

 CC-BY 4.0
CC-BY 4.0
1. Introduction
[6]Helicenes, helical molecules comprising six ortho-fused aromatic rings, have attracted considerable attention due to their planar chirality. Since the synthesis and resolution of the parent carbo[6]helicene [1], several helical scaffolds have been obtained via oxidative photocyclodehydrogenation of stilbene derivatives [2, 3, 4, 5, 6, 7, 8, 9, 10] or by other nonradiative methodologies [11, 12, 13, 14, 15, 16, 17, 18]. In fact, numerous synthetic and spectroscopic studies have been conducted to assess assignment of the absolute configuration of [6]helicenes [19, 20, 21], which was unequivocally established on the basis of the combined use of optical rotation (OR), electronic circular dichroism (ECD), and X-ray diffraction methods [22]. We wish to draw attention to the fact that helicenes have large optical [23, 24, 25], electronic [26, 27, 28], and enhanced physical-organic properties [29, 30, 31, 32, 33]. In addition, these organic compounds have been regarded as promising candidates for chiral catalysts and ligands in asymmetric synthesis [34, 35, 36]. Moreover, six-membered helicenes have shown importance in biological chemistry for their capability to bind selectively to DNA and are employed for molecular chiral recognition in polymers chemistry [37].
Continuous interest of our group aims to design and synthesize easily and in an acceptable quantity a new helical conjugated hexacyclic framework with useful functional groups. In our synthesis we are presently seeking the use of 2-bromobenzo[c]-phenanthrene-6-carbonitrile (1) as a main building block to achieve the target helical compound. The racemic hexacyclic helicene was separated by chiral HPLC into two enantiomers whose chiroptical and photophysical properties were experimentally examined and a remarkable feature was noticed.
2. Experimental section
2.1. Material and physical measurements
Synthetic procedures were carried out under an inert atmosphere of argon and were monitored by TLC. Solvents have been carefully dried and freshly distilled according to common laboratory techniques. Melting points were determined on an Electrothermal 9002 apparatus and were reported uncorrected. Column chromatography was performed on silica gel using a cyclohexane and ethyl acetate mixture as an eluent. NMR spectra were measured using a Bruker AC 300. Photocyclizations were performed in a water-cooled Quartz photoreactor equipped with a high-pressure mercury immersion lamp (Heraeus TQ 150). UV–Vis spectrum was recorded in CHCl3 at room temperature using a spectrophotometer UV-1600PC. Cyclic voltammetric experiments were performed using a three-electrode cell, consisting of a glass carbon working electrode, a platinum counter electrode, and an Ag∕Ag+ 0.1 M AgNO3 in acetonitrile nonaqueous reference electrode. Investigations have been carried out in millimolar anhydrous acetonitrile solutions containing 0.1 M of tetrabutylammonium tetrafluoborate ((n‐Bu)4NBF4) as a supporting electrolyte. The reference electrode Ag∕Ag+ was calibrated with respect to the formal potential of the ferrocenium/ferrocene (Fc+/Fc) couple in MeCN. All measurements were scanned in negative and positive directions at a scan rate of 50 mV/s, which are controlled by a PGSTAT30 Autolab potentiostat. Optical rotations (in deg⋅cm2⋅g−1) were measured in a 10-cm thermostated quartz cell using a Perkin Elmer-241 polarimeter. The circular dichroism spectra were acquired on a JASCO J-815 spectrometer equipped with a JASCO Peltier cell holder PTC-423 to maintain the temperature at 25.0 ± 0.2 °C in acetonitrile using a 1-mm quartz sample cell.
2.2. Experimental procedure for the Heck coupling
700 mg of 2-bromobenzo[c]phenanthrene-6-carbonitrile 1 (2.1 mmol), 190 mg of dry sodium acetate (2.3 mmol), and 3 mL of DMA were introduced into a double-necked flask fitted with a septum and was repeatedly degassed and purged with argon. Then, 0.45 mL of p-methoxystyrene (3.3 mmol) were added, and the mixture was heated to 100 °C. When reaching this temperature, a 10-mg solution of Herrmann’s catalyst (0.5 mol%) in 2 mL of DMA was added, and the reaction mixture was heated to 140 °C for about two days. After cooling to room temperature, 30 mL of distilled water were added and the organic phase was extracted three times with dichloromethane (3 × 30 mL). The combined organic layers were dried over MgSO4, and the solvent was evaporated to dryness. The crude residue was subjected to a flash column chromatography, using cyclohexane/EtOAc (90:10) as the mobile phase, to provide the desired diarylethene 2.
