



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Carbohydrates are defined as “compounds such as aldoses and ketoses having the stoichiometric formula Cn(H2O)n” (IUPAC definition) [1] including derivatives. When they are attached by a glycosyl group to a non-acyl moiety forming a mixed acetal, they are called “glycosides” (IUPAC definition) [2]. They generally contain a six-membered (pyranoside) or a five-membered (furanoside) cyclic structure. They are key biomolecules found in living cells and ubiquitously involved in numerous biological processes [3, 4]. They adorn cell surfaces in the form of oligomers linked in a covalent manner via O- or N-glycosidic bonds to proteins or lipids anchored in cell membranes. Their interactions with enzymes are responsible for cell adhesion, cell recognition, and immunological processes [5]. In the furanose series, they constitute the skeleton of crucial biopolymers, namely RNA and DNA, composed of ribose or 2-deoxyribose units. Therefore, from a therapeutic point of view, these compounds are interesting targets as drug candidates. However, to attain a suitable administration–distribution–metabolism–elimination (ADME) profile, the development of analogues possessing unnatural structures is often a profitable approach [6]. Indeed, many unnatural saccharide-type drugs have been already commercialized such as dapagliflozin for type II diabetes, miglustat for type I Gaucher disease, zanamivir for influenza A or B infections, and zidovudine for HIV infection, and so on (Scheme 1).
A few examples of carbohydrate mimics with therapeutic interest.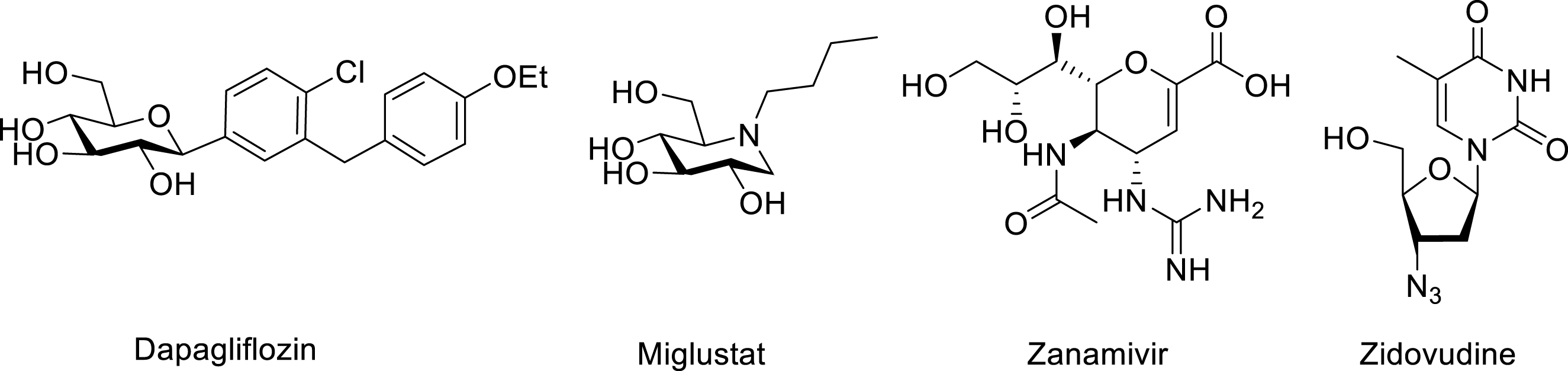
Unnatural chemical bonds may possess a superior chemical and enzymatic stability compared to their natural counterparts while retaining a similar conformation. The development of unnatural biomolecule analogues aims not only at increased biostability but also at increased specificity or biological activity compared to the natural substrate [7, 8]. In addition to medicinal applications, glycoside analogues have attracted interest from cosmetic and biotechnology fields as biosourced materials [9, 10]. Despite their huge potential, the application of such analogues is limited due to their poor availability, which frequently requires significant synthetic effort [11, 12, 13]. Indeed, the discovery of new glycosides of interest is conditioned by the chemical space accessible by using the current synthetic toolbox.
As such, the development of innovative synthetic methods is of great importance. In recent decades, among the synthetic tools developed in organic synthesis, metal-catalyzed reactions, such as cross-coupling or C–H functionalization, have gained huge popularity but have been mainly explored on aryl and heteroaryl compounds [14, 15, 16]. Indeed, the application of these powerful reagents on glycosides is an emerging field [17, 18]. This observation can be explained by the fact that the application of new synthetic methods on complex biomolecules is generally challenging and needs double expertise (e.g., glycochemistry and organometallic chemistry).
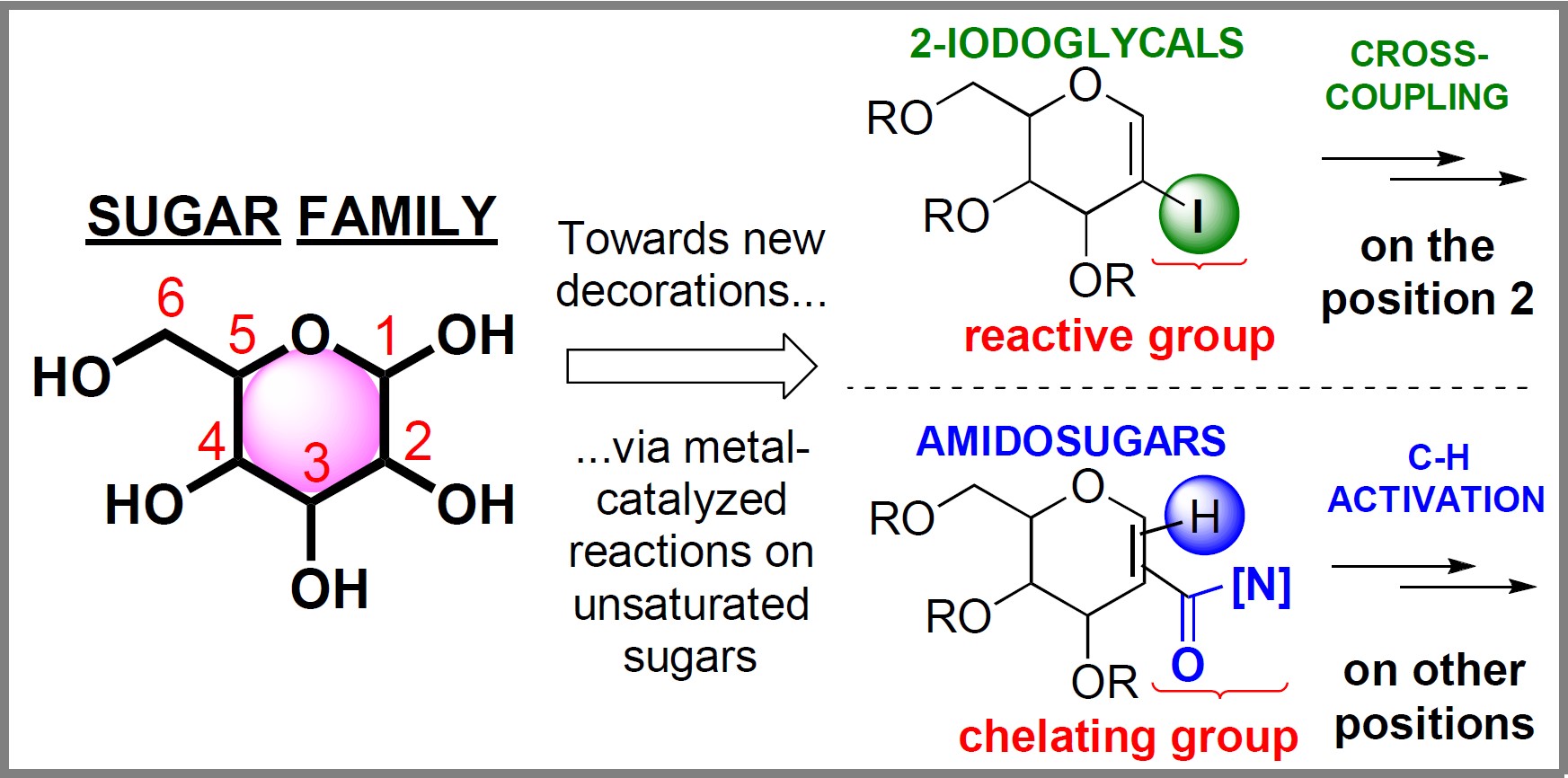
This account presents the contribution of our group to the development of metal-catalyzed methods on glycals (unsaturated carbohydrates) to generate unnatural analogues. Two types of reactivity are presented, namely, C–H functionalization and cross-coupling reactions, to produce substituted glycals. Glycals represent all carbohydrates possessing a double bond between the C1 position (pseudo-anomeric position, close to the endocyclic oxygen) and the C2 position. The reactivity of glycals is diverse, allowing the modification of several positions of the ring (C1, C2, and C3 positions) [19]. For instance, the so-called Ferrier rearrangement allows the introduction of a substituent at the C1 position by using different nucleophiles. In this reaction, a glycal bearing a leaving group at the C3 position can be activated by an acidic reagent and then be attacked at the C1 position by a nucleophile, inducing the shift of the double bond between the C2 and C3 carbons with the elimination of the leaving group at C3 [20, 21]. In addition to standard reactivity, the unsaturation was shown to be particularly suitable in metal-catalyzed reactivity. The two developed methods allow the functionalization of the two positions of the endocyclic double bond. As the accessibility and the reactivity of C1 and C2 positions of the glycal double bond are different, the synthetic strategy to target one specific position as well as the design of the starting substrates has to be soundly chosen (Scheme 2).
