



 CC-BY 4.0
CC-BY 4.0
Dedicated to Prof. Rick D. Adams, who has been a leader in the field of organometallic chemistry for over forty years, in particular in cluster chemistry, where he has made numerous outstanding contributions. He has beautifully served as Editor-in-Chief for the Journal of Organometallic Chemistry for twenty-seven years.
1. Introduction
After the pioneering report of Nyholm and coworkers of a heterometallic complex containing a direct metal–metal bond between a group 11 metal in the +I oxidation state (namely Cu(I), Ag(I), or Au(I)) and another transition metal in 1964 [1], a new research area developed in organometallic chemistry dedicated to the study of heterometallic bonds, and numerous relevant transition metal dinuclear and cluster complexes have since been synthesised and characterised with 1D, 2D, or 3D structures [2, 3, 4, 5, 6, 7]. Notably, several metal clusters were found to display short intermetallic contacts between group 11 centres that challenged the conventional understanding of metal–metal bonding and were interpreted as evidence for d10–d10 metallophilic interactions, a phenomenon that continues to attract significant attention due to its implications in bonding theory, structural diversity, and photophysics, with potential applications in material sciences [8, 9, 10, 11, 12, 13, 14, 15, 16, 17]. Longoni and collaborators have described octanuclear, tetra-anionic clusters [NMe3CH2Ph]4[Ag4{Fe(CO)4}4] [18] and [NEt4]4[Au4{Fe(CO)4}4] [19], in which all Ag–Ag and Au–Au bonds are bridged by the formally dianionic metalloligand μ-[Fe(CO)4] (Scheme 1). The inner core of the Ag4Fe4 cluster adopts a square geometry, with Ag–Ag distances ranging from 3.036(1) to 3.334(1) Å. In contrast, the corresponding Au4Fe4 clusters display either a square (2.973(2), 2.831(2) Å) or a rectangular arrangement (2.932(2) and 3.400(2) Å) of gold atoms.
Square or rectangular M4 core in Ag4Fe4 and Au4Fe4 clusters [6, 7, 17, 19].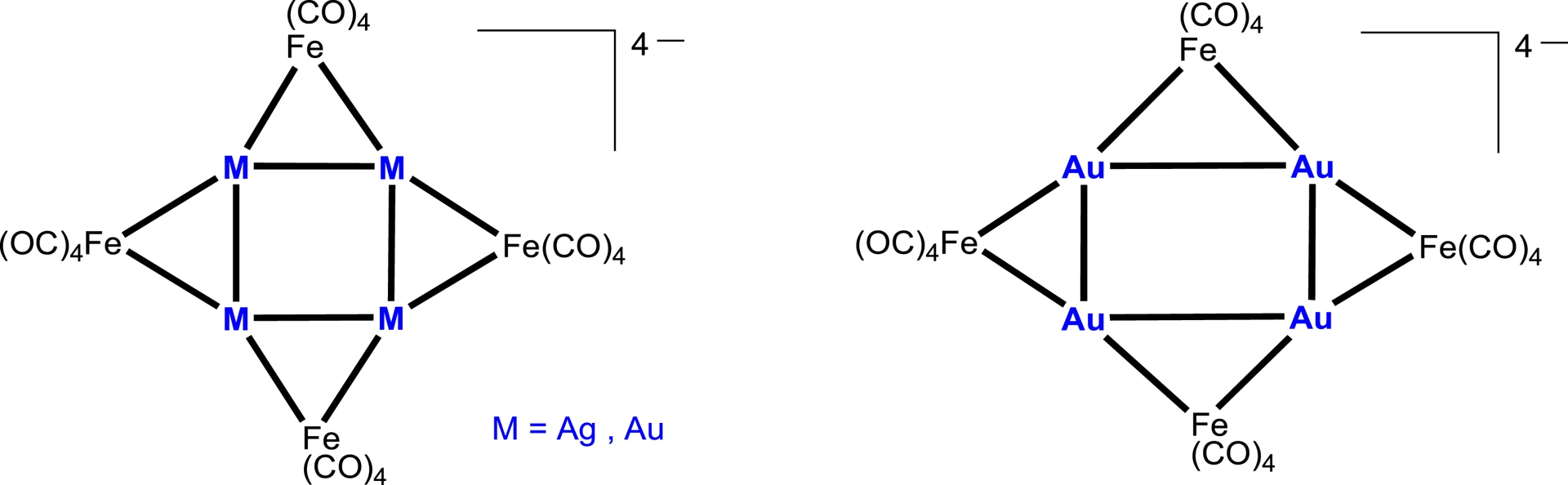
Furthermore, Klüfers et al. reported a series of clusters in which a central square of silver or copper atoms is stabilised by the formally monoanionic bridging metalloligand μ-[Co(CO)4] [20, 21, 22]. These diverse structures illustrate the ability of group 11 metals to assemble into highly symmetric frameworks when supported by carbonyl-based metalloligands. In contrast to these square arrangements, a triangular Cu3 core, with an average Cu–Cu separation of 2.602 Å, was later characterised in the hexanuclear, trianionic cluster [NEt4]3[Cu3{Fe(CO)4}3] [23], in which the copper atoms are interconnected by μ-[Fe(CO)4] units (Scheme 2). The characterisation of the triangular Cu3 and square Ag4/Cu4 systems highlights the structural versatility of group 11 closed-shell clusters, underscoring the delicate balance between electronic and steric effects that determines their nuclearity and overall structure.
Triangular structure of the Cu3Fe3 cluster [23].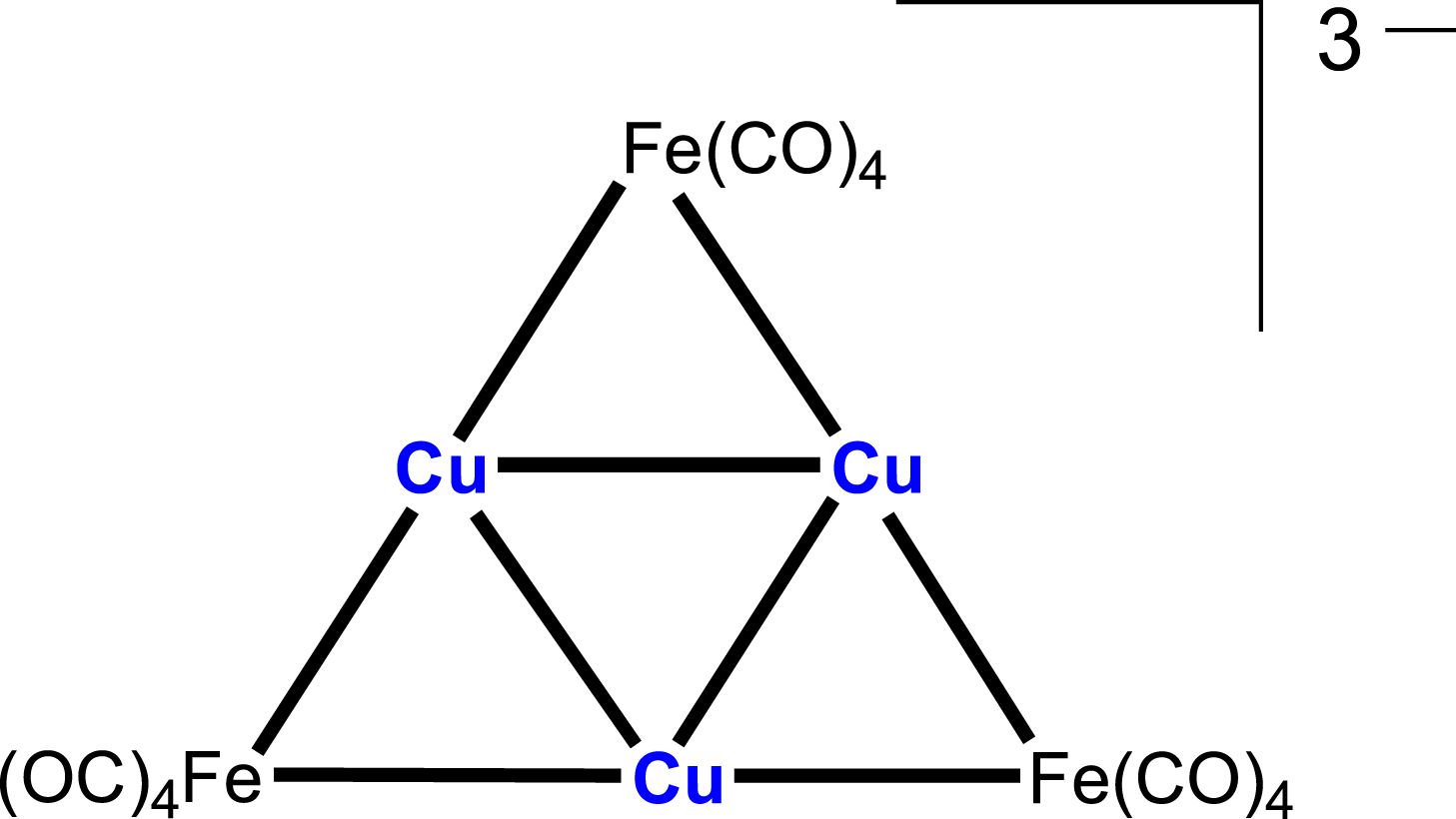
Recent work has reported an unprecedented case of polymerisation isomerism in heterobimetallic carbonyl clusters [{MFe(CO)4}n]n− (M = Cu, Ag, Au; n = 3, 4), where triangular and square nuclearities coexist for the same elemental composition. Depending on the synthetic protocol, Ag and Au systems selectively form either trinuclear or tetranuclear clusters, whereas Cu stabilises only the triangular form. This behaviour has been rationalised by a balance between Fe–M bonding and metallophilic interactions, as revealed by structural and AIM analyses [24].
Our group reported a series of hexa- and octanuclear heterometallic clusters with a central core displaying d10–d10 interactions, obtained by reaction of the carbonylmetallate [Mo(C5H4NMe2)(CO)3]− with [Cu(NCMe)4]PF6, AgBF4, or (NBu4)[AuBr2], respectively. These clusters were shown by X-ray diffraction to possess a triangular copper core in [Cu3{Mo(C5H4NMe2)(CO)3}3] A1, and a square silver or gold core in [Ag4{Mo(C5H4NMe2)(CO)3}4] A2 and [Au4{Mo(C5H4NMe2)(CO)3}4] A3, respectively (Scheme 3) [17, 25, 26, 27].
Clusters [MMo(C5H4NMe2)(CO)3]n with a triangular copper (n = 3) or square silver or gold central core (n = 4).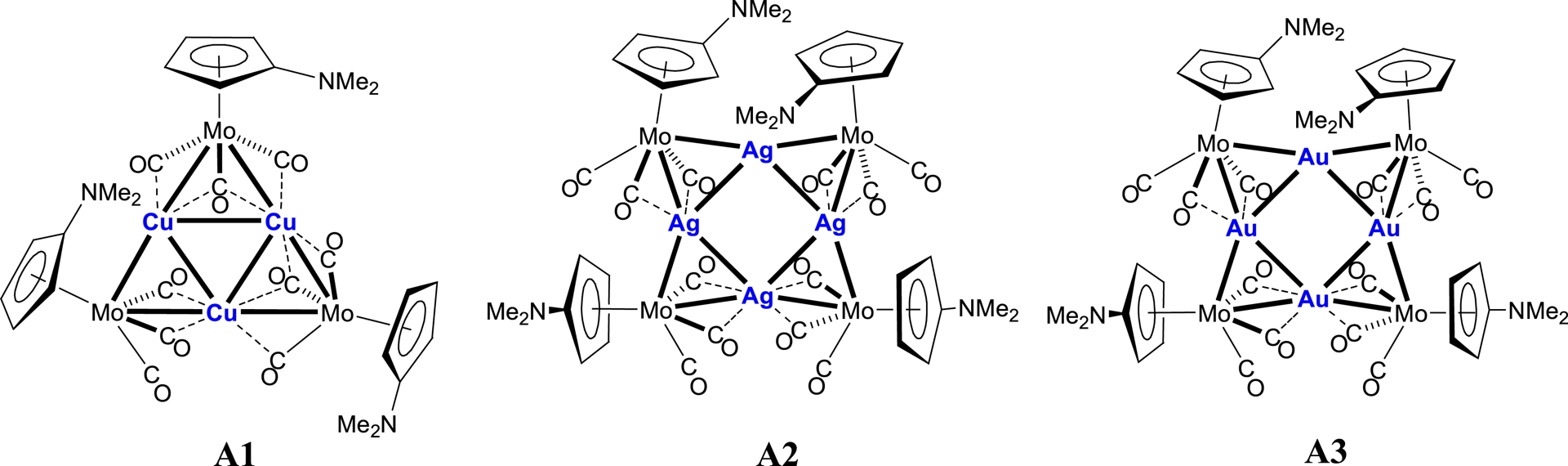
Using the anion [MoCp(CO)3]− (Cp = η5-C5H5) had previously yielded similar clusters, with a triangular copper core in [Cu3{MoCp(CO)3}3] A4, and a square silver or gold core in [Ag4{MoCp(CO)3}4] A5, and [Au4{MoCp(CO)3}4] A6, respectively (Scheme 4) [25, 26].
