


 CC-BY 4.0
CC-BY 4.0
1. Introduction
The synthesis and functionalization of porphyrinoid macrocycles are still the ongoing subjects of intense research activities because these exciting compounds are applied in numerous areas such as catalysis, biology or materials science [1,2,3,4,5]. One of the reasons for this major interest lies in the easy preparation of meso-substituted derivatives from the simple condensation of pyrrole and aldehydes which can produce, after oxidation and depending on the experimental conditions, a huge number of polypyrrolic and aromatic macrocycles such as porphyrins, corroles, contracted and expanded porphyrins [1,2,3,4,5,6,7,8]. The physico-chemical properties of these macrocycles can be tuned by the introduction of different meso-substituents that often require the prior synthesis of oligopyrrole precursors such as the widely used meso-substituted dipyrromethanes (DPMs). Though various synthetic procedures of meso-substituted-DPMs are described [9,10,11,12], very few are devoted to or can be applied to the preparation of derivatives bearing a meso-poly-halogenoalkyl group or a meso-perfluoroalkyl chain [13,14,15,16,17,18,19,20,21,22,23]. These specific substituents are however of major interest because, beyond their high electron-withdrawing character, they can confer to molecules a certain lipophilicity and/or an increased metabolic stability [24,25]. Moreover meso-perfluoroalkyl chains were shown to induce an important non-planar distortion [26,27,28,29] and a high photo-stability [30] when they occupy the meso-positions of porphyrins.
To the best of our knowledge, the DPMs 3a and 3b bearing respectively a meso-CF3 group or a meso-C3F7 group are the only DPMs substituted by a meso-perfluoroalkyl chain for which synthetic procedures are described (Scheme 1). The common preparation of meso-substituted DPMs relies on the acid-catalyzed condensation of an aldehyde and pyrrole [9,10,11,12]. In boiling tetrahydrofuran (THF) and concentrated aqueous HCl (Scheme 1, route a), the condensation of stoichiometric amounts of pyrrole 1 and aldehydes 2a or 2b (as hemiacetal or as hydrate) gave for the first time the meso-perfluoroalkyl DPMs 3a and 3b with high yields (50–70% for 3a and 35–50% for 3b) [13]. Similar yields of 3b were obtained by repeating this procedure [14,15,16,17] or by replacing the acid and the refluxing solvent by p-toluene sulfonic acid (PTSA) and CHCl3, respectively (Scheme 1, route b) [18]. It was later shown that milder experimental conditions (Scheme 1, route c) also give high yields of 3a (20–40%) and 3b (35%) [19,20]. An original preparation of 3a was reported by Dmowski et al. who used the sodium dithionite coupling of 1-bromo-1-chloro-2,2,2-trifluoroethane with 1 (Scheme 1, route d) [21,22].
Previous preparations of meso-perfluoroalkyl DPMs 3a and 3b (PTSA: p-toluene sulfonic acid).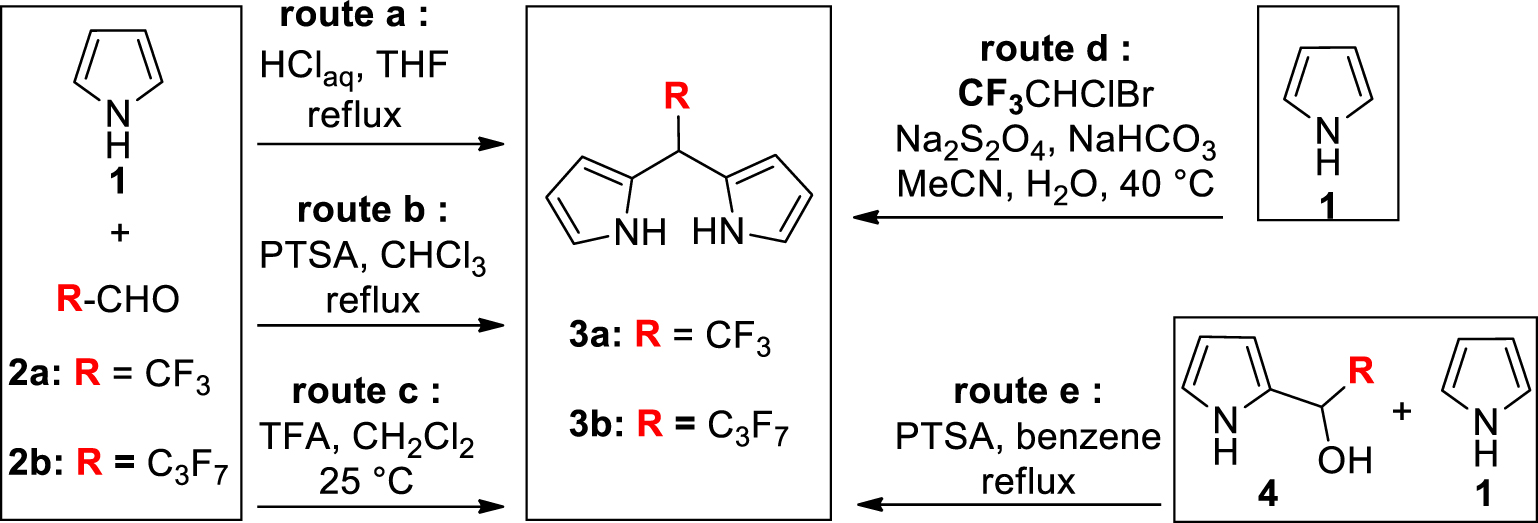
If this simple and inexpensive procedure gives high yields of 3a (49–58%), it can only be applied to incorporate this specific meso-substituent.
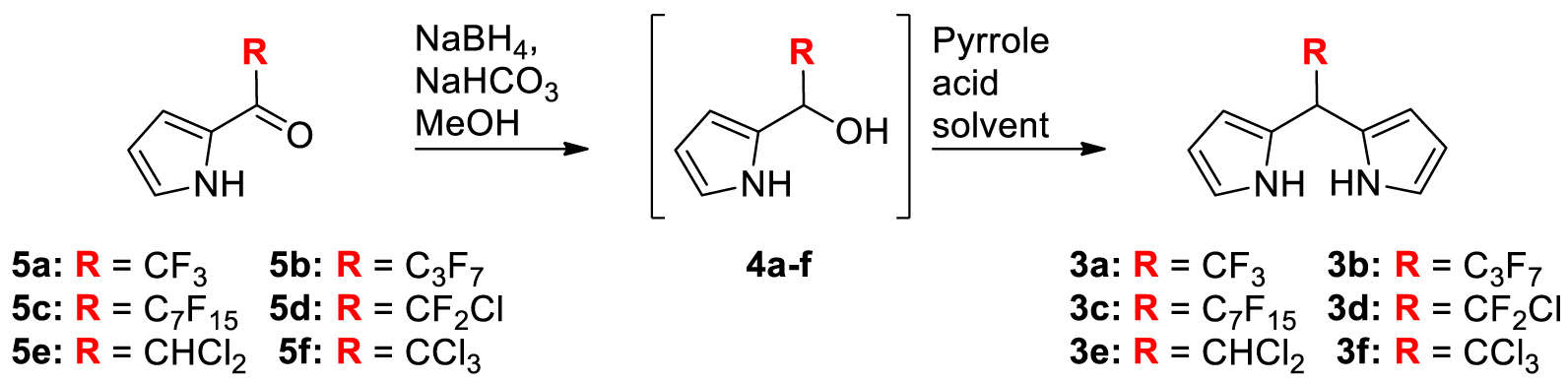
A key intermediate during the aldehyde-pyrrole condensation is the pyrrole-carbinol derivative 4 (Scheme 1). Such an intermediate bearing a heptafluoropropyl chain was prepared and dissolved with an excess of pyrrole in refluxing benzene containing PTSA (Scheme 1, route e) [23]. This stepwise strategy produced 3b with a high yield (77%). Having in mind that several 2-(polyhalogeno)acyl-pyrroles are easily prepared (or commercially available) and knowing that they are quantitatively reduced into the corresponding pyrrole-carbinol derivatives, we report herein how this latter strategy can be adapted, leading to a variety of meso-polyhalogenoalkyl DPMs. For this purpose, a set of three experimental conditions was selected and/or optimized to prepare the known 3a and 3b but also the unprecedented DPMs 3c–f bearing on the meso position a perfluoroheptyl (3c), a chlorodifluoromethyl (3d), a dichloromethyl (3e) or a trichloromethyl group (3f).