The two developed strategies to target the different positions of the glycal double bond.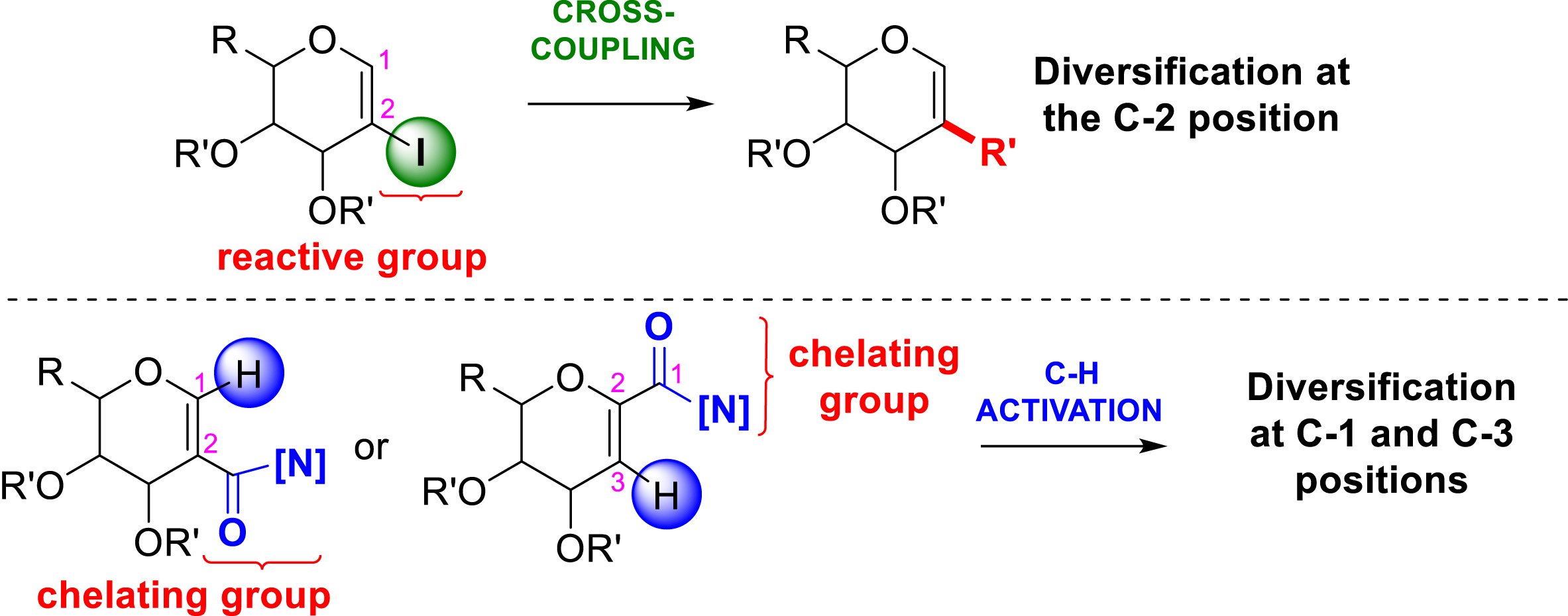
2. Metal-catalyzed cross-couplings on 2-iodoglycals
2.1. State of the art
Iodoglycals are glycosidic compounds containing a vinylic carbon–iodine bond with huge potential for functionalization using metal-catalyzed reactions. 1-Iodoglycals have been investigated as partners in metal-catalyzed cross-coupling but have faced two major problems, namely, the instability of the structure and the necessity to work on TIPS-protected compounds as the usual way to synthesize such molecules requires difficult conditions such as the use of t-BuLi [22, 23, 24, 25, 26]. On the contrary, the development of easily accessible and bench-stable 2-iodoglycals by Vankar et al. [27] in 2014 presented to the glycochemist a very interesting platform to plan diverse metal-catalyzed reactivities at the C2 position and thus to extend the chemical space with potential glyco-analogues [28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59]. Before this pioneering publication, only standard cross-couplings have been investigated on 2-iodoglycals, namely Suzuki–Miyaura, Sonogashira, and Heck reactions. Since 2015, numerous carbonylative methods have been described. A few examples of carbon–heteroatom bond formation (C–S, C–N, and C–P) have also been reported (Scheme 3).
State of the art of 2-iodoglycal functionalization using metal-catalyzed cross-couplings.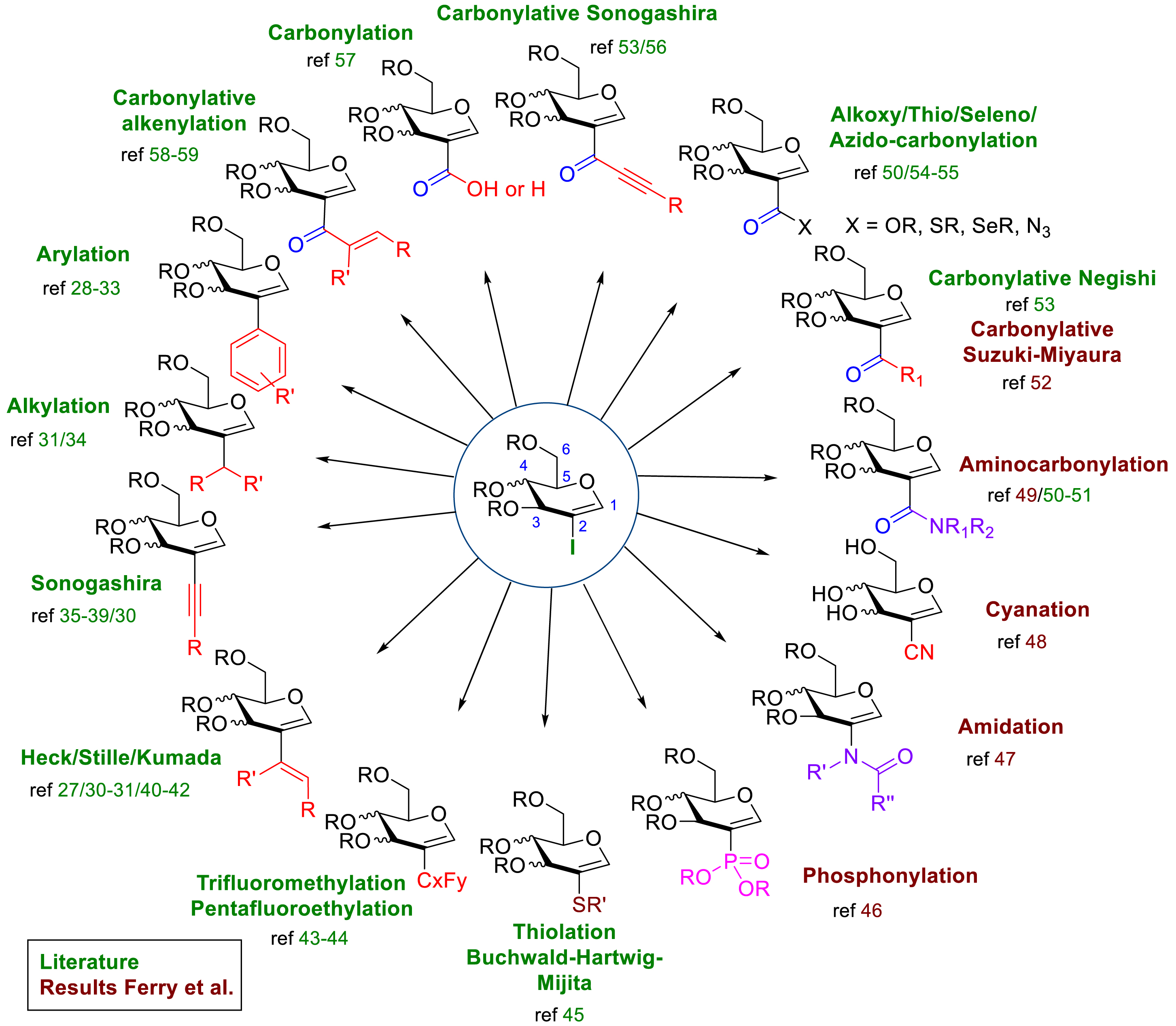
2.2. Contribution to the field
Our first contribution to the field of metal-catalyzed cross-coupling on 2-iodoglycals started in 2016 in a context where cross-coupling reactions on these substrates were an underexplored field.
2.2.1. Carbon–carbon bond formation
Our first objective was to propose a way to link a glycoside with an amine via a stable C–C bridge, and we turned our attention to an aminocarbonylation process. To transform 2-iodoglycals into 2-amidoglycals, palladium catalysis in the presence of “CO” and amines as partners was used (Scheme 4). The optimized setup involved a Pd(II) catalyst [Pd(OAc)2] and a phosphine ligand (PPh3) under basic conditions (K2CO3) and the use of an easy-to-handle solid “CO” source (e.g., Mo(CO)6) in the presence of 2 equiv of amine in dioxane at 80 °C. The palladium catalyst appeared to be crucial since its absence failed to form the desired C2-amidoglycal. To determine the role of the metallic “CO” source (Mo(CO)6) in the catalytic cycle, a CO gas atmosphere was used instead of Mo(CO)6, and the desired compound was formed with a similar yield. These results suggest that palladium is the species involved in the catalytic cycle and that Mo(CO)6 acts only as a “CO” generator. Under these conditions, diverse amine derivatives (aliphatic and cyclic amines, anilines, sulfonamides) have been successfully introduced, and the amido derivatives have been isolated in 36–94% yield. Glycosylated amino acids were also obtained (yields = 33–60%). Different protecting groups on the glycal, such as acetyl, benzyl, and isopropylidene as well as two glycal examples bearing a d-galactal configuration completed the scope of the study. Four obtained perbenzylated or peracetylated examples have been finally deprotected by hydrogenolysis or Zemplen-type conditions (MeONa in MeOH) to produce unprotected C2-amidoglycals where the glycal double bond remained intact (Scheme 4) [49].
