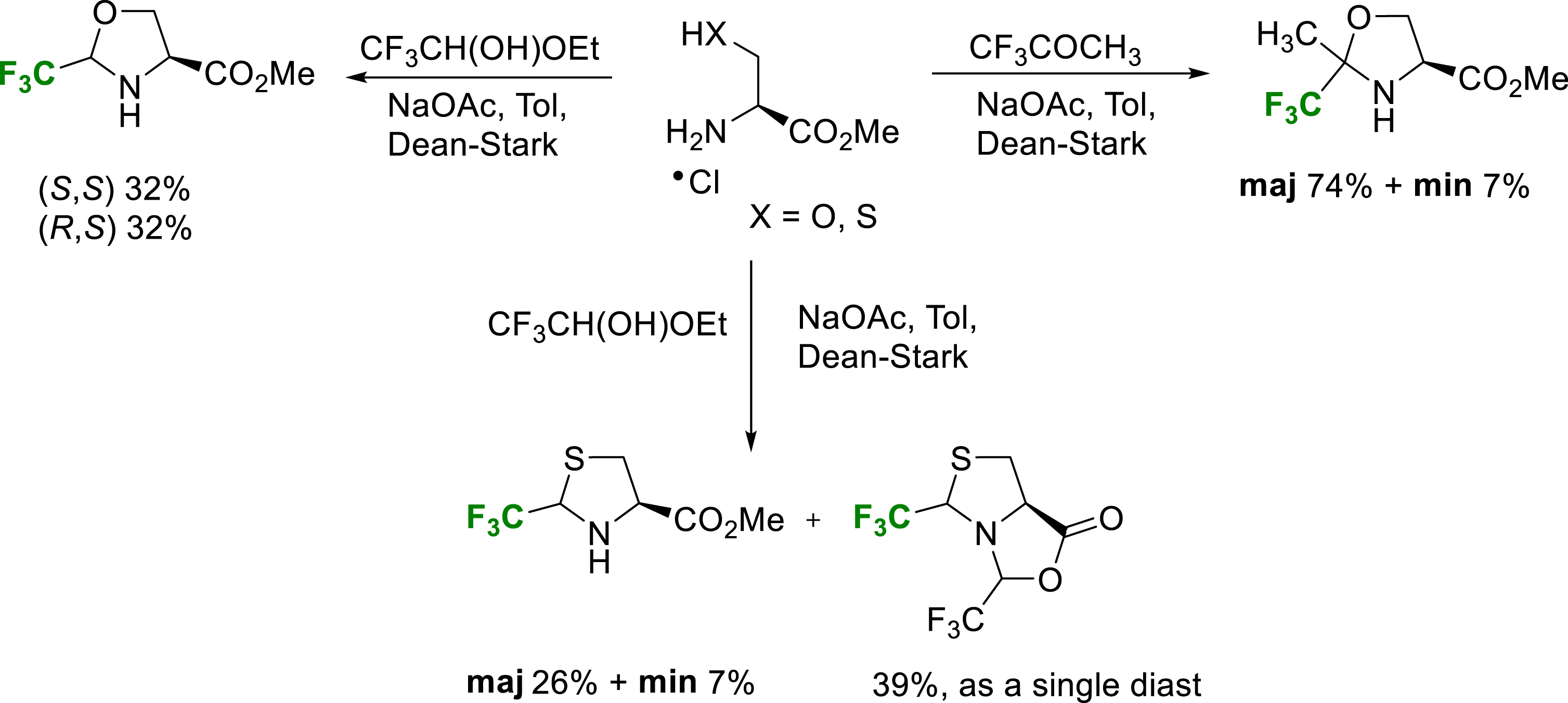
Synthesis of TfmΨpro esters.
Similarly, in 2019, original enantiopure (S,S) and (R,S)-CF2H-pseudoprolines were synthesized as stable 5-substituted proline substitutes that could be incorporated into a peptide chain (Scheme 22) [29].
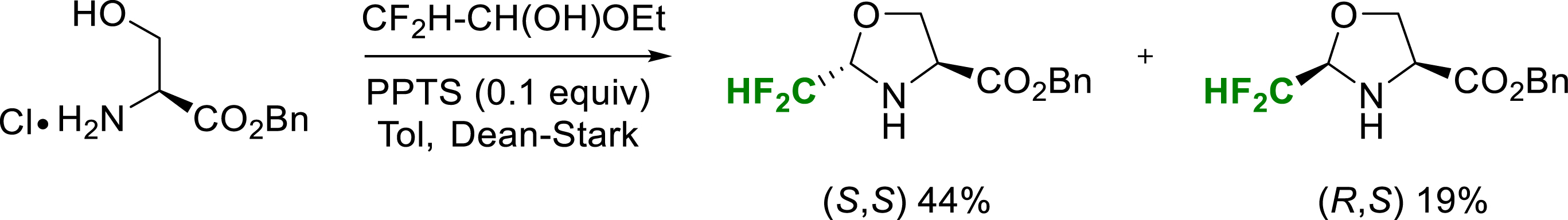
Synthesis of CF2H-Ψpro esters.
2.2.3. SCF3-amino acids
Among the fluorinated noncanonical amino acids, L-trifluoromethionine (TFM) and L-(S-trifluoromethyl)cysteine (Tfm-Cys), fluorinated analogues of methionine and cysteine, are of particular interest because of their ability to locally increase the hydrophobicity of peptides [30]. In 2017, our group reported the synthesis of tert-butoxycarbonyl/benzyl-protected TFM and Tfm-Cys using the cheap and user-friendly radical trifluoromethylation reagent previously developed by Langlois and co-workers [31]. This early-stage strategy allowed us to obtain the fluorinated noncanonical amino acids on gram scales, ready to use in liquid phase peptide synthesis (Scheme 23) [32]. In addition to this strategy, we reported a late-stage strategy inspired by the pioneer work of Seebach and Togni based on the electrophilic trifluoromethylation of a dimeric disulfide precursor. After the reduction of the precursor with tris-(2-carboxyethyl)phosphine (TCEP), electrophilic trifluoromethylation gave the expected peptides in low to moderate yield after purification, demonstrating that this strategy, already known for Tfm-Cys, was transposable to the synthesis of TFM-containing peptides.
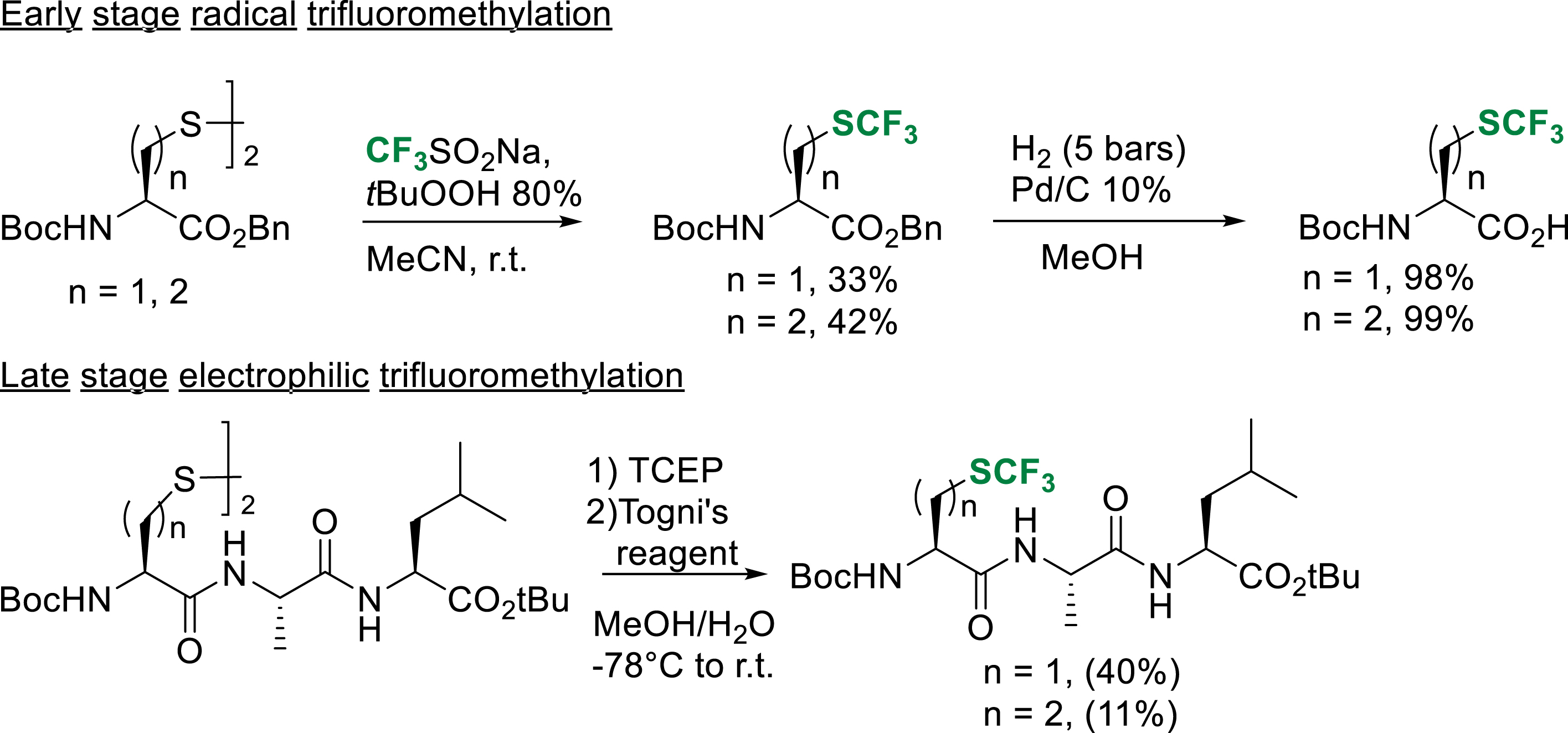
Early and late-stage access of TFM and TFM containing peptides.
Recently, we extended a methodology developed for the trifluoromethylthiolation of aromatic substrates [33], to the synthesis of Fmoc-protected trifluoromethylthiolated tyrosine (CF3S-Tyr) and tryptophan (CF3S-Trp) analogues on a gram scale (77–93% yield) [33]. The developed conditions are also efficient on tryptamine, tyramine and L-DOPA (Scheme 24).
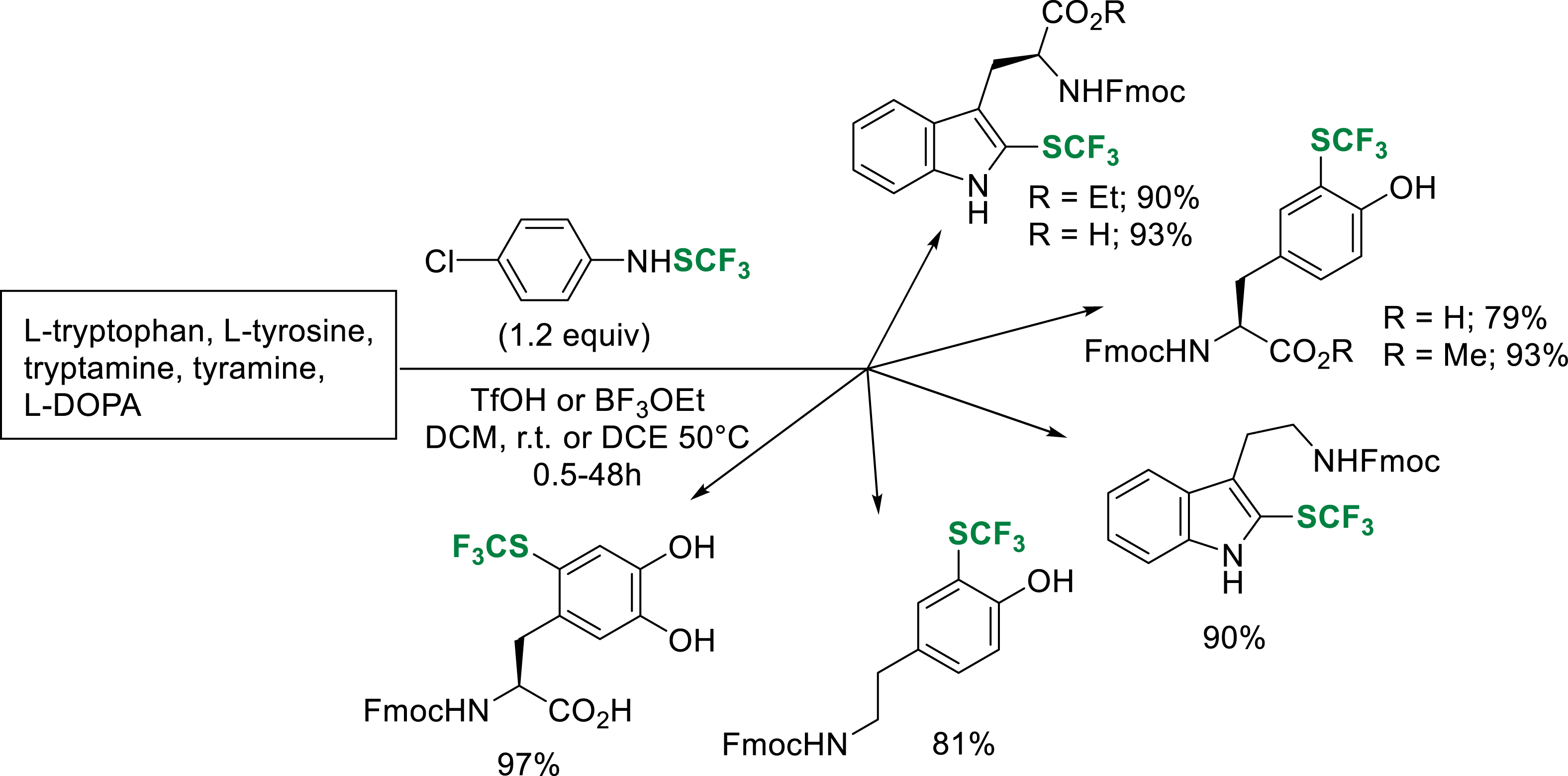
Trifluoromethylthiolation of substrates of interest.
In addition to the use of Fmoc-protected trifluoromethylthiolated tryptophanes and tyrosines in classical Solid Phase Peptide Synthesis (SPPS), the developed methodology was successfully applied to the late-stage regioselective trifluoromethylthiolation of Trp residues in short peptides. To prove the concept and explore the regioselectivity of the reaction, Fmoc-(CF3S)-Tyr and Fmoc-(CF3S)-Trp were incorporated into the endomorphin-1 chain (EM-1) and into model tripeptides obtained by SPPS (Scheme 25) [33].