2.2.1. Spectroscopic data of (E)-2-(p-methoxytstyryl) benzo[c]phenanthrene-6-carbonitrile (2)
Yield = 70%, yellow solid, m.p = 217–219 °C; 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 3.88 (s, 3H, OCH3), 6.95 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 16.2 Hz, 1H, Hvinyl), 7.28 (d, J = 16.2 Hz, 1H, Hvinyl), 7.55 (d, J = 8.7 Hz, 2H), 7.70–7.99 (m, 2H), 7.90 (d, J = 8.7 Hz, 1H), 8.01–8.10 (m, 3H), 8.22 (d, J = 8.7 Hz, 1H), 8.30 (s, 1H), 9.01 (s, 1H), 9.04 (d, J = 8.1 Hz, 1H); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 55.38 (OCH3), 108.37 (C), 114.31 (2CH), 118.37 (CN), 123.16 (CH), 124.24 (CH), 126.15 (CH), 126.54 (CH), 126.93 (2CH), 127.80 (CH), 127.83 (C), 128.13 (2CH), 128.82 (CH), 129.26 (CH), 129.49 (CH), 129.64 (2C), 129.80 (C), 130.57 (C), 130.78 (CH), 132.24 (C), 133.68 (C), 134.59 (CH), 138.67 (C), 159.86 (C); HRMS (MALDI-TOF) calculated for (C28H19NONa): [M + Na]+: 408.1364; found: 408.1360.
2.3. Experimental procedure for the photocyclization reaction
To a 150-mg solution of olefin 2 (0.38 mmol) in 1 L of toluene, 151 mg (0.42 mmol) of iodine was added under vigorous stirring. The solution was degassed and purged with argon for about 10 minutes, and then 1.3 mL of propylene oxide were added. The resulting solution was irradiated using a falling-film photoreactor and a high-pressure Hg-vapor lamp (150 W, Heraeus) while maintaining the argon flow throughout the irradiation. When completing, the reaction mixture was concentrated in vacuum, and the crude product was purified by flash silica gel column chromatography using cyclohexane/EtOAc (95:05).
2.3.1. Spectroscopic data of 7-cyano-15-methoxy[6] helicene (3)
Yied = 77%, yellow solid, 1H NMR (300 MHz, CDCl3): 𝛿 (ppm): 2.93 (s, 3H, OCH3), 6.76–6.82 (m, 2H), 6.90 (dd, J1 = 2.4 Hz, J2 = 8.7 Hz, 1H), 7.28 (td, J1 = 7.8 Hz, J2 = 0.9 Hz, 1H), 7.58 (d, J = 8.7 Hz, 1H), 7.75 (d, J = 8.7 Hz, 1H), 7.83 (d, J = 8.7 Hz, 1H), 7.90–7.98 (m, 3H), 8.04–8.09 (m, 2H), 8.37 (d, J = 8.7 Hz, 1H), 8.45 (s, 1H); 13C NMR (75 MHz, CDCl3): 𝛿 (ppm): 53.74 (OCH3), 106.77 (CH), 107.97 (C), 117.66 (CN and CH), 122.35 (CH), 123.08 (CH), 125.16 (CH), 125.83 (CH), 126.08 (C), 126.44 (C), 126.55 (CH), 126.81 (CH), 127.01 (CH), 127.54 (C), 128.34 (CH), 128.54 (C), 128.80 (CH), 128.37 (CH), 128.89 (CH), 129.25 (C), 130.33 (C), 130.63 (C), 131.27 (C), 133.01(C), 133.44 (CH), 157.07 (2C); HRMS (MALDI-TOF) calculated for (C28H17NONa): [M + Na]+: 406.1207; found: 406.1202.
Crystal data for compound 3 (C28H17NO) were recorded on a D8 VENTURE Bruker AXS diffractometer, M = 383.42, triclinic, space group P − 1. a = 8.1587(12) Å, b = 10.2469(18) Å, c = 11.6859(18) Å, V = 952.7(3) Å3, Z = 2, 𝜌calcd = 1.337 g⋅cm−3, X-ray source Mo-K𝛼, 𝜆 = 0.71073 Å, T = 150(2) K; observed reflections 4366; refinement method Full-matrix Least-squares on F2; parameters refined 272; R(F) = 0.0781, 𝜔R(F2) = 0.1747.
2.4. Enantiomeric separation of racemic 7-cyano-15-methoxy[6]helicene (3)
The enantiomeric separation of racemic 3 was conducted by HPLC on a Chiralpak IA column (250 × 10 mm) thermostated at 303 K using n-hexane/2-propanol/dichloromethane (80:10:10) as a mobile phase. The flow rate was 5.0 mL⋅min−1. UV–Vis detection was performed at 280 nm. The enantiomeric excess (ee) was then determined by chiral HPLC using the same condition under a flow rate of 1.0 mL⋅min−1.