Aminocarbonylation.
In 2019, our team took advantage of carbonylative reactions to propose the formation of 2-ketoglycals via a carbonylative Suzuki–Miyaura reaction between 2-iodoglycals and boronic acids as nucleophiles. The optimized conditions were quite similar to those employed in the aminocarbonylation method except for the palladium source and the solvent (Pd2dba3 instead of Pd(OAc)2 and DMF instead of dioxane). Despite similar reactivity, the mechanism seems to be different since the unique presence of Mo(CO)6 as the metallic species in the medium (in the absence of the palladium catalyst) still generated small amounts of the desired compound whereas the replacement of the solid “CO” source by “CO” gas failed to form the 2-ketoglycal. The formation of a new active species formed between Mo(CO)6 and boronic acid may be possible but remains to be proven. Different arylboronic acids bearing electron-donating or electron-withdrawing groups have been used. Whereas electron-donating groups led to good yields, electron-withdrawing groups furnished lower yields. Heteroaryl boronic acids (furan, thiophene, isoquinoline, and benzofuran types) showed good reactivity in addition to two alkenylboronic acids giving rise to unsymmetrical dialkenylated ketones. Three other 2-iodoglycals, a d-galactal, a d-xylal, and a d-lactal derivative, bearing benzyl or acetyl protecting groups proved to be also compatible with the reaction conditions. Due to the introduction of a ketone, conditions other than hydrogenolysis had to be considered to deprotect the compounds. Thus, two obtained perbenzylated compounds have been deprotected by applying BCl3 conditions. The products were obtained in moderate to good yields. However, the presence of this ketone allowed post-functionalizations. Indeed, the introduced carbonyl function has been successfully reduced to the corresponding mixture of two diastereomeric alcohols in good yield, which have then been engaged in an indium-mediated allylic substitution in the presence of oxygenated nucleophiles. The corresponding exocyclic double bond has been finally reduced under hydrogenolysis conditions (H2 (1 atm), Pd/C (20 wt%), NH4OH (2.5–5 equiv), MeOH, rt) to furnish C2-α-glucopyranosides (50–92%) (Scheme 5) [52].
Carbonylative Suzuki–Miyaura reaction.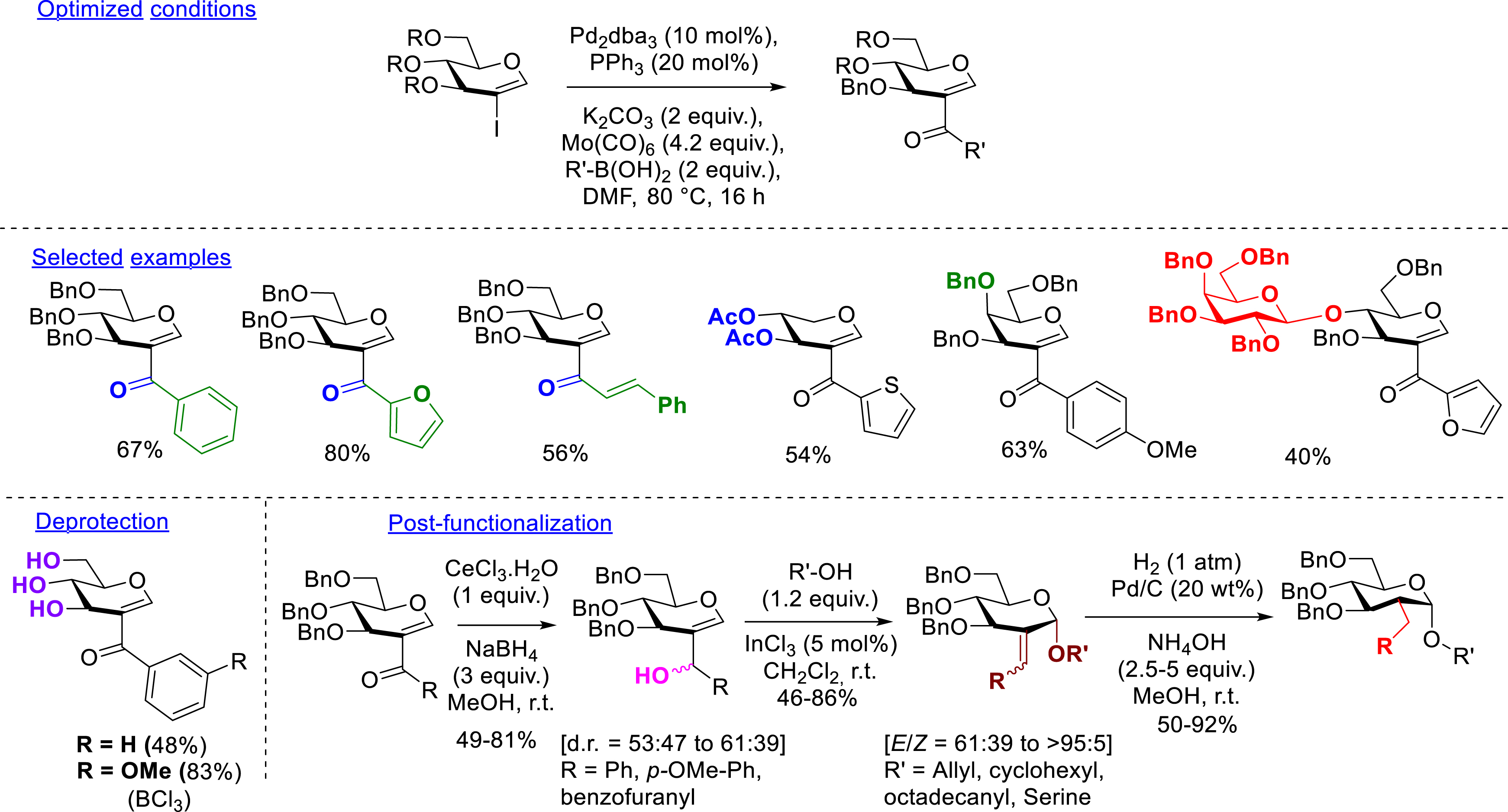
Most of the results reported in metal-catalyzed cross-couplings applied to 2-iodoglycals do not deal with the deprotection of the obtained structures. The deprotection step can be problematic depending on the sensitivity of the function introduced at the C2 position, which has been already pointed out for the carbonylative Suzuki–Miyaura reaction. In the case where the introduction of a sensitive functional group is planned, the synthetic strategy has to be rethought. In this context, we considered working on unprotected 2-iodoglycals in order to develop a cyanation reaction. Thus, the deprotection step was achieved before the cross-coupling reaction in order to directly generate unprotected cyano derivatives at the end of the metal-catalyzed reaction. Unprotected 2-iodoglycals can be easily obtained by deacetylation of the corresponding peracetylated 2-iodoglycals. From these unprotected 2-iodoglycals, a palladium-catalyzed cyanation in aqueous medium using K4[Fe(CN)6] as a safe and non-toxic “CN−” source has been developed leading to the desired 2-cyanoglycals. The developed conditions took advantage of the reactive palladium precatalyst, which allowed the use of mild temperatures. The crucial parameter was the quantity of base (CH3COOK), which has to be in slight excess compared to the palladium species. The reaction was performed in a mixture of t-BuOH/H2O. Diverse pyranosidic glycals (2-iodo-d-galactal, 2-iodo-d-xylal, 2-iodo-l-rhamnal), a disaccharide-type glycal (2-iodo-d-lactal), a furanosidic glycal (2-iodo-d-ribal) as well as diversely protected 2-iodo-d-glucals (perbenzylated or persilylated) have been successfully converted to the corresponding cyano derivatives in excellent yields (44–100%). The nitrile group can be a versatile platform to generate other functional groups. With this objective in mind, the obtained 2-cyanoglycals were transformed into five glyco-analogues bearing an amine (with and without the glycal double bond), an amide, a triazole, or a ketone at the C2 position of the glycal (Scheme 6) [48].