Clusters [MMoCp(CO)3]n with a triangular copper (n = 3) or square silver or gold central core (n = 4).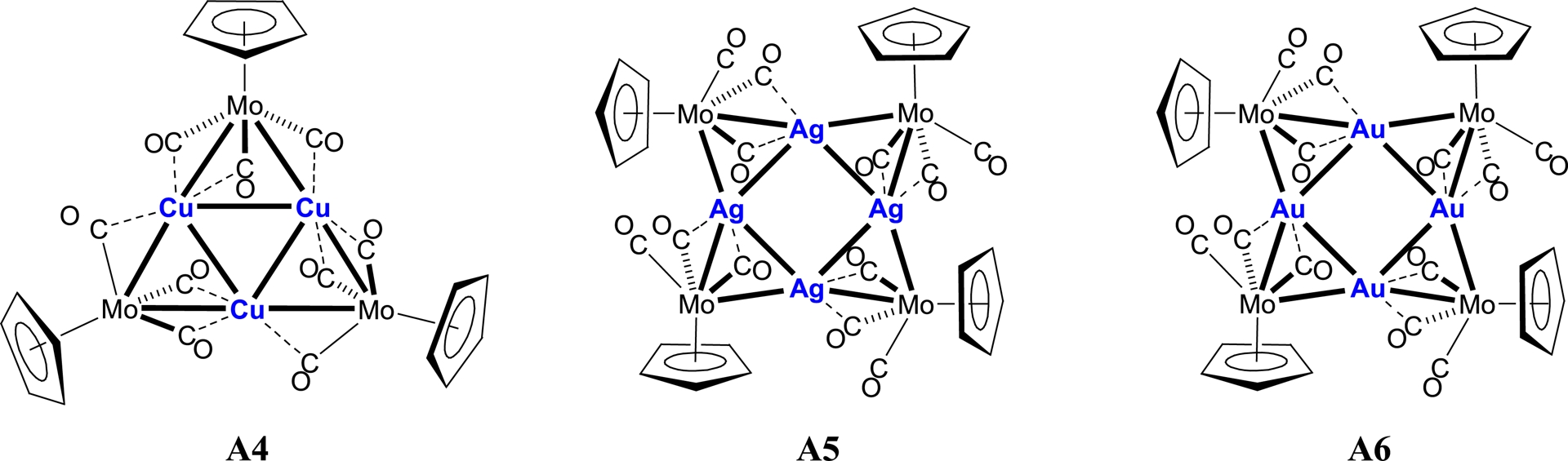
Combined experimental and theoretical investigations on clusters A4–A6 demonstrated that the structural preference arises from the delicate balance between steric effects, ligand coordination modes, and d10–d10 metallophilic interactions between the closed-shell metal centres [25, 26]. These findings provided important insight into the role of metallophilic interactions in governing the geometry and stability of such heterometallic carbonyl clusters. In the present work, we extend this investigation to related systems incorporating the substituted metalloligand [Mo(C5H4NMe2)(CO)3]− in order to evaluate the influence of ligand functionalisation on the structure and electronic properties of the resulting clusters.
The reason for introducing the C5H4NMe2 ligand instead of Cp in the metal carbonyl fragment was to examine its potential influence on the nature and/or structure of the resulting clusters. Because a bridging bonding mode of the ligand C5H4NMe2 has been demonstrated earlier in the heterodinuclear complex [Pt{Mo(μ-C5H4NMe2)(CO)3}(NCPh)Cl] (Mo–Pt) B [28] (Scheme 5), it was conceivable that the NMe2 donor group could engage in bonding with an adjacent group 11 metal centre in the clusters of type A investigated here. Although this situation was not observed, the C5H4NMe2 ligand in clusters A1–A3 was found to influence the orientation of the metalloligand with respect to the central metal core and the overall molecular structure.
Complex B with a bridging C5H4NMe2 ligand [28].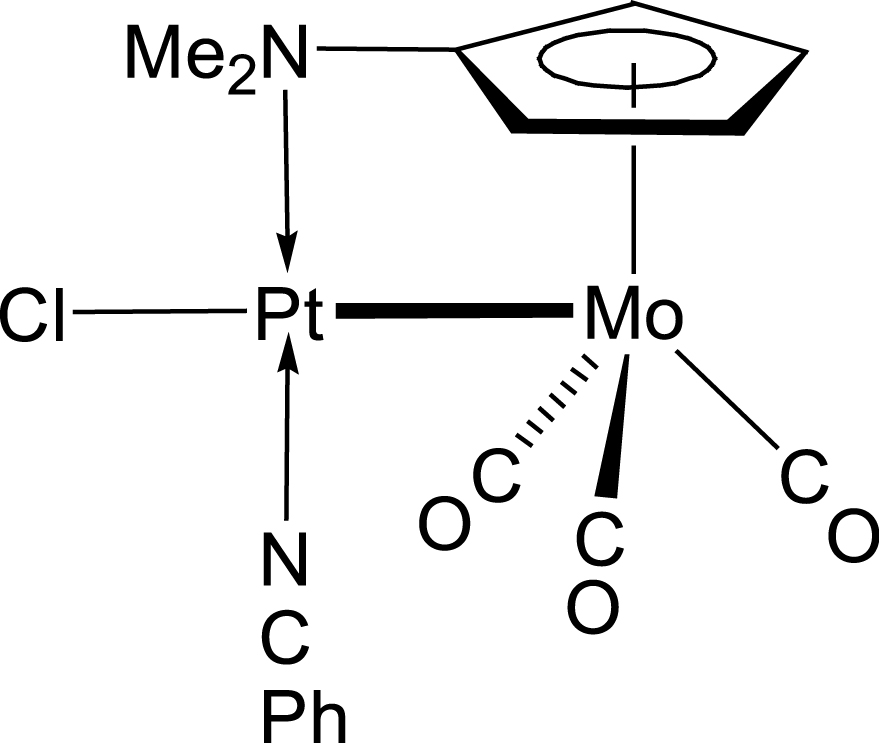
In clusters A1–A6, the bonding behaviour of the metalloligands [MoCp(CO)3]− and [Mo(C5H4NMe2)(CO)3]− with respect to the d10–d10 metal–metal bond is that of an anionic μ2-unit bridging the M(I)–M(I) centres, acting formally as four-electron donors, which confers the usual 14-electron count to the coinage metals. This bridging bonding mode was observed for the first time in the centrosymmetric butterfly clusters [Pd2(or Pt2){MoCp(CO)3}2(PR3)2] [29, 30, 31] (Scheme 6).
Centrosymmetric clusters of type C with centrosymmetric Mo2Pd2 or Mo2Pt2 core.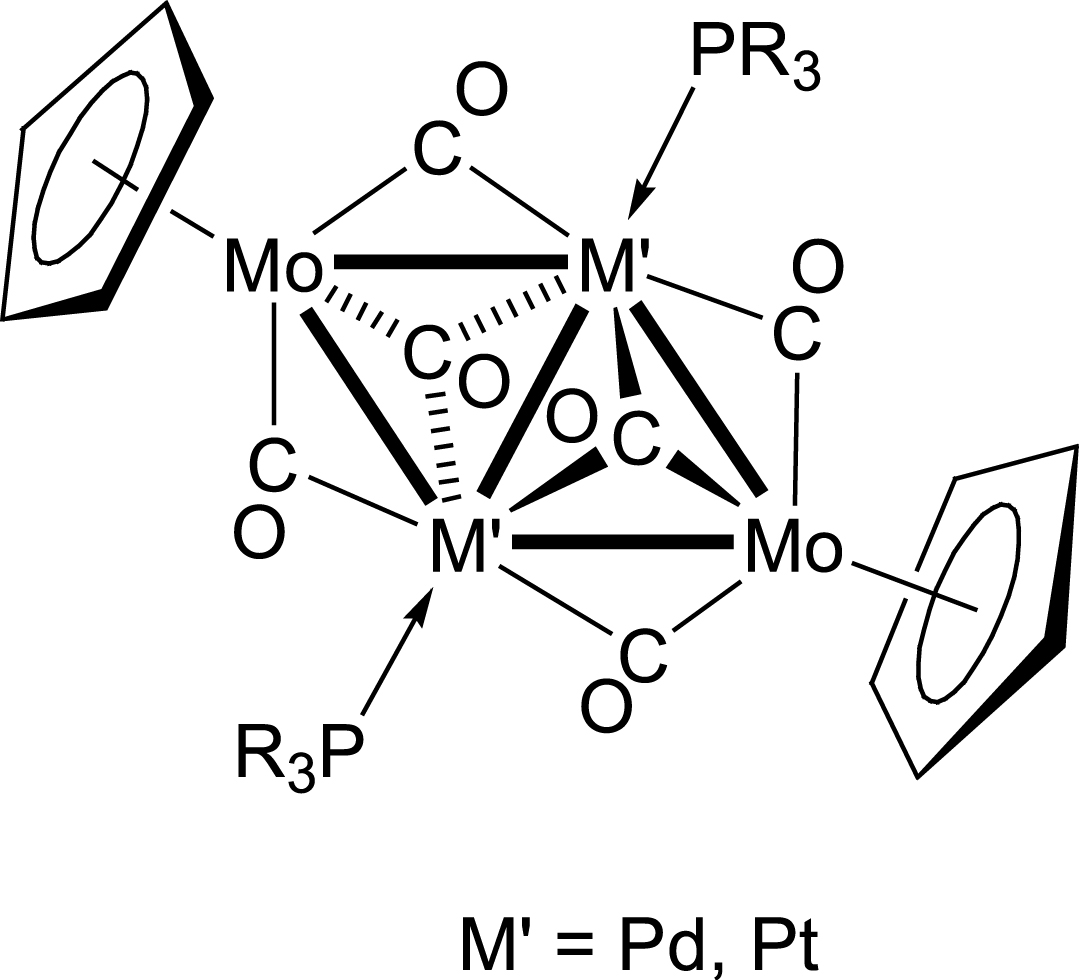
Although the structures of these centrosymmetric metal clusters did not follow the original Wade–Mingos rules [32, 33], they could be readily explained by considering that the d9–d9 M′–M′ bond was doubly bridged by the carbonylmetallates formally acting as four-electron donor anions, like μ2-halides. This analogy was extended to the even rarer μ3-bonding mode, first encountered in a Pd3Mo cluster, where the carbonylmetallate could be viewed as an anionic six-electron donor, like a μ3-halide [34] This electron-counting approach was subsequently successfully extended to isoelectronic or isolobal analogues of these carbonylmetallates [35].
In each of the clusters A1–A6, the short M–M distances provide clear evidence for the occurrence and stabilising role of metallophilic interactions between the closed-shell metal centres. To gain a deeper insight into the bonding in these systems, we carried out a detailed theoretical investigation aimed at elucidating the reasons for the stabilisation of specific structural motifs. Our study was designed to address two key aspects: (i) to rationalise the preference for the observed geometries of the six clusters under consideration, and (ii) to evaluate the relative stability of structures with different possible symmetries. DFT calculations have allowed us not only to reproduce the structural features observed experimentally but also to better understand the electronic factors underpinning their stability and structural diversity.
2. Computational details
All quantum chemical calculations were carried out using the TURBOMOLE software package [36]. The BP86 density functional (Becke–Perdew86) was selected because of its proven reliability in describing diverse bonding situations, including those involving transition-metal complexes [37, 38, 39]. This choice further enabled the use of the efficient RI–J approximation for the treatment of Coulomb two-electron integrals [40, 41, 42]. For geometry optimisations, SV(P) basis sets were employed [43] consisting of a single basis function for core orbitals, a double-ζ description for the valence shells, and one set of polarisation functions for all atoms except hydrogen. To refine the electronic energies, single-point calculations were performed using the larger triple-ζ valence plus polarisation (TZVP) basis sets [44]. Stationary points on the potential energy surface were verified through vibrational frequency analyses at the BP86/SV(P) level: minima were confirmed by the absence of imaginary frequencies, whereas the presence of a single imaginary frequency identified transition states [45, 46, 47]. In addition to Born–Oppenheimer potential energies (E, corrected for zero-point vibrational effects), Gibbs free energies (G) were also evaluated. Unless otherwise specified, all reported G values correspond to standard conditions of 25 °C and 1 bar CO, consistent with the experimental setup [26]. Energy decomposition analysis (EDA), originally developed by Morokuma [47], was employed to investigate the nature and origin of the interactions between the metal cores Mn (M = Cu, Ag, Au; n = 3, 4) and the metalloligand fragments [MoCp(or C5H4NMe2)(CO)3]n in the triangular (n = 3) and square (n = 4) heterometallic clusters. The EDA calculations were carried out using the Amsterdam modeling suite (AMS) [48]. Single-point EDA calculations were performed at the BP86/TZP level on geometries optimised at the BP86/SVP level with Turbomole. Within this formalism, the total interaction energy ΔEint between the interacting fragments is decomposed into physically meaningful contributions according to Equation (1):
| \begin {equation}\label {eq1} \Delta E_{\mathrm {int}}= \Delta E_{\mathrm {Pauli}}+ \Delta E_{\mathrm {elstat}}+ \Delta E_{\mathrm {orb}} \end {equation} | (1) |
3. Results and discussion
3.1. Optimised geometries of the metal cores
The main results from the computationally optimised geometries and the structures determined by X-ray diffraction of clusters A1–A6 are presented and compared below. A detailed compilation of structural parameters, including bond lengths and bond angles, available in Tables S1–S6 in the Supporting Information, provide insights into the reliability of the theoretical methods employed.