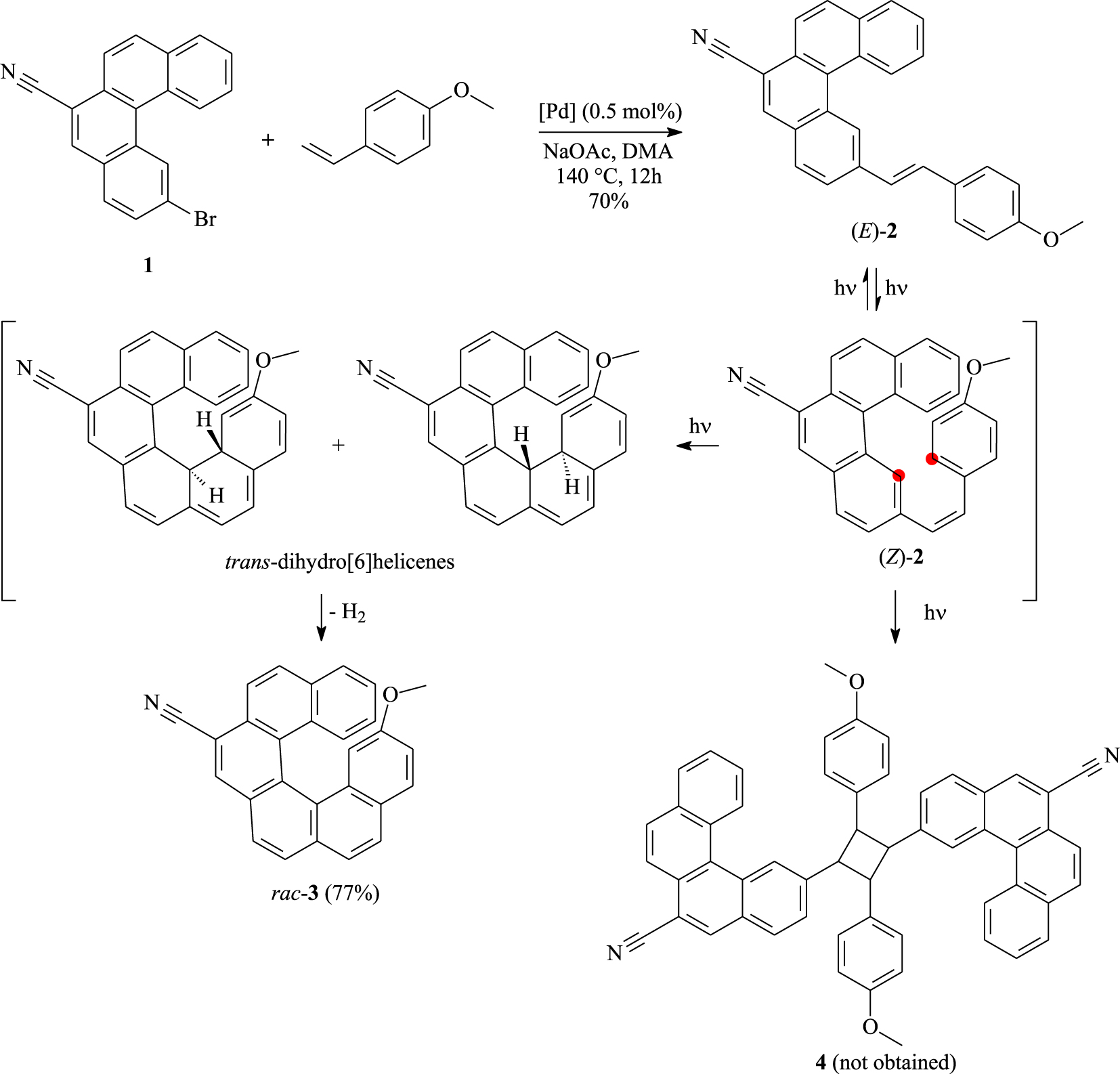
Synthetic pathway giving 7-cyano-15-methoxy[6]helicene (3).
2.4.1. Selected data for (P)-(+)-7-cyano-15-methoxy [6]helicene (3)
Yield = 46%; ee = 100%; yellow solid; mp = 244–246 °C;
2.4.2. Selected data for (M)-(−)-7-cyano-15-methoxy [6]helicene (3)
Yield = 48%; ee = 100%; yellow solid; mp = 245–247 °C;
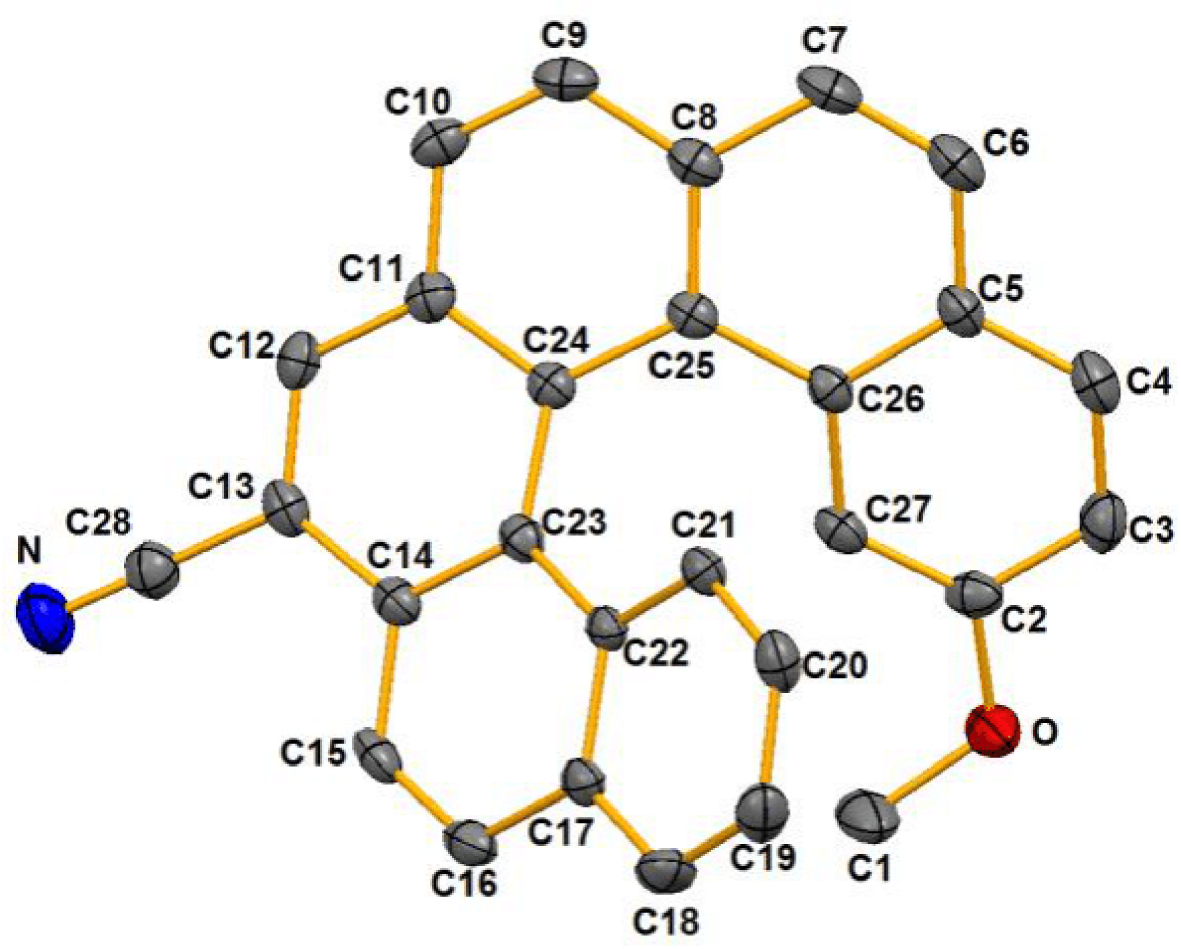
X-ray crystal structure of 7-cyano-15-methoxy[6]helicene (3). Hydrogen atoms have been omitted for clarity.
3. Results and discussion
The experiments below summarize a convenient method based on the oxidative photocyclization of a convenient diarylethene for the synthesis of an interestingly functionalized [6]helicene in an appreciable amount from inexpensive reagents (Scheme 1). To start the synthesis, we prepared 2-bromobenzo[c]phenanthrene-6-carbonitrile (1) according to a literature procedure [38] and reacted it with p-methoxystryrene to produce the corresponding helicene-precursor 2 in 70% yield following the Heck coupling [39], which is carried out in the presence of 0.5 mol% of Herrmann’s catalyst {trans-di(𝜇-acetato)-bis[o-(di-o-tolylphosphino)benzyl]dipalladium} and sodium acetate in DMA. Afterward, the suitable building block 2 must afford the [6]helicene scaffold upon a photochemical ring closure reaction. It has been dissolved in toluene and subjected to UV-light irradiation for approximately 3 hours in the presence of a stoichiometric amount of iodine as an oxidizing agent and excess of propylene oxide as a hydrogen iodide scavenger [40]. The photocyclization took place in a regioselective way and the target helicene 3 was isolated in 77% yield as a yellow solid stable in air and to light. Thus, no other regioisomer like an S-shaped compound was identified nor isolated from the reaction