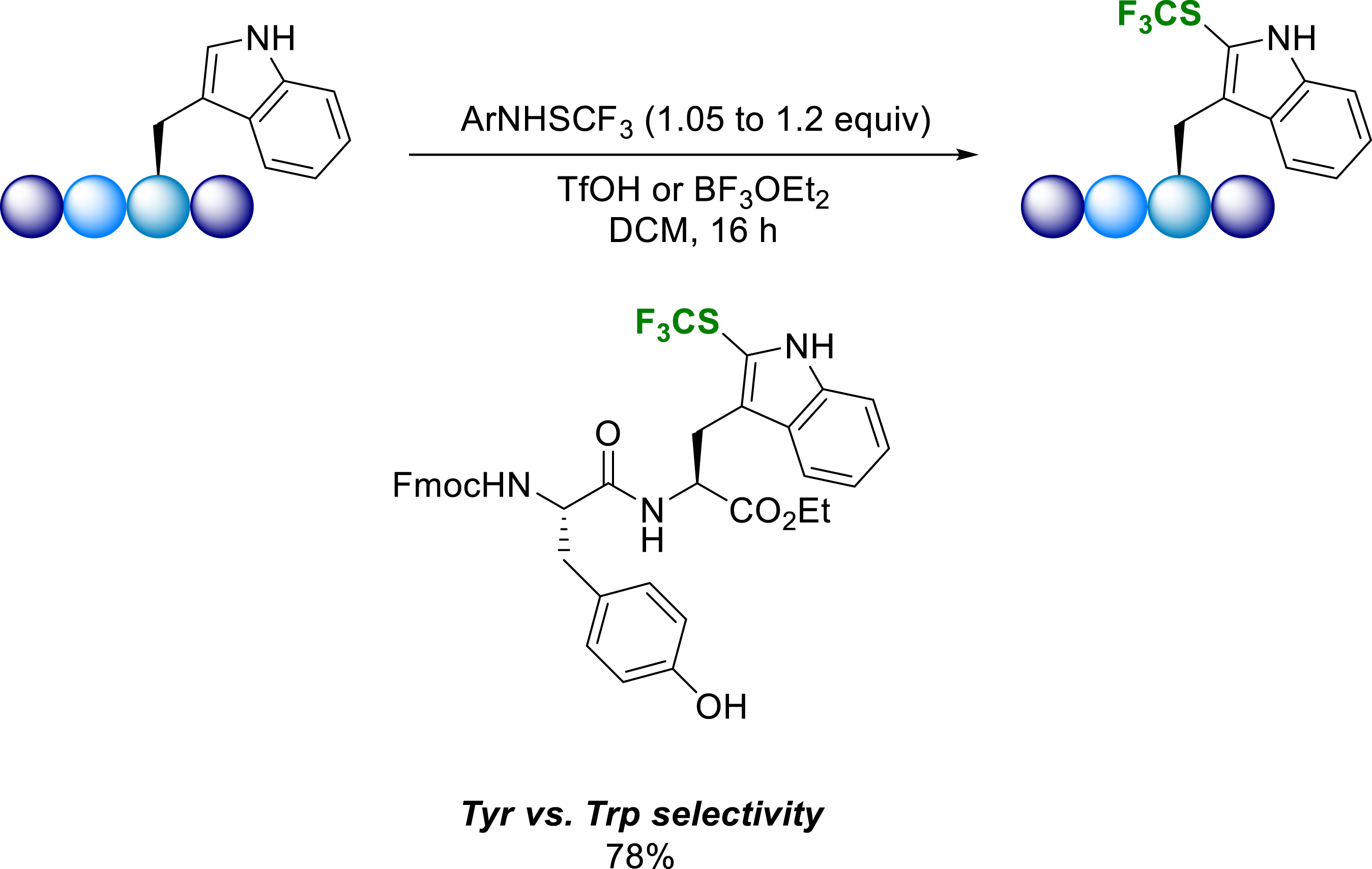
Trifluoromethylthiolation of short peptides.
3. Applications
3.1. Asymmetric synthesis
In 2006, we described the use of the enantiopure trifluoromethylated oxazolidine derived from phenylglycinol (trans-Fox) as a very efficient chiral auxiliary [34]. The starting point of this study was the excellent configurational stability of 2-trifluoromethyloaxazolidines derived from enantiopure aminoalcohols. Once acylated, unlike their non-fluorinated counterparts, the two positions of the fluorinated oxazolidines did not epimerize neither in acidic nor basic conditions. This property allowed us to isolate the trans substrates derived from the acylation of the (4R)-2-trifluoromethyl-4-phenyl-1,3-oxazolidine (trans-Fox amide, Scheme 26). We hypothesized that the configurationally stable trifluoromethyl group could be a very efficient chiral inducer due to its steric hindrance comparable to the iso-propyl or tert-butyl group. Indeed, the alkylation of amide enolates derived from various alkylated substrates proved to be completely diastereoselective (>99% de) when performed in THF at −78 °C using various alkylated electrophiles (R2 = Me, Et, Allyl, Bn, i-Pr, t-Bu). These results highlight the great potential of this (R)-phenylglycinol-based fluorinated oxazolidine as chiral auxiliary for asymmetric alkylation reactions and good alternative to the classical Evan’s oxazolidinones. Removal of the chiral auxiliary could be performed using reductive conditions. Treatment of the alkylated product with LiAlH4 afforded the corresponding aldehydes together with the deacylated oxazolidine that could be further recovered and reused. Enantiopure alcohols or carboxylic acids could be obtained from the aldehydes using a reductive or oxidative sequential two steps procedure [34].
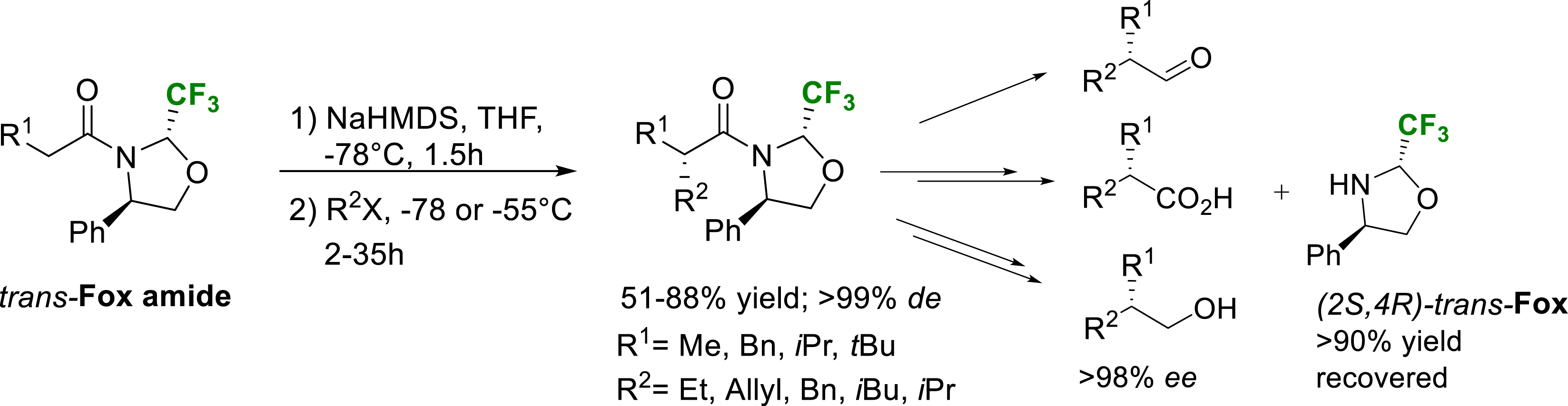
Diastereoselective alkylation of trans-Fox derived amide enolates.
Disappointing results obtained for the alkylation reactions of the cis-Fox amides, confirmed that a pseudo-C2-symmetry of the oxazolidine is mandatory to reach high levels of diastereoselectivity. The origin of the excellent stereocontrol was investigated by an experimental and theoretical (DFT) study [35]. We showed that, in the presence of the trans-Fox auxiliary, both F⋯Na+ and π⋯Na+ interactions compete to give the same diastereomer through Re face alkylation of the enolate. A 5.5 kcal⋅mol−1 energy difference found between the Re face and the Si face attack transition states is consistent with the complete diastereoselectivity that has been experimentally achieved (Figure 1). In the case of the cis-Fox chiral auxiliary, the competition between the F⋯Na+ and π⋯Na+ led to nearly isoenergetic (0.3 kcal⋅mol−1) Re and Si face attacks which are in good agreement with the very low diastereoselectivity experimentally observed.
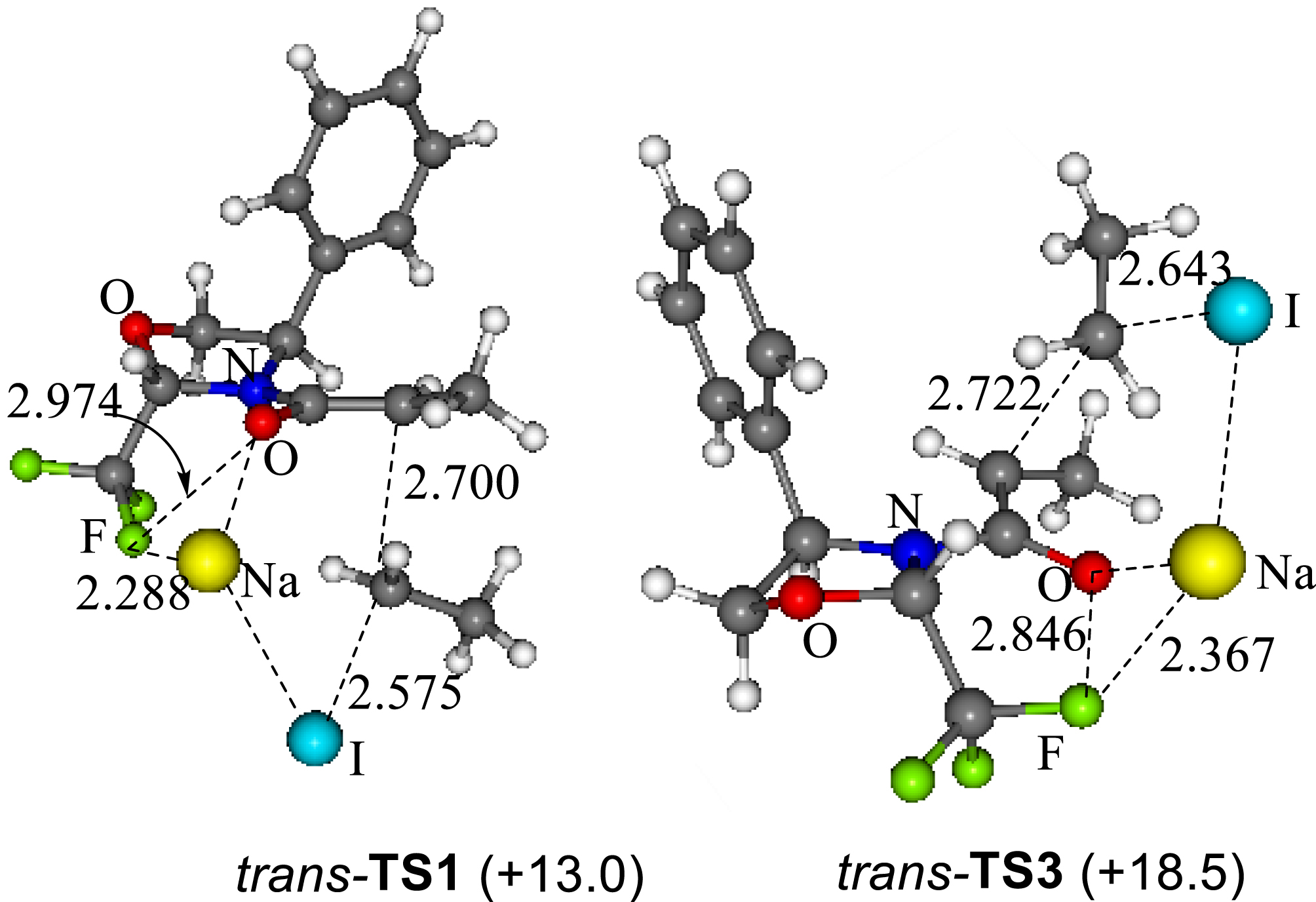
Theoretical Re face (TS1) and Si face (TS2) transition states with trans-Fox chiral auxiliary.
The alkylation reaction of trans-Fox amides with various halogenated electrophiles was later extended to β-amino substrates [36]. The alkylation with benzyl bromide, allyl bromide, and ethyl iodide was performed in good yield and with complete diastereoselectivity. Interestingly, the reaction with more hindered halogenated compounds such as isobutyl iodide and isopropyl iodide giving rise to β-2-homoleucine and β-2-homovaline precursors were also completely diastereoselective. As previously described, removal of the chiral auxiliary was achieved in a two-step procedure involving a LiAlH4 reduction of the alkylation product followed by the oxidation or the NaBH4 reduction of the intermediate amino aldehyde (Scheme 27).
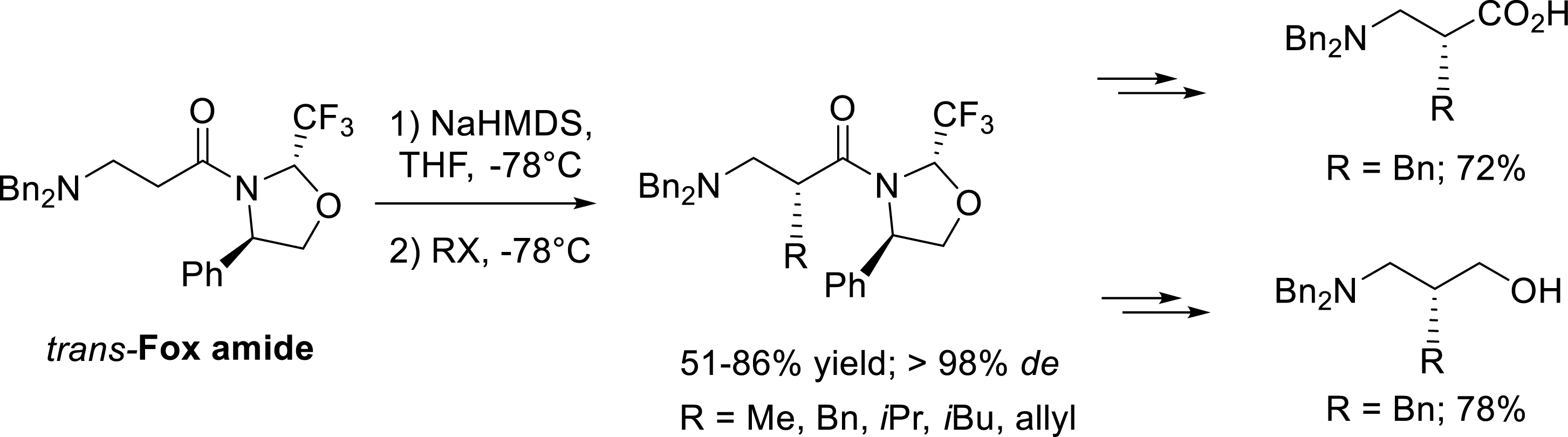
Diastereoselective alkylation of trans-Fox derived β-amino substrates.
In the course of previous studies, α-hydroxylated or α-hydroperoxylated amide were occasionally observed as side products of the alkylation reactions. We hypothesized that these oxygenated compounds probably resulted from the enolate oxidation by residual molecular oxygen. Submitted to a stream of bubbling molecular oxygen at −78 °C, trans-Fox amide enolate led to the corresponding hydroperoxide with good diastereoselectivity. When the hydroperoxide is reduced in situ with triethylphosphite, α-hydroxy amides were obtained in good yield and with excellent diastereoselectivity [37]. After benzyl protection of the newly formed alcohol, LiAlH4 reduction followed by subsequent sodium chlorite oxidation or NaBH4 reduction gave the corresponding enantiopure carboxylic acids and alcohols (Scheme 28).
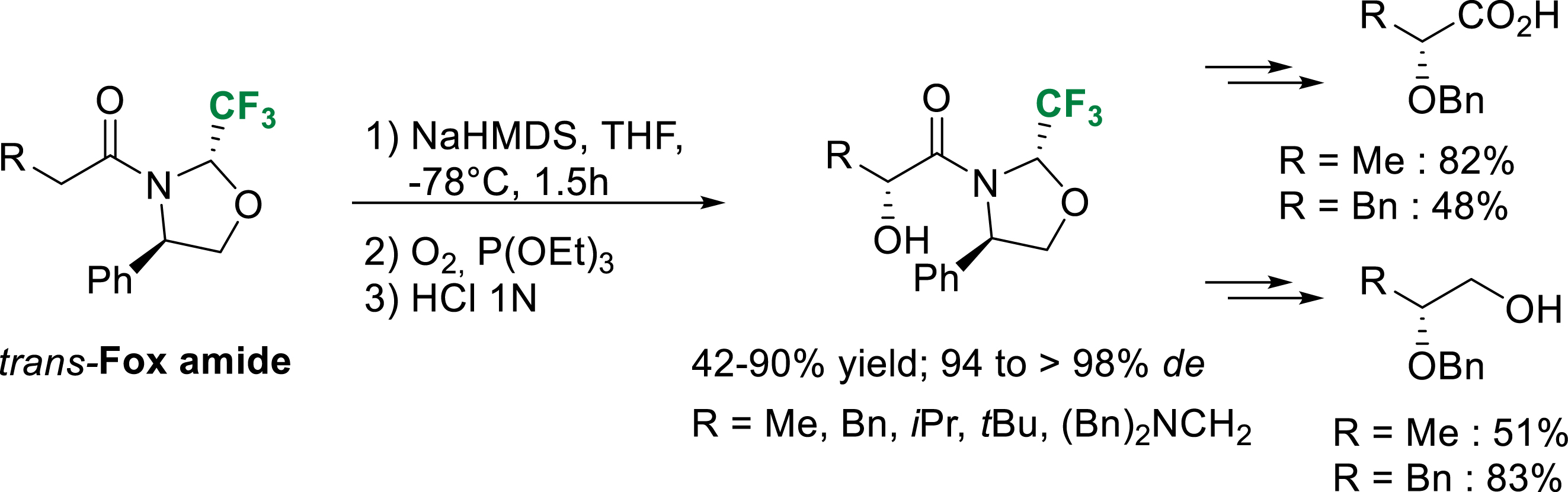
Diastereoselective α-hydroxylation of trans-Fox derived amide enolates.