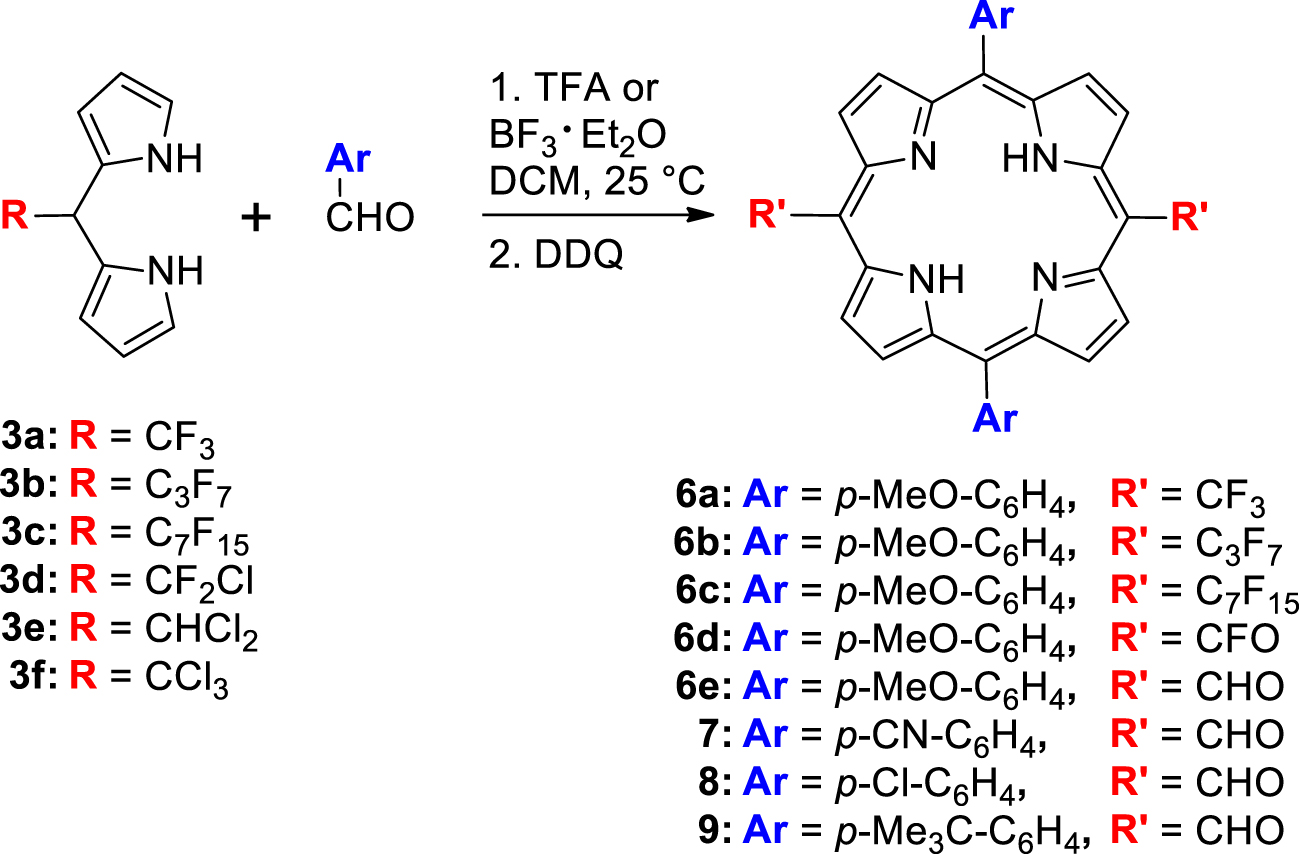
These DPMs were involved in the synthesis of trans-A2B2-meso-substituted porphyrins through their acid-catalyzed condensation with p-anisaldehyde. The DPMs 3a–c gave the expected porphyrins 6a–c whereas the condensation of DPMs 3d and 3e led surprisingly to porphyrins bearing two meso-fluoroacyl groups (6d) and two meso-formyl groups (6e), respectively (Table 2). Other benzaldehydes were used to illustrate the straightforward access to 5,10-bis-(formyl)-15,20-diarylporphyrin because it is really competitive compared to previously described ones that were based on multistep elegant strategies such as: (a) the Vilsmeier formylation of copper porphyrins and their subsequent demetallations [31]; (b) the palladium catalyzed formation of meso-bis-(trimethyl)silylmethylporphyrins followed by their oxidation [32]; (c) the preparations and/or derivations of protected formyl groups (dithiane or acetal moieties) before the DPM-aldehyde condensation and their deprotections after the porphyrin ring formation [33,34].
Because the physico-chemical properties of meso-perfluoro alkyl and meso-formyl porphyrins have been rarely described, those of the eight new free base porphyrins 6a–e, 7, 8 and 9 will be investigated in this contribution by a combination of photophysical, electrochemical and theoretical studies.
2. Results and discussion
2.1. Synthesis
The reaction of a slight excess of acyl-chloride or acid anhydride and pyrrole gave the 2-acyl pyrroles 5a–f according to previously reported procedures (Table 1) [27,31]. These compounds were at first reduced to the corresponding carbinol derivatives 4a–f using a slight excess of NaBH4 (2 equiv.) in MeOH containing 2 equivalents of NaHCO3 [35]. Because TLC analyses showed a complete conversion of the acylpyrroles 5a–f and the formation of the alcohols 4a–f with no observable side products, the pyrrole-carbinols 4a–f were not isolated (nor characterized) but were always freshly prepared and used directly in the next condensation step (Table 1).
Optimization of the synthesis of DPMs 3a–f
| Entry | Procedure | Reactant | R | [4a–f] (M) | Solventa | Acid | Pyrrole/4a–f/acid | t (h) | Product | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | A | 4a | CF3 | 0.07 | DCM | P2O5 | 2:1:2 | 16 | 3a | 60 |
| 2 | 4a | CF3 | 0.23 | DCM | P2O5 | 2:1:2 | 16 | 3a | 39 | |
| 3 | 4a | CF3 | 7.28 | — | P2O5 | 2:1:2 | 16 | 3a | 44 | |
| 4 | 4a | CF3 | 0.58 | Pyrrole | P2O5 | 25:1:2 | 16 | 3a | 0 | |
| 5 | 4a | CF3 | 3.64 | Pyrrole | Eaton’s reagentf | 4:1:1 | 1 | 3a | 0b | |
| 6 | 4a | CF3 | 0.2 | THFc | HClaq | 2:1:1.5 | 2 | 3a | 26 | |
| 7 | 4a | CF3 | 0.15d | H2O | HClaq | 2:1:1.2 | 4 | 3a | 22 | |
| 8 | B | 4a | CF3 | 0.15d | H2O | HClaq | 2:1:1.2 | 16 | 3a | 24 (28)e |
| 9 | 4a | CF3 | 0.15d | H2O | HClaq | 2:1:1.2 | 40 | 3a | 35 | |
| 10 | 4a | CF3 | 0.15d | H2O | HClaq | 4:1:1.2 | 16 | 3a | 20 | |
| 11 | A | 4b | C3F7 | 0.07 | DCM | P2O5 | 2:1:2 | 16 | 3b | 52 (41)e |
| 12 | B | 4b | C3F7 | 0.15d | H2O | HClaq | 2:1:1.2 | 16 | 3b | 0 |
| 13 | A | 4c | C7F15 | 0.07 | DCM | P2O5 | 2:1:2 | 16 | 3c | 65 (87)e |
| 14 | B | 4c | C7F15 | 0.15d | H2O | HClaq | 2:1:1.2 | 16 | 3c | 0 |
| 15 | A | 4d | CF2Cl | 0.07 | DCM | P2O5 | 2:1:2 | 16 | 3d | 0–2 |
| 16 | B | 4d | CF2Cl | 0.15d | H2O | HClaq | 2:1:1.2 | 16 | 3d | 18 (18)e |
| 17 | B | 4e | CHCl2 | 0.15d | H2O | HClaq | 2:1:1.2 | 16 | 3e | 60 (67)e |
| 18 | A | 4f | CCl3 | 0.07 | DCM | P2O5 | 2:1:2 | 16 | 3f | 0 |
| 19 | B | 4f | CCl3 | 0.15d | H2O | HClaq | 2:1:1.2 | 16 | 3f | 0 |
| 20 | 4f | CCl3 | 0.2 | DCM | TFA | 1:1:1.2 | 16 | 3f | 0 | |
| 21 | 4f | CCl3 | 0.2 | THFc | HClaq | 1:1:1.2 | 2 | 3f | 3 | |
| 22 | C | 4f | CCl3 | 0.2 | THFc | HClg | 11:1:0.9 | 2 | 3f | 9 |
aAll reactions were performed at room temperature unless otherwise noted. bPyrrole polymerization occurs. cReactions at reflux. dResulting concentrations if pyrrole carbinol 4 was soluble in water. eGram-scale yields are noted in brackets. fEaton’s reagent: P2O5 (7.7 wt%) in CF3SO3H. gHCl in Et2O (1 M).
In 2011, a pyrrole-carbinol derivative analogous to 4a but bearing an aryl group on the second α-position was condensated with pyrrole (2 equiv.) in dichloromethane (DCM) during 16 h at room temperature and using P2O5 (1 equiv.) as the activating agent [35]. These conditions gave very high yields of an asymmetrical meso-CF3 DPM. Consequently, they were applied, in the present study, at the millimolar scale (condensation at 0.07 M in DCM) and using pyrrole 5a to give 3a with a yield of 60% (Table 1, entry 1). Higher concentrations or using pyrrole as the solvent did not improve the reaction yield (Table 1, entries 2–5). This procedure (Table 1, procedure A) was extended to pyrroles 5b,c and afforded high amounts of 3b (52%) and 3c (65%) but only traces of 3d (0–2%) and no 3f (Table 1, entries 11, 13, 15 and 18). The preparation of 3b and 3c were repeated on a gram scale and gave DPMs with high yields.
To ensure better yields of 3d, new experimental conditions were applied in the preparation of 3a from 4a (used as a model compound). The utilization of refluxing THF and concentrated aqueous HCl was moderately efficient (Table 1, entry 6) but proved that the presence of water in the solvent was not detrimental to get meso-polyhalogeno alkyl DPMs. Several works have shown that meso-aryl-oligopyrrole syntheses can be performed in water and using concentrated aqueous HCl as catalyst [36,37,38]. The application of the conditions of Dehaen et al. [39] ([HCl] = 0.18 M, [4a] = 0.15 M, 2 equiv. of pyrrole, room temperature, 4 h) afforded 3a with a yield of 22% that increases with the reaction time whereas higher amounts of pyrrole do not have any beneficial effect (Table 1, entries 7–10). We selected a reaction time of around one night (16 h) and applied these conditions (procedure B) to get DPMs 3b–f (Table 1, entries 12, 14, 16 and 19). This procedure failed to give DPMs 3b,c and 3f, but produced 3d and 3e with up-scalable yields of 18% and 66% respectively.