3.1.1. Clusters with a copper core
In the structure of cluster A1, optimised in C1 symmetry, the calculated Cu–Cu bond distances are 2.642, 2.580, and 2.531 Å, shorter than the experimental values by only 0.013, 0.036, and 0.048 Å, respectively, while the calculated Cu–Mo distances are longer by about 0.03 Å. A noticeable difference concerns the bridging carbonyl groups, where the calculated Cu–C bond lengths are shorter by up to 0.117 Å compared to the experimental values. The calculated bond angles match well the experimental data, with only small differences of 3.94°, 4.12°, 3.80°, and 3.01° for Mo(2)–C(1)–O(1), Mo(2)–C(11)–O(4), Mo(3)–C(21)–O(7), and Mo(3)–C(23)–O(9), respectively.
The X-ray diffraction study revealed that cluster A4 with a triangular copper core crystallises in two different systems, triclinic and orthorhombic. The main difference between them lies in the presence of a triply bridging carbonyl group in the orthorhombic form. For both systems, geometry optimisation yielded similar structures, with the Cp and carbonyl ligands arranged in an overall C3-symmetric orientation. For our study, we considered cluster A4 in its orthorhombic crystalline form. The optimised Cu–Cu distances are, on average, shorter by 0.05 Å compared to those determined by X-ray diffraction. As in the case of cluster A1, slight deviations from the experimental values are observed in the calculated bond angles Mo(1)–C(3)–O(3), Mo(2)–C(7)–O(7), Mo(3)–C(4)–O(4), and Mo(3)–C(6)–O(6) of 3.36°, 3.49°, 3.95°, and 4.86°, respectively.
3.1.2. Clusters with a silver or gold core
The geometries of clusters A2 and A3 with a silver and gold square core, respectively, were optimised in C2 symmetry. Compared to the experimental values, the calculated metal–metal distances are longer by about 0.04 Å in the silver square and by 0.12 Å in the gold square. Furthermore, the diagonal distances in the gold cluster are clearly overestimated, with a calculated Au(1)–Au(3) distance of 4.151 Å against 3.875 Å determined experimentally. In contrast, the M–Mo distances are in good agreement, with a maximum deviation of 0.04 Å for silver and 0.10 Å for gold. For the bridging carbonyl ligands, more pronounced discrepancies are observed between the calculated and experimental values. For instance, in the case of silver, the Ag(2)–C(1) distance is longer by 0.16 Å while the Ag(2)–C(2) distance is shorter by 0.05 Å. This effect is even more pronounced in the gold square, where the Au(2)–C(2) distance decreases by 0.12 Å, while another Au–C bond length increases by 0.37 Å (Table S3). A significant structural difference between the silver- and gold-containing clusters lies in the Au–CO bonds being shorter than the corresponding Ag–CO bonds. Nevertheless, the calculated bond angles are in good agreement with the experimental values. The differences in bond lengths and angles sometimes observed between calculated and experimental values involving the coinage metals and CO ligands can be explained by the relative weakness of these interactions and their soft energy profile compared to the Mo–CO interactions; it is mostly the orientation of the Mo(CO)3 cone that defines the position of the CO ligands with respect to the coinage metals [53, 54].
After discussing the C5H4NMe2 derivatives (A2 and A3), we now turn to their Cp analogues, A5 and A6, which display notable structural differences in the arrangement of their metal cores.
The silver square in A5 and the gold square in A6 exhibit significant differences in the arrangement of their atoms. For the geometry optimisation of cluster A5, it was necessary to impose a constraint, under C2 symmetry, by arbitrarily fixing the torsion angle Ag(1)–Ag(2)–Ag(1a)–Ag(2a) at 21°, since unconstrained optimisation led to a planar square structure like that of the gold cluster, thereby causing convergence issues. The calculated Ag–Ag distances show a maximum elongation of about 0.07 Å. A significant difference is also observed for the diagonal distances, with calculated Ag(1)–Ag(1a) of 4.00 Å compared to 3.50 Å in the experimental structure, while calculated Ag(2)–Ag(2a) is shorter by 0.35 Å. Moreover, a pronounced discrepancy is found in the bond angles, with calculated values for Ag(1)–Ag(2)–Ag(1a) and Ag(2)–Ag(1)–Ag(2a) of 87.41° and 88.76°, compared to the experimental values of 75.92° and 100.23°, respectively.
In the structure of cluster A6, the four gold atoms are coplanar, and the diagonal distances are nearly identical, with Au(1)–Au(3) at 3.940 Å and Au(2)–Au(4) at 3.904 Å (Table S6). In contrast, the diagonal distances are very different in A5 for Ag(1)–Ag(1a) and Ag(2)–Ag(2a) at 3.530 and 4.405 Å, respectively. Like in A5, the four Cp ligands and the carbonyl groups in A6 adopt a similar orientation, consistent with an approximate S4 symmetry (Table S5). When the geometry optimisation of cluster A6 was started from C1 symmetry, it converged to a geometry close to S4 symmetry. The calculated Au–Au bond lengths are overestimated by up to ca. 0.15 Å compared to the experimental data. Nevertheless, the overall set of calculated distances is in satisfactory agreement with the experimental data, thereby validating the DFT method and basis set employed.
3.2. Relative stabilities of the various models as a function of their molecular symmetry
To better understand the relative stabilities of the different cluster architectures, we carried out geometry optimisations using DFT at the BP86 level of theory combined with the SV(P) basis set and explored, for each system, several possible symmetries, namely C2, D2, S4, C1, and Cs, to identify the most stable configuration.
The optimised geometries of the clusters containing the C5H4NMe2 ligands are presented in Figure 1, while the corresponding structures with unsubstituted Cp ligands are displayed in Figure 2. This distinction allows a direct comparison of the effect of the NMe2 substituent on the cluster stability and geometry.
Optimised DFT/BP86 structures of the [M4{Mo(C5H4NMe2)(CO)3}4] and [M3{Mo(C5H4NMe2)(CO)3}3] clusters (M = Cu, Ag, Au) under different symmetry constraints (C1, C2, D2, S4, and Cs).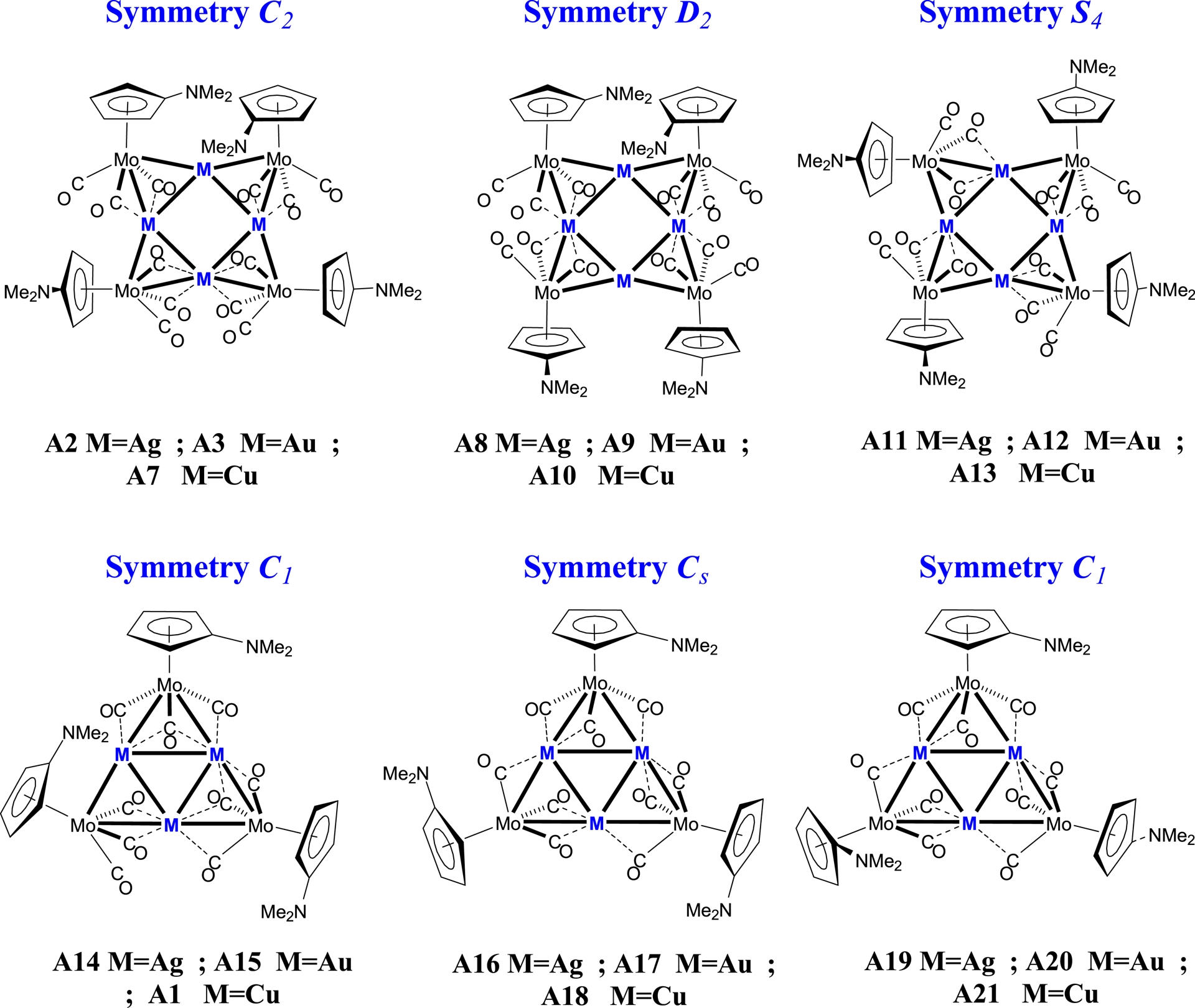
Optimised DFT/BP86 structures of [M4{MoCp(CO)3}4] and [M3{MoCp(CO)3}3] clusters (M = Cu, Ag, Au) under different symmetry constraints (C1, C2, D2, S4, and Cs).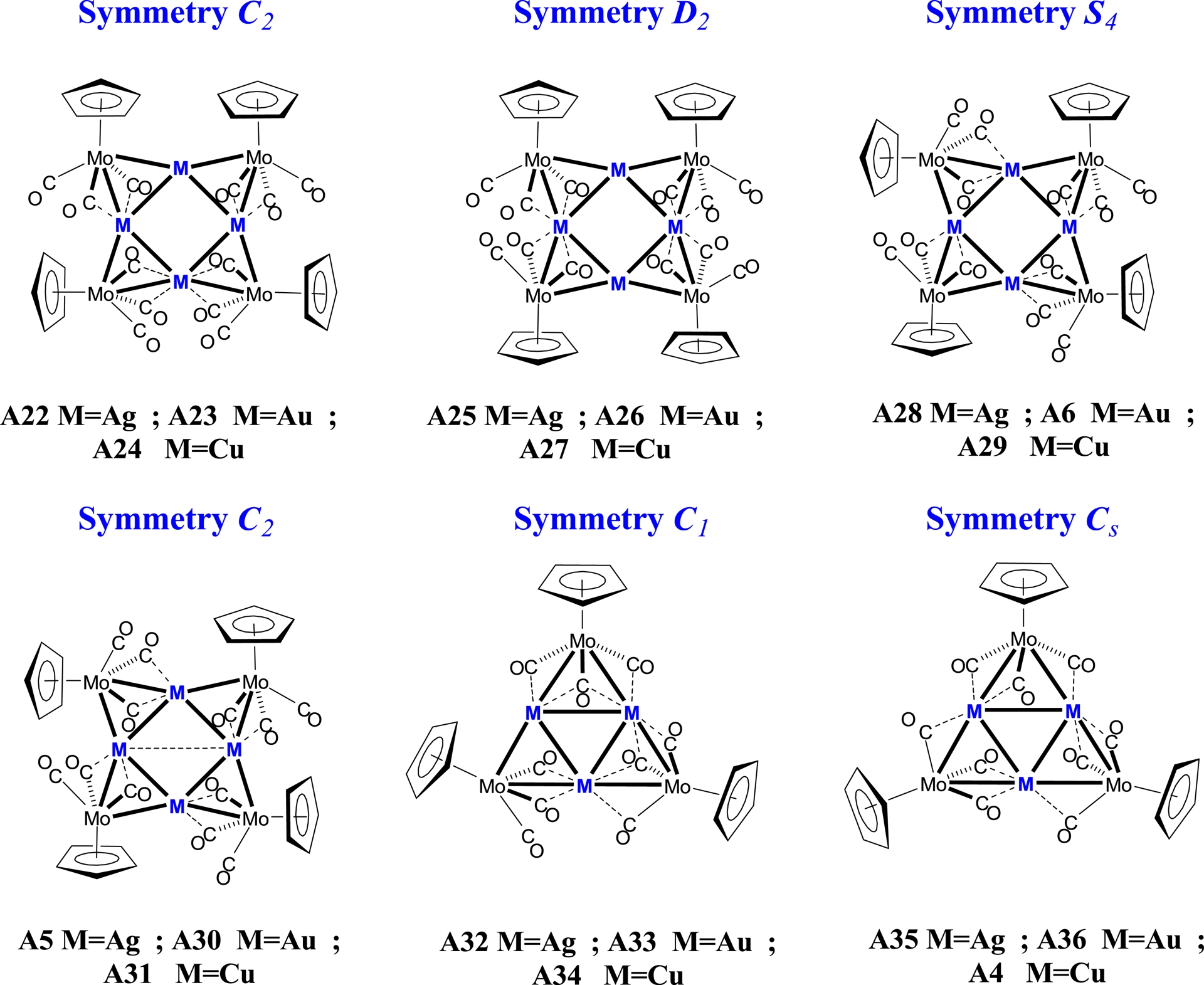
For the eight-metal-atom clusters, the initial geometries were constructed on the basis of the experimental data for the related silver and gold square-type clusters. Specifically, we started from C2 symmetry for the clusters containing the C5H4NMe2 ligands, whereas we started from S4 symmetric geometries for those containing the Cp ligands. To complement these models, we also examined hypothetical structures of D2 symmetry (see structures A8–A10 in Figure 1 and A25–A27 in Figure 2), which provided useful comparisons.