mixture [41, 42, 43]. Photodimers such as 4 have been observed during the photocyclization step [44] but not in this study. The whole photocyclodehydrogenation process proceeded via isomerization of (E)-2 to (Z)-2 followed by an intramolecular electrocyclization leading to trans-dihydro[6]helicenes that are oxidized into the [6]helicene.
It is noteworthy that single crystals were obtained from racemic 3 by slow evaporation of a dichloromethane solution at room temperature and the molecular structure of the helicene was determined by X-ray diffraction analysis. It crystallizes in the triclinic system with the space group P − 1. The structure of helicene 3 is illustrated in Figure 1, and the torsion angles (Table 1) which vary from 29.40° to 12.10° gave evidence of a helical unit as compared to those of the carbo[6]helicene [45]. Inner and outer C–C bonding distances vary from 1.345 Å to 1.457 Å.
Having obtained 3 in racemic form, it was then interesting to examine its separation into enantiomers. We find that efficient separation was ensured by high performance liquid chromatography (HPLC) using a Chiralpak IA column (250 × 10 mm) eluted with the mobile phase n-hexane/2-propanol/dichloromethane (80/10/10) under a UV–Vis detector. The sample was dissolved in dichloromethane then injected on the column at a flow rate of 5 mL⋅min−1. When starting from 87 mg of racemic helicene 3, a total amount of 82 mg of pure product was separated which are equivalent to 94% yield (Scheme 2). The faster-eluting fractions gave 40 mg (46% yield) of the dextrorotatory enantiomer (+)-3 in 100% ee. The slower-eluting fractions provided 42 mg (48% yield) of the levorotatory enantiomer (−)-3 in 100% ee. The enantiomeric purity of each helical enantiomer was checked by HPLC using the above-mentioned conditions.