Syntheses of porphyrins 6a–e, 7, 8 and 9
| DPM | R | Acid | Porphyrin | Ar | R′ | Yield (%) |
|---|---|---|---|---|---|---|
| 3a | CF3 | BF3 | 6a | p-MeO–C6H4– | CF3 | 9 |
| 3b | C3F7 | TFA | 6b | p-MeO–C6H4– | C3F7 | 4 |
| 3c | C7F15 | TFA | 6c | p-MeO–C6H4– | C7F15 | 4 |
| 3d | CFCl2 | BF3 | 6d | p-MeO–C6H4– | CFO | 2 |
| 3e | CHCl2 | TFA | 6e | p-MeO–C6H4– | CHO | 11 |
| 3e | CHCl2 | TFA | 7 | p-CN–C6H4– | CHO | 9 |
| 3e | CHCl2 | TFA | 8 | p-Cl–C6H4– | CHO | 2 |
| 3e | CHCl2 | TFA | 9 | p-Me3C–C6H4– | CHO | 12 |
R′ groups are at the 5,15 positions, Ar are at the 10,20 positions.
Since mild conditions did not afford the DPM 3f, and because the only example of a 𝛽-substituted DPM bearing a meso-CCl3 group was obtained through harsh conditions (TFA as solvent, 40 °C) [40], we used again refluxing THF and HCl(aq) during the condensation step (Table 1, entry 21). A low yield of 3f was obtained (3%) but reached 9% when using the experimental conditions described to synthesize a meso-CF3 tripyrromethane from a DPM-mono-carbinol (Table 1, entry 22) [41]. Because no porphyrin could be, up to now, obtained from this particular DPM (vide infra), we have not pursued this optimization.
The DPMs 3a–f were used as building blocks in the synthesis of porphyrins (Table 2). For this purpose, we chose to apply the conditions of the acid-catalyzed condensation of the DPMs 3a,b with various aromatic aldehydes that were shown to be effective in the preparation of trans-A2B2 porphyrins bearing two meso-perfluoroalkyl groups (Table 2) [19]. The reaction of p-anisaldehyde (0.01 M) with DPMs 3a–f catalyzed by TFA (10 equiv.) or by BF3⋅OEt2 (0.33 equiv.) in DCM was stopped after the complete disappearance of the DPMs (TLC analysis). The two catalysts have been tested and this contribution reports only the conditions giving the highest yields.
After oxidation by DDQ (1.5 equiv.) and purification by chromatography, modest yields (4–9%) of the trans-A2B2 porphyrins 6a–c were obtained from the DPMs 3a–c although, as previously described, no scrambling occurred during the condensation step [19].1 It has to be noted that, in addition to the expected porphyrin, each experiment led to the formation of several other colored chromophores but in too low yields to ensure their characterization. On the one hand, no porphyrin was detected from the reactions involving the DPM 3f bearing a sterically hindered trichloromethyl group, while on the other hand, the less chlorinated DPMs 3d and 3e gave quite unexpected results (Table 2).
Unlike the common purple spots on TLC and purple solutions observed during the purification of porphyrin 3a–c, green colored TLC spots and solutions were obtained while running the chromatography on the crude mixture from 3e. The red-shifted UV–visible absorption of the isolated compound combined with its 1H NMR spectrum featuring a downfield singlet located at 12.54 ppm were the first evidence of the exclusive formation of the bis-formyl porphyrin 6e which was further confirmed by its HRMS analysis and its IR spectrum displaying an intense carbonyl stretching band at 1672 cm−1 (see the Supporting Information). The hydrolysis of the two CHCl2 groups remains unexplained because it was neither observed during the preparation of DPM 3e though being performed in acidic water, nor during its purification, which was also performed on silica gel. Suspecting that this hydrolysis would not have been quantitative, we performed another synthesis of 6e and washed the reaction mixture with water after the oxidation step. This supplementary treatment led to an identical yield of 6e. The CHCl2 groups of the intermediate porphyrinogen have probably stability close to the CHCl2 group of DPM 3e. Their hydrolysis could occur during the oxidation step as it was previously observed after the condensation of a meso-nitromethyl DPM with an aromatic aldehyde leading to meso-formyl porphyrins [42]. Such hydrolysis was also observed during the metal-assisted cyclization of a meso-CHCl2-𝛽-substituted-a,c-biladiene into meso-formyl metallocorroles [43]. Because the simple use of DPM 3e gives straightforward access to trans-meso-bis(formyl) porphyrin, it was extended to p-cyano-, p-chloro and p-tert-butyl-benzaldehydes and led in the same way to porphyrins 7, 8 and 9 with respective yields of 9%, 2% and 12% (Table 2). The low isolated yield of 8 comes partly from its delicate purification due to its very low solubility in common organic solvents. In the same way, pure and solid 7 has only a slightly higher solubility. Consequently, a protonation–deprotonation sequence was used to ensure the complete dissolution of 7, 8 and 9 when recording their physico-chemical properties (see the experimental part).
As observed during the porphyrin synthesis starting from DPM 3e, the acid-catalyzed condensation of DPM 3d with p-anisaldehyde gave, after oxidation, a green spot on TLC and a green solution during the purification on silica. After purification, the red-shifted UV–visible spectrum of the isolated porphyrin 6d together with the presence of a single signal at − 62.63 ppm in its 19F NMR spectrum indicated that the former CF2Cl group borne by DPM 3d had probably disappeared in 6d. The unexpected structure of the bis-fluoro-acyl derivative 6d was elucidated thanks to its HRMS analysis and its IR spectrum featuring a carbonyl stretching band at 1796 cm−1 (see the Supporting Information). Although the yield of this transformation is low (⩽2%), it gave repeatedly the first example of a porphyrin-bearing meso-fluoro acyl groups. As for 6e, we are not able yet to give the causes and/or the mechanism of the peculiar hydrolysis that has been rarely observed [44] although it has been reported that C–X bonds of meso-perfluoroalkyl groups borne by DPMs or porphyrins are prone to be involved in eliminations, intramolecular cyclization or solvolysis processes [45,46,47,48,49,50].
Half-wave and peak potentials (V versus SCE) of porphyrins 6a–e, 9, (CF3)4PH2 and (C3F7)4PH2 measured in CH2Cl2 containing 0.1 M of [(nBu4N)PF6] (scan rate of 100 mV/s)
| R10,20 | R5,R15 | Reduction | Oxidation | 𝛥E (V)a | |||
|---|---|---|---|---|---|---|---|
| Second | First | First | Second | ||||
| 6a | 4-OCH3–C6H4 | CF3 | − 1.33b | − 0.81 (100)c | 1.32b | 1.79b | 2.11 |
| 6b | 4-OCH3–C6H4 | C3F7 | − 1.34b,d | − 0.80 (80)c | 1.31b | 1.75b | 2.09 |
| 6c | 4-OCH3–C6H4 | C7F15 | − 0.81 (100)c | 1.29b | 1.78b | 2.08 | |
| 6d | 4-OCH3–C6H4 | CFO | − 0.79 (107)c | − 0.54 (72)c | 1.39b | 1.93 | |
| 6e | 4-OCH3–C6H4 | CHO | − 0.89 (74)c | − 0.57 (64)c | 1.33b | 1.81b | 1.90 |
| 9 | 4-Me3C–C6H4 | CHO | − 0.90 (73)c | − 0.58 (69)c | 1.40b | — | 1.98 |
| 10d | 4-PO3Et2–C6H4d | C3F7 | − 0.81 | 1.43 | 2.24 | ||
| (CF3)4PH | CF3 | CF3 | − 0.48f (160)c | ||||
| (C3F7)4PH | C3F7 | C3F7 | − 0.44f (158)c | ||||
aHOMO–LUMO electrochemical gap calculated by 𝛥E = E1∕2(ox1) − E1∕2(red1) or by 𝛥E = Epa(ox1) − E1∕2(red1). bIrreversible peak potential at a scan rate of 100 mV/s. c𝛥Ep = Epa − Epc in mV. dA third irreversible reduction peak is placed at Epc = −1.56 V versus SCE. dFrom Ref. [17]. eFrom Ref. [51]. fMeasured in benzonitrile.
2.2. Electrochemical analysis of porphyrins 6a–e and 9
The cyclic voltammograms (CVs) of 6a–e and 9 were recorded in DCM containing 0.1 M of [(nBu4N)PF6] (Figure 1 and Figure S1 in the Supporting Information). The corresponding oxidation and reduction half-wave and peak potential values are listed in Table 3 together with those of the tetra-meso-CF3 porphyrin (CF3)4PH2 [51], the tetra-meso-C3F7 porphyrin (C3F7)4PH2 [51], and of the porphyrin 10 analogous to 6b but where the p-anisyl groups are replaced by p-diethylphophonate-phenyl substituents [17]. Each CV features a first reversible reduction and a first irreversible oxidation centered on the porphyrin ring. For the perfluoro derivatives 6a–c, this oxidation seemed to be reversible at a scan rate of 100 mV/s but proved to be irreversible when increasing the scan rate up to 800 mV/s. Depending on the substituents, a second oxidation and/or a second reduction were also enlightened by the CVs of 6a–e.