3.2.1. Cu-containing clusters
3.2.1.1. With the C5H4NMe2 ligands
For the hexanuclear clusters, the optimisations were guided by experimental observations. The geometries of the C5H4NMe2-containing clusters were optimised starting from C1 symmetry, while for the Cp analogues (see below), we adopted a slightly distorted C1 geometry close to Cs symmetry. Furthermore, a hypothetical Cs-like model was considered, where the C5H4NMe2 ligands are positioned on opposite sides of the metal plane (structures A19–A21, Figure 1). This configuration was introduced to probe the effect of the orientation of the aromatic ligand on the overall cluster stability and electronic distribution.
Overall, this systematic optimisation approach, covering multiple symmetries and both Cp and C5H4NMe2 ligand types, provided a coherent framework for evaluating how the metal (Cu, Ag, Au) and the π-ligand substitution (Cp or C5H4NMe2) affect the structural preferences and relative stabilities of these polymetallic clusters.
The relative energies and the calculated metal–metal bond distances for the different clusters optimised under the various symmetry constraints are compiled in Tables 1–4, which allows a direct comparison between the alternative structural models, highlighting the effect of symmetry on both the energetic stability and the geometric features of the clusters.
Main geometrical parameters (distances in Å) and relative energies (in Hartree and ΔE in kJ/mol) for the models [Cu4{Mo(C5H4NMe2)(CO)3}4] and [Cu3{Mo(C5H4NMe2)(CO)3}3], optimised under different symmetry constraints (C1, C2, D2, S4, and Cs)
| Clusters | Symmetry | d10–d10 Distances (Å) | E (Hartree) | ΔE (kJ/mol) |
|---|---|---|---|---|
| [Cu4{Mo(C5H4NMe2)(CO)3}4] | ||||
| A7 | C2 | 2.55; 2.65 | −9508.231 | 0 |
| A10 | D2 | 3.32 | −9508.249 | −47.40 |
| A13 | S4 | 3.34 | −9508.252 | −53.37 |
| [Cu3{Mo(C5H4NMe2)(CO)3}3] | ||||
| A1 | C1 | 2.58, 2.60, 2.64 | −7131.182 | 0 |
| A18 | Cs | 2.62 | −7131.178 | +10.18 |
| A21 | C1 | 2.60 | −7131.187 | −12.72 |
Main geometrical parameters (distances in Å) and relative energies (in Hartree and ΔE in kJ/mol) for the models [Cu4{MoCp(CO)3}4] and [Cu3{MoCp(CO)3}3], optimised under different symmetry constraints (C1, C2, D2, S4, and Cs)
| Clusters | Symmetry | d10–d10 Distances (Å) | E (Hartree) | ΔE (kJ/mol) |
|---|---|---|---|---|
| [Cu4{MoCp(CO)3}4] | ||||
| A24 | C2 | 2.58, 2.68 | −8972.149 | 0 |
| A27 | D2 | 2.52 | −8972.140 | +23.52 |
| A29 | S4 | 2.75 | −8972.159 | −25.45 |
| A31 | C2 | 2.70, 2.74 | −8972.154 | −13.74 |
| [Cu3{MoCp(CO)3}3] | ||||
| A34 | C1 | 2.54, 2.55, 2.64 | −6729.124 | 0 |
| A4 | Cs | 2.60 | −6729.127 | −9.55 |
Main geometrical parameters (distances in Å) and relative energies (in Hartree and ΔE in kJ/mol) for the models [M4{Mo(C5H4NMe2)(CO)3}4] and [M3{Mo(C5H4NMe2)(CO)3}3] (M = Ag or Au), optimised under different symmetry constraints (C1, C2, D2, S4, and Cs)
| Clusters | Symmetry | d10–d10 Distances (Å) | E (Hartree) | ΔE (kJ/mol) |
|---|---|---|---|---|
| [Ag4{Mo(C5H4NMe2)(CO)3}4] | ||||
| A2 | C2 | 2.84, 2.94 | −3533.290 | 0 |
| A8 | D2 | 2.91 | −3533.286 | +10.44 |
| A11 | S4 | 2.96 | −3533.294 | −10.92 |
| [Ag3{Mo(C5H4NMe2)(CO)3}3] | ||||
| A14 | C1 | 2.89, 2.95, 2.99 | −2649.958 | 0 |
| A16 | Cs | 3.01 | −2649.958 | +1.81 |
| A19 | C1 | 2.98 | −2649.962 | −9.26 |
| [Au4{Mo(C5H4NMe2)(CO)3}4] | ||||
| A3 | C2 | 2.88, 2.93 | −3488.520 | 0 |
| A9 | D2 | 2.95 | −3488.513 | +17.25 |
| A12 | S4 | 2.90 | −3488.522 | −7.063 |
| [Au3{Mo(C5H4NMe2)(CO)3}3] | ||||
| A15 | C1 | 2.96, 3.12, 3.18 | −2616.377 | 0 |
| A17 | Cs | 3.17 | −2616.378 | −2.25 |
| A20 | C1 | 3.12, 3.13, 3.14 | −2616.379 | −6.20 |
Main geometrical parameters (distances in Å) and relative energies (in Hartree and ΔE in kJ/mol) for the models [M4{MoCp(CO)3}4] and [M3{MoCp(CO)3}3] (M = Ag or Au), optimised under different symmetry constraints (C1, C2, D2, S4, and Cs)
| Clusters | Symmetry | d10–d10 Distances (Å) | E (Hartree) | ΔE (kJ/mol) |
|---|---|---|---|---|
| [Ag4{MoCp(CO)3}4] | ||||
| A22 | C2 | 2.88, 2.95 | −2997.209 | 0 |
| A25 | D2 | 2.85 | −2997.209 | +0.92 |
| A28 | S4 | 2.93 | −2997.216 | −18.14 |
| A5 | C2 | 2.88, 2.92 | −2997.212 | −8.72 |
| [Ag3{MoCp(CO)3}3] | ||||
| A32 | C1 | 2.88, 2.93, 3.02 | −2247.901 | 0 |
| A35 | Cs | 2.99 | −2247.903 | −4.38 |
| [Au4{MoCp(CO)3}4] | ||||
| A23 | C2 | 2.91, 2.92 | −2952.437 | 0 |
| A26 | D2 | 2.90 | −2952.434 | +8.18 |
| A6 | S4 | 2.91 | −2952.442 | −14.16 |
| A30 | C2 | 2.89, 2.91 | −2952.439 | −7.12 |
| [Au3{MoCp(CO)3}3] | ||||
| A33 | C1 | 2.94, 3.07, 3.22 | −2214.317 | 0 |
| A36 | Cs | 3.14 | −2214.318 | −1.56 |
The optimisation of the three models with a copper square core led to distinct geometries. Cluster A7, with C2 symmetry, converges to the same structure as that found for the silver square, displaying short Cu–Cu distances of 2.55 and 2.65 Å. In contrast, for clusters A10 and A13, with D2 and S4 symmetry, longer Cu–Cu distances of 3.32 Å and 3.34 Å were obtained, respectively. Taking the energy of isomer A7 as a reference, isomers A10 and A13 were found to be energetically more stable by 47.40 and 53.37 kJ/mol, respectively.
For the clusters with a triangular core A1, A18, and A21, geometry optimisation led to the same structural motif, with Cu–Cu distances in the 2.58–2.62 Å range. This results in a stabilisation of isomer A21 by 12.72 kJ/mol and a destabilisation of isomer A18 by 10.18 kJ/mol, relative to the experimentally observed structure of cluster A1 (the energy of isomer A1 being taken as the reference).
3.2.1.2. With the Cp ligands
In the case of the square copper clusters containing the Cp ligands, the Cu–Cu bond lengths are noticeably shorter than those obtained for the systems containing the C5H4NMe2 ligand. For instance, cluster A27 with D2 symmetry exhibits Cu–Cu distances of approximately 2.52 Å, which are even shorter than the experimental values determined by X-ray diffraction. By contrast, cluster A29 with S4 symmetry displays d10–d10 separations very similar to those observed in cluster A31 with C2 symmetry. Cluster A29 was found to be more stable by 25.45 kJ/mol relative to A24 taken as a reference and by nearly 50 kJ/mol compared to cluster A27. These results clearly indicate that the S4-symmetric square arrangement represents the energetically most favourable configuration among the clusters containing a square copper core.
For the triangular copper clusters, the optimised geometries reveal that the Cu–Cu bond lengths vary slightly depending on the considered symmetry. In the case of cluster A4 with Cs symmetry, the calculated Cu–Cu distances are uniform, around 2.60 Å. For cluster A34 with C1 symmetry, the Cu–Cu separations are more dispersed, ranging from 2.54 to 2.64 Å. From an energetic standpoint, cluster A4 is more stable than cluster A34 by 9.5 kJ/mol, suggesting that the Cs-symmetric triangular arrangement is energetically preferred over the C1 variant. This stabilisation highlights the role of symmetry in determining the most favourable structural configuration for copper clusters containing a triangular core.
The energies calculated for clusters A2 (M = Ag) and A3 (M = Au), optimised from the experimental data, are used as reference values for the square systems, while the energies of clusters A14 (M = Ag) and A15 (M = Au) are arbitrarily chosen as reference values for the triangular systems.
3.2.2. Ag- and Au-containing clusters
3.2.2.1. With the C5H4NMe2 Ligands
In contrast to what was observed for the square copper clusters, the geometry optimisation of the silver and gold square clusters yielded calculated distances close to the experimental values for all three symmetries, C2, D2, and S4.
Cluster A11 with S4 symmetry, featuring an Ag–Ag distance of 2.96 Å, is more stable by 10.92 kJ/mol than isomer A2, whereas cluster A8 with D2 symmetry is destabilised by 10.44 kJ/mol relative to A2. For the triangular clusters A14, A16, and A19, the calculated Ag–Ag distances range from 2.89 to 3.00 Å for A14 and are around 3.00 Å for both A16 and A19. Cluster A19 is more stable than A14 by 9.26 kJ/mol.
For the triangular and square gold clusters, results similar to those found for the silver clusters were obtained. The Au–Au distances in the triangular clusters are significantly longer than those in the square clusters, as exemplified by cluster A15, where the distances range from 2.96 to 3.18 Å. Among the square systems, the S4-symmetric cluster A12 is the most favourable, while in the triangular systems, the Cs-like symmetric cluster A20 is preferred.
3.2.2.2. With the Cp ligands
We obtained similar results for the square silver clusters containing Cp or C5H4NMe2 ligands. Cluster A28 with S4 symmetry is preferred by 18.14 kJ/mol over cluster A22 with C2 symmetry, and by 10.58 kJ/mol over cluster A5, also with C2 symmetry.
For the triangular silver clusters, A32 with C1 symmetry exhibits different Ag–Ag distances of 2.88, 2.93, and 3.02 Å, while cluster A35 with Cs symmetry shows Ag–Ag separations of about 2.99 Å. The latter is more stable by 4.38 kJ/mol compared to cluster A32. Similar observations can be made for the gold-containing clusters, where cluster A6 with S4 symmetry is the most favourable among the square systems. In contrast, the triangular clusters A33 and A36 are nearly isoenergetic; however, their optimisation leads to longer Au–Au distances, reaching up to 3.22 Å.