Cyanation of unprotected 2-iodoglycals.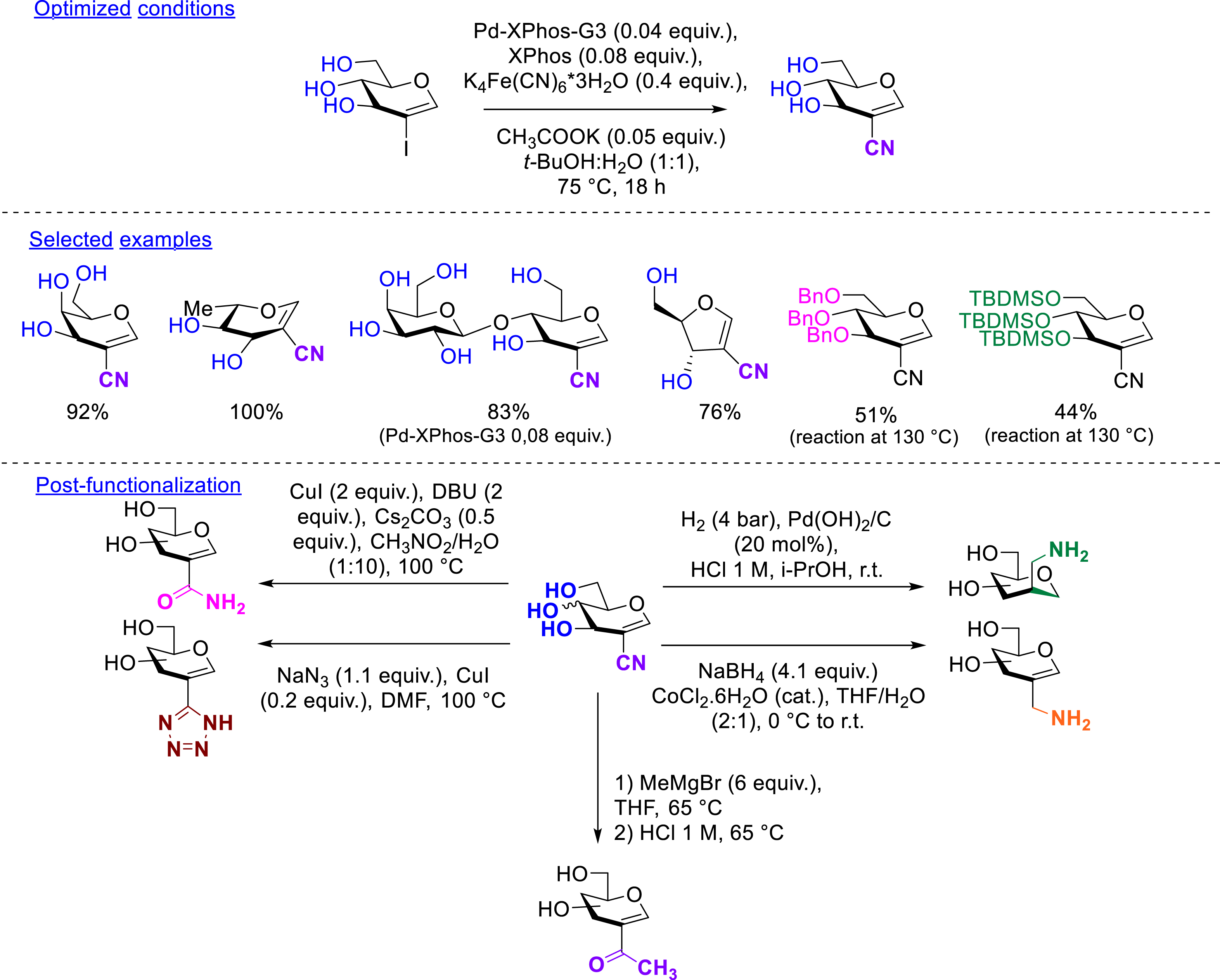
2.2.2. Carbon–heteroatom bond formation
Before 2020, the formation of C2–heteroatom bonds on 2-iodoglycals were underreported in the literature (only one example of a C–S bond formation). The creation of the C–N bond was first explored by our team due to the importance of N-acetylglycosamine structures in biology. With this aim, an amidation reaction has been developed using dual catalysis combining a nickel and a copper salt catalyst in the presence of a ligand and a base, in N,N-dimethylacetamide at 120 °C, with an excess of amides (2 equiv) as the coupling partners. Cyclic amides such as pyrrolidinone, morpholinone, and oxazolidinone as well as diversely substituted benzamides have been tolerated. The resulting coupling products were isolated in moderate to excellent yields (20–82%). Diverse perbenzylated as well as unprotected 2-iodo-d-glucals and 2-iodo-d-galactals have been considered. Aliphatic linear and primary amides unfortunately failed to form the desired compounds as did peracetylated 2-iodoglycals. The hydrogenolysis of the obtained structures led to the removal of the benzyl protecting groups as well as to the reduction of the glycal double bond, forming a unique isomer: d-mannosamide structures from d-glucals and d-talosamides from d-galactals (Scheme 7) [47].
Amidation of 2-iodoglycals.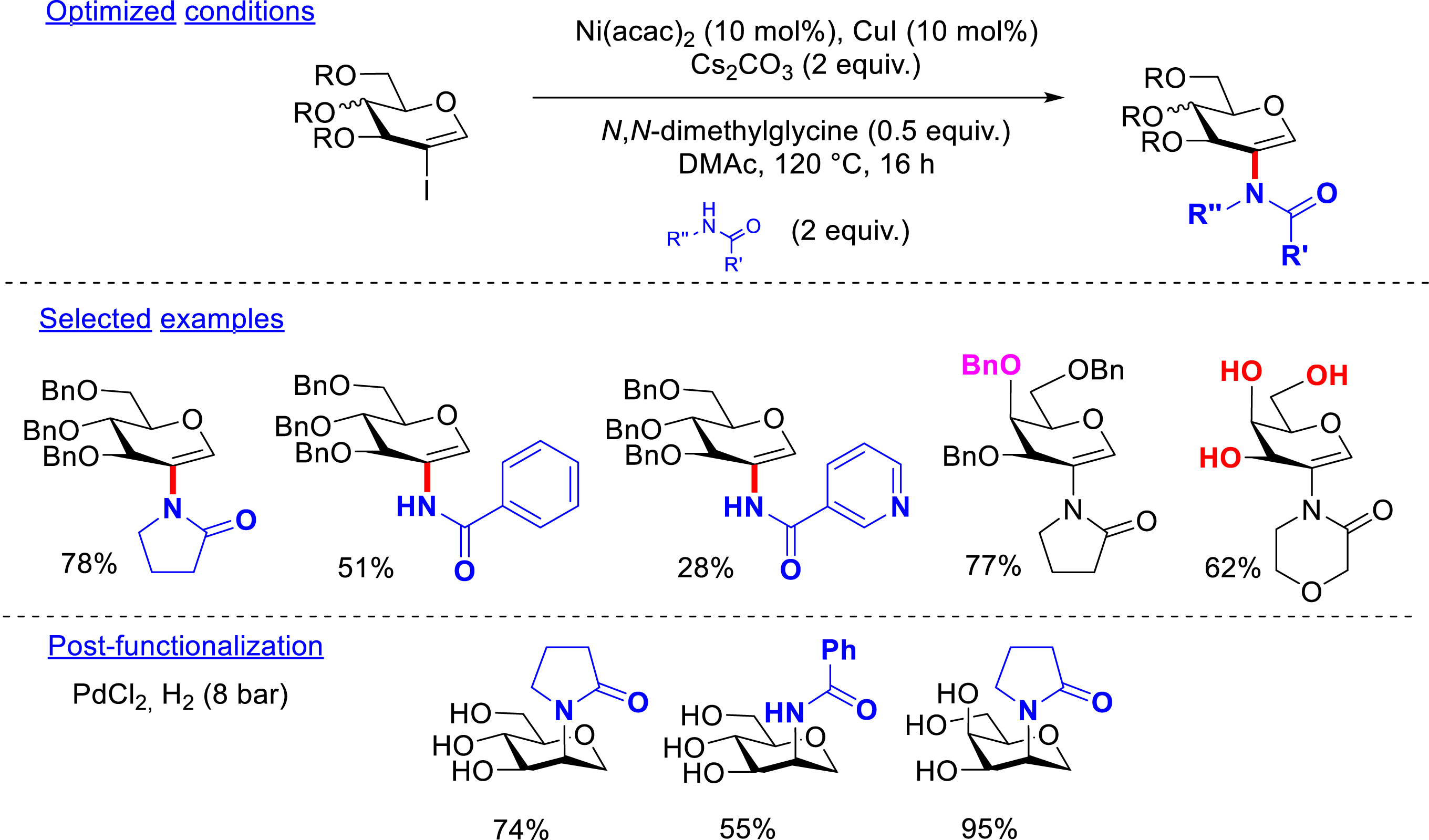
The possibility of inserting a phosphonate group at the C2 position appeared to be interesting from a biological point of view considering the ubiquity of phosphorylation in cells. The creation of a C–P bond has been explored via a palladium-catalyzed Hirao-type reaction. The use of trialkyl P(III) phosphite partners instead of a dialkyl P(V) phosphite led to excellent yields in only 1 h. From a mechanistic point of view, after the standard oxidative addition of palladium to the C–I bond of the glycal, a ligand exchange allowed the chelation of the nucleophilic P(III) form of the phosphite at the palladium center. Thanks to a nucleophilic species present in the medium, an Arbuzov rearrangement occurs to form the P(V) intermediate. The desired product is finally generated after reductive elimination. The developed conditions showed wide compatibility not only with numerous 2-iodoglycals (with a d-glucal, a d-galactal, a l-rhamnal, or a d-lactal configuration) bearing benzyl, acetyl, or para-methoxybenzyl protecting groups but also with unprotected 2-iodoglycals (Scheme 8). Different phosphites (trimethyl, triethyl, triisopropyl phosphites) have been successfully coupled. The protic cosolvent has to be adapted to the phosphite reagents (MeOH when trimethyl phosphite is used, EtOH when triethyl phosphite is used, etc.). The deprotection of a perbenzylated and a peracetylated compound has been also successfully demonstrated [46].