In 2013, our group reported a highly efficient crystallization-induced dynamic resolution (CIDR) of trans-Fox. Taking advantage of the rapid equilibrium between cis and trans conformers of fluorinated oxazolidines in acidic medium, pure para-toluene sulfonic acid salt of trans-Fox could be crystallized from a mixture of cis–trans diastereomers in ethyl acetate. This dynamic resolution allowed us to prepare the trans-Fox chiral auxiliary on multigram scale avoiding subsequent tedious separation of cis and trans amides [38]. The electrophilic fluorination of trans-Fox amides with N-fluorobenzenesulfonimide (NFSI) proceeded smoothly at low temperature to give the α-fluoro amides with excellent diastereoselectivity. Corresponding α-fluoro carboxylic acids and β-fluoro alcohols could be obtained using the previously described procedure (Scheme 29).
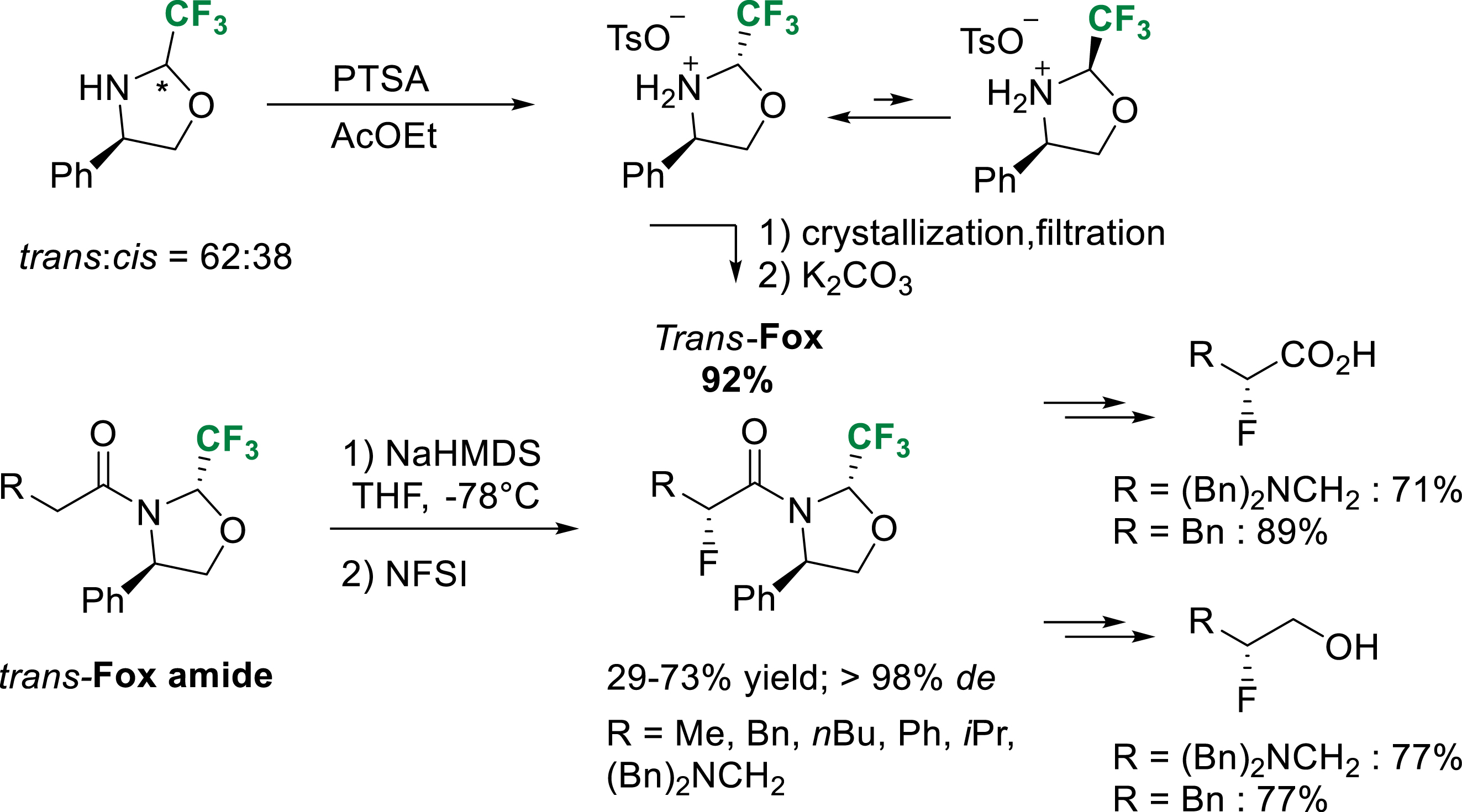
CIDR of trans-Fox and diastereoselective α-fluoration of trans-Fox amides.
Aldol reactions of sodium amide enolates derived from trans-Fox chiral auxiliary occurred in good yields but moderate anti diastereoselectivity. Syn diastereoselectivity was obtained using boron enolates. After removal, the Fox chiral auxiliary is very conveniently and efficiently recovered in basic conditions (Scheme 30) [39].
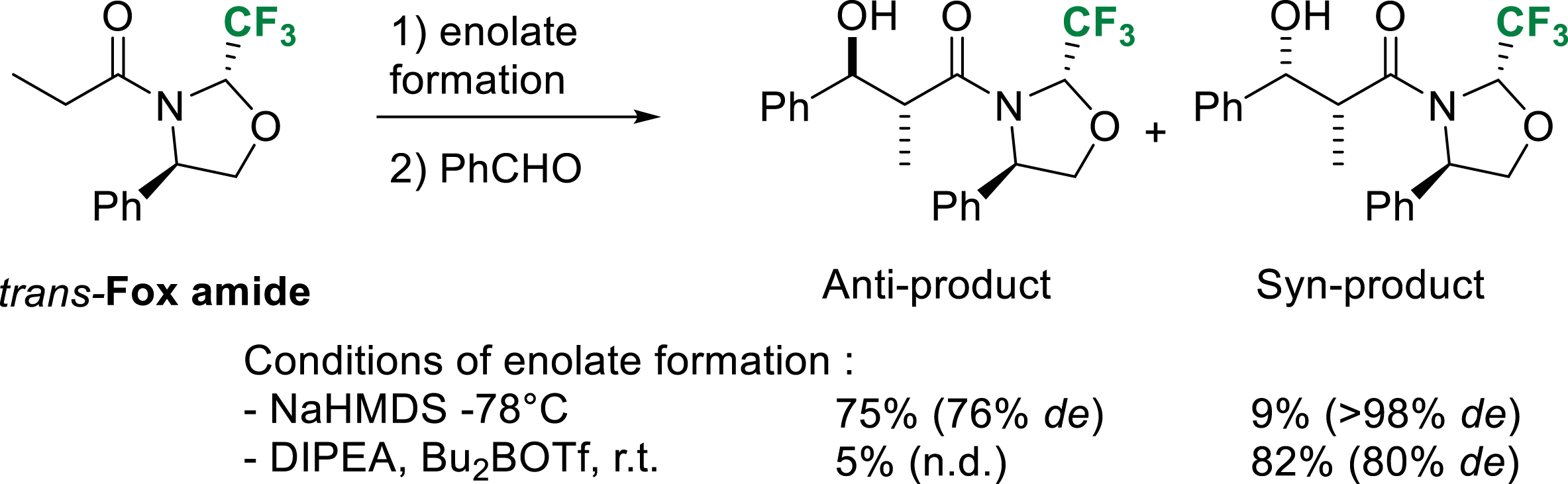
Diastereoselective aldol reaction of trans-Fox amide.
3.2. Incorporation in peptides and modulation of the biophysical and biological properties of peptides
3.2.1. Incorporation in peptides
The incorporation of α-amino acids bearing a fluorinated side chain in peptides does not present any particular difficulties when the fluorinated group is not deactivating too much the amino group of the amino acid. This is the case for difluoroalanine and SCF3 group containing amino acids (Scheme 31). Standard peptide coupling reaction conditions can be used for solution phase or SPPS incorporation of these amino acids in peptides [21, 32, 33].
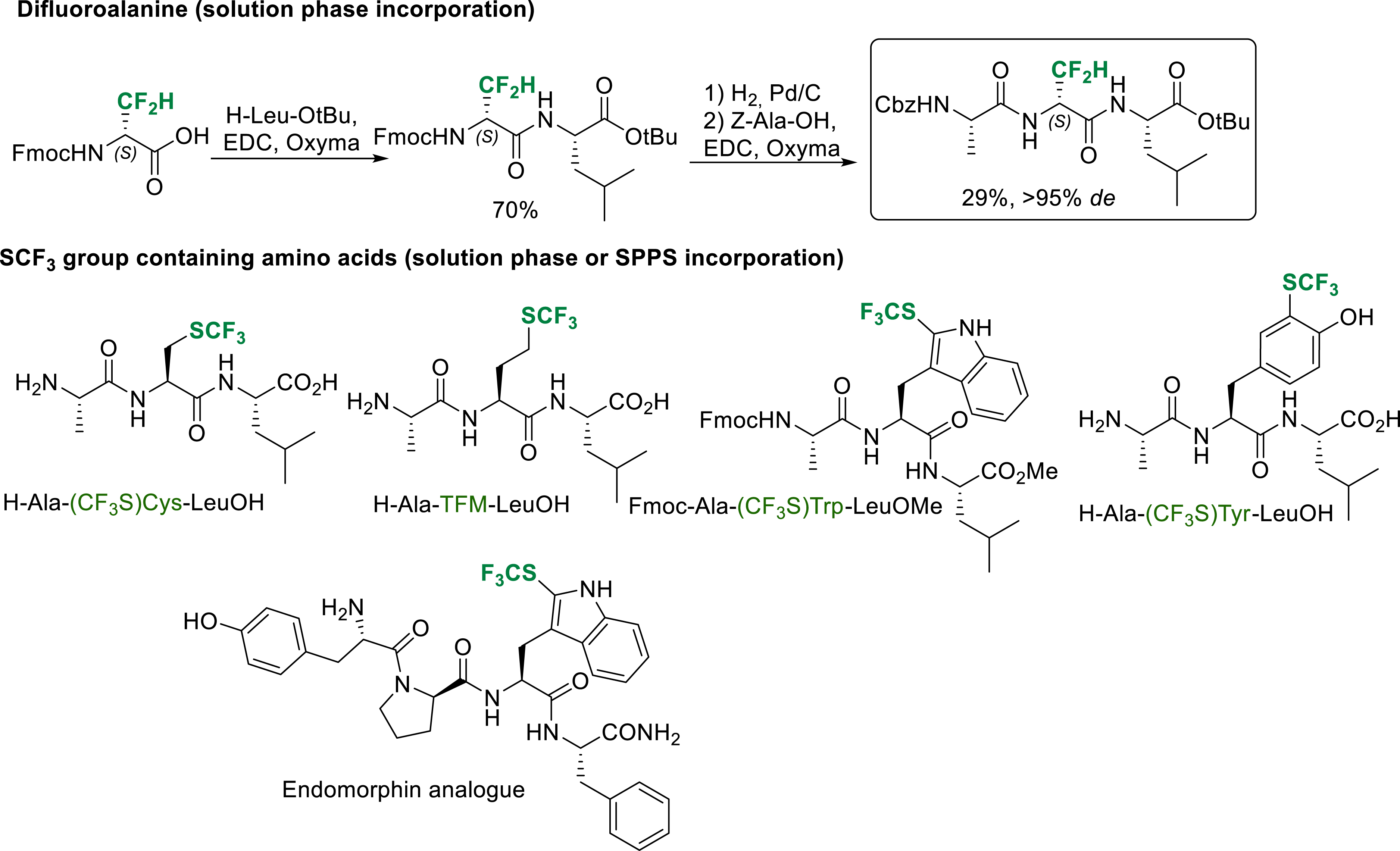
Selected examples of solution phase or SPPS synthesis of peptide incorporating difluoroalanine and SCF3 group containing amino acids.
The peptide coupling reactions at the C-terminal position of α-fluoroalkyl-α-amino acids, 5-Tfm-prolines and Tfm-pseudoprolines can also be achieved in good yields by standard procedures (Scheme 32) [22, 27, 40, 41, 42]. It should be noticed that because of the strong deactivation of the nitrogen atom of the α-trifluoromethyl-α-amino acids, the protection of its amino group is not required.
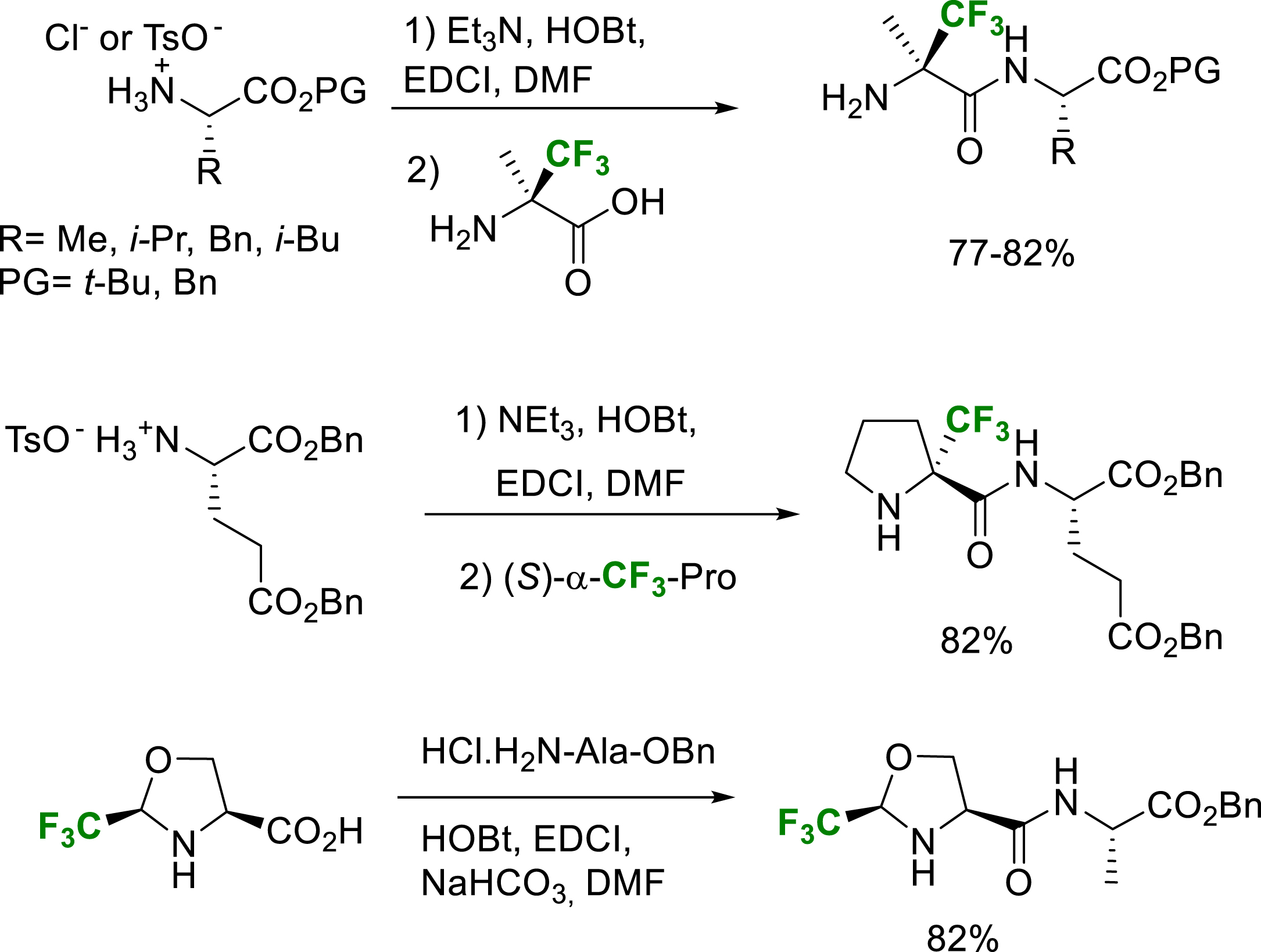
Selected examples of C-terminal position coupling peptides with solution phase or SPPS.