Overall, the stability of the clusters studied in this work is strongly influenced by both the nature of the coinage metal and the adopted symmetry, which is itself influenced by the NMe2-substituent at the organic π-ligand. In the case of copper, short M–M contacts are favoured in square geometries, while triangular motifs show moderate stabilisation depending on symmetry. In the case of silver and gold, the optimised structures generally reproduce the experimental distances more closely, with S4 symmetry emerging as the most stable for square clusters. For triangular clusters, the relative stabilisations are smaller, but optimisation often leads to elongated M–M distances, especially for gold. These trends highlight the key role of symmetry in dictating both geometry and relative stability across the series.
3.3. Optimised geometries starting from elongated metal–metal distances
Geometry optimisations were initiated from deliberately elongated metal–metal distances to avoid any predefined interaction. This approach allows an unbiased evaluation of whether a metal–metal bond is intrinsically favoured, with any contraction arising solely from the system’s electronic effects. After examining the stability of the different models (A1–A36), we determined that the square copper clusters A10 (D2 symmetry), A13 (S4 symmetry), and A29 (S4 symmetry) are the most stable. The optimised structures of A10 and A13 display large Cu–Cu separations of 3.32 and 3.34 Å, respectively, and the molybdenum and copper atoms are coplanar. In these systems, each Cu–Mo–Cu unit contains one triply bridging carbonyl and two doubly bridging carbonyls (see Figure 3). In contrast, structure A29 exhibits a shorter Cu–Cu distance of 2.75 Å, with the molybdenum atoms displaced out of the plane defined by the copper square, and the carbonyl ligands adopting only doubly bridging coordination modes. Furthermore, the orientation of the C5H4NMe2 ligands in A10 and A13 differs markedly from that of the Cp ligands in A29 (Figure 3).
Optimised DFT/BP86 structures of A10, A13, and A29.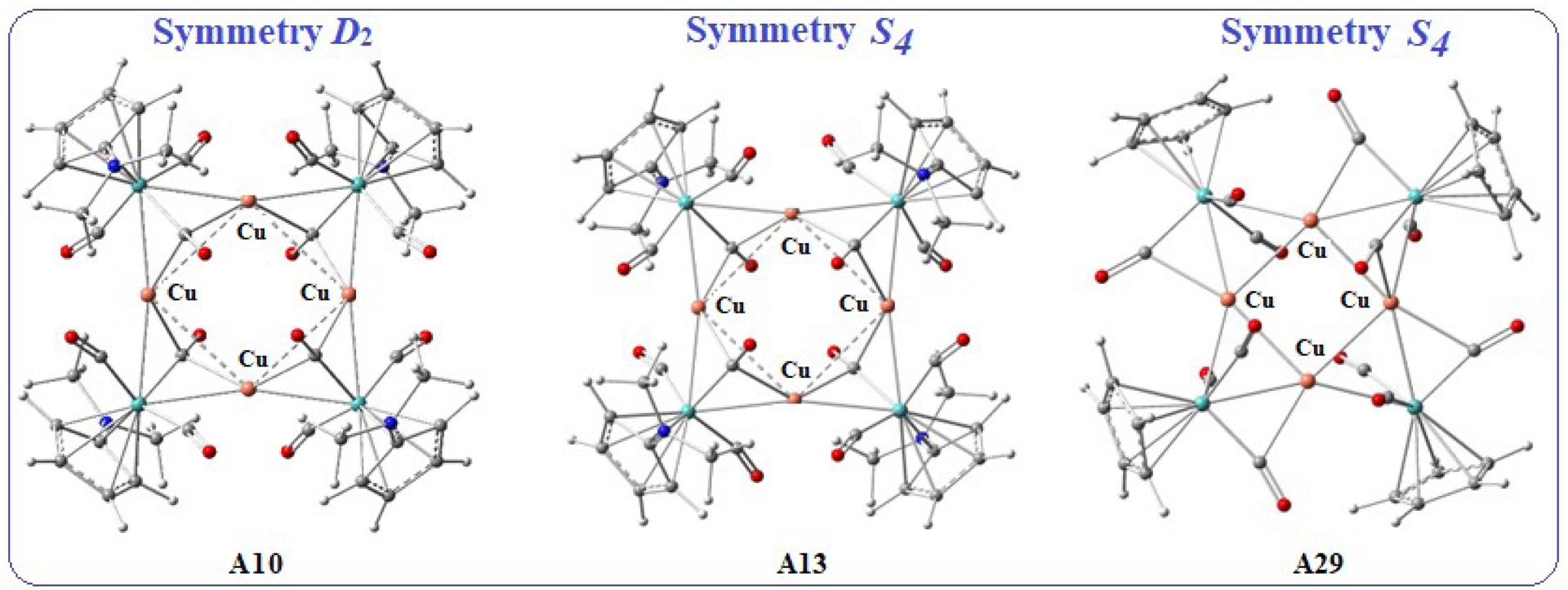
We sought to optimise structures derived from models A10 and A13 with D2 and S4 symmetries, which exhibit large metal–metal distances for the three coinage metals, and Cp or C5H4NMe2 ligands. In the case of the copper square, we successfully obtained two optimised structures, A41 and A42 (Figure 4), showing large Cu–Cu distances for the D2 and S4 symmetries with the Cp ligand. These results are comparable to those for structures A10 and A13 presented in Figure 3. In contrast, for silver and gold, the optimisation of the S4 symmetric structures with both types of ligands yielded geometries with short M–M distances of about 2.75 Å for silver and 2.90 Å for gold. However, optimisation of the D2 symmetric structures with both Cp or C5H4NMe2 ligands led to geometries with large metal–metal separations, 3.38 Å and 3.44 Å for the silver squares A37 and A39, and 3.54 Å and 3.56 Å for the gold squares A38 and A40 (Figure 4).
Clusters with silver, gold, and copper squares A37–A41 with D2 symmetry and A42 with S4 symmetry.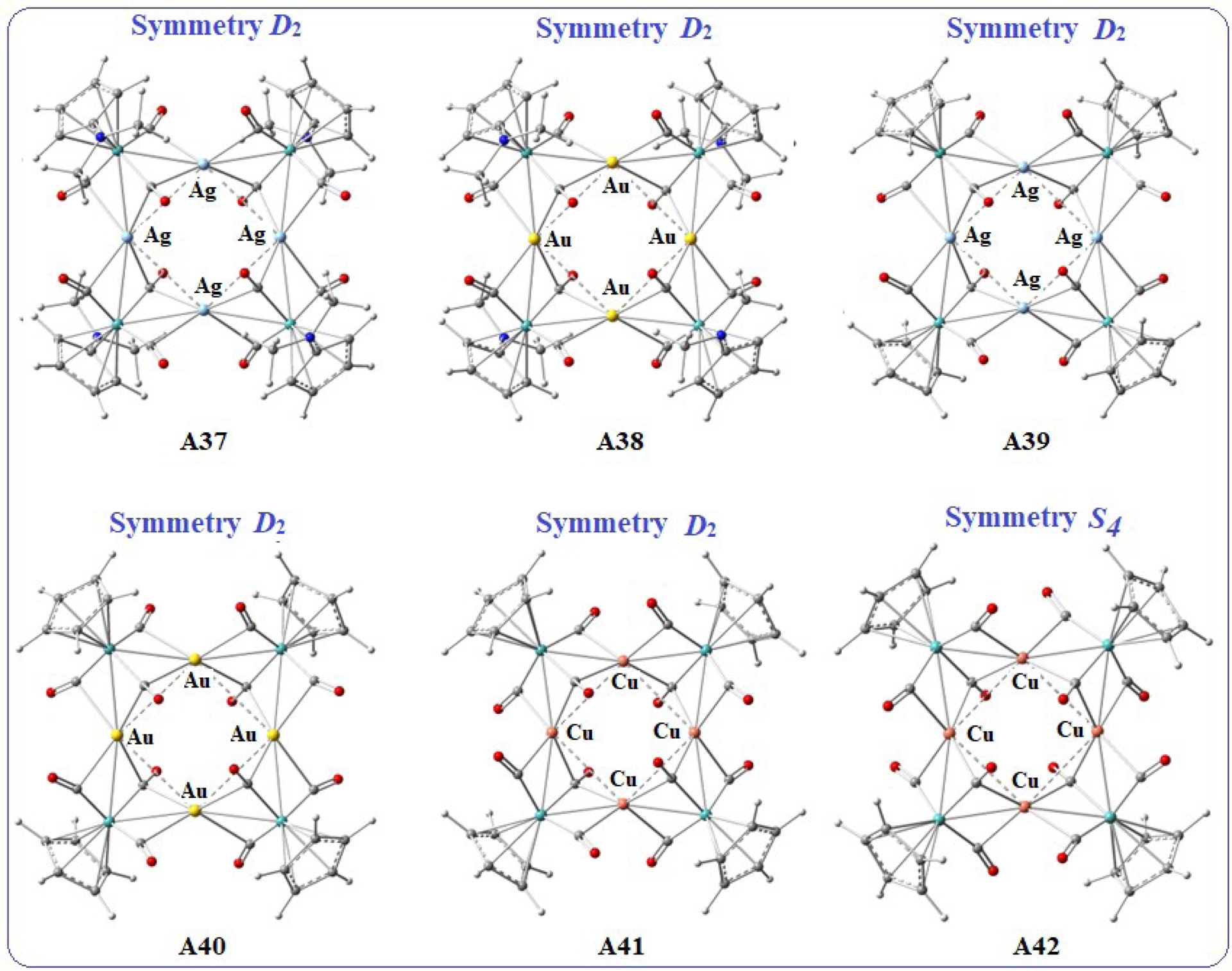
Optimisation attempts starting from elongated metal–metal separations show that only square copper clusters can retain such large Cu–Cu distances, while the silver or gold square clusters predominantly relax to shorter M–M bonds in S4 symmetry. However, D2 symmetric Ag and Au squares can retain elongated M–M distances, highlighting a clear dependence of the final geometry on both the metal type and cluster symmetry.
3.4. Energies of the individual fragments MMo(C5H4NMe2)(CO)3 and MMoCp(CO)3 (M = Cu, Ag, Au)
To complement the previous results and clarify further the structural preferences and relative stabilities of triangular and square clusters, we conducted a comparative analysis of the relative energies of the fragments MMo(C5H4NMe2)(CO)3 and MMoCp(CO)3 (M = Cu, Ag, Au), which constitute building blocks in the final clusters. For the square-type clusters, we normalised the total electronic energy by dividing it by four, considering the four constitutive fragments, while for the triangular-type clusters, the total energy was divided by three. This normalisation procedure, which allows a direct comparison between clusters with different nuclearities, follows the approach previously introduced in our earlier study [25]. As reference points, we used A2, A3, and A7 for the MMo(C5H4NMe2)(CO)3 systems, and A22–A24 for the MMoCp(CO)3 systems. The relative energies thus obtained provide a reliable basis for evaluating which geometries and symmetries are energetically more favourable. The resulting energy differences were plotted as energy diagrams (Figures 5–8), clearly illustrating stability trends across the coinage metals. These diagrams not only emphasise the energetic preferences of each system but also enable visualisation of the effect of ligand substitution (Cp or C5H4NMe2) on cluster stability. Overall, this comparative study offers a consistent picture of how the interplay between the nature of the metal centre, the π-ligand, and the symmetry influences fragment stabilisation. The analysis of these relative energies is key to understanding the structural preferences of the copper-, silver-, and gold-containing clusters and will underpin discussions of their electronic properties in the following sections.