Phosphonylation of 2-iodoglycals.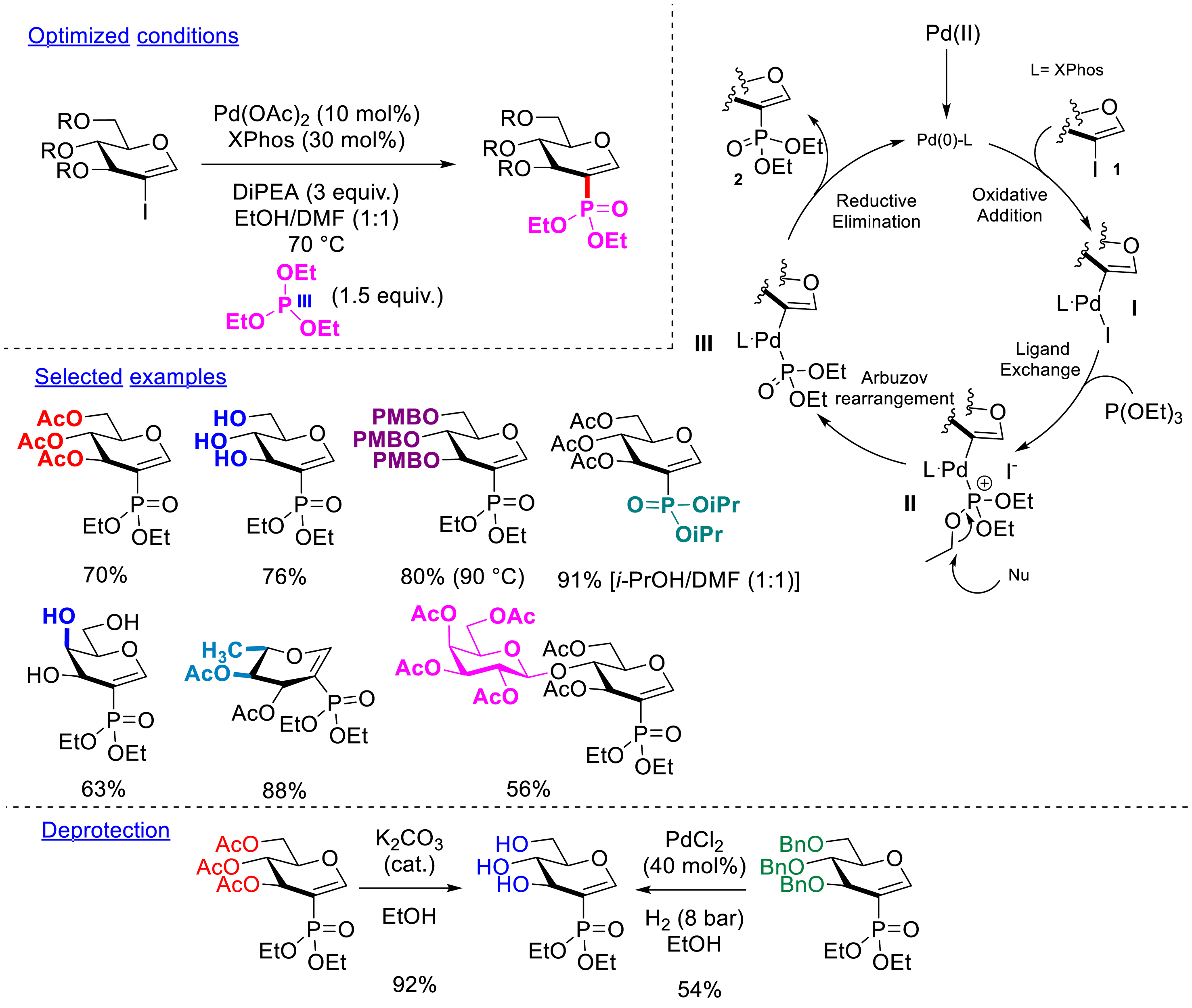
In 2022, in a collaboration with Malinowski et al. (Warsaw University of Technology, Poland), the Sonogashira reaction on peracetylated 2-iodoglycals was revisited with alkynyl partners ready to be used in porphyrin heterocycle synthesis. This reaction was developed to create unnaturally glycosylated porphyrins. The Sonogashira cross-coupling was realized with a dual palladium/copper catalyst (Scheme 9) [39].
Synthesis of unnatural glycoporphyrins via a Sonogashira reaction.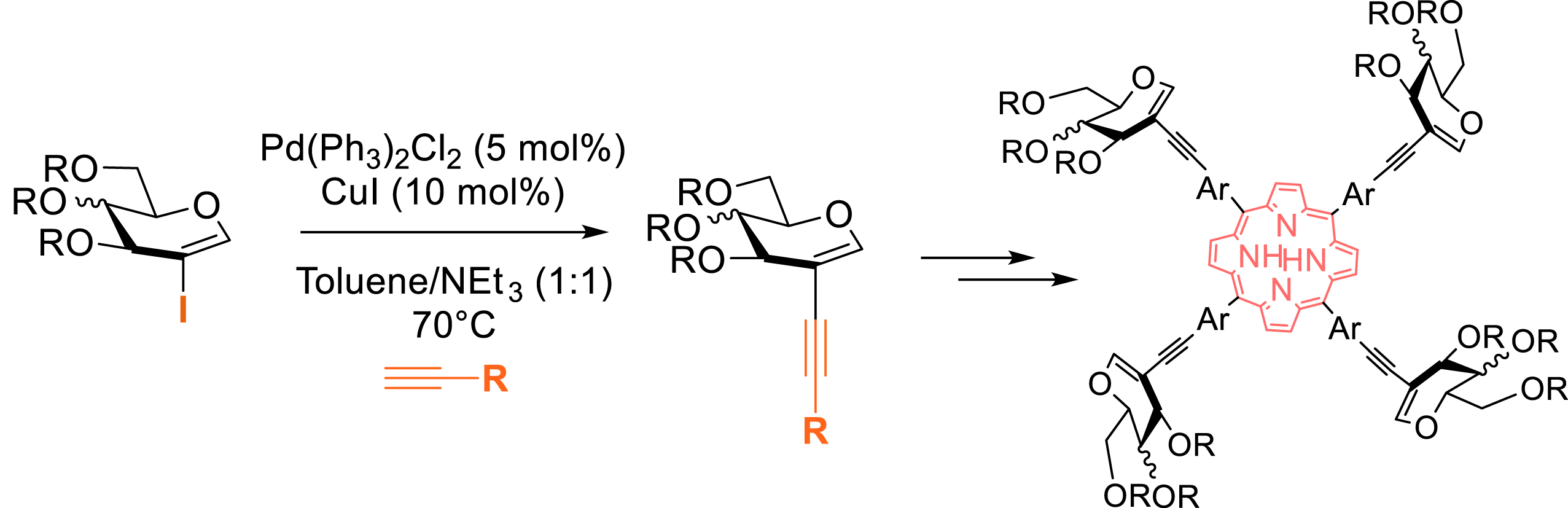
3. Directed metal-catalyzed C–H functionalization of unsaturated carbohydrates
3.1. State of the art
The goal here is to directly functionalize a C–H bond while avoiding the necessity to synthesize reactive intermediates [14, 15, 16]. The application of such reactivity to carbohydrates is therefore highly challenging, and the number of such examples in the literature is scarce but slowly increasing [17]. The main challenge is to selectively mono-functionalize complex structures having several similar C–H bonds. A few metal-catalyzed C–H functionalizations of Csp3–H bonds of saturated carbohydrates have been developed in the last few decades by Compain et al., Lecourt et al., and more recently by Messaoudi et al. using rhodium- or palladium-catalyzed processes [17, 22, 60, 61, 62, 63, 64, 65, 66, 67, 68, 69]. The strategy consists of introducing a reactive group (intramolecular reactivity) or a metal-chelating directing group (DG) (intermolecular reactivity) on the carbohydrate, close enough to target a C–H bond specifically and more generally the anomeric position (C1 position) or the C2 or C5 position (Scheme 10). However, these methods depend on a fine design of the starting carbohydrate, which frequently needs to be synthesized via a multi-step sequence, and suffer generally from a lack of versatility. On the contrary, the C–H activation of unsaturated glycosides bearing differentiated Csp2–H bonds appeared to be a good option to easily functionalize a specific position [17, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81]. In the absence of a DG, the reactivity of glycals operates generally at the nucleophilic C2 position (Scheme 10). However, the numerous options to easily fill the C2 position of the glycal by a cross-coupling reaction allows the addition of a DG at this strategic position, which further allows the possibility of functionalizing other positions. By using this strategy, several C–H functionalizations of glycals have been developed using a pre-installed amide-type DG (Scheme 10).