However, when fluorine atoms are located at the β-position of the amino group of the amino acid, their strong electron-withdrawing effect dramatically decreases the nucleophilicity of the nitrogen atom and specific activation methods are generally required. The peptide coupling reactions at the N-terminal position of α-Tfm-amino acids and especially the hindered quaternarized ones, required the activation of the amino acid as a mixed anhydride or an acyl halide (Scheme 33). The mixed anhydride was prepared by reaction of the amino acid with isobutyl chloroformate (IBCF) in the presence of N-methylmorpholine (NMM). The amino acyl chlorides were conveniently prepared by treatment of the amino acid with an excess of thionyl chloride and evaporation. The coupling reaction of Fmoc-amino acid chlorides were conveniently achieved at the N-terminal position of α-Tfm-alanine [43], α-Tfm-proline [26], CF3- and CF2H-pseudoprolines [26, 29, 42, 44]. It should be mentioned that the N-terminal position coupling reaction on CF3-pseudoprolines requires the use of amino acids chlorides without bases. In these conditions, the amide bond formation is generally associated with an epimerization of the chiral center bearing the trifluoromethyl group through a dynamic kinetic resolution process [42]. Starting from cis or trans CF3-pseudoprolines, the only cis CF3-pseudoprolines dipeptides are obtained. In the CF2H-pseudoproline series, the coupling reaction can be performed in the presence of pyridine and the difluorinated pseudoprolines are obtained in good yield without epimerization. Several examples of N-terminal position peptide coupling conditions are presented in Scheme 33.
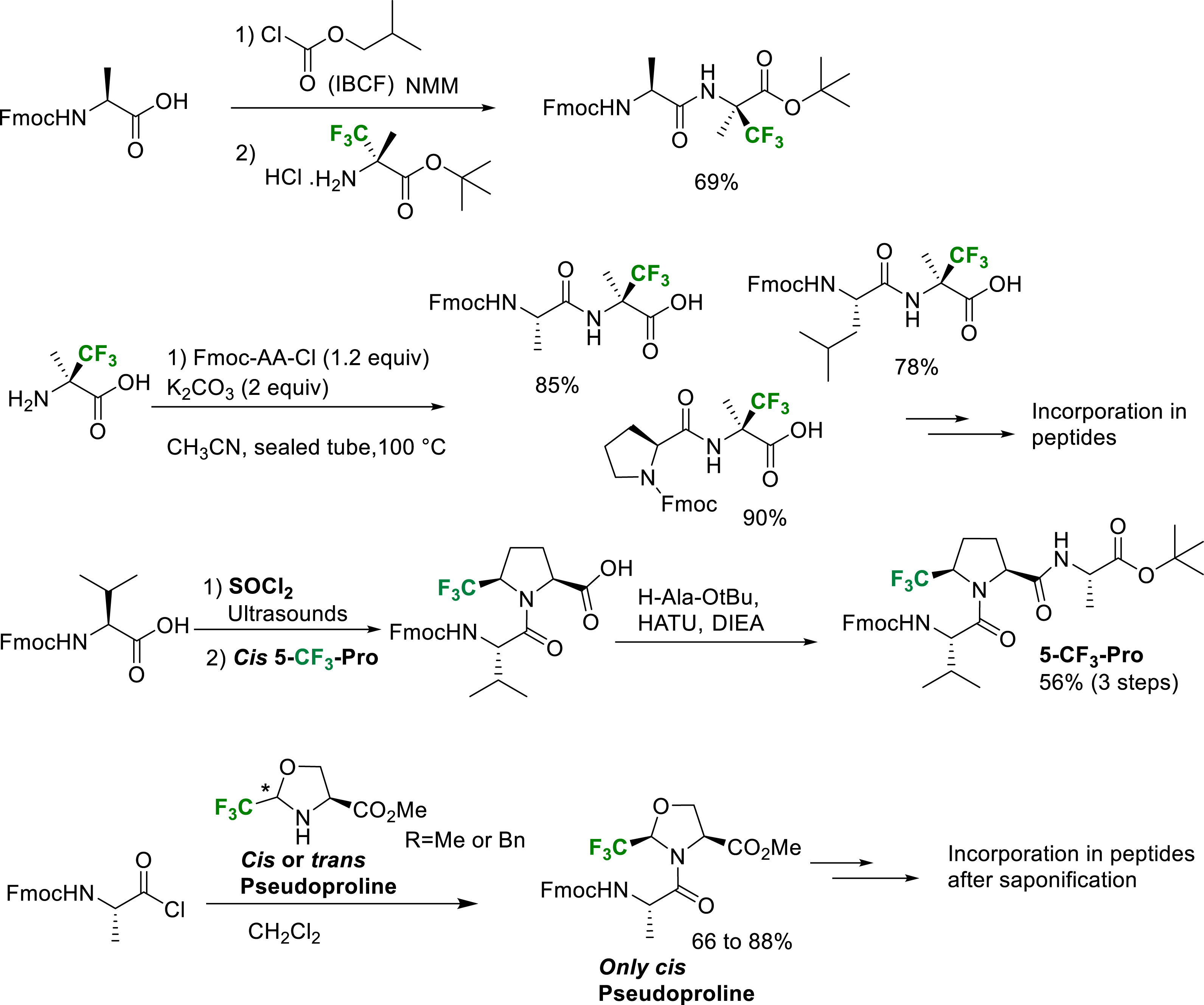
Selected examples of N-terminal position peptide coupling reactions.
The direct incorporation of α-Tfm-AAs (α-trifluoromethyl amino acids) in the middle of a peptide chain is still a challenge and all attempts to perform on resin the coupling of an amino acid at the N-terminal position of the deactivated fluorinated amino acid failed so far. The alternative strategy implemented for the incorporation of such amino acid in long peptides by SPPS is the incorporation of Fmoc-protected dipeptides building blocks resulting from the solution phase coupling (Scheme 34) [43, 45]. According to this strategy, numerous fluorinated labeled peptides of the peptaibol series, such as Harzianin [45], have been prepared notably for 19F NMR studies of interactions with membranes. Interestingly, the 5-CF3 proline can be conveniently incorporated in peptides by SPPS [26].
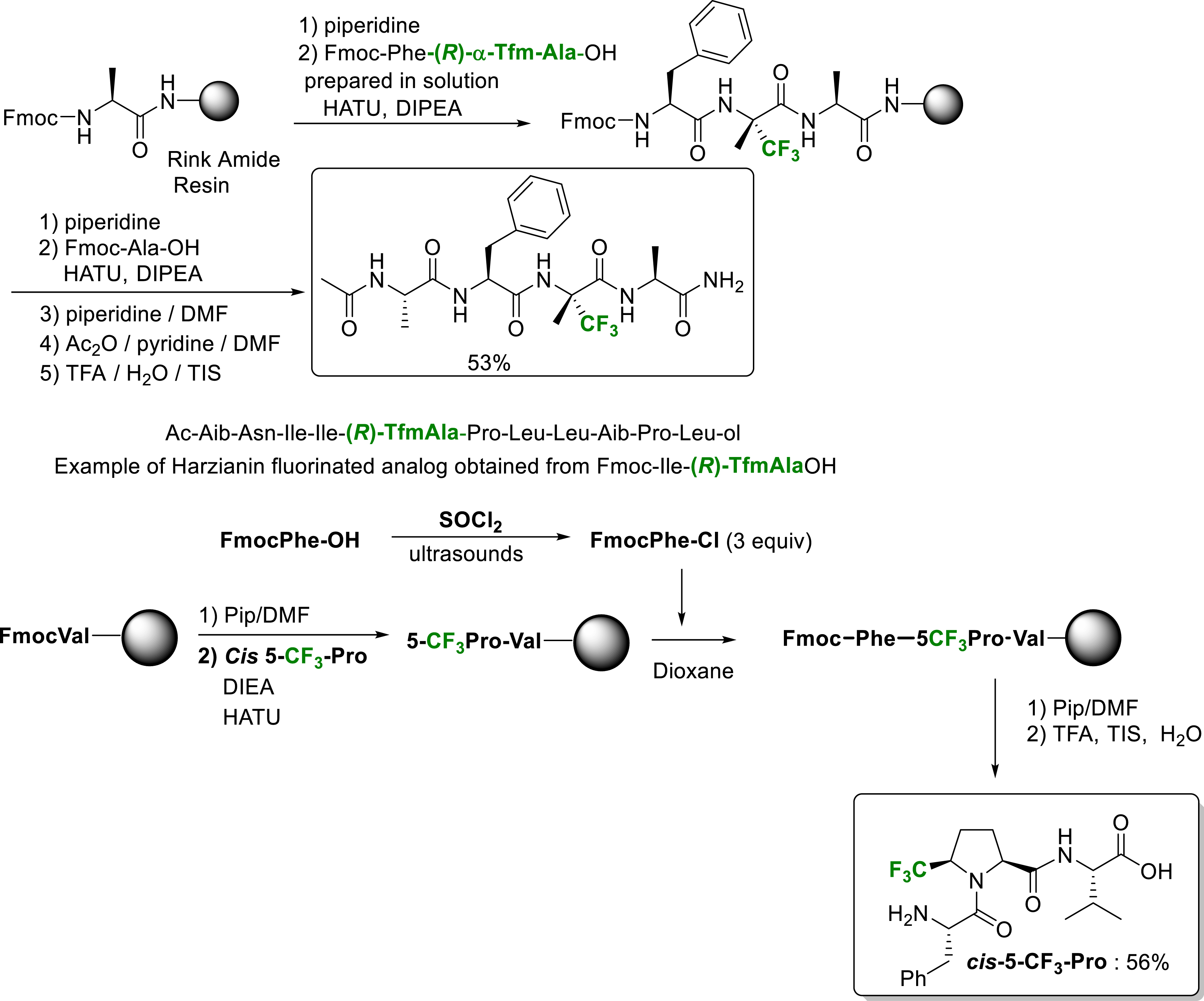
Selected examples of peptides incorporating α-Tfm-alanine and 5-Tfm-proline synthesized by SPPS.
3.2.2. Hydrophobicity modulation and biological applications
The modulation of the hydrophobicity of peptides by the incorporation of fluorinated amino acids is mostly reported and well documented in the γ- or ω-fluoro substituted series. Generally, the monofluorination lowers the hydrophobicity compared to the non-fluorinated analogues while the di- and trifluorination increase it. Because of their difficult synthesis, the hydrophobicity modulation assessment of peptides incorporating α-fluoroalkyl chains has been investigated only recently. The approach we adopted consists in the HPLC Hydrophobic Indexes (CHI) determination of peptides. According to this method, we measured the CHI of peptides incorporating a α-fluoroalkyl amino acid by comparison with their non-fluorinated analogues [30]. This method allows an accurate assessment of the local hydrophobic contribution of the fluorinated side chain compared to an aliphatic side chain (Figure 2). Di- and trifluorinated amino acids, such as difluoroalanine (DfAla) and trifluoromethylalanine (Tfm-Ala), provided an increased local hydrophobicity compared to alanine and aminoisobutyric acid (Aib). The hydrophobicity index shift dCHI (CHIDfAla − CHIAla = 9.9) constitutes a quantitative assessment of the increased hydrophobic contribution of the CF2H substituent compared to the methyl group of alanine [21]. It is very interesting to note that despite its smaller van der Waals volume, the CF2H group provides nearly the same hydrophobic contribution than the side chain of isoleucine (Ile), which is known to be the most hydrophobic aliphatic amino acid (CHIDfAla − CHIIle = 0.2). Similarly, trifluoromethylalanine provides an increased hydrophobicity compared to Aib (CHITfmAla − CHAib = 6.7) (Figure 2) [30]. SCF3 group containing amino acids are providing a dramatic increase of hydrophobicity [30, 33].
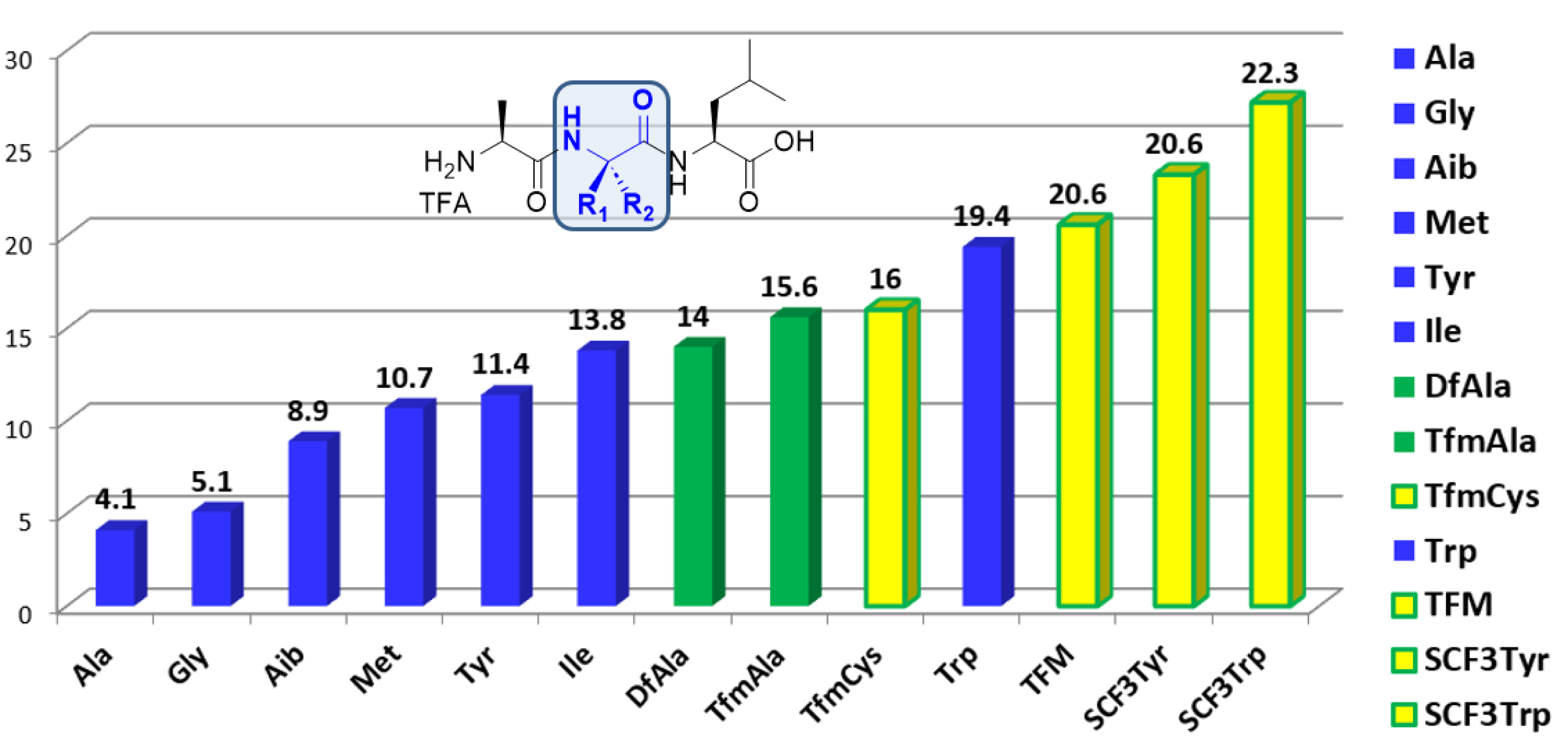
Hydrophobic contribution of fluorinated amino acids compared to their non-fluorinated analogues using the Chromatographic Hydrophobic Indexes (CHI) method [21, 30, 33].