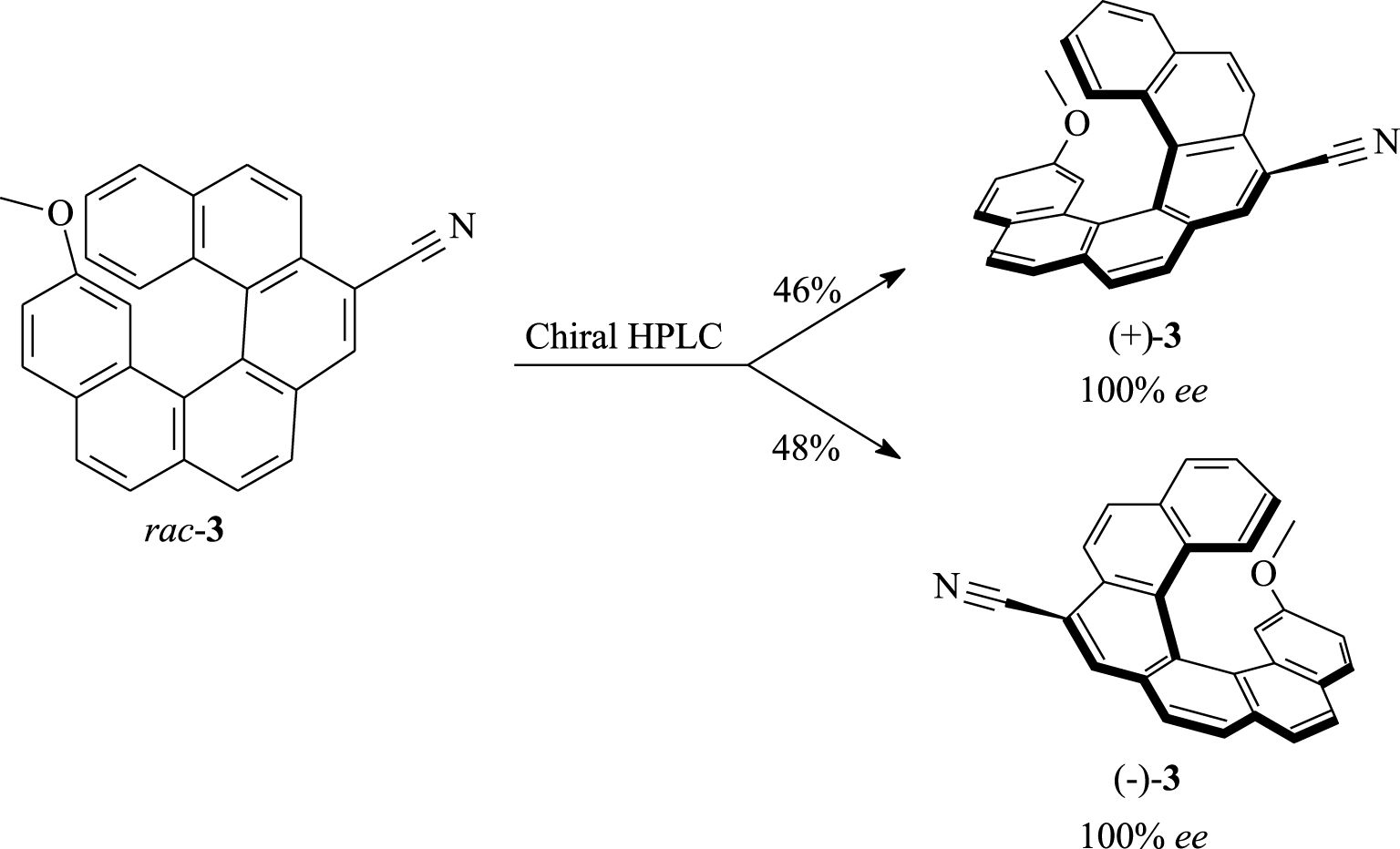
Enantiomeric separation of (±)-7-cyano-15-methoxy[6]helicene (3).