State of the art of metal-catalyzed C–H functionalization of glycosides.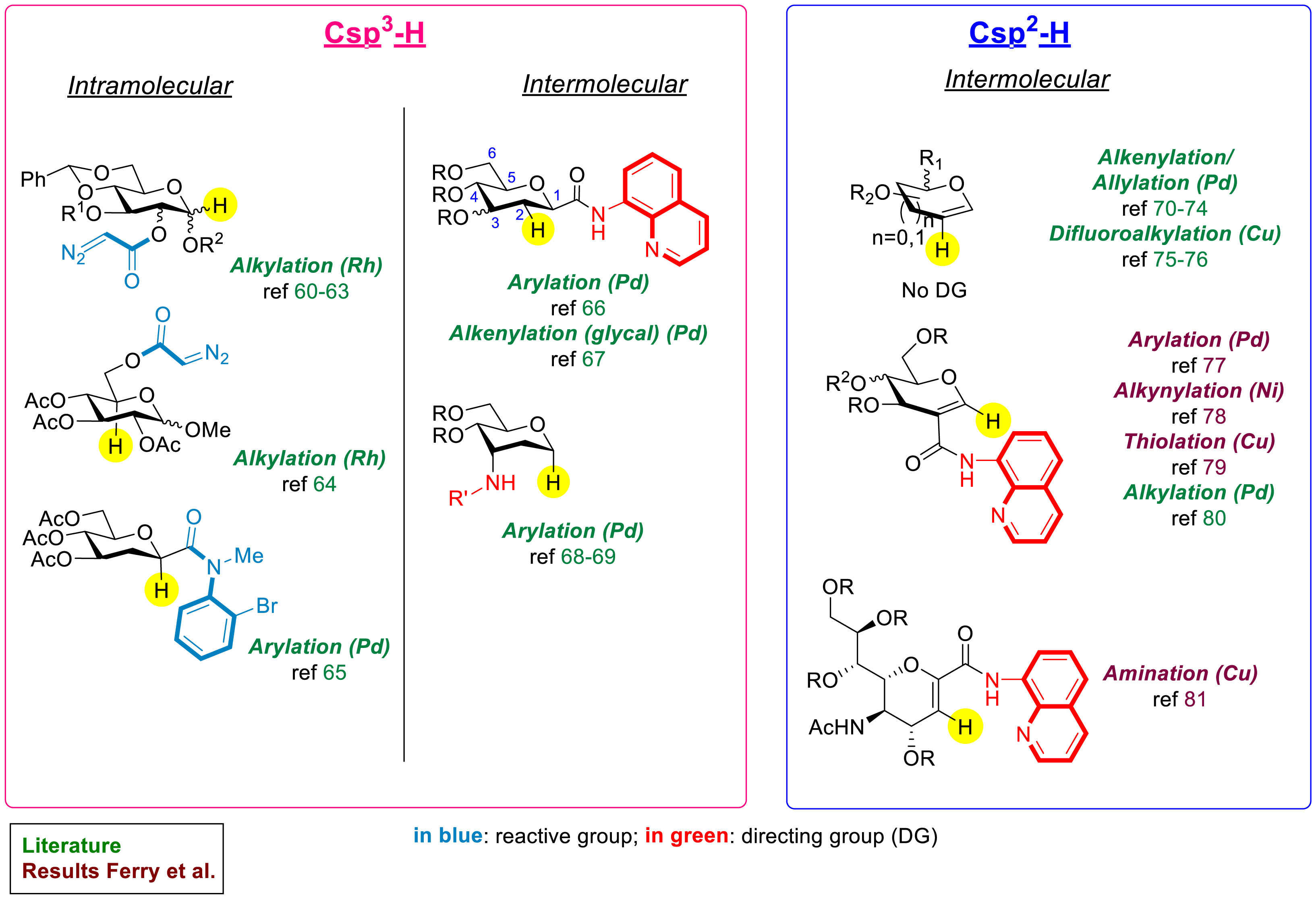
The C–H activation of carbohydrates was sporadic until 2019. Since then, several studies have been reported, but the complexity of the sugars makes the C–H activation somewhat elusive.
3.2. Contribution to the field
3.2.1. Simple pyranosidic glycals
First, we considered the functionalization of the C–H bond at the C1 position of the glycals. However, in the absence of a DG, the C–H functionalization operates almost exclusively at the C2 position, which is the nucleophilic position of the enol ether. In order to guide the reactivity to the C1 position, the introduction of a metal-chelating DG at the C2 position was envisaged. The C2-amidoglycals obtained by the aminocarbonylation method (see Section 2.2.1; Scheme 4) appeared to be ideal starting substrates for the C–H functionalization at the C1 position of glycals thanks to the metal-chelating amide function at C2 (Scheme 11).
Directed C–H functionalization strategy on glycals.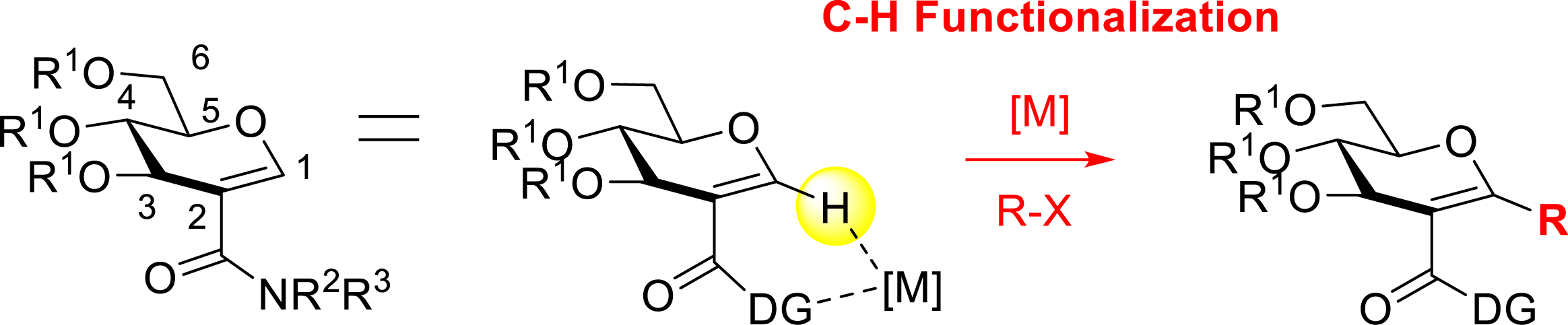
A starting C2-amidoglycal possessing a strong metal-chelating bidentate group, namely 8-aminoquinolide, has been designed and synthesized via aminocarbonylation. For a proof of concept, we turned our attention to the development of palladium-catalyzed intermolecular C–H arylation (Scheme 12). This method uses aryl or alkenyl iodides as electrophilic partners. The reaction was catalyzed by palladium in the presence of silver salts and was achieved in the presence of a weak base and citric acid in toluene at 130 °C. Aryl iodides bearing electron-withdrawing and electron-donating groups were successfully coupled in good yields (38–78%). A heteroaryl and an alkenyl iodide have also been introduced under these conditions. Different types of C2-amidoglycals (d-galactal and d-lactal) protected by benzyl and isopropylidene groups have been synthesized. Finally, this method was applied for the synthesis of glycosylated amino acids (mono- and disaccharide) as well as for the synthesis of an analogue of the dapagliflozin drug. However, the removal of the DG failed due to the poor reactivity of the conjugated amide function. Similarly, the deprotection of benzyl groups has not been achieved so far [77].