CVs of porphyrins 6a (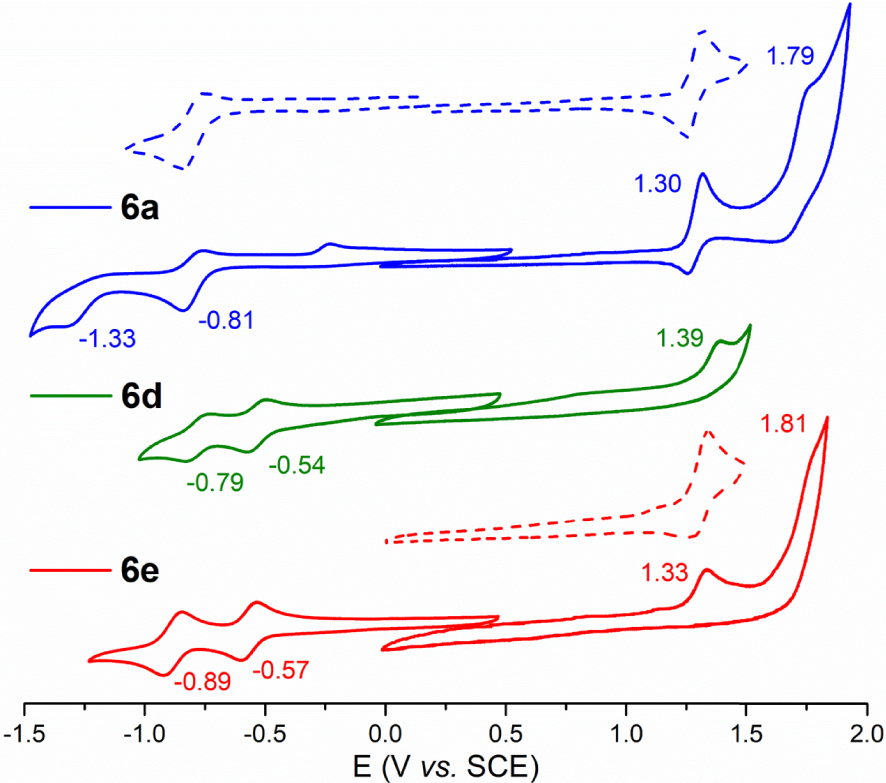
 ), 6d (
), 6d ( ) and 6e (
) and 6e ( ) in DCM containing 0.1 M of [(nBu4N)PF6] (scan rate of 100 mV⋅s−1). The dotted traces emphasize the reversibility of selected redox processes.
) in DCM containing 0.1 M of [(nBu4N)PF6] (scan rate of 100 mV⋅s−1). The dotted traces emphasize the reversibility of selected redox processes.
No oxidation could be observed previously when recording the CVs of (CF3)4PH2 and (C3F7)4PH2 (Table 3) [51]. The replacement of two of these chains by p-anisyl groups in 6a and 6b leads to a significant negative shift of the first reduction potential (∼300 mV) and allows access to the data corresponding to the macrocycle oxidation (E1∕2 ∼ 1.3 V versus SCE).
Whereas the electrochemical properties of porphyrins are substantially varying with the number of meso-perfluoroalkyl chain, they are not notably tuned by the length of these lipophilic chains because the first oxidation/reduction potential values of 6a, 6b and 6c are nearly the same (Table 3). Interestingly, the comparison between 6b and 10 shows that decreasing the electron richness of the 10,20-aryl groups shifts, as expected, the first oxidation process to higher potential values but leaves unchanged the energy of the first reduction. Therefore, varying the nature of 10,20-aryl groups has probably little influence on the energy of the LUMO of such trans-A2B2 porphyrins which is controlled by the meso-perfluoroalkyl chains.
The important anodic shift (+240 mV) affecting the first reduction process when replacing CF3 moieties in 6a by formyl groups in 6e cannot be explained by a field/inductive effect because the latter is stronger for CF3 than for CHO (Hammett parameters: σp (CF3) = +0.54, σp (CHO) = +0.42) [52]. A positive shift is also observed between the two corresponding macrocycle-centered oxidations but has a little amplitude (∼30 mV) (Table 3). As for the CF3 porphyrins 6b and 10, modifying the aryl groups in the formyl-substituted macrocycles 6e and 9 produces only a slight modification of the first oxidation potential whereas the reduction potentials remain unchanged. It has to be noted that the solubility of 7 and 8 was too low in DCM to record accurate electrochemical data.
Supplementary anodic shifts are affecting the first reduction and potential processes when two CFO groups are appended by the porphyrin ring in 6d. Therefore, the remarkable reduced HOMO–LUMO electrochemical gaps of 6d and 6e compared to those of derivatives 6a–c result from a π-conjugation between the carbonyl groups and the aromatic porphyrin ring that are put into evidence by theoretical calculations.
2.3. Spectral properties of the porphyrins
The absorption and corrected emission spectra of porphyrins 6a–e, 7, 8 and 9 were recorded in DCM at room temperature and are shown in Figure 2 and Figure S2 in the Supporting Information. The molar absorption coefficients and absorption maxima of these seven novel compounds are gathered in Table 4 with their emission maxima (excitation at 𝜆 = 590 nm) and fluorescence quantum yields. In order to give structure–properties relationships, selected photophysical data of 10 [17], (CF3)4PH2 [53] and of (C3F7)4PH2 [53] are also given in Table 4 together with those of the porphyrins 11 [19] and 12 [32] analogous to 6a and 6e but where the p-anisyl groups are replaced by simple phenyl substituents.
Electronic absorption spectra and normalized corrected emission spectra of porphyrins 6a (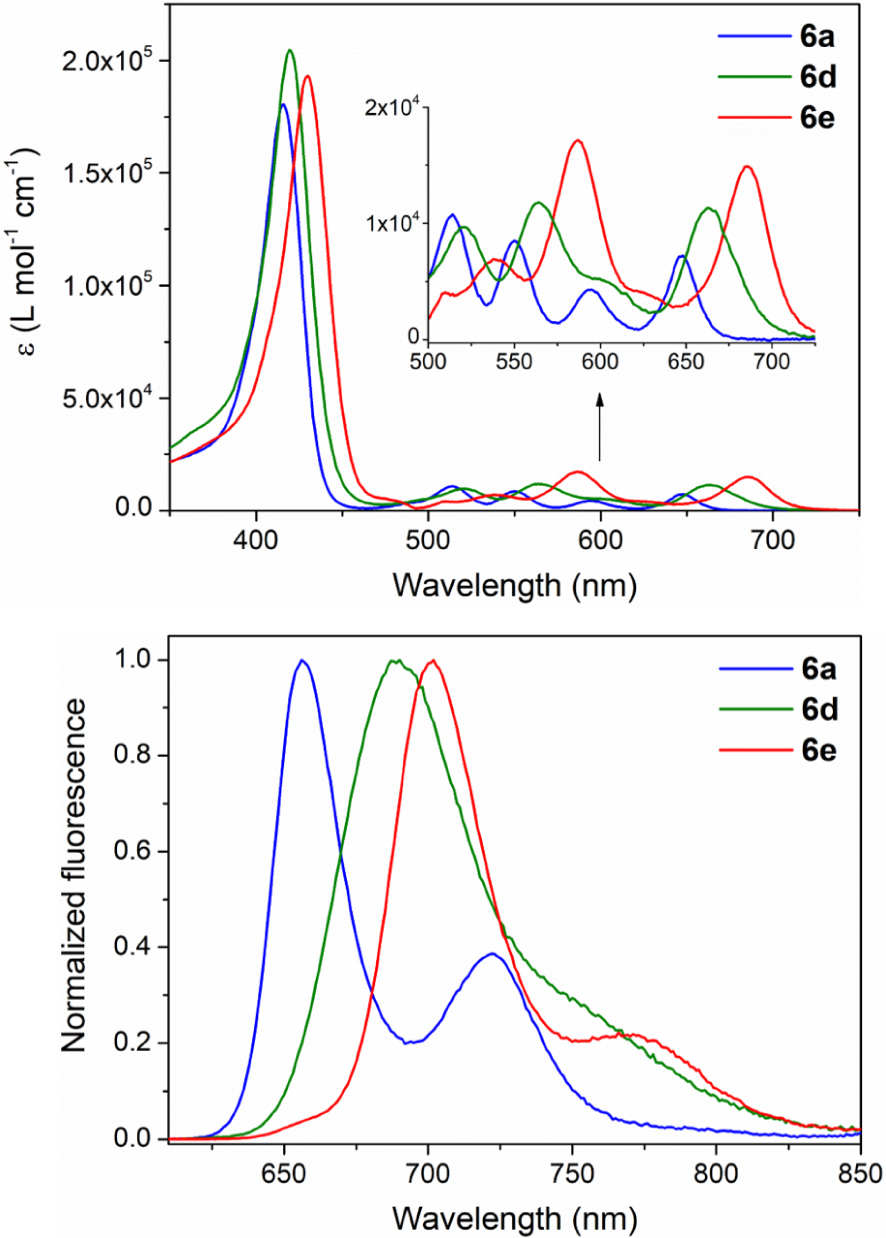
 ), 6d (
), 6d ( ) and 6e (
) and 6e ( ) in aerated dichloromethane at room temperature.
) in aerated dichloromethane at room temperature.