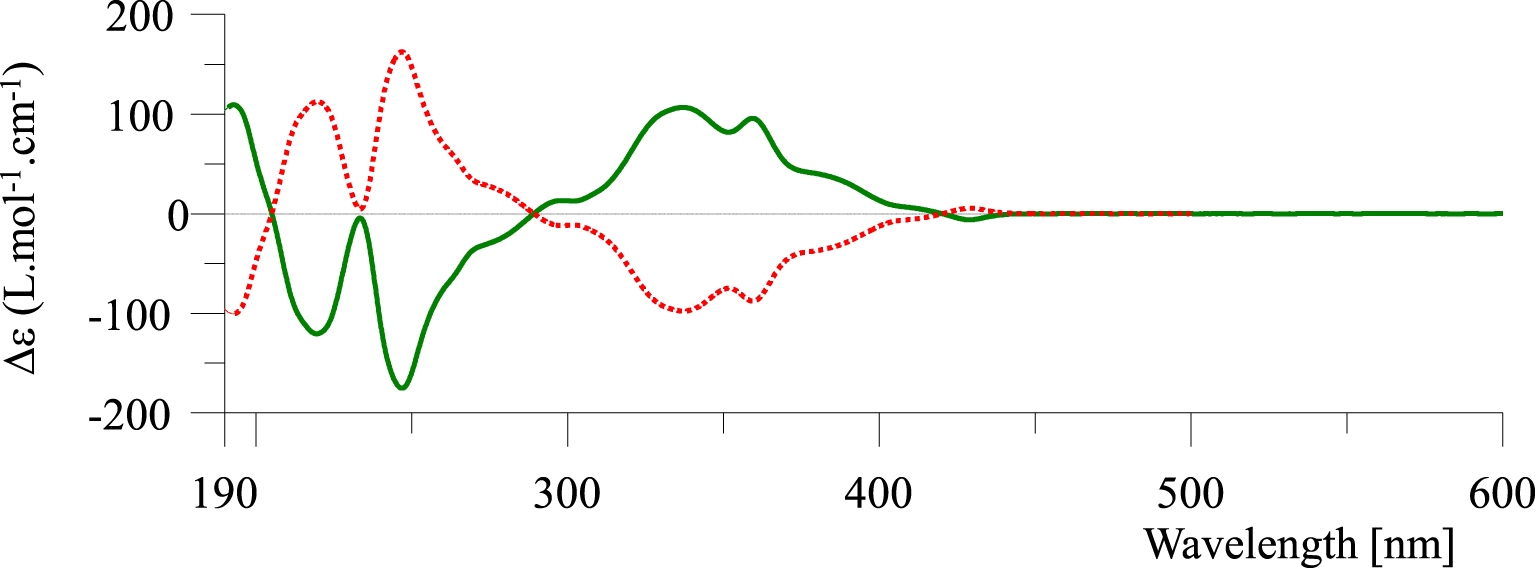
Electronic circular dichroism (ECD) spectra of (P)-(+)-3 (green line) and (M)-(−)-3 (red dotted line) in acetonitrile (2.2 × 10−4 M).
The optical rotations of the enantiopure helical species (−)-3 and (+)-3 were measured in dichloromethane solutions at different wavelengths by using sodium (589 nm) and mercury (578 nm and 546 nm) lines of a Jasco P-2000 polarimeter thermostated at 25 °C. As shown in Table 2, the specific and molar rotation values noted for both enantiomers are extremely high, quite opposite and represent one of the common features of helical backbones [46, 47].
Dihedral angles, inner and outer bonding distances in 7-cyano-15-methoxy[6]helicene (3)
| Dihedral angles (°) | |||||
|---|---|---|---|---|---|
| C21–C22–C23–C24 | 15.00 | ||||
| C22–C23–C24–C25 | 29.40 | ||||
| C23–C24–C25–C26 | 26.10 | ||||
| C24–C25–C26–C27 | 12.10 | ||||
| Inner bonding distances (Å) | |||||
| C20–C21 | 1.374(4) | C24–C25 | 1.456(3) | ||
| C21–C22 | 1.419(4) | C25–C26 | 1.455(4) | ||
| C22–C23 | 1.456(4) | C26–C27 | 1.408(4) | ||
| C23–C24 | 1.457(3) | C27–C2 | 1.379(4) | ||
| C20–C21 | 1.374(4) | C24–C25 | 1.456(3) | ||
| Outer bonding distances (Å) | |||||
| C2–C3 | 1.408(4) | C8–C9 | 1.420(4) | C14–C15 | 1.435(4) |
| C3–C4 | 1.366(4) | C9–C10 | 1.351(4) | C15–C16 | 1.349(4) |
| C4–C5 | 1.412(4) | C10–C11 | 1.425(4) | C16–C17 | 1.430(4) |
| C5–C6 | 1.419(4) | C11–C12 | 1.420(4) | C17–C18 | 1.413(4) |
| C6–C7 | 1.345(4) | C12–C13 | 1.363(4) | C18–C19 | 1.367(4) |
| C7–C8 | 1.443(4) | C13–C14 | 1.437(4) | C19–C20 | 1.405(4) |
Optical rotations for both enantiomers of helicene 3 in CH2Cl2
| 𝜆 (nm) | First eluted enantiomer (+)-3 (c = 0.098, CH2Cl2) | Second eluted enantiomer (−)-3 (c = 0.094, CH2Cl2) | ||
|---|---|---|---|---|
| 589 | + 3100 | + 11 873 | − 3100 | − 11 873 |
| 578 | + 3300 | + 12 639 | − 3300 | − 12 639 |
| 546 | + 4200 | + 16 086 | − 4200 | − 16 086 |
Assignment of the absolute configurations P and M for the enantiomers of 3 has been investigated on the bases of the electronic circular dichroism (ECD) spectroscopy [48, 49]. As shown in Figure 2, the ECD spectrum of (+)-3 gives rise to two distinct negative bands at 220 (𝛥𝜀= −120 L⋅mol−1⋅cm−1) and 248 nm (𝛥𝜀= −175 L⋅mol−1⋅cm−1) followed by two strong positive ones at 335 (𝛥𝜀= +100 L⋅mol−1⋅cm−1) and 365 nm (𝛥𝜀= +90 L⋅mol−1⋅cm−1) and a very weak band at 430 nm (𝛥𝜀= −6 L⋅mol−1⋅cm−1). In the same way, it is clear that the ECD spectrum of the enantiomer (−)-3 is a perfect mirror image to that of the dextrorotatory enantiomer. Moreover, the ECD spectral characteristic of (+)-3 displays similar signature to those of known (P)-helicenes [50, 51, 52, 53], with respect to the intensities of the absorption bands. Notably, the helicene scaffold with P-helicity (right-handed helix) has dextrorotatory specific rotations (+), and that with M-helicity (left-handed helix) has levorotatory specific rotations (−). When comparing the ECD spectra of (+)- and (−)-3 with other [6]helicenes [50], we noticed the presence of a low intense band around 430 nm (𝛥𝜀= −6 L⋅mol−1⋅cm−1), which may be due to the electron-withdrawing cyano group.