Relative energies (ΔE in kJ/mol) for the fragment Cu[Mo(C5H4NMe2)(CO)3].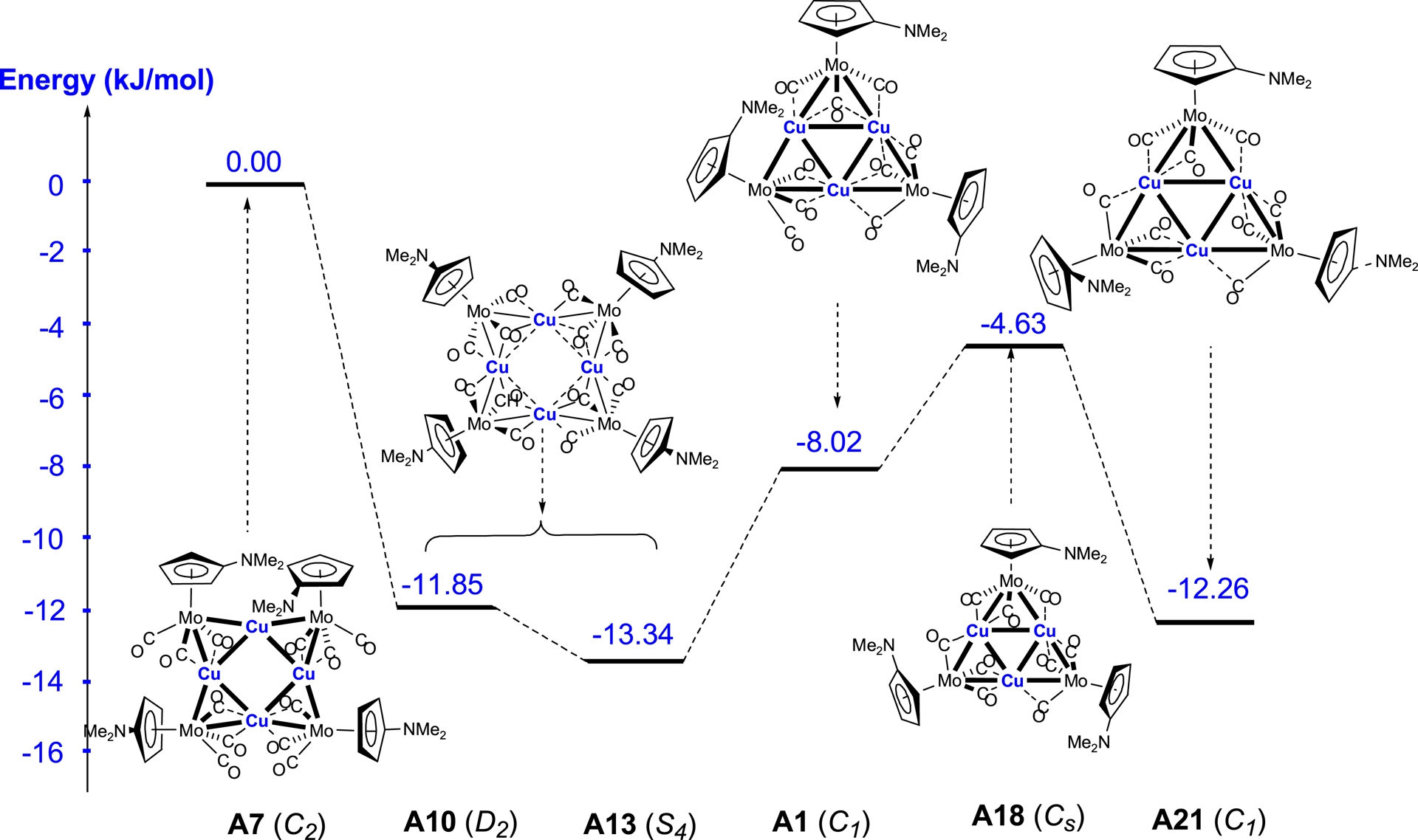
Relative energies (ΔE in kJ/mol) for the fragments M[Mo(C5H4NMe2)(CO)3] (M = Ag and Au).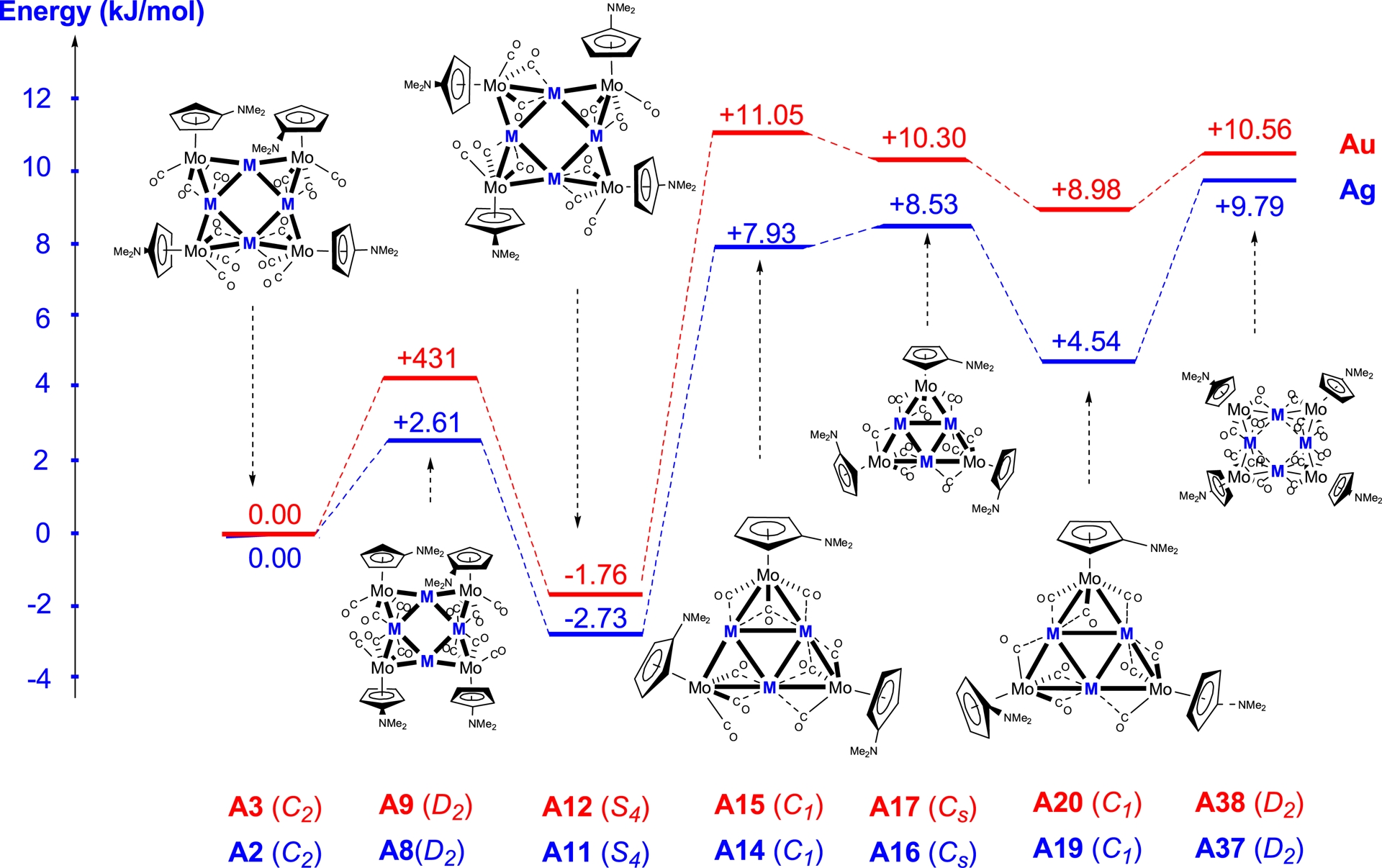
Relative energies (ΔE in kJ/mol) for the fragment Cu[MoCp(CO)3].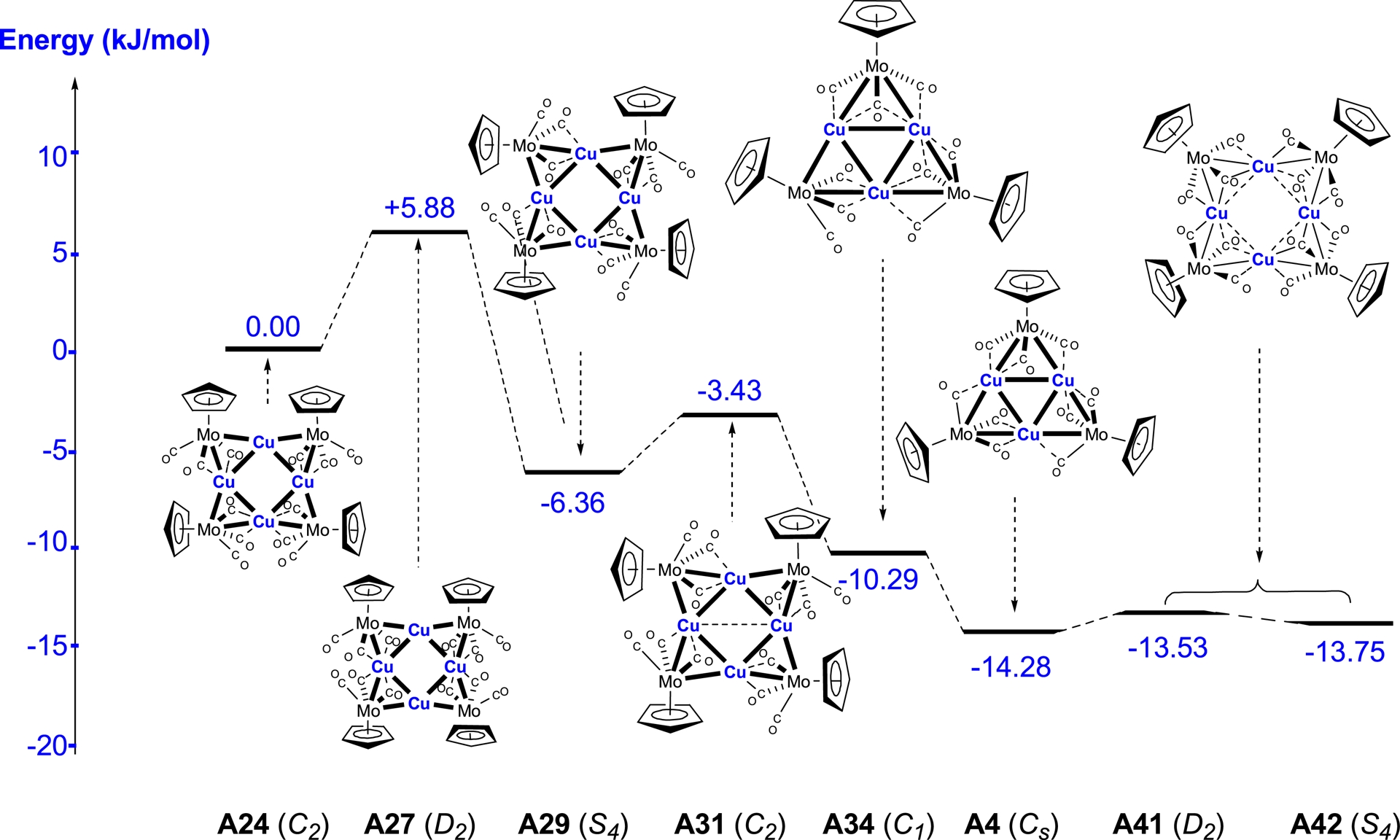
Relative energies (ΔE in kJ/mol) for the fragments M[MoCp(CO)3] (M = Ag and Au).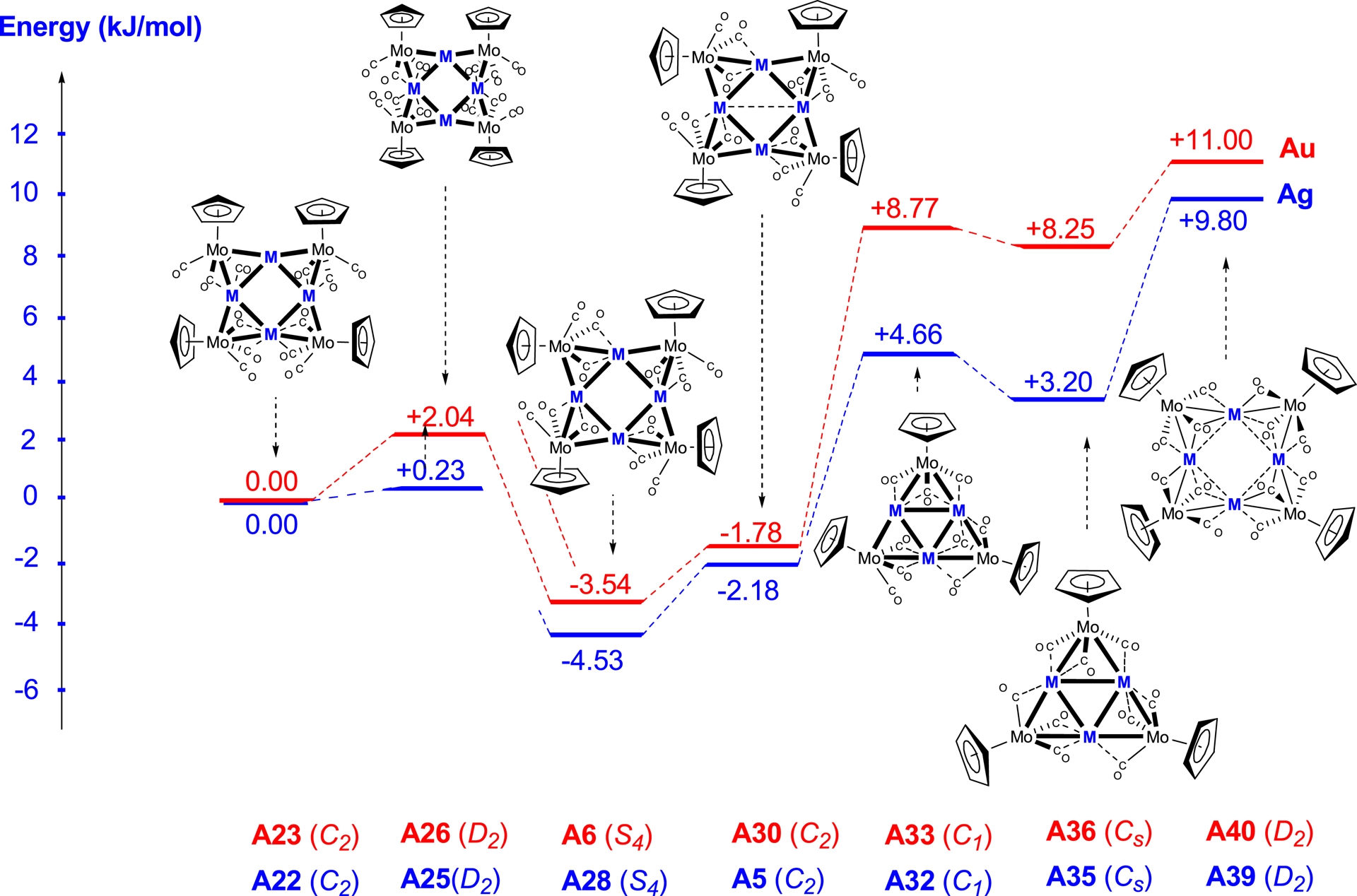
3.4.1. Fragment Cu[Mo(C5H4NMe2)(CO)3]
The analysis shows that the square cluster A13 with S4 symmetry and the triangular cluster A21 are energetically more favourable compared to the experimentally observed structure A1. This energetic preference highlights an intrinsic tendency of copper to prefer the square rather than the triangular arrangement. However, this stabilisation is only achieved when the Cu–Cu separations are significantly elongated, with optimised distances around 3.30 Å. Such elongated Cu–Cu distances reduce the repulsive interactions within the metallic core and allow a better electronic distribution between the copper and molybdenum centres, thereby favouring the square geometry.