UV–Vis and fluorescence data of porphyrins 6a–e, 7, 8, 9–12, (CF3)4PH2 and (C3F7)4PH2 recorded in aerated dichloromethane at 298 K
| Absorption | Fluorescence | |||||
|---|---|---|---|---|---|---|
| R10,20 | R5,15 | Soret band | Q bands | (nm) | ||
| 𝜆max (nm) (ε (104 M−1⋅cm−1)) | 𝜆max (nm) (ε (104 M−1⋅cm−1)) | |||||
| 6a | 4-OCH3–C6H4 | CF3 | 416 (18.05) fwhm (28)c | 514 (1.07), 550 (0.85), 595 (0.43), 648 (0.72) | 658, 722 | 0.063 |
| 6b | 4-OCH3–C6H4 | C3F7 | 414 (18.90) fwhm (28)c | 514 (0.99), 551 (1.14), 595 (0.46), 646 (1.07) | 656, 719 | 0.086 |
| 6c | 4-OCH3–C6H4 | C7F15 | 415 (19.81) fwhm (27)c | 514 (1.04), 552 (1.27), 596 (0.51), 647 (1.06) | 655, 720 | 0.089 |
| 6d | 4-OCH3–C6H4 | CFO | 419 (20.46) fwhm (31)c | 521 (0.97), 564 (1.18), 663 (1.13) | 690 | 0.100 |
| 6e | 4-OCH3–C6H4 | CHO | 430 (19.32) fwhm (31)c | 510 (0.40), 538 (0.69), 587 (1.71), 686 (1.49) | 707, 771 | 0.079 |
| 7d | 4-CN–C6H4 | CHO | 427 (10.30) fwhm (28)c | 508 (0.23), 535 (0.38), 579 (0.85), 680 (0.75) | 688, 757 | 0.057 |
| 8d | 4-Cl–C6H4 | CHO | 427 (11.69) fwhm (30)c | 535 (0.36), 583 (1.03), 682 (0.91) | 692, 763 | 0.077 |
| 9d | 4-Me3C–C6H4 | CHO | 427 (20.64) fwhm (30)c | 539 (0.62), 585 (1.75), 684 (1.54) | 696, 767 | 0.081 |
| 10e | 4-PO3Et2–C6H4 | C3F7 | 410 (19.1) | 510 (1.1), 546 (1.2), 590 (0.5), 643 (1.1) | 647, 715 | |
| 11f | C6H5 | CF3 | 414 (16.50) fwhm (27)c | 511 (1.01), 546 (1.01), 592 (0.53), 647 (0.77) | 650, 720f | 0.044 |
| 12g | C6H5 | CHO | 426 (15.85) | 535 (0.50), 586 (1.26), 685 (1.00) | ||
| (CF3)4PH | CF3 | CF3 | 408 (9.55) | 512 (0.87), 546 (0.88), 596 (0.43), 651 (0.96) | 654, 721 | 0.016 |
| (C3F7)4PH | C3F7 | C3F7 | 409 (9.23) | 513 (0.92), 547 (0.75), 596 (0.43), 649 (0.97) | 651, 718 | 0.021 |
| TPPi | C6H5 | C6H5 | 419 (47.00) | 514 (1.87), 549 (0.77), 591 (0.54), 647 (0.34) | 650, 715j | 0.15j |
a𝜆max for the bands derived from corrected emission spectra. bLuminescence quantum yields in air-equilibrated dichloromethane by comparing corrected emission spectra and using tetraphenylporphyrin (TPP) in aerated acetonitrile as a standard (𝛷fl = 0.15 [54]). Excitation at 𝜆 = 590 nm. cFull width of half maximum in nm. dA protonation–deprotonation sequence was used to ensure a complete dissolution. eFrom Ref. [17], measured in CHCl3/MeOH. fFrom Ref. [19], 𝜆abs and 𝛷fl measured in toluene, 𝜆em measured in CH2Cl2/EtOH (3:1). gFrom Ref. [32], measured in CHCl3. hFrom Ref. [53], measured in benzene. iFrom Ref. [19], measured in benzene. jRecorded in acetonitrile.
(a) Bond distances (Å), angle values (°) and external dihedral angles 𝛼 and 𝛽 of the optimized structure of 6a; views of the single X-ray structures of (b) 6a and (c) 6b (solvent molecules are omitted for the sake of clarity).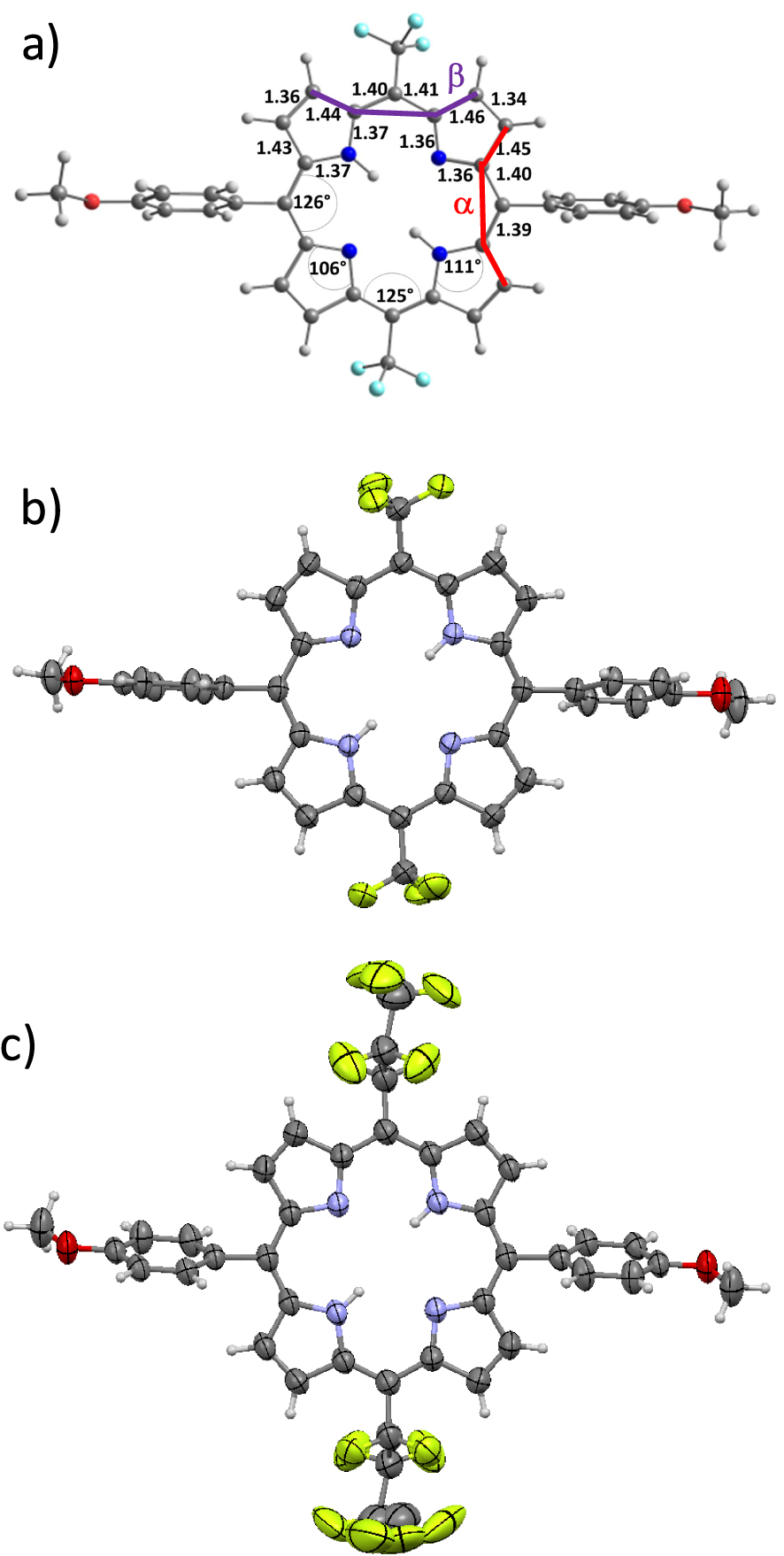
As reported for the meso-bis-CF3 porphyrin 11 [19], porphyrins 6a–c exhibit absorption spectra that are distinct from that of the well known tetra-phenylporphyrin (TPP). Compared to TPP, their Soret bands are blue-shifted and broadened (fwhm 27–28 nm) but significantly red-shifted compared to those of the tetra-perfluoroalkyl derivatives (CF3)4PH2 and (C3F7)4PH2.
In the same way, as shown for 11, the Q bands of 6a–c have absorption maxima close to those of TPP but with a different pattern. For example, the UV–visible spectra of the trans-A2B26a–c feature, as 10 and 11, a noticeable increased intensity for the 0–0 component corresponding to a band at ca. 646 nm (Table 4). The length of the perfluoro alkyl chain has only a minor impact on the light-absorption and light-emission properties of 6a–c that give similar fluorescence spectra and fluorescence quantum yields of ca. 7–9%. When comparing on the one hand the analogous 6a and 11, and on the other hand the analogous dyes 6b and 10, it can be noted that enhancing the electron richness of the aromatic substituents induces a little red shift of the absorption and fluorescence maxima.
As observed when recording the electrochemical data, introducing meso-formyl groups produces a noticeable lower HOMO–LUMO optical gap. The UV–visible absorption and fluorescence bands of 6e are importantly red-shifted. For example, the Soret band maximum of 6e is at 430 nm and the 0–0 component of its Q bands can be found at 686 nm. Its quantum yield is as for 6a–c of ca. 8%. Replacing electron-withdrawing aryl groups in 7 and 8 by electron-donating ones in 9 and 6e produces a supplementary red shift of the absorption and emission bands.
For the unique 6d-bearing meso-fluoro acyl moieties, it can be noted that its light-absorption and emission bands are also red-shifted compared to those of 6a–c and of TPP but with a lower amplitude. Its fluorescence spectrum has also a unique pattern with a single and broadened band at 690 nm corresponding to a quantum yield of 10%.