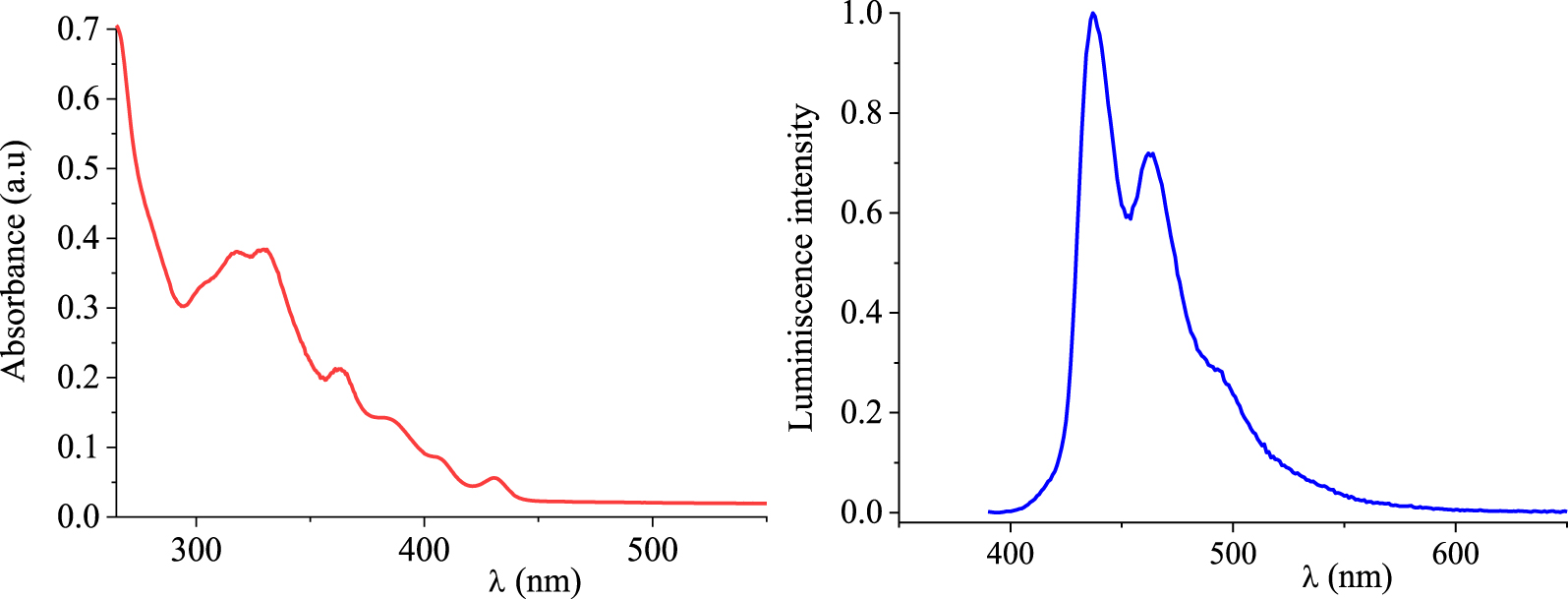
UV–Vis absorption (red line) and PL (blue line) spectra of compound 3 in dilute chloroform solutions (c ≈ 1 × 10−5 M).
Photophysical properties of 7-cyano-15-methoxy[6]helicene (3)
| Compound | Absorption | Photoluminescence | |||
|---|---|---|---|---|---|
| 𝜆onset (nm) | Eg‐opb (eV) | 𝜆emsc (nm) | FWHMd (nm) | ||
| 3 | 330 | 444 | 2.79 | 437 | 45 |
a Absorption maxima measured in CHCl3 solution (c ≈ 1 × 10−5 mol⋅L−1) at room temperature.
b The optical gap (Eg‐op) was estimated from the onset point of the absorption spectrum: Eg‐op = 1240∕𝜆onset.
c Emission maxima measured in CHCl3 solution (c ≈ 1 × 10−5 mol⋅L−1) at room temperature; 𝜆ext = 380 nm.
d Spectrum full width at half maximum.