3.4.2. Fragments M[Mo(C5H4NMe2)(CO)3] (M = Ag or Au)
In contrast to the case of copper, the most stable silver and gold clusters with the C5H4NMe2 ligands are the square clusters A11 and A12, respectively, both adopting S4 symmetry with short M–M distances.
The triangular structures A14–A17, A19, and A20, as well as the square structures A37 and A38 with elongated M–M separations, are less stable than A2 and A3. The preference for shorter M–M distances in the case of silver and gold can be attributed to electronic and steric factors: shorter metal–metal distances maximise metal–metal interactions and allow a more favourable overlap with the CO ligands. Overall, these results highlight that, unlike with copper, silver and gold square arrangements with S4 symmetry and short M–M separations are energetically preferred, emphasising the role of the intrinsic electronic properties of the coinage metal in determining the cluster stability.
3.4.3. Fragment Cu[MoCp(CO)3]
In this case, the theoretical results are in excellent agreement with the experimental structures determined by X-ray diffraction, confirming that the triangular cluster A4 indeed corresponds to the most stable arrangement for the copper-containing clusters. We also successfully obtained an optimised S4-symmetric structure A29, featuring short Cu–Cu distances of approximately 2.75 Å. In this structure, the carbonyl ligands occupy positions very similar to those observed experimentally in A4, demonstrating that the stereoelectronic environment is properly captured by the DFT calculations. The fragment corresponding to structure A29 exhibits a stability comparable to that of the fragment from structure A4, and it is also more stable than the square structure A42, which has elongated metal–metal distances. These observations highlight that triangular arrangements are energetically favoured for copper when metal–metal distances are moderate, while square geometries with long Cu–Cu separations can also be stabilised under specific conditions. Overall, the results provide a coherent picture of the interplay between geometry, metal–metal distances, and ligand orientation in determining the relative stability of the copper-containing clusters (Figure 7).
Overall, the calculations confirm that the triangular copper cluster A4 is the most stable, with a S4-symmetric structure like A29 where shorter Cu–Cu distances provide additional stabilisation, while square clusters with elongated metal–metal separations are less favoured.
3.4.4. Fragments M[MoCp(CO)3] (M = Ag or Au)
In the case of silver-containing clusters, our calculations reveal a clear discrepancy with the experimental observations. Indeed, the fragment corresponding to the experimentally determined structure A5 does not correspond to the most stable structure, since the fragment of the square structure A28 with S4 symmetry is more stabilised.
This suggests that, although the triangular geometry is accessible experimentally for silver, the square arrangement with short Ag–Ag distances is intrinsically more favourable from an energetic point of view. In contrast, for the gold cluster A6, the theoretical calculations are in perfect agreement with the experimental data obtained by X-ray diffraction. This concordance strongly supports the reliability of our computational approach and confirms that, unlike for silver, the triangular geometry experimentally observed for gold indeed corresponds to the most stable form.
3.5. Bonding analysis
Tables S6–S11 summarise the energy decomposition analysis (EDA) results for the interactions between the metal cores and the metalloligand fragments [MoCp(CO)3]n, calculated at the BP86 level. Here, ΔEint denotes the total interaction energy, while ΔEelstat and ΔEorb correspond to the electrostatic and orbital (covalent) contributions, respectively. Energy decomposition analysis at the BP86 level was performed to elucidate the nature of the interactions between the Cun, Agn, and Aun metal cores and the metalloligand fragments [MoCp(CO)3]n and [Mo(C5H4NMe2)(CO)3]n for all investigated clusters. In every case, the strongly negative ΔEint values indicate that cluster formation is governed by highly stabilising metal–ligand interactions.
The BP86 EDA results show that all Cu-based complexes are strongly stabilised by their interaction with the metalloligand fragments. The Cu3 systems (A1, A18, and A21) exhibit interaction energies of around −4.93 × 103 kJ/mol, whereas the Cu4 clusters (A7, A10, and A13) exhibit much larger stabilisations of −7.28 to −7.60 × 103 kJ/mol, highlighting the stronger binding in the square architectures. This difference mainly arises from the electrostatic term, which increases markedly when going from Cu3 (−4.46 to −4.56 MJ/mol) to Cu4 (−6.38 to −6.95 MJ/mol). The orbital contribution also becomes significantly more stabilising in the Cu4 systems, indicating improved Cu–Mo covalency and more efficient metal–ligand orbital mixing. Although Pauli repulsion is larger in the more compact Cu4 cores, it is clearly outweighed by the enhanced electrostatic and orbital attractions, resulting in superior overall stability of the tetranuclear complexes.
All Ag-containing systems display highly favourable interaction energies. The Ag3 complexes (A14, A16, A19) show ΔEint values close to −4.45 MJ/mol, while the Ag4 species (A2, A8, A11, and A37) are much more stabilised, with values approaching −7.0 MJ/mol. This enhanced binding in the Ag4 series is primarily driven by the electrostatic term, which becomes substantially more negative upon increasing the nuclearity of the Ag core. In parallel, the orbital interaction also strengthens, reflecting improved Ag–Mo covalent interactions and electronic delocalisation within the square Ag4 rings. Despite the rise in Pauli repulsion, especially in A37, the dominant attractive contributions lead to a pronounced stabilisation of the tetranuclear assemblies.
For all gold complexes, the interaction between the Au cores and the metalloligand is highly stabilising. The Au3 species (A15, A17, and A20) display interaction energies around −5.0 MJ/mol, whereas the Au4 derivatives (A3, A9, A12, and A38) reach significantly larger values between −7.4 and −7.7 MJ/mol. Electrostatic attraction is the leading stabilising factor in both series, but it is strongly reinforced in the Au4 clusters. The orbital term also increases markedly with nuclearity, confirming a substantial covalent contribution associated with Au–Mo bonding and metal–metal cooperation. Although Pauli repulsion rises in the Au4 species, the net effect remains highly favourable, yielding the most stable complexes in this family.
The Cu3 complexes A4 and A34 exhibit interaction energies of about −4.87 MJ/mol, whereas the Cu4 derivatives display a much broader but generally more stabilising range extending down to −7.51 MJ/mol. In all cases, electrostatic interactions dominate the attractive forces, supported by sizable orbital contributions that point to significant Cu–Mo covalent bonding. The larger Pauli repulsion observed in some Cu4 systems is counterbalanced by the stronger electrostatic and orbital stabilisation, leading overall to enhanced binding in the tetranuclear clusters. Although the EDA results indicate that the tetranuclear systems generally exhibit stronger interaction energies with the metalloligand fragments, this does not exclude the existence of trinuclear Cu3 rafts. The EDA analysis mainly describes the metal–ligand interactions and does not fully account for other factors such as geometric constraints and intrinsic Cu–Cu bonding preferences. These effects can stabilise the triangular Cu3 arrangement, allowing it to exist as a viable and locally stable structural motif despite the greater stabilisation predicted for the tetranuclear systems.
The Ag3 complexes A32 and A35 show interaction energies around −4.40 MJ/mol, while the Ag4 clusters (A5, A22, A25, A28, and A39) are substantially more stabilised, with ΔEint values approaching −6.9 MJ/mol. As in the other series, electrostatic attraction is the principal stabilising factor and increases significantly with the size of the silver core. This is accompanied by a marked rise in orbital stabilisation, evidencing enhanced Ag–Mo covalent interactions. Even though Pauli repulsion becomes larger in the Ag4 clusters, the combined electrostatic and orbital effects clearly favor the tetranuclear species.
The Au4 complexes (A6, A23, A26, A30, and A40) exhibit much more negative interaction energies (−7.3 to −7.6 MJ/mol) than the Au3 species A33 and A36, confirming the strong stabilising effect of increasing gold nuclearity. This enhanced binding is the result of both a stronger electrostatic attraction and a reinforced orbital interaction, indicating more effective Mo→Au donation and Au→Mo back-donation in the square Au4 cores. Among them, A23 and A26 stand out by their particularly large orbital contributions, reflecting pronounced covalent character and efficient metal–metal electronic communication. Although Pauli repulsion is higher in the Au4 systems, it is more than compensated by the dominant attractive interactions.
The EDA results also rationalise the higher stability of the tetranuclear Ag4 and Au4 clusters compared with their trinuclear analogues. The square M4 cores provide a more favourable electronic environment that enables a more efficient charge redistribution between the metal core and the metalloligand fragments. Consequently, both the electrostatic ΔEelstat and orbital ΔEorb contributions become more stabilising when moving from M3 to M4 species. Although Pauli repulsion increases in the more compact M4 structures, it is largely compensated by the stronger attractive interactions.
A clear and systematic trend is observed with increasing nuclearity of the metal core. For all three metals, the tetranuclear M4 systems are significantly more stabilised than their trinuclear M3 analogues. For Cu and Ag, the ΔEint values increase from approximately −4.4 to −5.0 MJ/mol in the M3 species to about −6.6 to −7.6 MJ/mol in the M4 clusters, while for Au, the stabilisation increases from around −5.0 MJ/mol in Au3 to nearly −7.7 MJ/mol in Au4. This pronounced enhancement reflects the cooperative effect of adding a fourth metal centre, which enables a more efficient distribution of charge and stronger metal–ligand and metal–metal interactions within the square cores.
In all complexes, the electrostatic term ΔEelstat represents the dominant attractive contribution, accounting for the largest fraction of the stabilisation. This highlights the strong Coulombic attraction between the positively charged coinage-metal cores and the anionic Mo-based metalloligand fragments. Importantly, ΔEelstat becomes markedly more negative when moving from M3 to M4 systems, indicating that the square arrangements provide a more favourable electrostatic environment for binding the four metalloligands around the metal core. The orbital interaction term ΔEorb is also very large and systematically increases with nuclearity, revealing a substantial covalent component in the metal–ligand bonding. The much stronger ΔEorb values in the M4 complexes indicate enhanced Mo→M σ-donation and M→Mo π-back-donation, as well as more effective metal–metal electronic communication within the cyclic M4 frameworks. This effect is particularly pronounced for the gold clusters, consistent with the well-known high polarisability and relativistic stabilisation of Au orbitals, which promote strong covalent and aurophilic interactions. Pauli repulsion ΔEPauli increases with the size and compactness of the metal core, especially in tetranuclear clusters, as a consequence of greater overlap between filled orbitals in crowded square geometries. Nevertheless, this destabilising contribution is systematically outweighed by the much larger electrostatic and orbital stabilisations. The steric term ΔEster, which combines Pauli repulsion and electrostatic interactions, remains overall favourable, further supporting the structural viability of these clusters.
Taken together, the EDA results demonstrate that the superior stability of the M4 assemblies relative to the M3 analogues arises from the cooperative interplay of strong electrostatic attraction, enhanced covalent metal–ligand bonding, and efficient metal–metal interactions within the square cores. This synergy explains the marked preference for tetranuclear Cu, Ag, and Au architectures in these Mo-based metalloligand systems and underlines the key role of nuclearity in tuning the bonding and stability of coinage-metal clusters.
3.6. Frontier molecular orbital analysis
The frontier molecular orbitals (FMOs), namely the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), are fundamental to the electronic structure, chemical stability, and reactivity of molecular systems [50, 51, 55, 56]. The HOMO reflects a molecule’s electron-donating capability, whereas the LUMO indicates its electron-accepting potential. The energy separation between these two orbitals, commonly referred to as the HOMO–LUMO gap (ΔE), serves as an important descriptor of molecular stability and chemical reactivity. To visualise the electronic structure of clusters A1–A42, the HOMO and LUMO energy levels for the hydride complexes were plotted. The corresponding FMO distributions are presented in Figure 9 (see also Supplementary Figures S1–S6).