2.4. Theoretical and structural studies of selected porphyrins
DFT and Time-dependent DFT (TD-DFT) calculations were performed to analyze the effects of the meso-substituents CF3, C3F7, C7F15, CFO and CHO on the structural, electronic and optical properties of porphyrins 6a–e. The structures of 6a–e were fully optimized and gave values of geometrical parameters such as bond distances, angles and dihedral angles that are illustrative of the effect of the meso substitutions on their core structures (Figure 3 and Figure S3 in the Supporting Information). Although there were no symmetry constraints in the calculation procedures, results did provide symmetric structures. Therefore, only parameters corresponding to one-half of the porphyrin macrocycles are detailed in Figures 3 and S3. Very slight differences are observed between the bond distances and angle values of 6a–e despite the variation of the nature of 5,15 meso-substituents (Figures 3 and S3). In fact, most relevant differences in the geometrical parameters are related to the values of the calculated external dihedral angles 𝛼 and 𝛽 which are listed in Table 5 and represented in Figure 3. These dihedral angles stand for the plane torsion regarding two adjacent pyrrole units of the porphyrin core center. As observed in Table 5, the 𝛼 values are about 5.0° for 6a-bearing CF3 moieties and increase up to 10.6° for 6b and 6c substituted respectively by the more bulky C3F7 and C7F15 substituents. The 𝛽 values are quite low with values in the range of ∼0.7°. On the other hand, the presence of CFO and CHO as highly acceptor moieties in 6d and 6e induces decreased 𝛼 values of 7.8° and 7.2°, respectively, while significantly increased 𝛽 values of 12.4° (6d) and 8.5° (6e) are observed for these systems. These results indicate that the introduction of CHO or CFO groups on the meso-positions produce a noticeable supplementary out-of-plane distortion of the macrocycles in 6d and 6e compared to those of the alkyl appended porphyrins 6a–c.
Calculated dihedral angles (°), experimental dihedral angles (°) and orbital energies (eV) of porphyrins 6a–e
| R5,15 | α (°) | β (°) | HOMO−1 (eV) | HOMO (eV) | LUMO (eV) | LUMO+1 (eV) | |
|---|---|---|---|---|---|---|---|
| 6a | CF3 | 4.3 3.3a | 0.7 6.4a | − 5.36 | − 5.25 | − 3.47 | − 3.16 |
| 6b | C3F7 | 10.6 13.4a | 0.7 6.5a | − 5.37 | − 5.23 | − 3.49 | − 3.16 |
| 6c | C7F15 | 10.6 | 0.6 | − 5.38 | − 5.24 | − 3.50 | − 3.16 |
| 6d | CFO | 7.8 | 12.4 | − 5.44 | − 5.27 | − 3.69 | − 3.23 |
| 6e | CHO | 7.2 | 8.5 | − 5.40 | − 5.26 | − 3.78 | − 3.18 |
aExperimental dihedral angle values from the single crystal X-ray diffraction structures of 6a and 6b.
During the final editing stages of this contribution, we were glad to get diffracting single crystals of porphyrins 6a and 6b (Figures 3(b) and (c)). As for the calculated geometries, the corresponding experimental structures feature a small out-of-plane distortion of the porphyrin 6a and 6b which is higher for 6b and correspond to a slight ruffling of the macrocycles.
The frontier molecular orbitals (FMOs) of 6a–e were also studied by DFT calculations. The energies of the two HOMOs and of the two LUMOs are listed in Table 5 and their plots are displayed in Figure 4 and Figure S4 in the Supporting Information. The calculated HOMO–LUMO gaps are in accordance with the experimental data and decrease in importance when fluoro acyl or formyl groups are borne by the 5 and 15 meso-positions. As observed during the UV–Vis absorption and electrochemical experiments, the lowest HOMO–LUMO gap is obtained for the bis-formyl derivative 6e. For 6a–e, the HOMO appears mainly to be located at the p-anisyl substituents, the core-N atoms and the meso-bridging carbon atoms at the 5,15 positions. Therefore, the lack of any apparent contribution from the 5,15 substituents results in similar HOMO energy values differing by less than 1% for the 6a–e series. Similarly, the five HOMO-1 and the five LUMO+1 of 6a–e have energy values and electron density distributions that are only slightly varying with the nature of the 5,15 meso-substituents.
Plots of the calculated frontier orbitals and values of the HOMO–LUMO gaps (in eV) of 6a, 6d and 6e.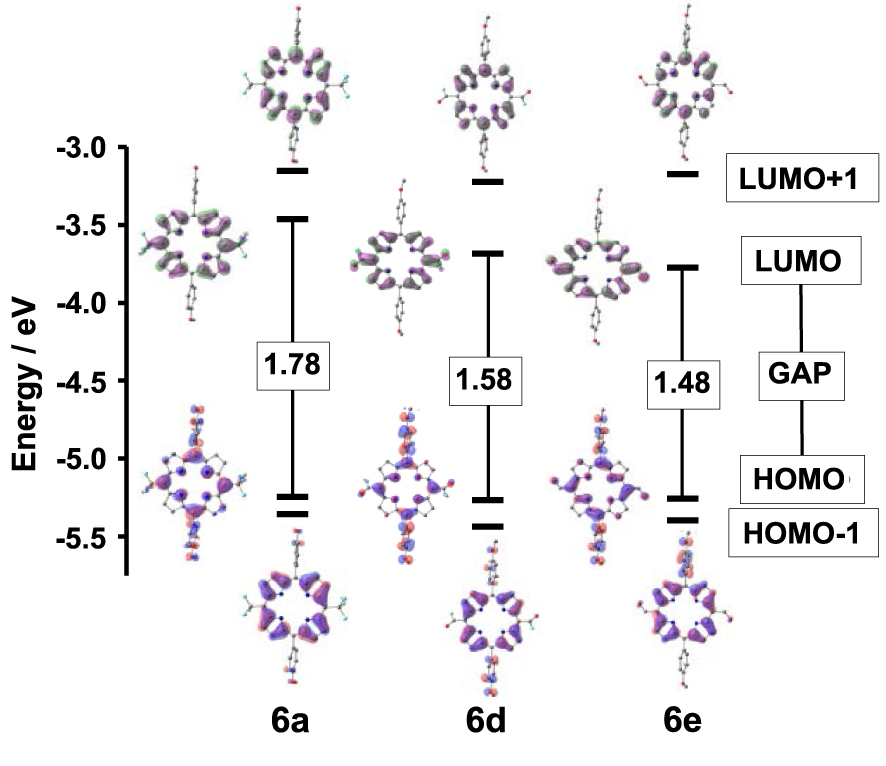
On the contrary, substantial differences appear when comparing the energies of the LUMOs. Those of 6a–c have electron density distributions barely affected by the length of their perfluoroalkyl chains. Consequently, 6a, 6b and 6c have quite identical FMOs energies and HOMO–LUMO gap values in accordance with the corresponding photophysical and electrochemical data. The LUMO iso-surfaces of 6d and 6e feature a remarkable participation of the π-conjugated meso-electron-withdrawing groups CFO and CHO that are responsible for the concomitant lower LUMO energies and lower HOMO–LUMO gaps. The simulation of the UV–visible spectra of 6a–e was obtained from further TD-DFT calculations and is detailed in the Supporting Information together with the corresponding compositions, vertical excitation energies, oscillator strengths and hole–electron excited states’ surface distributions. For 6a–e, theory gives Soret bands between 380 and 450 nm and these are red-shifted for 6d and 6e as observed experimentally. On the contrary, few differences are obtained when comparing the locations and intensities of the calculated Q bands of 6a–e, because the different 5,15 meso substituents have only a slight impact on the corresponding electronic transitions.
3. Conclusions
Meso-polyhalogeno alkyl dipyrromethanes were prepared by the reduction of 2-acyl pyrroles and their subsequent acid-catalyzed reaction with pyrrole. Three sets of experimental conditions were optimized depending on the starting compound. This procedure lead firstly to the known DPMs 3a and 3b substituted by a trifluoromethyl group and a heptafluropropyl chain, respectively. New derivatives were also obtained, bearing on the meso-position a perfluoroheptyl (3c), a chlorodifluoromethyl (3d), a dichloromethyl (3e) or a trichloromethyl group (3f). Dipyrromethanes are useful compounds in the synthesis of various chromophores including BODIPYs, corroles or expanded porphyrins. Herein, they were used to build trans-A2B2 meso-substituted porphyrins through their condensation with aromatic aldehydes. The perfluoro DPMs produce the expected bis-alkylporphyrins while exciting meso-functionalized analogs were obtained from DPMs 3d and 3e-bearing chlorine atoms. Indeed, several bis-formyl porphyrins were prepared directly from 3e and illustrate an original and straightforward strategy to get such derivatives. Similarly, the use of 3d leads to 6d as a unique example of a porphyrin-bearing meso-fluoro acyl moieties. The electron-withdrawing character of the perfluoralkyl chains and of the π-conjugated formyl and fluoro acyl groups were investigated and enlightened thanks to photophysical and electrochemical analyses supported by theoretical calculations. These studies revealed, for example, that when appended by porphyrins, formyl and fluoro acyl functional groups give to the macrocycles reduced HOMO–LUMO electrochemical gaps and red-shifted absorption and emission properties.
4. Experimental section
4.1. Materials
All reagents were used as received. Pyrrole was distilled before use. The distillation of THF was performed on sodium/benzophenone. Flash column chromatography was performed on silica gel 60 (230–400 mesh). Visualization of DPM 3a–f TLC spots was achieved by staining the TLC plates with a solution of phosphomolybdic acid (2.5 g) in ethanol and by subsequent heating.