Photophysical properties of the [6]helicene 3 were examined in dilute chloroform solutions (c ≈ 1 × 10−5 M) and results are gathered in Table 3. The UV–Vis absorption spectrum (Figure 3) reflects two strong absorption bands located at 317 (𝜀≈ 37870 cm−1) and 330 nm (𝜀≈ 38700 cm−1) followed by one medium band at 363 nm (𝜀≈ 21360 cm−1), which are characteristic of π–π∗ and n–π∗ electronic transitions of the helicene unit [54, 55]. Three shoulder peaks, with decreasing intensity, are also depicted in 307, 377, and 401 nm. By examining the low-energy region, only one absorption band which is weaker in absorbance is depicted at 431 nm (𝜀≈ 5700 cm−1) and may be ascribed to the intramolecular charge transfer (ICT) [56] from the electron-donating group, i.e., methoxy groups, to the electron-withdrawing group, i.e., cyano group. The onset of absorption occurs at 𝜆onset = 444 nm and the optical band gap energy (Eg‐op) was found to be 2.79 eV as it has been estimated according to the empirical formula:
A bathochromic shift of about 15 nm is observed for the low-energy band (>400 nm) on the UV–Vis spectrum of compound 3 in comparison to that of the carbo[6]helicene [57].
The photoluminescence spectrum of compound 3 has been recorded in a chloroform solution (c ≈ 1 × 10−5 mol⋅L−1) at room temperature upon excitation at 380 nm (Figure 3). The helicene shows an emission in the blue region with two bands at 437 and 462 nm, the first of which is the most intense. These bands are followed by a shoulder peak located at 490 nm. The full width at half maximum (FWHM) was found to be of 45 nm.
Finally, with the new helical molecule 3 in hand, we next investigated its electrochemical properties by using cyclic voltammetry (CV). The highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels of the obtained helicene have been experimentally estimated. As indicated in Figure 4, the cyclic voltammogram recorded at a scanning rate of 50 mV/s shows that the hexacyclic system 3 exhibits reversible cathodic and irreversible anodic peaks. Furthermore, the onset potential of oxidation (Vonset‐ox) was found to occur at 0.97 V, while the onset potential of reduction (Vonset‐red) was at − 1.14 V [vs saturated calomel electrode (Ag/AgCl)]. In fact, HOMO and LUMO energy levels of the hexahelicene 3 were determined from the onset potentials of the oxidation and reduction processes, according to the following empirical method [58, 59]:
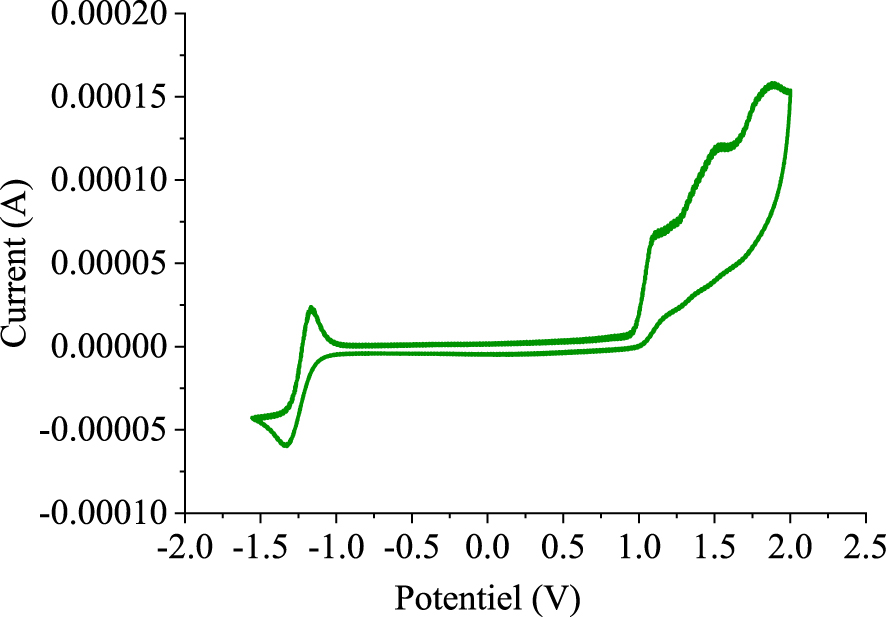
Cyclic voltammogram of 7-cyano-15- methoxy[6]helicene (3) in 0.1 M (n‐Bu)4NBF4/ acetonitrile at a scan rate of 50 mV/s.
4. Conclusion
We have prepared racemic 7-cyano-15-methoxy[6] helicene, in a good overall yield, through a short photochemical procedure based on the use of a suitable tetracyclic building block and requiring inexpensive reagents and mild conditions. The racemic helicene was successfully separated into optically pure P- and M-enantiomers (100% ee) for which chiroptical properties were experimentally investigated. Absorption and photoluminescence properties of the target [6]helicene were examined in solutions and an emission in the blue region was noticed. The electrochemical behavior of this hexacyclic scaffold was evaluated, showing a notable charge transfer interaction due to its π-conjugated electronic system, and HOMO and LUMO energy levels were estimated making the new helicene a good candidate for optoelectronic applications.
Vous devez vous connecter pour continuer.
S'authentifier