C–H arylation.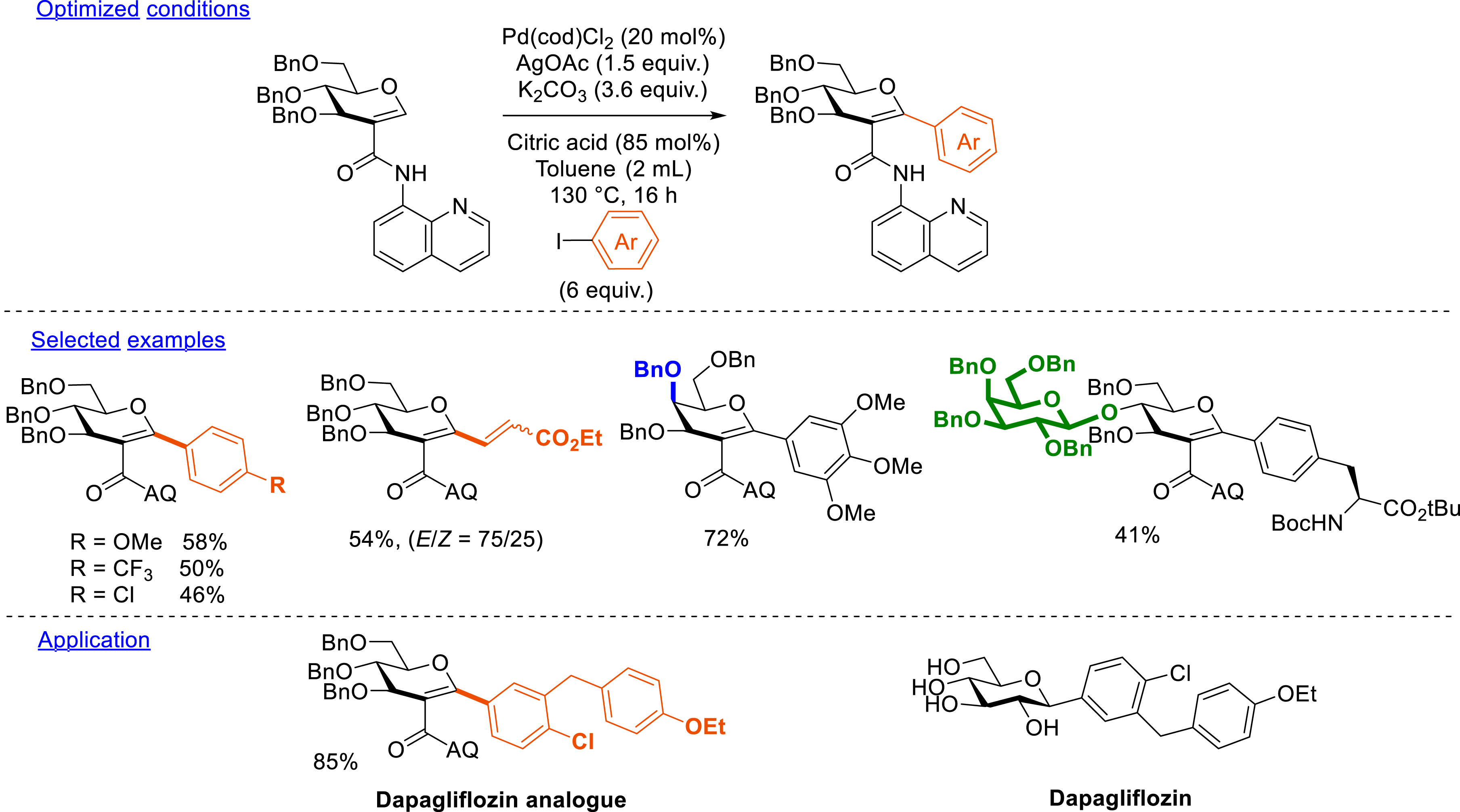
With the C2-amidoquinolide glycals in hand, we took advantage of these substrates to introduce alkynyl groups by C–H alkynylation (Scheme 13). Indeed, the presence of an alkynyl group allows access to further compounds via click chemistry. However, the introduction of an alkynyl substituent failed under palladium catalysis. Inspired by the work of Li et al. [82], we turned our attention to nickel catalysis utilizing alkynyl bromide partners. The conditions involve a nickel salt in the presence of a base in toluene at 140 °C. The key parameter was the introduction of nickel powder after 4 h of preheating with the rest of the reagents. The reason behind this observation is still unclear. Silylated alkynes were also well tolerated and led to moderate to good yields of the coupling products: 22–64%. More interestingly, cyclopropylacetylenyl bromide allowed the formation of the corresponding product in moderate yield (29%). Diverse C2-amidoglycals (d-galactal and l-rhamnal) were involved successfully (21–62%) in the C–H functionalization with alkynes. The post-functionalization of the obtained alkynyl glycosides was finally explored. A preliminary Boc protection of the amide was necessary to successfully remove the silylated group, and the corresponding compound was involved in a one-pot sequence to a copper-catalyzed click reaction with azide partners. This led to several complex glycoconjugates. A glycosylated amino acid, a disaccharide, and a glycosylated biotin were obtained by applying these conditions (Scheme 12). The removal of the DG using pyrrolidine was equally described, and the product was obtained in moderate yield. However, the deprotection of the benzyl groups of these structures was not successful [78].
C–H alkynylation.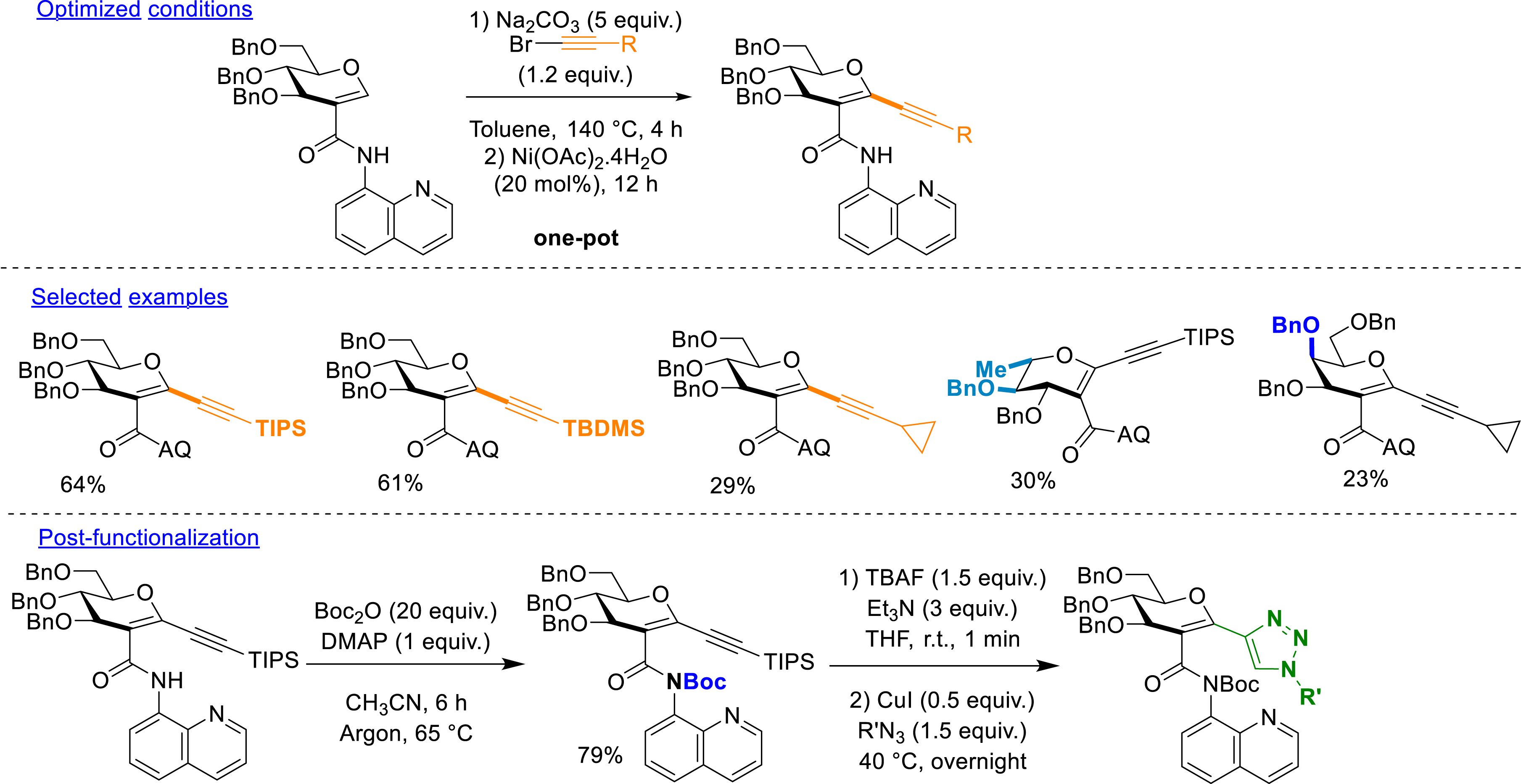
In 2022, the starting C2-amidoquinolide glycal was used in C–H thiolation to furnish trehalose analogues using the reactivity of thiosugars in metal-catalyzed reactions developed by Messaoudi et al. [83, 84, 85, 86, 87]. The utilization of copper catalysis in the presence of silver salts and 2 equiv of thiosugars allowed the formation of several trehalose analogues in moderate to good yields (21–67%). The active species appeared to be thiol since the use of the corresponding disulfide failed to form the desired compound. Diverse C2-amidoglycals (d-glucal, d-galactal, and l-rhamnal) and diverse thiosugars (with glucose, galactose, and disaccharide types) protected by benzyl, acetyl, benzoyl, and para-methoxybenzyl protecting groups were coupled. It is interesting to point out here the importance and influence of the protecting groups on the reactivity of each partner. Indeed, benzyl groups were crucial to the reactivity of the glycal partner whereas peracetylated thiosugars were the best option for thiol. These conditions have been also extended to thiophenols, aliphatic thiols, and to protected cysteine partners with success. The oxidation of the sulfur function into sulfoxide was demonstrated, leading to a mixture of two separable diastereoisomers (Scheme 14). However, for the same reasons as those mentioned previously, the removal of the DG as well as the deprotection of the molecules was unsuccessful [79].