4.2. Physical measurements
1H, 13C and 19F nuclear magnetic resonance (NMR) spectra were recorded on a JEOL ECS400 NMR spectrometer at room temperature. NMR chemical shifts are given in ppm (𝛿) relative to Me4Si using solvent residual peaks as internal standards (CDCl3: 𝛿 = 7.26 ppm for 1H and 77.2 for 13C; Acetone-d6: 𝛿 = 2.05 ppm for 1H and 29.8 for 13C; DMSO-d6: 𝛿 = 2.50 ppm for 1H and 39.5 for 13C). IR spectra were recorded on an Agilent Cary 630 FTIR equipped with an attenuated total reflectance (ATR) sampling. Melting points (M.P.) were measured in open capillary tubes with a STUART SMP30 melting points apparatus and are uncorrected. High resolution mass spectrometry (HRMS-ESI) analyses were performed on a QStar Elite (Applied Biosystems SCIEX) spectrometer or on a SYNAPT G2 HDMS (Waters) spectrometer by the “Spectropole” of Aix-Marseille University. These two instruments are equipped with an electrospray ionization (ESI) or a MALDI source and a TOF analyser.
4.3. Electronic absorption and fluorescence
UV–Vis absorption spectra were recorded in spectrophotometric grade solvents (ca. 10−6 M) on a Varian Cary 50 SCAN spectrophotometer. Ten equivalents of TFA were added to the suspension of the porphyrins 7, 8 and 9 in order to ensure their complete dissolution as protonated and non-aggregated dyes. A subsequent addition of twelve equivalents of iPr2NEt afforded back the non-protonated free bases soluble in solution. Emission spectra were obtained using a Horiba-Jobin Yvon Fluorolog-3 spectrofluorimeter equipped with a three-slit double-grating excitation and a spectrograph emission mono-chromator with dispersions of 2.1 nm⋅mm−1 (1200 grooves per mm). A 450 W xenon continuous wave lamp provided excitation. Fluorescence of diluted solutions was detected at right angle using 10 mm quartz cuvettes. Fluorescence quantum yields 𝛷 were measured in diluted dichloromethane solutions with an optical density lower than 0.1 using the following equation:
4.4. Electrochemistry
Cyclic voltammetric (CV) data were acquired using a BAS 100 Potentiostat (Bioanalytical Systems) and a PC computer containing BAS100W software (v2.3). A three-electrode system with a Pt working electrode (diameter 1.6 mm), a Pt counter electrode and a leak-free Ag/AgCl reference electrode (diameter 5 mm) was used. [nBu4N]PF6 (0.1 M in CH2Cl2) served as an inert electrolyte while the concentration of the electro-active compound is of ca. 5 × 10−4 M. Cyclic voltammograms were recorded at a scan rate of 100 mV⋅s−1. Ferrocene (0.46 V/SCE) was used as internal standard [55]. Solutions were degassed using argon and the working electrode surface (Pt) was polished before each scan recording. The solubility of porphyrins 7 and 8 in the solvent is too low to get accurate electrochemical values.
4.5. Computational details
All (TD-)DFT calculations have been performed using the Gaussian 16 program [56]. The geometry optimizations were carried out without symmetry constraints to ensure the minima energy. Calculations were performed using the CAM-B3LYP exchange functional (since this level of theory have been widely used in this type of compounds and also because this functional reproduces correctly the corresponding geometrical parameters) and the 6-311+G(d,p) basis set for all atoms [57,58]. Frequency calculations were also included to corroborate the optimized structures, showing only positive values in all vibrational modes. TD-DFT calculations were performed for the first 80 vertical excitations, in vacuum and in dichloromethane (DCM) as solvent, using the conductor-like polarizable continuum model (CPCM) [59]. Here, the BLYP functional was implemented in conjunction with the 6-311+G(d,p) basis set for all atoms. The BLYP functional was chosen because this level of theory accurately reproduces the experimental UV–Vis results [60].
4.6. Single crystal X-ray diffraction
Suitable diffracting single crystals of 6a and 6b were obtained by slow diffusion of n-pentane into concentrated solutions of the porphyrins in dichloromethane. The intensity data for compound 11a were collected on a Rigaku Oxford Diffraction SuperNova diffractometer using CuKα radiation (𝜆 = 1.54184 Å) at 293 K. Data collection, cell refinement and data reduction were performed with CrysAlisPro (Rigaku Oxford diffraction). Using Olex2 [61], the structures were solved with shelXT [62] and shelXL [62] was used for full matrix least squares refinement. CCDC-2074306 (6a) and CCDC-2074307 (6b) contain the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request:cif.
4.7. Synthetic methods
The 2-acylpyrroles 5a (R = CF3) [63], 5b (R = C3F7) [26], 5c (R = C7F15) [64], 5f (R = CCl3) [65] and 5e (R = CHCl2) [66] were prepared as previously reported.
4.7.1. 2-(Chlorodifluoroacetyl)pyrrole (5d)
This compound was prepared following a protocol adapted from the synthesis of 2-(trifluoroacetyl)pyrrole 5a [63]. A solution of chlorodifluoroacetic anhydride (4.15 mL, 23 mmol, 1.1 equiv.) in anhydrous CH2Cl2 (15 mL) was cooled to −15 °C under an argon atmosphere. A solution of pyrrole (1.44 mL, 20.9 mmol, 1 equiv.) in anhydrous CH2Cl2 (15 mL) was then added dropwise under firm stirring. The mixture was stirred at −15 °C for 1.5 h, then at room temperature during an additional hour. The organic phase was washed with water, dried over Na2SO4, and the solvent removed by evaporation. The resulting residue was purified by flash chromatography (CH2Cl2/petroleum ether 1:1) to afford 5d as a white solid (4.01 g, 22 mmol, 99%).
RF = 0.45 (silica, CH2Cl2/petroleum ether 1:1). Mp: 41.9 °C; 1H NMR (CDCl3) 𝛿 = 10.27 (br s, 1H, NH), 7.28 (m, 1H, α-H), 7.25 (m, 1H, β-H), 6.41 (m, 1H, β-H) ppm; 19F NMR (CDCl3) 𝛿 = −61.5 ppm; 13C {1H} NMR (CDCl3) 𝛿 = 172.2 (t, 2 JC,F = 30.5, C), 129.6 (CH), 124.7 (C), 122.1 (t, 4 JC−−F = 4.7, CH), 120.5 (t, 1 JC,F = 302.5, C), 112.7 (CH) ppm; IR:v∼ = 3329 (vs, N–H), 1638 (vs, C =O) cm−1; HMRS-ESI: calcd. for C6H3NOF2Cl [M–H]− 177.9877. Found: 177.9877.
4.7.2. 5-(Trifluoromethyl)dipyrromethane (3a)
Procedure A: to a solution of 2-(trifluoroacetyl)pyrrole 5a (500 mg, 3.07 mmol, 1 equiv.) and NaHCO3 (514 mg, 6.13 mmol, 2 equiv.) suspended in methanol (46 mL) under firm stirring was added NaBH4 (232 mg, 6.12 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was dissolved in Et2O. This organic phase was washed with water, dried over Na2SO4 and evaporated at 30 °C. The resulting residue and pyrrole (0.42 mL, 6.12 mmol, 2 equiv.) were dissolved in CH2Cl2 (40 mL) and under argon atmosphere. P2O5 (87 mg, 6.13 mmol, 2 equiv.) was then added and the mixture was stirred at room temperature for 16 h. A saturated aqueous NaHCO3 solution (10 mL) was added, and the stirring maintained for 1 h. The crude product was filtered and washed with DCM on a Büchner funnel equipped with sintered glass. The filtrate was dried on Na2SO4. The subsequent flash chromatography (CH2Cl2/petroleum ether 1:1, then 2:1) afforded 3a as a white solid (395 mg, 1.8 mmol, 60%).
Procedure B: to a solution of 2-(trifluororoacetyl)pyrrole 5a (250 mg, 1.53 mmol, 1 equiv.) and NaHCO3 (257 mg, 3.06 mmol, 2 equiv.) suspended in methanol (4.6 mL) under firm stirring was added NaBH4 (116 mg, 3.06 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was extracted with Et2O, then washed with water and dried over Na2SO4. The organic phase was evaporated at 30 °C then dissolved with pyrrole (0.21 mL, 3.06 mmol, 2 equiv.), in an aqueous HCl solution (0.18 M, 5 mL). The mixture was stirred at room temperature for 16 h. The mixture was then extracted with CH2Cl2. This organic phase was washed with water, dried on Na2SO4 before removal of the solvent by evaporation. The residue was finally purified by flash chromatography (CH2Cl2/petroleum ether 2:1) to afford 3a as a gray solid (79 mg, 0.37 mmol, 24%). Scale up: starting from 2-(trifluororoacetyl)pyrrole 5a (2.5 g, 15.3 mmol, 1 equiv.), 3a was obtained as a gray solid (938 mg, 4.4 mmol, 28%).
RF = 0.5 (CH2Cl2/petroleum ether 2:1). 1H NMR (CDCl3) 𝛿 = 8.10 (br s, 2H, NH), 6.77 (m, 2H, α-H), 6.25 (m, 2H, β-H), 6.22 (m, 2H, β-H), 4.85 (q, 3 JH,F = 9.0, 1H, meso-H) ppm; IR:v∼ = 3366, 3346 (vs, N–H), 1242, 1161, 1119, 1092, 1043 (s, C–F) cm−1. Other analytical data are consistent with literature values [13].