This trend has been applied by us for the design of small hydrophobic peptides for β-amyloid aggregation inhibition [46]. As hydrophobic amino acids and/or the α-aminoisobutyric acid (Aib) are known to act as β-sheet breakers, the H-Ala–Aib–Leu-OH and H-Ala–(R)-Tfm-Ala–Leu-OH peptides were investigated as inhibitors of Aβ42 aggregation. These peptides were evaluated by the ThioflavinT (ThT) binding assay and both showed an inhibition of the kinetics of fibril formation at high concentration. The Tfm group containing peptide gave significantly better inhibition results because of its increased hydrophobicity and then a good affinity with the hydrophobic region of Aβ.
Another method reported in the literature for the hydrophobicity evaluation of fluorinated peptides is the Parallel Artificial Membrane Permeability assay (PAMPA) [47]. This method has been implemented to investigate the ability of fluorinated analogues of the PLG peptide (MIF-1) to cross the blood brain barrier (BBB). The fluorinated analogue of PLG with an α-trifluoromethylproline in place of the proline showed a better analgesic activity [48, 49]. This improved biological activity has been related to an increased hydrophobicity and ability to cross the BBB through passive diffusion as shown by PAMPA (Figure 3) [47].
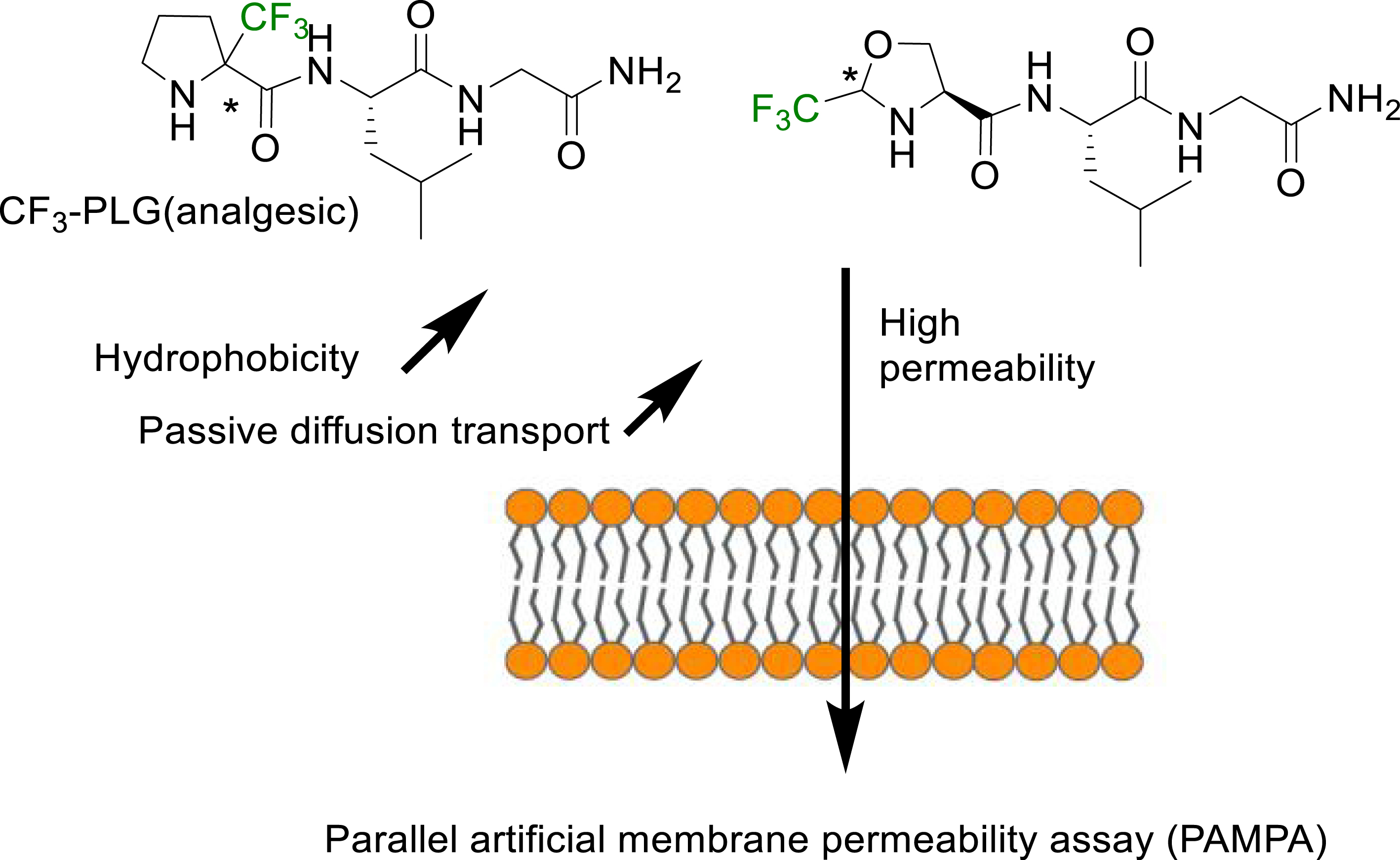
PAMPA method for measuring the increased passive diffusion of α-Tfm-AAs containing short peptides through membranes [47].
3.2.3. Increased resistance towards proteases and protease inhibitor design
The modulation of the resistance of peptides towards proteolysis have been mainly investigated with amino acids carrying fluorine atoms at the γ or ω position of the side chain [50]. Once again, because of their difficult synthesis and incorporation in peptides, α-fluoroalkyl amino acids have rarely been studied. After the pioneer works by Burger and Koksch [51], we recently revisited the high potential of these amino acids to circumvent peptides proteolysis. Pepsin is generally cleaving peptides at the C-terminus of a phenylalanine residue (Phe). By incorporating an α-Tfm-Ala adjacent to the Phe carboxylic group of the peptide A (Figure 4), the hydrolysis was almost inhibited and a slow cleavage occurred at another position [52]. A similar peptide incorporating an arginine (Arg) (peptide B in Figure 4) has been used as a fluorinated tag in a 3-FABS 19F NMR based activity assay to probe the IC50 of a trypsin inhibitor [52]. Very recently we incorporated (R)- and (S)-α-Tfm-Ala as well as (R)- and (S)-TFM (trifluoromethionine) in Pepstatin A based inhibitors of Cathepsin D and Pepsin (peptides C, D, E and F in Figure 4) [53]. The inhibitory potency of these Pepstatin analogues was measured towards isolated Cathepsin D and Pepsin using a classical FRET based assay. Both peptides presented nanomolar IC50. However, they were slightly less active and selective than Pepstatin. The (R)-TFM based analog of Pepstatin A (peptide E, Figure 4) returned a sub-nanomolar IC50 against CD and an increased selectivity. Molecular Docking experiments clearly demonstrated the stabilization of the inhibitor E in the catalytic pocket of Cathepsin D by strong hydrophobic interactions due to the long and flexible SCF3 containing side chain.
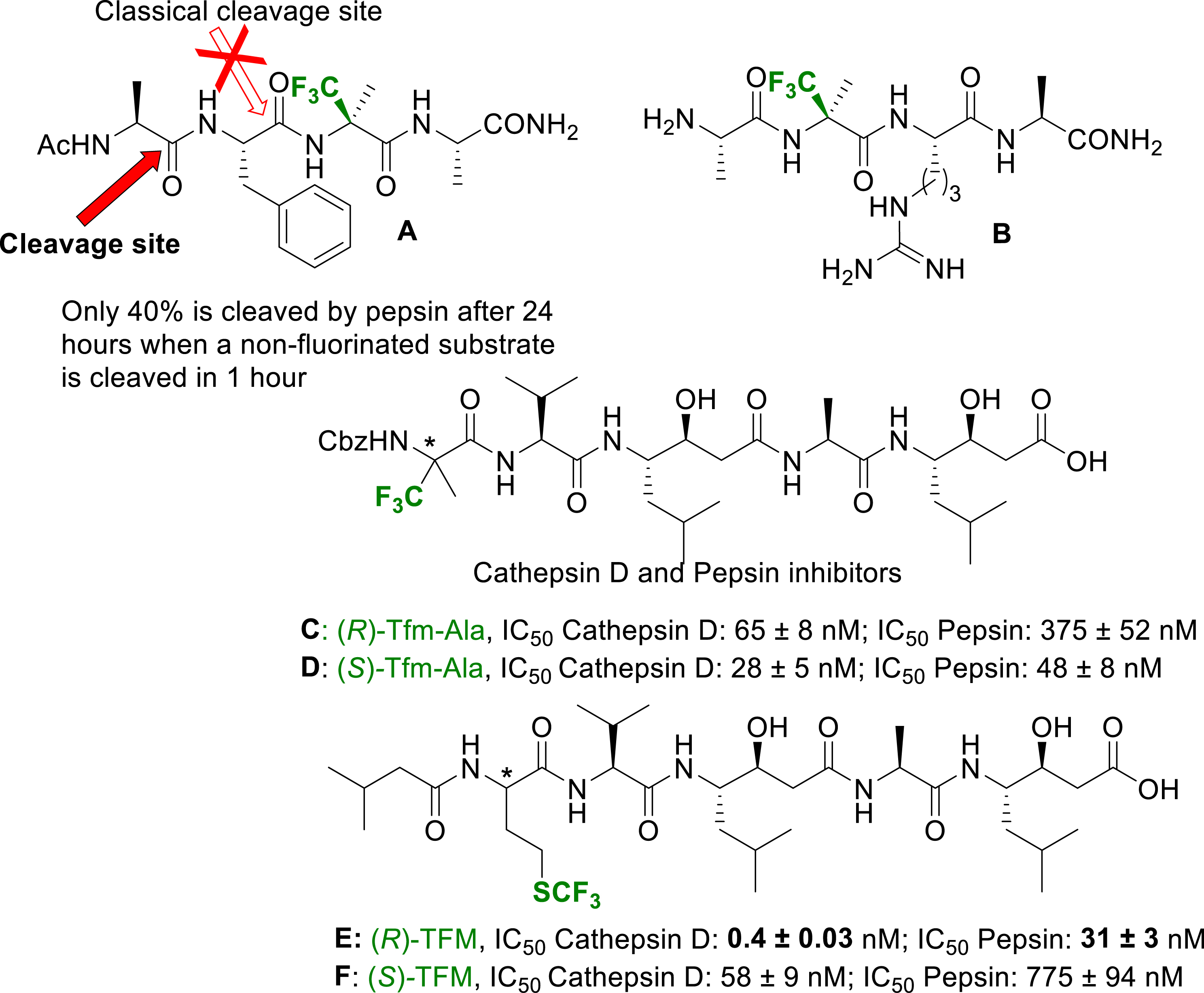
Tfm-Ala-containing peptides for increased resistance towards proteases and proteases inhibitors design.
Recently we also reported that a fluorinated polyproline PPII helix incorporating trifluoromethyl pseudoprolines exhibits a strong resistance in Pronase since no degradation was observed after two days [54].
3.3. Conformational aspects of α-Fluoroalkyl-α-amino acids and their peptides
3.3.1. Cyclic α-Fluoroalkyl-α-amino acids
Because of its cyclic structure, the proline residue is recognized to play a unique and crucial role in the peptide backbone conformation and biological properties. When included into a peptide, the pyrrolidine ring cannot act as a hydrogen bond donor and restrains the 𝜙 dihedral angle to about −60° (Figure 5). The Xaa–Pro peptide bond is characterized by a small free energy difference (
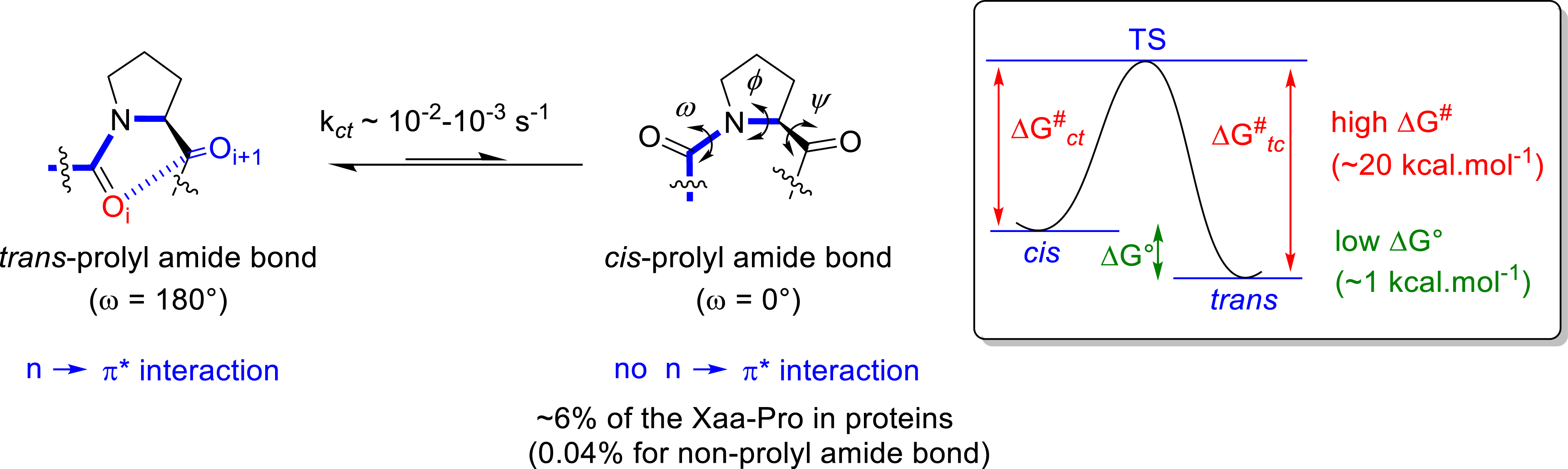
General features of proline residue when incorporated into peptides.
Many proline analogues have been reported in the literature to control the cis–trans isomerization of the peptidyl–prolyl amide bond. Our group investigated the ability of our CF3- and CF2H-substituted prolines and pseudoprolines to serve as attractive tools for the control of the peptide bond conformation. We first focused on fluorinated Ac–Pro–NHMe analogues, considered as tripeptide models (Figure 6). We demonstrated that the introduction of a CF3 group at the C𝛿 position of the pyrrolidine ring increased the cis rotamer population compared to the proline itself, the CF3-pseudoprolines 2-(R)-CF3ΨPro (TfmΨPro) and 2-(S)-CF3ΨPro (tfmΨPro) [55] exhibiting a higher cis conformer content than the 5-(R)-CF3-proline (Tfm-Pro) [26]. We also showed that our CF3-proline analogues with (R)-configuration at the C𝛿 position significantly decreased the trans to cis amide bond isomerization energy (ca. 3–5 kcal⋅mol−1 compared to proline).