C–H thiolation.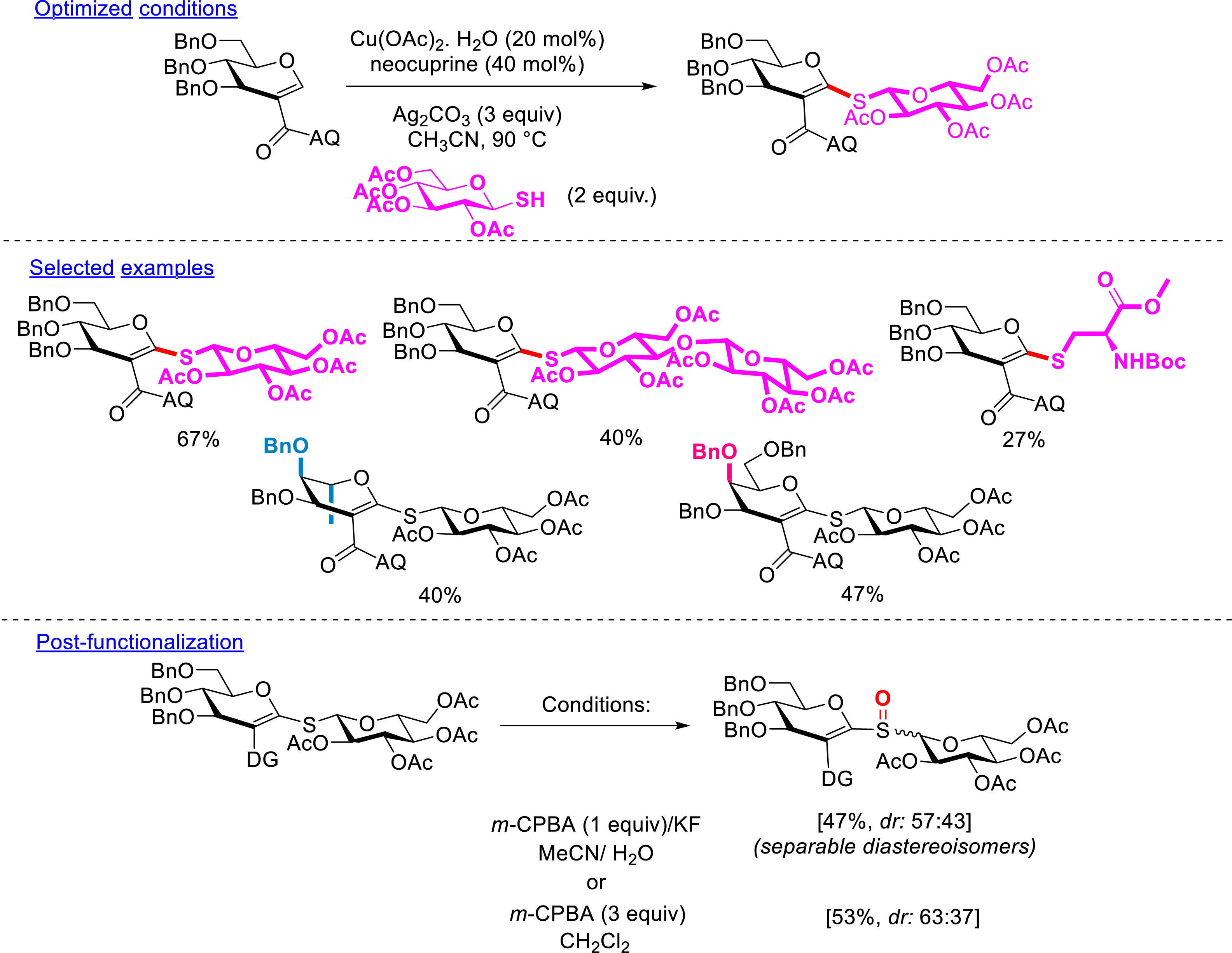
In 2023, the synthetic method involving C2-amidoquinolide glycals was used by Mandal et al. in palladium-catalyzed pseudo-anomeric C–H functionalization with para-quinone methide partners to generate unsymmetrical gem-diarylmethyl C-glycosides [80].
3.2.2. Unsaturated sialic acids
The directed C–H functionalization of unsaturated sugars has been then adapted to more complex structures, namely, unsaturated sialic acids. In collaboration with Messaoudi et al., the C–H amination at the C3 position of unsaturated sialic acids, bearing an amidoquinoline group at the C1 position, was developed. The bidentate amidoquinoline DG has been introduced by derivatization of the carboxylic acid function already present at C2. Thanks to copper catalysis, diverse nitrogen-containing heterocycles were introduced by C–H amination. Among the successful heterocycles, different substituted 7-azaindoles were used as well as diverse 5,7-diazaindoles, 6-azaindoles, 5-azaindoles, 4-azaindoles, indazoles, imidazoles, carbazoles, azacarbazoles, and indoles. Perbenzylated, per-para-methoxybenzylated, and peracetylated unsaturated sialic acid substrates were tolerated. The removal of the DG was successfully carried out by preactivation of the amide by a Boc protecting group followed by cleavage to the corresponding carboxylic acid under oxidative conditions. The deprotection of the benzyl groups has been successfully realized by applying BCl3 conditions, and the desired product was isolated in moderate yield. Finally, three different post-functionalizations of the obtained structure bearing a 4-iodo-7-azaindole moiety were developed using palladium-catalyzed cross-coupling reactions (thiolation and Suzuki–Miyaura reactions) (Scheme 15) [81].
C–H amination.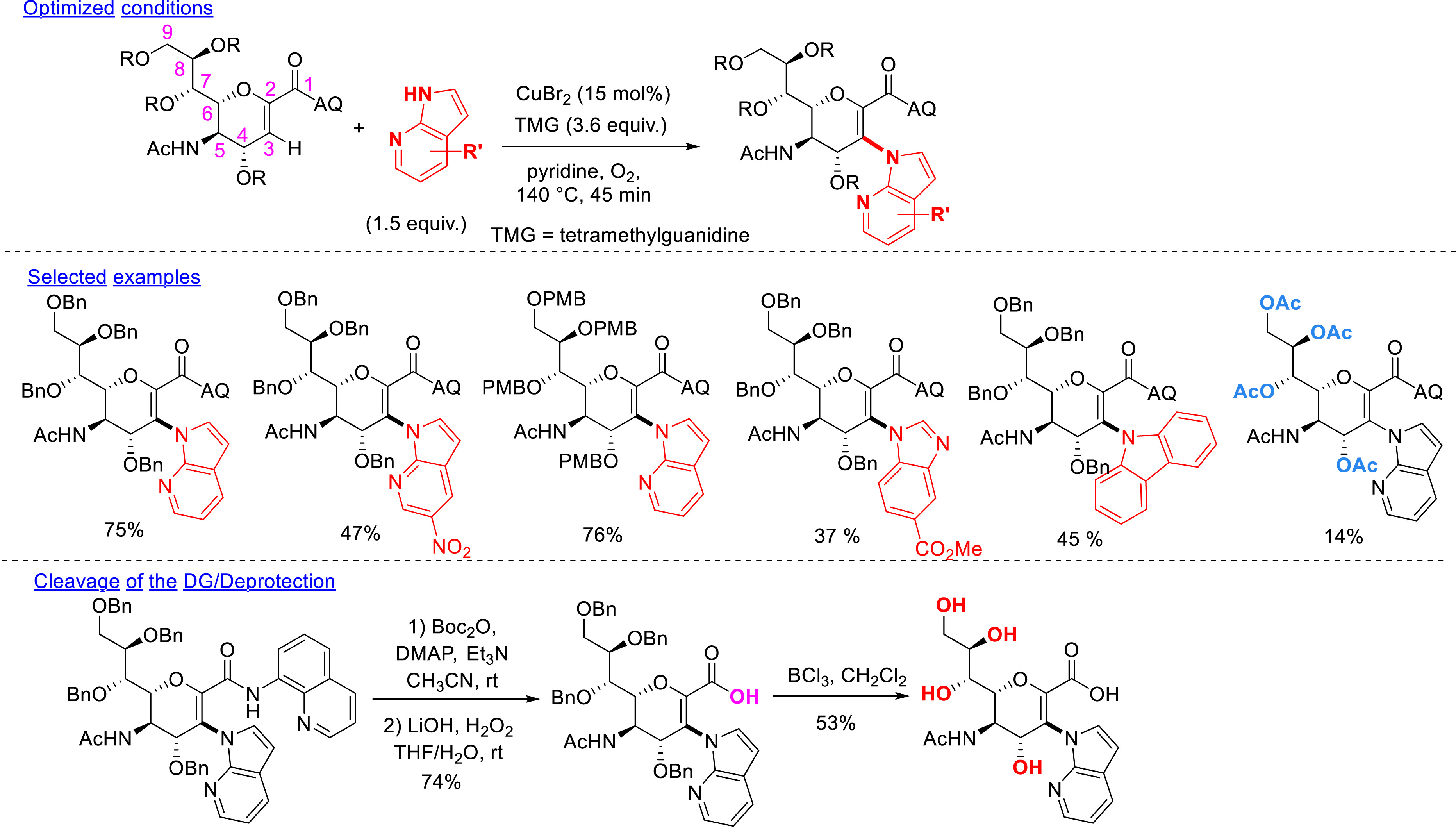
4. Conclusion
In the past decades, metal-catalyzed reactions made a real breakthrough, offering new possibilities to extend the glycoside chemical space. This review showed that cross-coupling reactions as well as C–H activation could be successfully applied to carbohydrates while being complementary. These two methods opened up the possibility of introducing substituents on glycals especially at the C1 and C2 positions, which are two key sites. These recent advances have pushed glycochemistry towards a new direction. This new field is just at its infancy but promises to expand in the near future. However, numerous aspects are still underexplored such as the use of unprotected starting sugars, which would avoid protection/deprotection steps. Indeed, the deprotection of the obtained structures is problematic, which limits potential therapeutic applications. In addition, the use of more environmentally friendly metals, such as iron, manganese, and so on, has to be considered as palladium catalysis dominates the literature even if some nickel or copper catalysis were used. The C–H activation methods also suffer, in some examples, from the inability of removing the DG. A better design of the DG or the use of transient DG (as it was proposed by Messaoudi et al. in only one paper [69]) should increase the attractiveness of the method. Another point of improvement could be the development of late-stage metal functionalization of complex oligosaccharides, which possess high therapeutic value but are currently absent from the literature. To conclude, in addition to metal-catalyzed reactions, in the last few years, glycochemists have shown that other emerging tools such as photochemistry and electrocatalysis can be useful in producing different types of sugars [17]. This heralds a fruitful future for glycochemistry and promises the discovery of original and innovative glyco-analogues of interest.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.