Molecular orbital energy (eV) of [CuMo(C5H4NMe2)(CO)3]n (n = 3 for A1, A18, A21, and n = 4 for A7, A10, A13).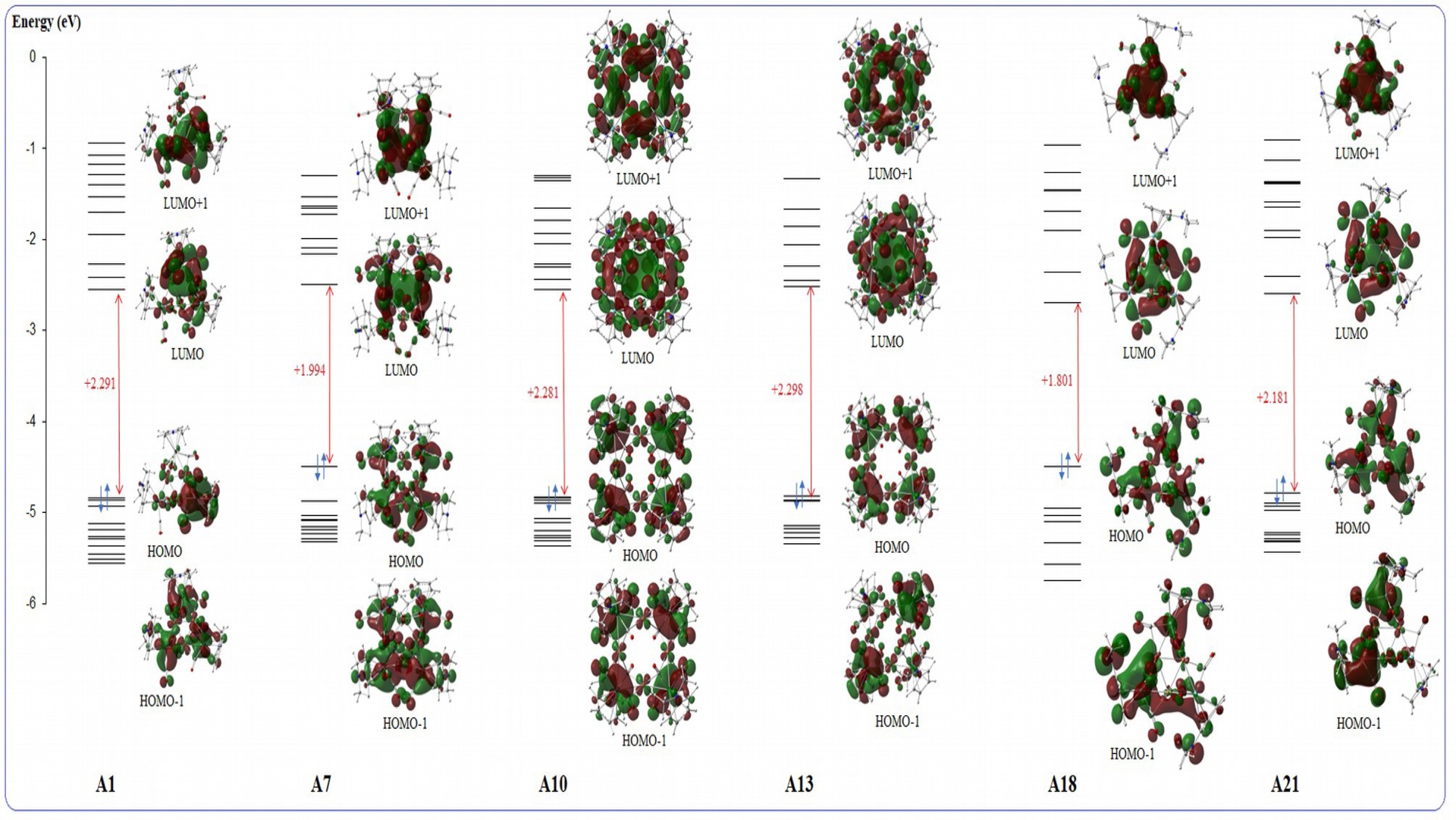
The FMOs provide essential insight into the electronic stability and reactivity of the Cu, Ag, and Au clusters investigated in this series (A1–A42). The calculated energy levels and HOMO–LUMO gaps clearly demonstrate that the nature of the metal, the presence of the electron-donating NMe2 ligand, and the cluster nuclearity jointly govern the overall electronic behaviour. For all complexes, the HOMO corresponds to the highest doubly occupied molecular orbital and is predominantly metal-centred, with major contributions from the d orbitals of the coinage metal (Cu, Ag, or Au) and Mo, as well as varying participation from the Cp and C5H4NMe2 ligands. The LUMO is systematically an unoccupied orbital dominated by Mo d and CO π∗ contributions. Accordingly, the lowest-energy electronic excitation in all clusters corresponds to a metal/ligand-to-carbonyl charge-transfer (MLCT) process. The energy separation between these two orbitals, ΔE = ELUMO − EHOMO, directly reflects the ease of electron promotion, redox activity, and electronic softness of the clusters. A dominant trend across all metal families is the influence of the NMe2 substituent. In the substituted series [MMo(C5H4NMe2)(CO)3]n (M = Cu, Ag, Au), the HOMO is significantly destabilised relative to the unsubstituted [MMoCp(CO)3]n analogues. This effect originates from the strong σ-donor character of the NMe2 group, which injects electron density into the metal–Mo–Cp framework and increases the population of metal–ligand bonding orbitals. Consequently, the HOMO energies are shifted upward and the HOMO–LUMO gaps are reduced. This behaviour is clearly reflected in the orbital plots (Figures S1, S3, and S5), where the HOMO of the NMe2-substituted clusters shows significant ligand participation in addition to metal d contributions. The resulting increase in electron density at the metal centres enhances back-donation to the carbonyl ligands and stabilises low-lying antibonding orbitals, bringing the LUMO closer in energy to the HOMO. As a result, the NMe2 substituted clusters are more polarisable, electronically softer, and chemically more reactive than their unsubstituted counterparts. Among the three coinage metals, Cu-based clusters exhibit the highest HOMO energies and the smallest HOMO–LUMO gaps, making them the most electronically active systems. In the substituted [CuMo(C5H4NMe2)(CO)3]n series, the HOMO lies in the range −4.49 to −4.84 eV, reflecting strong Cu–Mo and Cu–NMe2 interactions. The HOMO is fully occupied and delocalised over the Cu–Mo core with substantial ligand contribution, indicating efficient electron donation from NMe2 into metal-centred bonding orbitals. The corresponding LUMO is largely Mo–CO π∗ in character and remains relatively low in energy (−2.49 to −2.69 eV), resulting in small HOMO–LUMO gaps (1.80–2.30 eV). In particular, A18, which exhibits the smallest gap, features a strongly delocalised HOMO and a low-lying LUMO, enabling facile MLCT transitions and high redox activity. In the unsubstituted [CuMoCp(CO)3]n clusters, removal of the NMe2 donor stabilises the HOMO (−4.83 to −5.29 eV) and leads to a concomitant increase in the energy gap. The HOMO becomes more localised on the metal–CO framework, while the LUMO retains its Mo–CO π∗ character. This larger separation between occupied and virtual orbitals explains why these systems are more electronically robust but less reactive than their NMe2-substituted counterparts. Replacing Cu with Ag leads to a pronounced increase in electronic stability. In [AgMo(C5H4NMe2)(CO)3]n, the HOMO energies are lower than in the Cu analogues (−4.53 to −4.86 eV), while the LUMO levels remain relatively high (−2.19 to −2.43 eV), yielding HOMO–LUMO gaps of 2.14–2.64 eV. The HOMO in these systems is dominated by Ag–Mo d orbitals with limited ligand participation, reflecting weaker σ donation from NMe2 compared with the Cu system. In the unsubstituted [AgMoCp(CO)3]n series, the HOMO is further stabilised (−5.02 to −5.24 eV), whereas the LUMO remains Mo–CO π∗ in character, resulting in the largest HOMO–LUMO gaps of the entire dataset (up to 2.73 eV for A35). The diffuse nature of the Ag 4d orbitals reduces metal–ligand overlap and promotes electron localisation, which is clearly visible in the FMO plots. Consequently, Ag-based clusters are calculated to be the most electronically stable and least reactive systems in this study. Au clusters display an intermediate electronic character between Cu and Ag systems. In the NMe2-substituted [AuMo(C5H4NMe2)(CO)3]n series, the HOMO spans a broad energy range (−4.25 to −5.20 eV), reflecting sensitivity to nuclearity and coordination environment. Relativistic stabilisation of the Au 5d orbitals enhances Au–Mo and Au–ligand covalency, leading to efficient electronic delocalisation. The LUMO remains dominated by Mo–CO π∗ character, but for some complexes, notably A9, it lies unusually close to the HOMO, giving rise to a small gap (1.76 eV) and pronounced electronic softness. In the unsubstituted [AuMoCp(CO)3]n family, two distinct regimes emerge. The n = 4 clusters display relatively small gaps (1.95–1.99 eV), consistent with strong delocalisation over the extended Au–Mo framework, whereas the n = 3 species exhibit much larger gaps (>2.45 eV), indicative of more localised electronic structures and higher stability. Across all metal families, increasing the nuclearity from n = 3 to n = 4 systematically reduces the HOMO–LUMO gap. The larger metal framework allows greater delocalisation of the HOMO over multiple metal centres and stabilises low-lying charge-transfer states. This effect is particularly pronounced for Au clusters but is also evident for Cu and Ag systems. Taken together, the FMO analysis shows that the HOMO is always a fully occupied metal–ligand bonding orbital, while the LUMO is a Mo–CO antibonding orbital. The HOMO→LUMO transition therefore corresponds to a MLCT process, which is most facile in Cu- and Au-based NMe2-substituted clusters and in n = 4 species. These systems are thus predicted to be the most electronically soft, redox-active, and catalytically promising, whereas Ag-based and unsubstituted clusters, characterised by larger HOMO–LUMO gaps and more localised FMOs, are expected to be the most electronically stable and resistant to excitation.
4. Conclusion
Taking advantage of the full characterisation of a unique series of 2D raft-type heterometallic clusters, we could perform a systematic investigation of the energies and relative stabilities of Cu(I), Ag(I), and Au(I)-containing clusters with triangular and square core geometries under different symmetries (C1, Cs, C2, D2, and S4), which allowed a clear comparison with the experimentally determined X-ray structures. In the case of clusters bearing the Cp ligand, an excellent agreement was observed between theory and experiment, since the most stable computed structures corresponded closely to those reported crystallographically. With C5H4NMe2 as π-ligand, our calculations indicated that alternative arrangements more stable than those experimentally observed are possible, thereby highlighting the significant effect of the π-ligand substitution on the overall stability and geometry of the clusters, although no direct interaction was observed between the NMe2 group and the coinage metals. It is not too surprising to sometimes observe differences between calculated and experimental values involving the carbonyl ligands and the coinage metals, since the relevant interactions are much weaker than those involving the carbonyl ligands and molybdenum.
Furthermore, previously reported square copper clusters such as [Et4N]4[M4Cu4S12O4] (M = Mo, W) [56] and [(η5-C5Me5)WS3Cu]4 [57] exhibit elongated Cu–Cu distances, which are consistent with our theoretical findings since we found that square copper clusters with long Cu–Cu separations can indeed be stabilised. There is a subtle balance between stabilisation through electronic factors and destabilisation due to steric effects, while packing effects may also play a role.
The energy decomposition analysis provides a clear and quantitative understanding of the bonding mechanism in these coinage-metal clusters. In all systems, electrostatic attraction between the positively charged metal cores and the Mo-based metalloligands represents the dominant stabilising force, while large orbital interaction terms reveal a substantial covalent component associated with Mo→M donation and M→Mo back-donation. The markedly higher stabilisation observed for the tetranuclear M4 (M = Cu, Ag, Au) complexes compared with their M3 analogues demonstrates the cooperative nature of metal–metal and metal–ligand interactions within the square cores. These findings rationalise the enhanced stability of the M4 architectures and highlight nuclearity as a key factor governing the electronic structure and bonding of metalloligand-stabilised coinage-metal clusters. The electronic properties of these clusters are controlled by a balance between metal type, ligand donation, and nuclearity. Cu- and Au-based C5H4NMe2-substituted clusters, especially with n = 4, show enhanced delocalisation and superior charge-transfer capability. In contrast, Ag-based and unsubstituted systems are more electronically stable and better suited for applications requiring robust molecular frameworks.
This work demonstrates that DFT calculations not only satisfactorily reproduce experimental findings but can also predict new, potentially accessible geometries, thereby offering valuable guidance for future experimental investigations.
Acknowledgements
The authors are grateful to Dr. Peter Deglmann (University of Heidelberg, Germany; now BASF SE, Ludwigshafen, Germany), for providing computer resources and continuous encouragement.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Supplementary materials
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.448 or from the author.