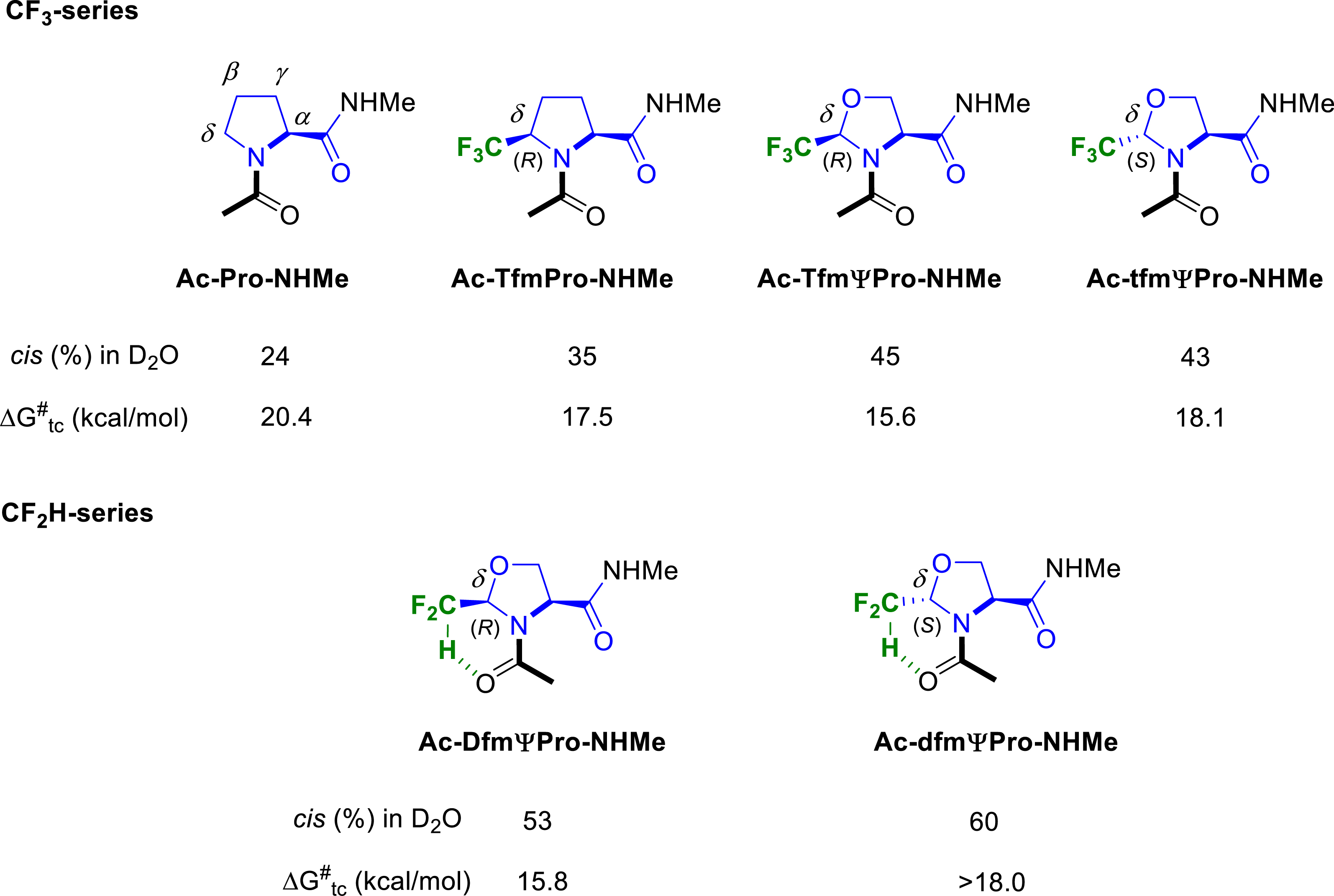
Cis–trans ratio and rotational energy barrier of fluorinated analogues of Ac–Pro–NHMe peptide models.
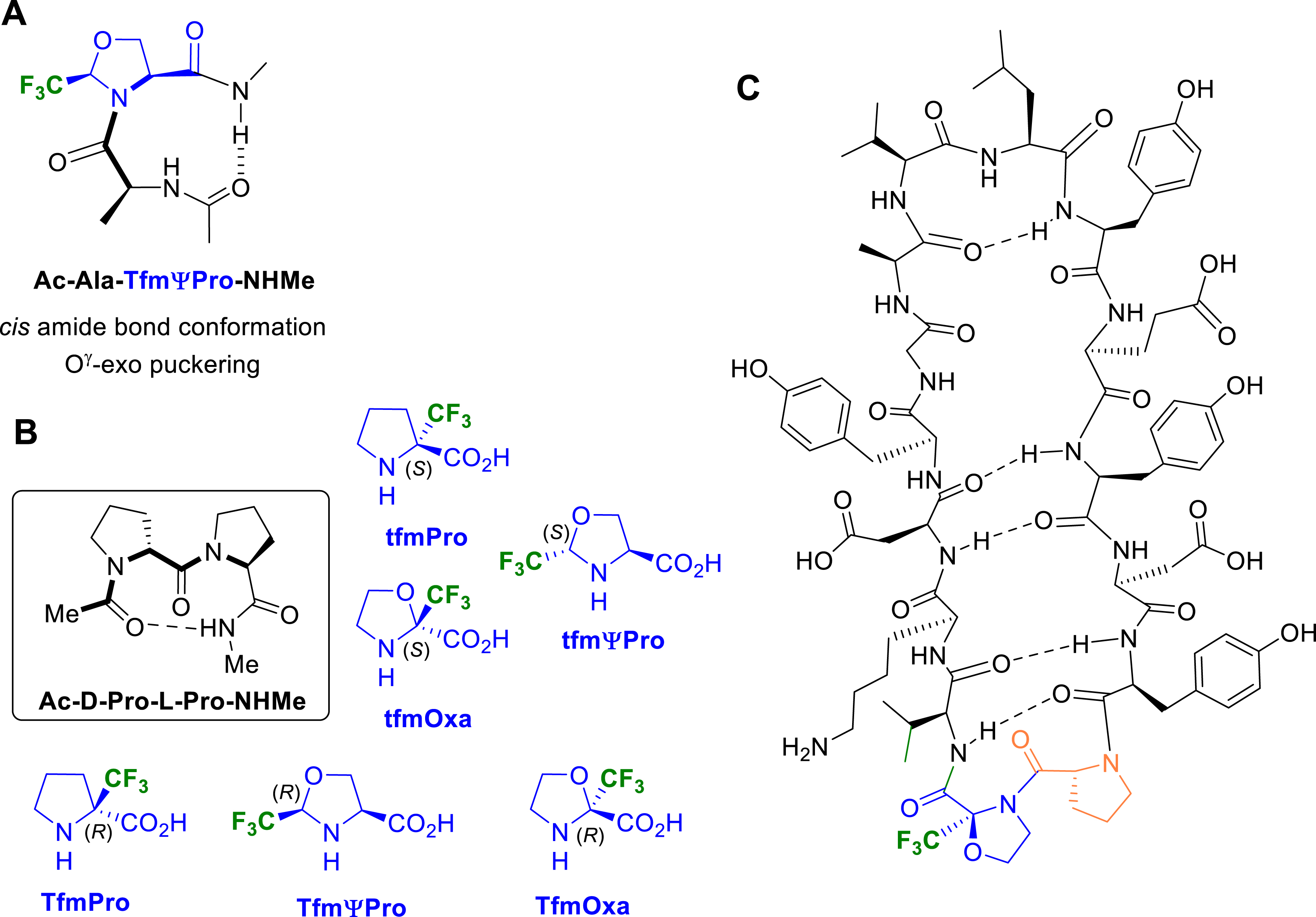
In collaboration with Prof. Y. K. Kang and Dr. E. Miclet, we conducted NMR studies and theoretical calculations on our model peptides Ac–CF3ΨPro–NHMe in chloroform, DMSO and water, allowing for characterizing the solvent effect on their conformational properties [55]. In particular, we showed the ability of the CF3 group to control the ψ-dihedral angle. In CHCl3, the 2-(R)-CF3ΨPro (TfmΨPro) significantly stabilized a γ-turn–like conformation, whereas 2-(S)-CF3ΨPro (tfmΨPro) stabilized polyproline I (PPI)/polyproline II (PPII)-like backbone conformations (Figure 7A). These same PPI/PPII conformations were highly dominating in water (>90%), whatever the CF3ΨPro configuration. The proline effect is therefore greatly enhanced using trifluoromethylated pseudoprolines since the PPI/PPII-like population in water is only ∼50% for the natural prolyl residue. We also observed that the C𝛿–CF3 configuration could be used to efficiently constrain the ring puckering without affecting the cis/trans population ratio. The 2-(R)-CF3ΨPro (TfmΨPro) and the 2-(S)-CF3ΨPro (tfmΨPro) exhibit a strong preference for the O𝛾-exo (up) and the O𝛾-endo (down) puckering, respectively. Minimized structures have proven that both electrostatic interactions and hyperconjugations involving the CF3 group were responsible for these unique structural properties (Figure 7B) [55]. Analysis of the transition states have shown that the favorable F⋯HN electrostatic contact observed in Ac–TfmΨPro–NHMe was replaced by an unfavorable H𝛼⋯HN contact in Ac–tfmΨPro–NHMe, providing a rationale for the low cis–trans energy barrier observed in Ac–TfmΨPro–NHMe (Figure 7C).
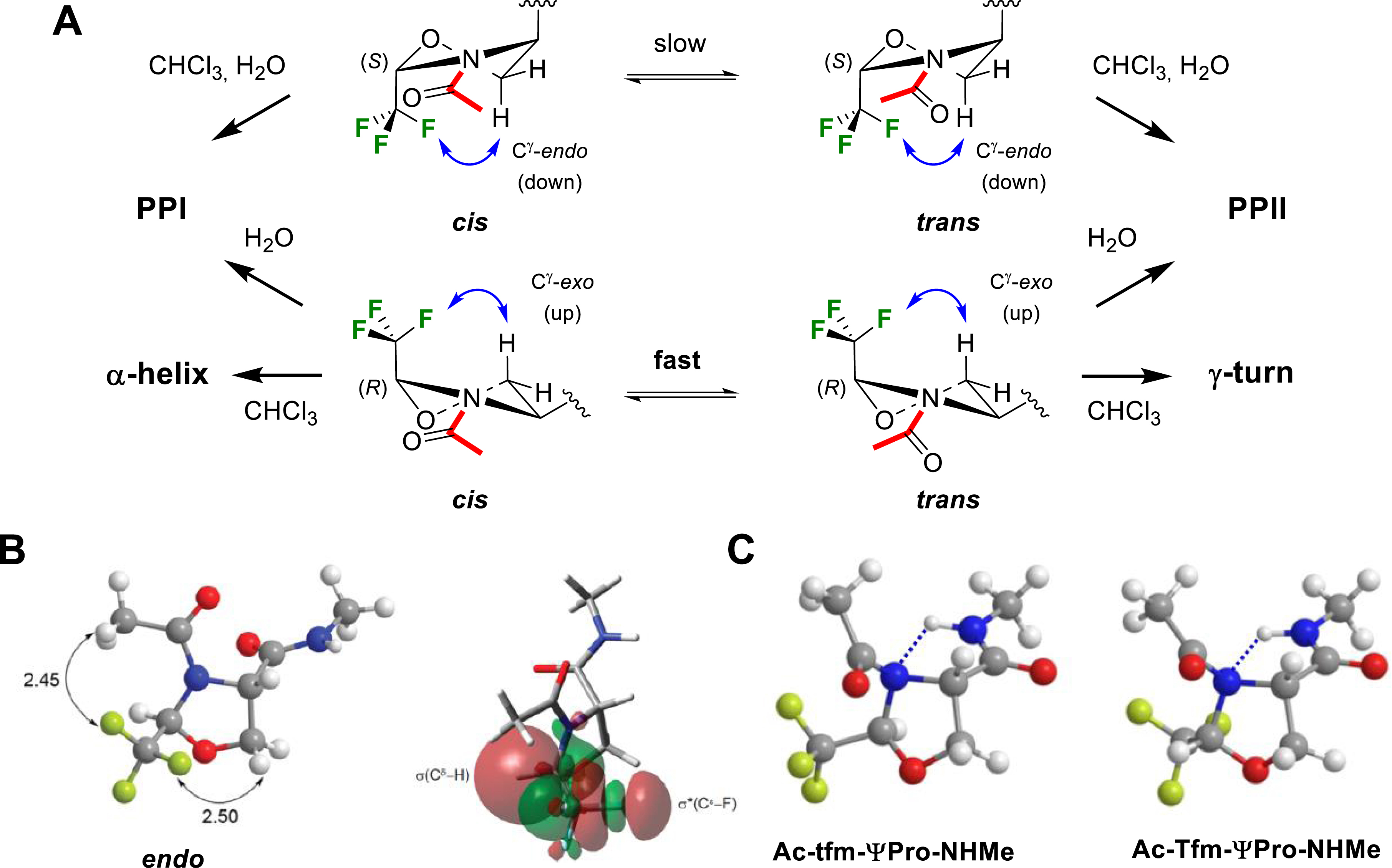
(A) Solvent effect on Ac–CF3ΨPro–NHMe peptide conformation. (B) Main electrostatic interactions and hyperconjugations observed in Ac–tfmΨPro–NHMe. (C) Electrostatic contacts in the transition states of Ac–tfmΨPro–NHMe and Ac–TfmΨPro–NHMe. Reprinted with permission from [55]. Copyright 2012 American Chemical Society.
Replacing the CF3 group by the CF2H on pseudoprolines promoted an amide bond conformation in favour of the cis rotamer in D2O regardless the CF2HΨPro configuration (Figure 8) [29]. This conformational feature was understood by analyzing the unusual NMR parameters of the CF2H groups (Figure 8A). The cis conformers of the 2-(R)-CF2HΨPro (DfmΨPro) and 2-(S)-CF2HΨPro (dfmΨPro) displayed high chemical shift difference (Δ𝛿) of the diastereotopic 19F as well as typical 1H vicinal couplings, both reflecting a very limited rotameric averaging of the CF2H group. Such a restricted rotation of the CF2H group likely arose from an intramolecular H-bonding, the oxygen of the preceding acetyl group facing the positively charged CF2H proton in the cis conformation. Given that the cis conformer remains the majority in D2O, we suggested that such an intramolecular H-bond is endowed with a hydrophobic character. While the 2-(S)-CF2HΨPro (dfmΨPro) displays the same preference for the O𝛾-endo (down) puckering as observed with its CF3 analogue, the 2-(R)-CF2HΨPro (DfmΨPro) shows different puckering preferences between the cis and the trans conformers (Figure 8B) [29]. This puckering switch seems to be required to allow the intramolecular NH⋯O=C H-bond in the trans form.
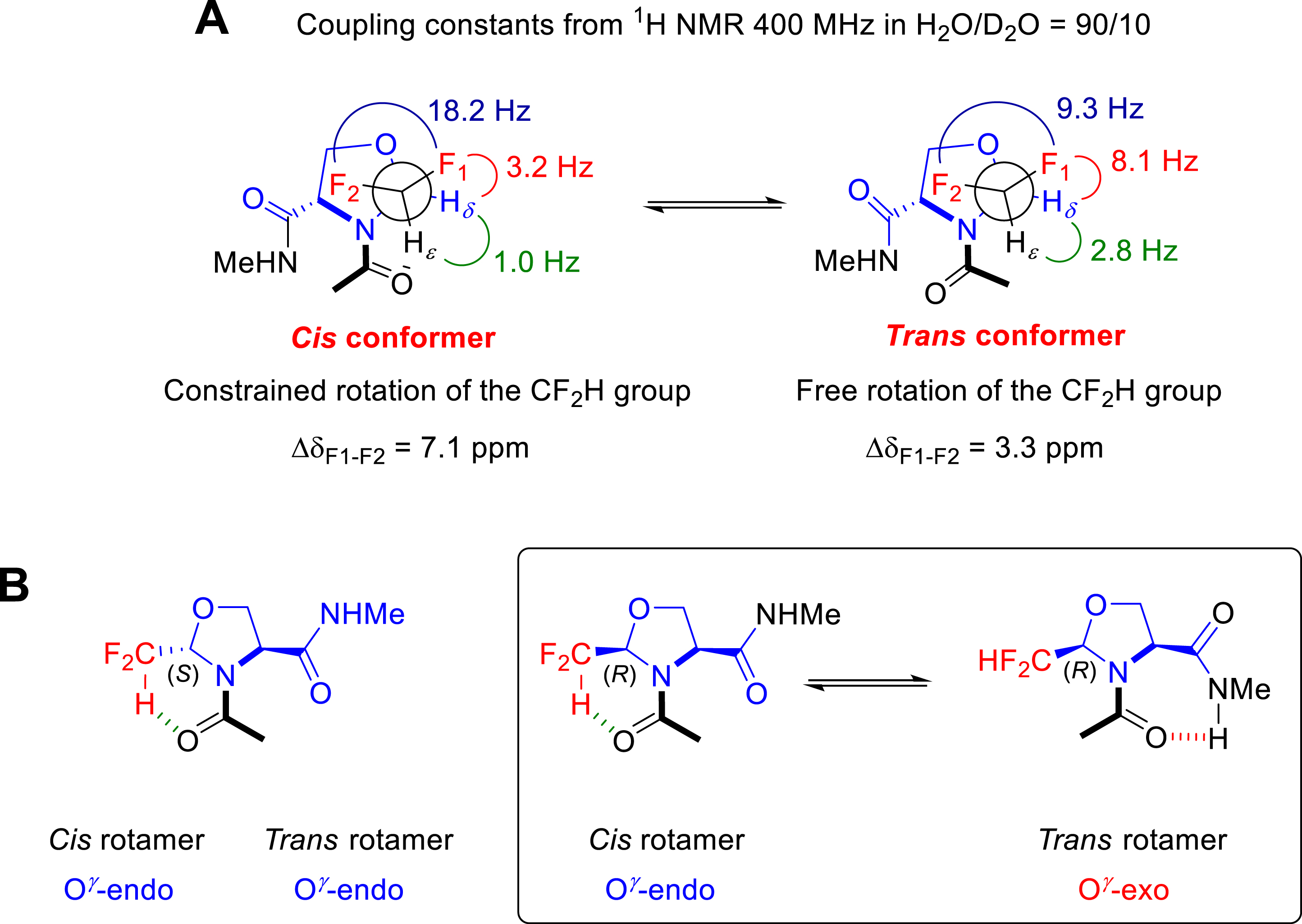
(A) NMR parameters and conformational features of Ac–dfmΨPro–NHMe. (B) Conformational features of Ac–dfmΨPro–NHMe and Ac–DfmΨPro–NHMe.
Later, we reported the conformational study realized on several heterochiral or homochiral Fmoc-Xaa–TfmΨPro-OMe dipeptides, where Xaa = Gly, Ala, Val, Pro, or Aib [44]. The results revealed an interdependence of the stereochemistry of the amino acid preceding the CF3ΨPro residue, the ring puckering and the cis–trans ratio of the prolyl bond (Figure 9). While homochiral dipeptides exhibit strong preference for the trans amide bond, heterochiral analogues favoured the cis amide bond. Interestingly, homo- and heterochiral dipeptides exhibit marked imbalance in conformer populations with opposite cis/trans ratio which is closely related with the steric hindrance of the amino acid side chain. The pyrrolidine puckering of each conformer was then investigated by NMR spectroscopy using the measurement of coupling constants 3 JHβ–Hα. It appeared that the puckering was strongly correlated with the prolyl amide bond since trans amide bond favored a O𝛾-exo puckering and cis amide bond stabilized a O𝛾-endo puckering.
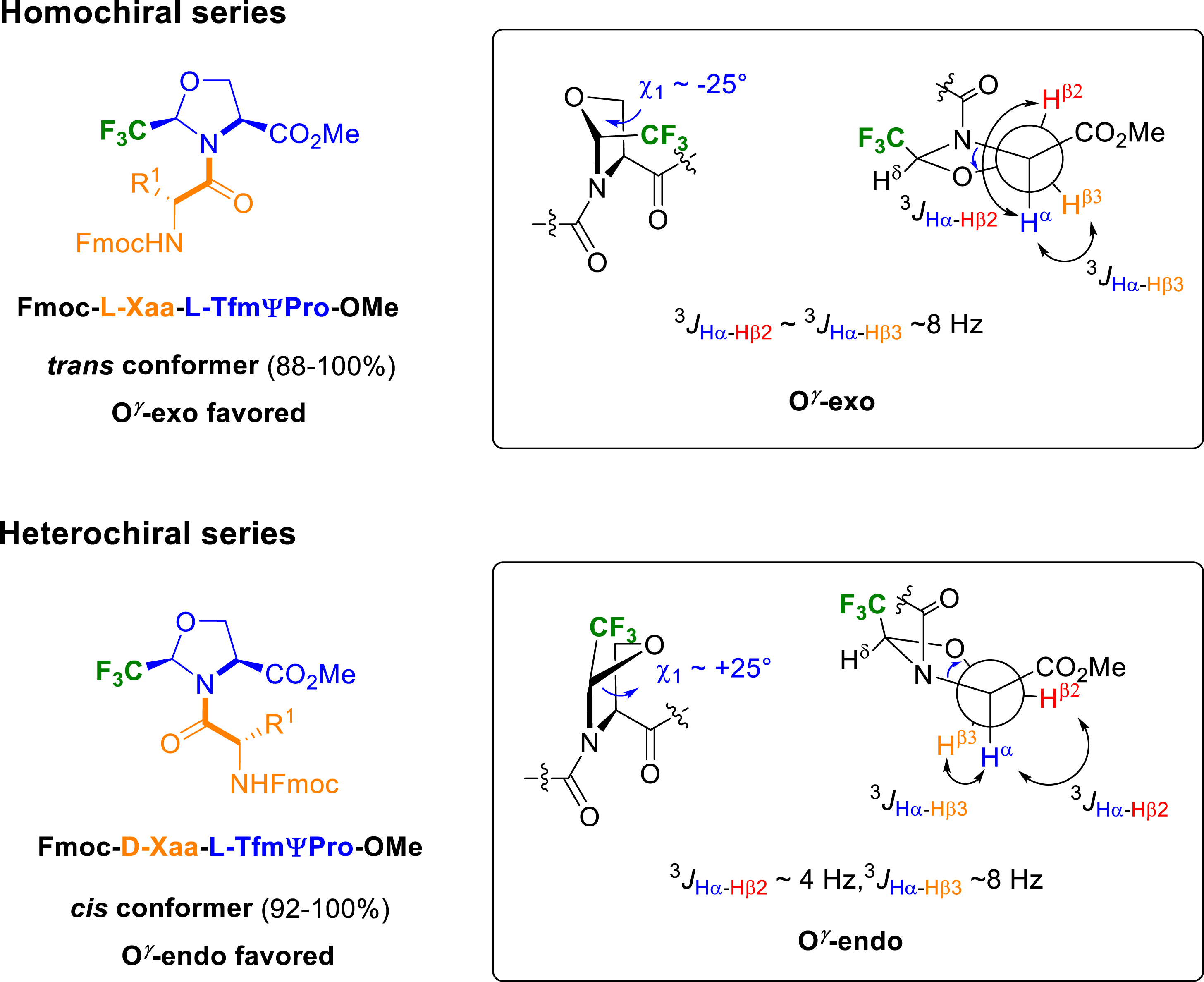
Cis/trans populations and puckering conformation of Fmoc-Xaa–TfmΨPro-OMe.
We then decided to demonstrate that our fluorinated pseudoprolines can serve as an efficient tool to control and increase the structural stability of biomolecules and as a first example, we focused on collagen, the most abundant protein in the animal kingdom. Because collagen can be engineered to serve as a valuable material for biomedical applications, numerous collagen mimetic peptides (CMPs) have been engineered. They are based on the repetition of the triplet Pro–Hyp–Gly, used to mimic the primary sequence of collagen, and contain proline derivatives substituted at their C𝛾 and/or C𝛽 position to stabilize or functionalize collagen triple-helix mimics. Using CD, NMR, and molecular dynamics (MD) studies, we first demonstrated on collagen triplet models that the 2-(R)-CF3ΨPro (TfmΨPro) and the 2-(S)-CF3ΨPro (tfmΨPro), when properly arranged, meet the structural requirements for a triple-helix assembly (Figure 10A) [56]. Despite small distortions of the canonical 𝜙 and 𝛹 dihedral angles, the incorporation of the two adjacent tfmΨPro and TfmΨPro residues maintained a high population of the all-trans conformation (tt), which is required for a triple-helix assembly (Figure 10B). Analysis of ring puckers for the tt conformers showed that tfmΨPro and TfmΨPro residues displayed the strongest propensities to stabilize the endo and exo conformations, respectively.
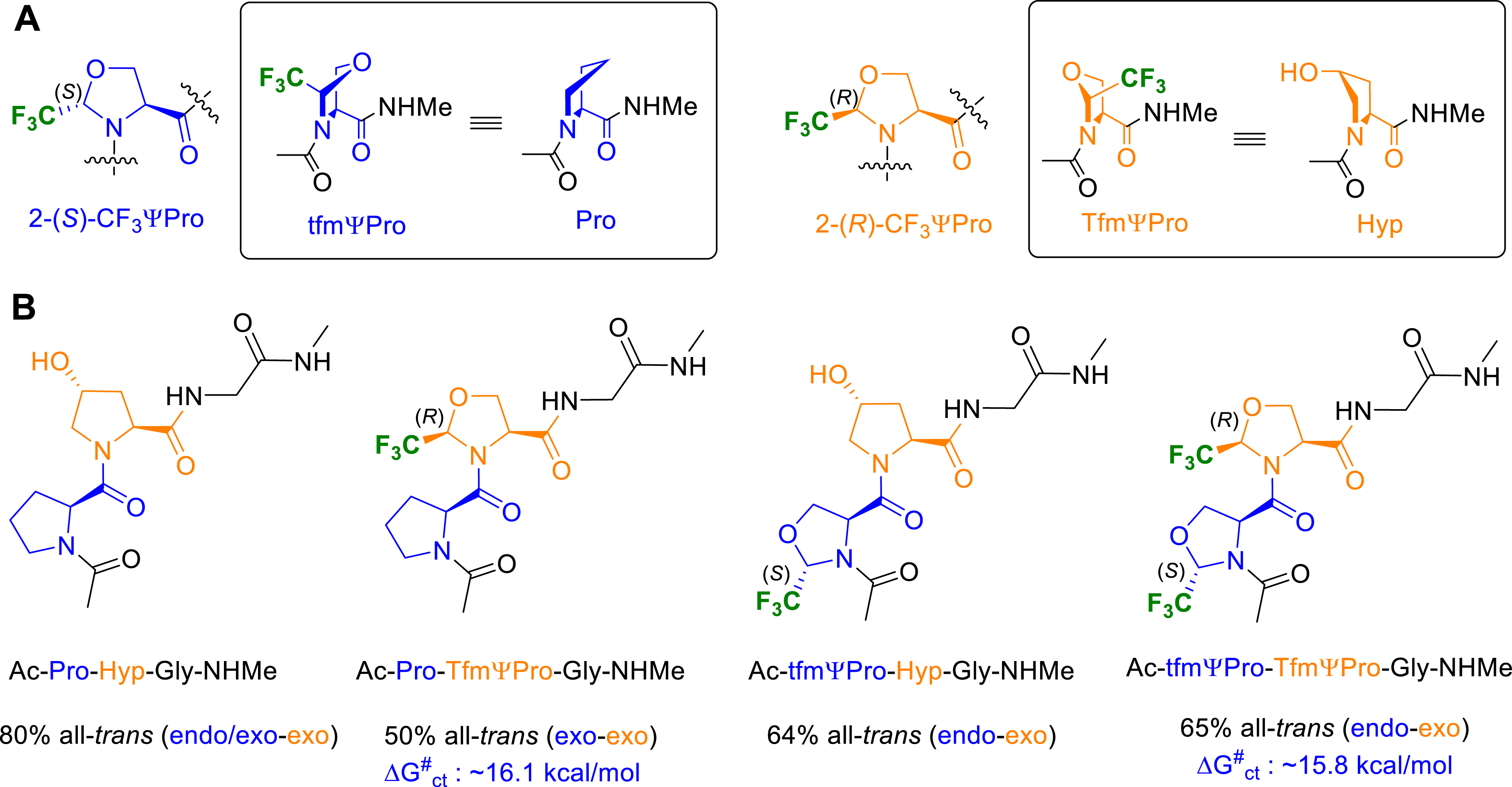
(A) Puckering preferences of Pro, Hyp, tfmΨPro and TfmΨPro residues. (B) Cis–trans populations (%) and puckering preferences for collagen triplet model peptides.
MD calculations showed that incorporation of the tfmΨPro and TfmΨPro in a host–guest CMP sequence slightly affects the hydrogen-bond network in the close neighborhood of the substitutions. Nevertheless, the triple-helix model remained stable with all six trifluoromethyl groups pointing outward from the triple helix (Figure 11A). Therefore, we decided to synthesize two host–guest CMPs (Figure 11B). Both CD and 19F NMR spectroscopy were used to demonstrate the existence of the fluorinated triple helix in solution, which, according to the literature, was, however, less stable than its parent CMP (Figure 11C). These results demonstrated for the first time that C𝛿-substituted proline analogues can be incorporated in a CMP sequence, even for relatively large groups as trifluoromethyl groups [56].
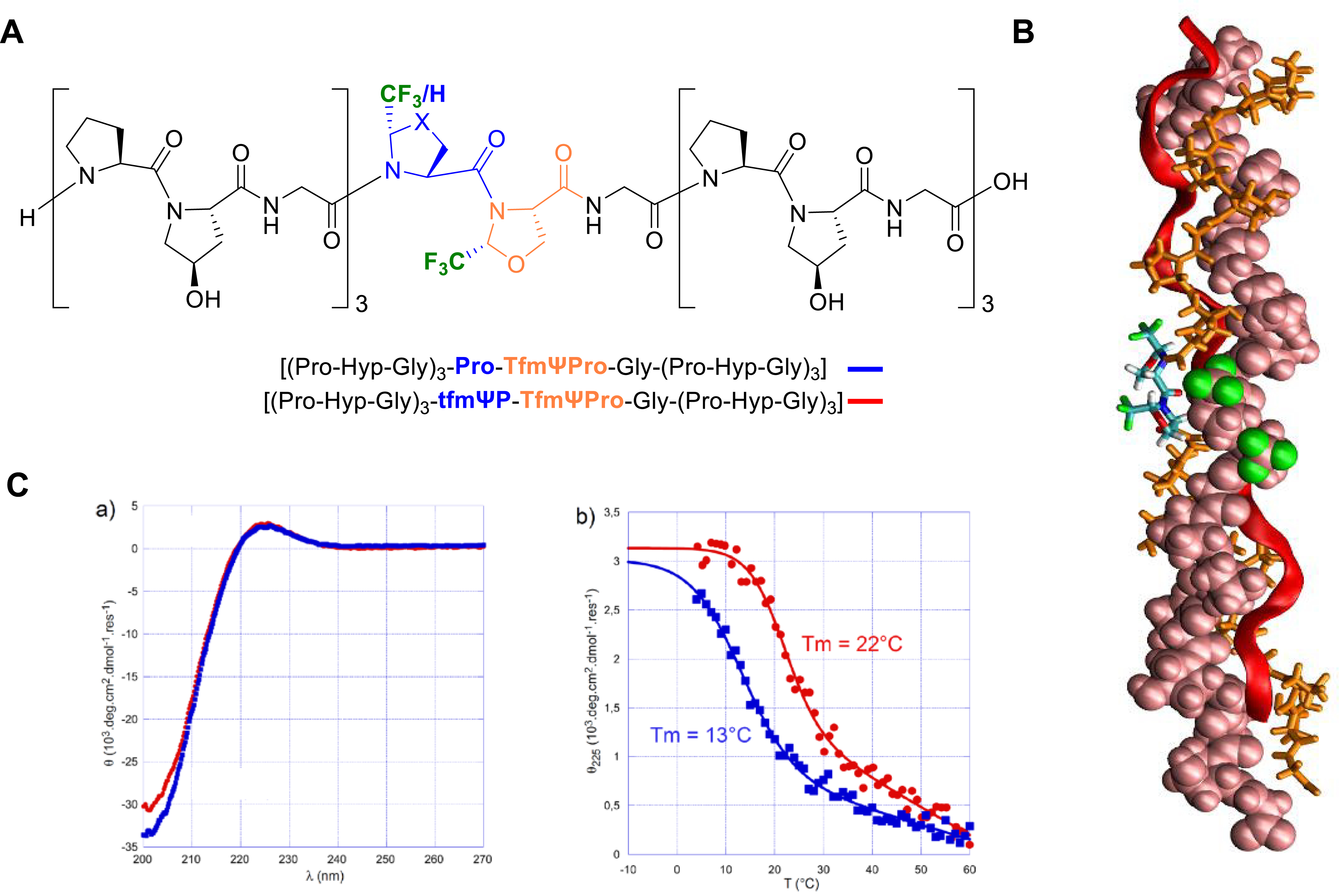
(A) Chemical structure of fluorinated host–guest CMPs. (B) Conformation of the homo-trimer helix of [(Pro–Hyp–Gly)3–tfmΨPro–TfmΨPro–Gly–(Pro–Hyp–Gly)3] after a 500 ns simulation in explicit solvent. The initial structure was derived from the 1K6F PDB structure. (C) Circular dichroism spectroscopy of fluorinated CMP [(Pro–Hyp–Gly)3–Pro–TfmΨPro–Gly–(Pro–Hyp–Gly)3] (blue squares) and [(Pro–Hyp–Gly)3–tfmΨPro–TfmΨPro–Gly–(Pro–Hyp–Gly)3] (red circles). (a) CD spectra recorded at 4 °C. (b) Thermal denaturation of CMPs followed by the molar ellipticity at 225 nm. (B) and (C) are adapted from [56]. Reprinted with permission from [56]. Copyright 2023 American Chemical Society. Masquer
(A) Chemical structure of fluorinated host–guest CMPs. (B) Conformation of the homo-trimer helix of [(Pro–Hyp–Gly)3–tfmΨPro–TfmΨPro–Gly–(Pro–Hyp–Gly)3] after a 500 ns simulation in explicit solvent. The initial structure was derived from the 1K6F PDB structure. (C) Circular dichroism spectroscopy of fluorinated CMP [(Pro–Hyp–Gly)3–Pro–TfmΨPro–Gly–(Pro–Hyp–Gly)3] (blue squares) ... Lire la suite
Very recently trifluoromethyl pseudoprolines have been incorporated into polyproline backbones and the result is that these incorporations do not affect the PPII helical conformations of the peptides. The trifluoromethyl groups provides an increased hydrophobicity of the peptides and can act as a 19F NMR probes to demonstrate the enzymatic stability of the PPII helix [54].
Proline is often found as a turn inducer in peptides or proteins. The ability of the CF3-pseudoprolines to stabilize β-turn was first investigated on the tetrapeptide model Ac–Ala–TfmΨPro–NHMe. MD calculations and extensive NMR analyses have been carried out in solution and revealed the presence of a stable type-VI β-turn which is characterized by a cis peptide bond conformation (Figure 12A) [57]. Later, we were interested in designing a cyclic peptide mimicking the β-hairpin structure of the CDR3 loop of Nb80. In this context, we investigated different fluorinated proline analogues of the well-established d-Pro–l-Pro segment (pP) segment as a β-turn promoter [58]. An in silico conformational study was first performed on a set of 10 fluorinated variants of the pP sequence incorporating the (S)- or (R)-α-trifluoromethylprolines (tfmPro and TfmPro, respectively), the (S)- or (R)-α-trifluoromethyl-oxazolidines (tfmOxa and TfmOxa, respectively), or the 2-(S)- or 2-(R)-trifluoromethyl-pseudoprolines (tfmΨPro and TfmΨPro, respectively) (Figure 12B). Each fluorinated variant of the d-Pro–Pro segment was based on the Ac–XX1–XX2–NHMe pseudotetrapeptide structure, providing therefore the required H-bond donor and acceptor to promote a type II′ β-turn. Out of the investigated combinations, only tfmPro–l-Pro and d-Pro–TfmOxa exhibited a strongly stabilized β-turn conformation relative to the parent pP segment. By grafting the d-Pro–TfmOxa sequence to the CDR3 loop of the nanobody Nb80, the corresponding cyclic peptide showed only one conformer, allowing unambiguous assignment of the NMR signals. This result appeared as an improvement knowing that the d-Pro–l-Pro bearing cyclic peptidomimetic presented a large conformational heterogeneity and multiple overlapping resonances between major and minor conformers (Figure 12C).
(A) Chemical structure of pseudotetrapeptide Ac–Ala–TfmΨPro–NHMe and overlay of the 5-lowest energy structures obtained by molecular dynamics under NMR restraints. (B) Ac–d-Pro–l-Pro–NHMe as a model for β-turn formation and set of CF3-prolines surrogates. (C) Chemical structure of structure of the fluorinated cyclic mimetic of the CDR3 loop of Nb80.
3.3.2. Acyclic α-Fluoroalkyl-α-amino acids
C𝛼-tetrasubstituted α-amino acids, such as α-aminoisobutyric acid (Aib), are conformationally constrained nonproteinogenic α-amino acids. They are commonly used in peptides as strong inducers of helical conformations, such as α-helix or 310-helix. We demonstrated that the (R)-and (S)-α-trifluoromethylalanine (Tfm-Ala), two chiral fluorinated analogues of Aib, can serve as efficient 19F-labels to elucidate by solid-state NMR spectroscopy the backbone conformation and the helix alignment in lipid bilayers of the harzianin HK-VI (HZ), a member of the antimicrobial peptaibol family (Figure 13A). Circular dichroism (CD) spectroscopy confirmed that the secondary structure of the fluorinated analogues of harzianin HK-VI wild type remained unperturbed [45]. Chiral Cα-tetrasubstituted α-amino acids also proved to be useful tools to control the handedness of helical structures, in particular when incorporating into achiral Aib oligomers. We reported the synthesis of short Aib-based foldamers bearing the (R)-Tfm-Ala or (S)-Tfm-Ala at different positions into the peptide chain [59, 60]. NMR studies, CD spectroscopy and X-ray crystallography confirmed the stabilization of the 310 helical structure either in solution or in the solid-state. We demonstrated that the chiral fluorinated Tfm-Ala residue can be employed not only as chiral controllers to induce the screw-sense preference of short Aib-based oligomers, but also as 19F NMR reporter to quantify the magnitude of the helical excess (Figure 13B). Interestingly, the selectivity of the screw-sense was found to be reversed compared to that induced by the non-fluorinated quaternary l-amino acid chiral inducers due to the electronic properties of the CF3 group that favors a dipole alignment of the CF3 with the helix macrodipole [60].
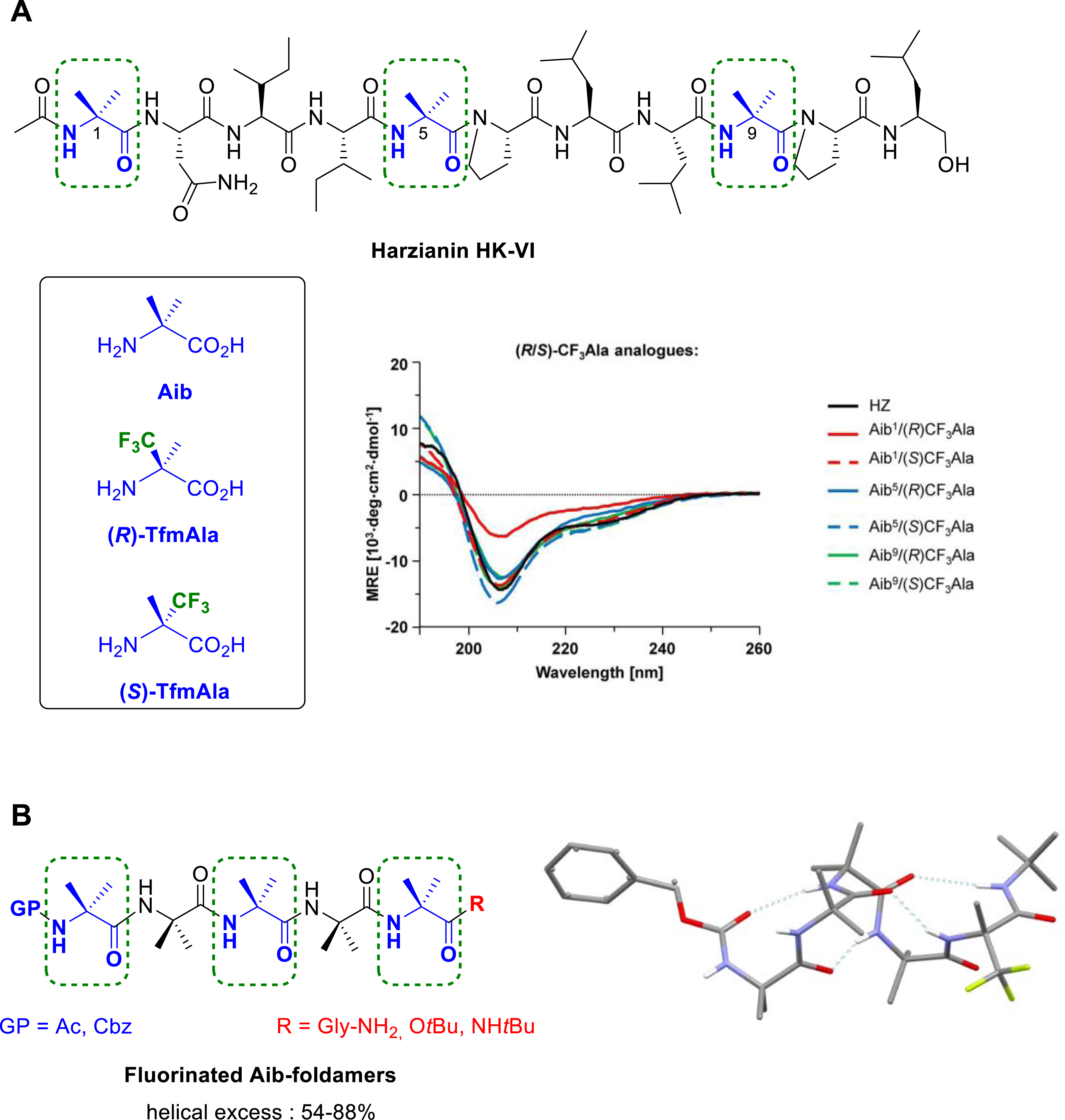
(A) Chemical structure of Harzianin HK-VI and representative CD spectra of selectively 19F-labeled HZ analogues in the presence of lysoMPC micelles. (B) Chemical structure of Aib-foldamers and solid-state structure of Cbz–(Aib)4–(R)-TfmAla-OtBu (M helix). Dashed boxes indicate the positions of the native Aib that have been replaced by (R)- or (S)-Tfm-Ala residue.
3.4. α-Fluoroalkyl-α-amino acids as 19F NMR probes
Orthogonal 19F-labeling for solid-state NMR reveals the conformation and orientation of short peptaibols in membranes [45]. Fluorinated peptides constitute valuable probes for investigating by liquid or solid-state 19F NMR spectroscopy the interactions of biological molecules since biological media are free of fluorine atoms avoiding background signal and because 19F isotropic chemical shift is very sensitive to small structural perturbations. In 2022 our group showed that a small peptide incorporating the non-natural α-quaternarized (R)-α-trifluoromethylalanine ((R)-α-Tfm-Ala) provides a convenient and accurate monitoring of trypsin activity [52]. The accuracy of the 3-FABS (3-Fluorine atoms for biochemical Screening) technique was demonstrated by the rapid inhibition potency (IC50) evaluation of a known trypsin inhibitor (Figure 14).
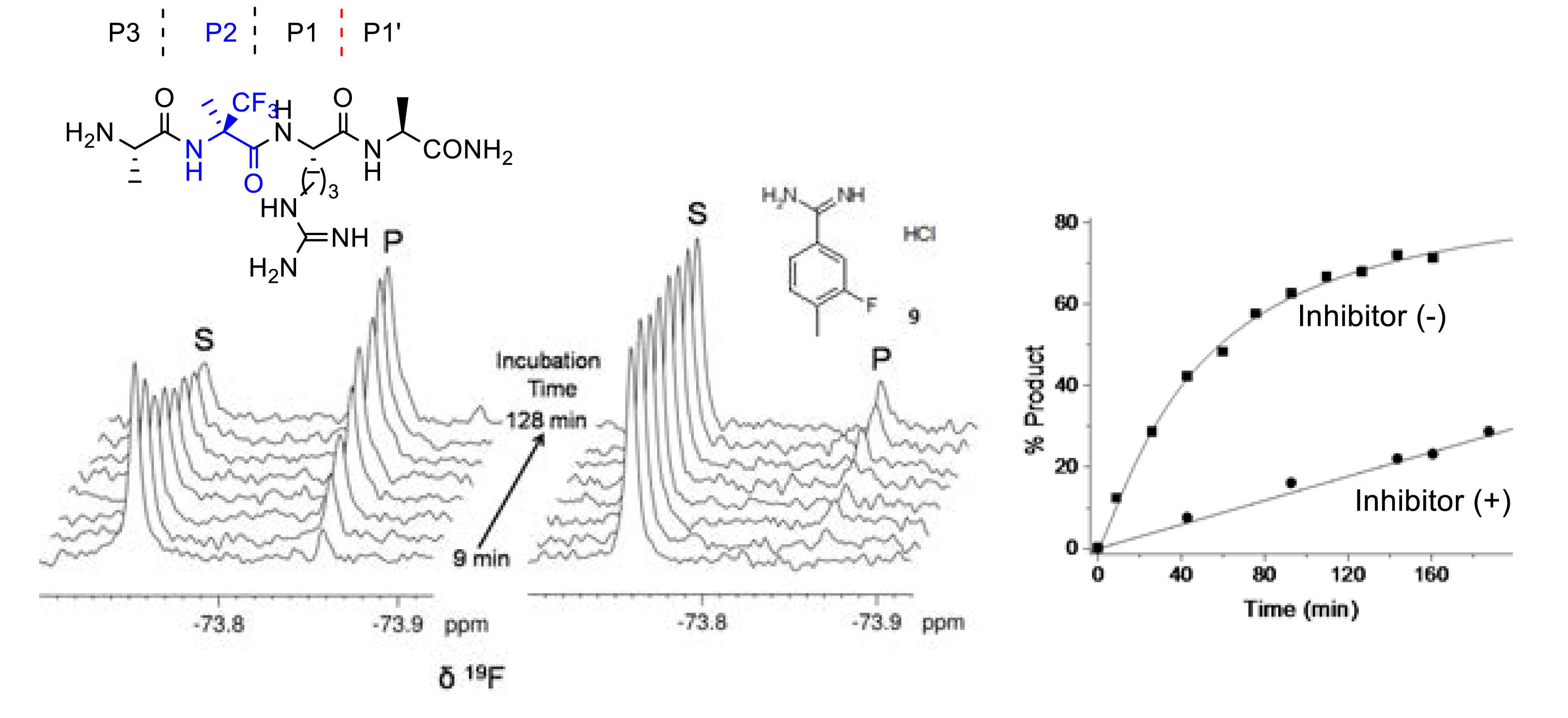
(R)-α-trifluoromethylalanine for 3-FABS monitoring of trypsin inhibition. Reprinted with permission of Wiley-VCH from [52], Copyright 2022.
4. Conclusion
In summary, we have developed over the past 20 years several highly efficient synthetic methods for the synthesis of enantiopure chiral fluorinated compounds mainly in the amino acids series and their derivatives. The synthesis of these compounds is still a highly challenging task and there is a great need of new robust and highly stereoselective methods for their preparation. We demonstrated that these compounds provided original advances in asymmetric synthesis and peptide chemistry and biology. It is anticipated that further development could display highly promising opportunities in organocatalysis, medicinal chemistry and chemical biology.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Acknowledgements
The authors warmly thank all the students, PhD students, post-docs and colleagues who contributed to these works. The French Ministry of Education and Research, CY Cergy Paris Université, the CNRS, the ANR, the CY Initiative of Excellence (grant “Investissements d’Avenir”) and the French Fluorine Network (GIS-FLUOR) are thanked for their financial support.