4.7.3. 5-(Perfluoropropyl)dipyrromethane (3b)
Procedure A: scale up: to a solution of 2-(perfluorobutyryl)pyrrole 5b (4.18 g, 15.9 mmol, 1 equiv.) and NaHCO3 (2.67 g, 31.8 mmol, 2 equiv.) suspended in methanol (50 mL) under firm stirring was added NaBH4 (1.20 g, 31.8 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was dissolved in Et2O. The organic phase was washed with water, dried over Na2SO4 and was evaporated at 30 °C. The resulting residue and pyrrole (2.21 mL, 31.8 mmol, 2 equiv.) were dissolved in CH2Cl2 (200 mL) under argon. P2O5 (4.51 g, 31.8 mmol, 1 equiv.) was then added and the mixture was stirred at room temperature for 16 h. NaHCO3 (3.47 g, 41.3 mmol, 2.6 equiv.) was added and the stirring maintained for 1 h. The mixture was filtrated and washed with DCM on a Büchner funnel equipped with sintered glass. The filtrate was dried on Na2SO4 before removal of the solvent by evaporation. The residue was finally purified by flash chromatography (CH2Cl2/petroleum ether 2:1) to afford 3b as a white solid (2.05 g, 6.5 mmol, 41%).
RF = 0.65 (silica, CH2Cl2/petroleum ether 2:1). 1H NMR (CDCl3) 𝛿 = 8.15 (br s, 2H, NH), 6.78 (m, 2H, α-H), 6.25 (m, 2H, β-H), 6.20 (m, 2H, β-H), 4.94 (t, 3 JH,F = 16.6, 1H, meso-H) ppm; IR:v∼ = 3368 (s, N–H), 1233, 1207, 1175, 1113, 1093 (s, C–F) cm−1. Other analytical data are consistent with literature values [13].
4.7.4. 5-(Perfluoroheptyl)dipyrromethane (3c)
Procedure A: scale up: to a solution of 2-(perfluoro-octanoyl)pyrrole 5c (2.01 g, 4.31 mmol, 1 equiv.) and NaHCO3 (722 mg, 8.60 mmol, 2 equiv.) suspended in methanol (16 mL) under firm stirring was added NaBH4 (325 mg, 8.59 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was dissolved in Et2O. The organic phase was washed with water, dried over Na2SO4 and was evaporated at 30 °C. The resulting residue and pyrrole (0.6 mL, 8.62 mmol, 2 equiv.) were dissolved in CH2Cl2 (65 mL) under argon. P2O5 (1.22 g, 8.65 mmol, 2 equiv.) was then added and the mixture was stirred at room temperature for 16 h. NaHCO3 (940 mg, 11.2 mmol, 2.6 equiv.) was added and the stirring maintained for 1 h. The crude product was filtrated and washed by DCM on a Büchner funnel equipped with sintered glass. The filtrate was dried on Na2SO4 before removal of the solvent by evaporation. The residue was finally purified by flash chromatography (CH2Cl2/petroleum ether) to afford 3c as a light purple solid (1.92 g, 3.73 mmol, 87%).
RF = 0.60 (silica, CH2Cl2/petroleum ether). 1H NMR (CDCl3) 𝛿 = 8.15 (br s, 2H, NH), 6.78 (m, 2H, α-H), 6.25 (m, H, β-H), 6.20 (m, 2H, β-H), 4.96 (t, 3 JH,F = 16.8, 2H, meso-H) ppm; 19F NMR (CDCl3) 𝛿 = −80.7 (CF3), − 112.2, − 120.5, − 121.5, − 121.9, − 122.6, − 126.0 ppm; 13C {1H} NMR (CDCl3) 𝛿 = 122.3 (C), 119.0 (CH), 109.7 (CH), 109.1 (CH), 108.3 (C), 41.2 (t, 2 JC−−F = 23.0, C) ppm, other carbon atoms could not be assigned due the low intensity of the perfluorinated chain peaks [67] and because of the rapid degradation of the compound in solution; IR:v∼ = 3405 (br, N–H), 1235, 1198, 1143, 1095 (vs, C–F) cm−1; HRMS-ESI: calcd for C16H8F15N2 [M–H]−: 513.0453. Found: 513.0461.
4.7.5. 5-(Chlorodifluoromethyl)dipyrromethane (3d)
Procedure B: to a solution of 2-(chlorodifluoroacetyl)pyrrole 5d (100 mg, 0.56 mmol, 1 equiv.) and NaHCO3 (94 mg, 1.12 mmol, 2 equiv.) suspended in methanol (1.7 mL) under firm stirring was added NaBH4 (42 mg, 1.1 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was dissolved with Et2O. This organic phase was washed with water, dried over Na2SO4 and evaporated at 30 °C. The resulting residue was then dissolved in pyrrole (0.78 mL, 1.1 mmol, 2 equiv.) before the addition of an aqueous HCl solution (0.18 M, 2 mL). The mixture was stirred at room temperature for 16 h. The mixture was then extracted with CH2Cl2, washed with water, and dried on Na2SO4 before removal of the solvent by evaporation. The subsequent flash chromatography (CH2Cl2/petroleum ether 2:1) afforded 3d as a white solid (23 mg, 0.1 mmol, 18%). Scale up: The same procedure at a larger scale and starting from 5d (2.74 g, 15.3 mmol) afforded 3d as a white solid (589 mg, 2.55 mmol, 18%).
RF = 0.65 (silica, CH2Cl2/petroleum ether 2:1). Mp: 75–80 °C; 1H NMR (CDCl3) 𝛿 = 8.19 (br s, 2H, NH), 6.79 (m, 2H, α-H), 6.27 (m, 2H, β-H), 6.22 (m, 2H, β-H), 4.96 (t, 3 JH,F = 11.1, 1H, meso-H) ppm; 19F NMR (CDCl3) 𝛿 = −53.3 (d, 3 JF,H = 11.1) ppm; 13C {1H} NMR (CDCl3) 𝛿 = 118.7 (CH), 117.8 (C), 109.6 (t, 2 JC,F = 1.4, CH), 109.1 (CH), 108.3 (CH), 49.8 (t, 1 JC,F = 26.0, C) ppm; IR:v∼ = 3351 (vs, N–H) cm−1; HRMS-ESI: calcd. for C10H8ClF2N2 [M–H]−: 229.0350. Found: 229.0351.
4.7.6. 5-(Dichloromethyl)dipyrromethane (3e)
Procedure B: to a solution of 2-(dichloroacetyl)pyrrole 5e (272 mg, 1.53 mmol, 1 equiv.) and NaHCO3 (257 mg, 3.06 mmol, 2 equiv.) suspended in methanol (4.6 mL) under firm stirring was added NaBH4 (116 mg, 3.06 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was dissolved in Et2O. This organic phase was washed with water, dried over Na2SO4 and evaporated at 30 °C. The resulting solid was then dissolved in pyrrole (0.21 mL, 3.06 mmol, 2 equiv.) before the addition of an aqueous HCl solution (0.18 M, 5 mL). The mixture was stirred at room temperature for 16 h. The mixture was then extracted with CH2Cl2. The organic phase was washed with water, dried on Na2SO4 and evaporated to dryness. The final purification by flash chromatography (CH2Cl2/petroleum ether 2:1) afforded 3e as a gray solid (212 mg, 0.9 mmol, 60%). Scale up: the same procedure at a ten-fold larger scale starting from 5e (2.72 g, 15.3 mmol) afforded 3e as a white solid (2.37 g, 10.3 mmol, 67%).
RF = 0.4 (silica, CH2Cl2/petroleum ether 2:1). Mp: > 60 °C (degradation) 1H NMR (CDCl3) 𝛿 = 8.26 (br s, 2H, NH), 6.75 (m, 2H, α-H), 6.25 (m, 2H, β-H), 6.21 (m, 3H, β-H and CHCl2), 4.83 (d, 3 JH,H = 3.5, 1H, meso-H) ppm; 13C {1H} NMR (CDCl3) 𝛿 = 126.8 (C), 118.3 (CH), 108.8 (CH), 108.7 (CH), 74.9 (CH), 49.0 (CH) ppm; IR:v∼ = 3344 (vs, N–H), 742 (vs, C–Cl) cm−1; HRMS-ESI: calcd. for C10H8Cl2N4 [M–H]−: 227.0148. Found: 227.0151.
4.7.7. 5-(Trichloromethyl)dipyrromethane (3f)
Procedure C: to a solution of 2-(trichloroacetyl)pyrrole 5f (1.63 g, 7.7 mmol, 1 equiv.) and NaHCO3 (1.28 g, 15.3 mmol, 2 equiv.) suspended in methanol (23 mL) under firm stirring was added NaBH4 (581 mg, 15.3 mmol, 2 equiv.) by portions. The mixture was stirred at room temperature for 30 min before removal of the solvent by evaporation. The crude product was dissolved in Et2O. This organic phase was washed with water, dried over Na2SO4 and evaporated at 30 °C. The resulting solid and pyrrole (6.0 mL, 86 mmol, 11 equiv.) were dissolved in distilled THF (40 mL) under argon atmosphere before the addition of a solution of HCl in Et2O (1 M, 6.9 mL, 6.9 mmol, 0.9 equiv.). The mixture was stirred at 85 °C for 2 h, quenched with a saturated aqueous NaHCO3 solution. The mixture was extracted by diethyl ether. The organic phase was washed with water, dried over Na2SO4 and evaporated to dryness. The final purification by flash chromatography (CH2Cl2/petroleum ether 2:1) afforded 3f as a white solid (181 mg, 0.68 mmol, 9%).
RF = 0.45 (silica, CH2Cl2/petroleum ether 2:1). Mp: > 70 °C (decomposition); 1H NMR (CDCl3) 𝛿 = 8.38 (br s, 2H, NH), 6.78 (s, 2H, α-H), 6.41 (s, 2H, β-H), 6.23 (s, 2H, β-H), 5.16 (s, 1H, meso-H) ppm; 13C {1H} NMR (CDCl3) 𝛿 = 126.3 (C), 118.3 (CH), 110.2 (CH), 108.8 (CH), 59.2 (CH), 31.0 (C) ppm; IR: v∼ = 3340 (s, N–H), 741 (vs, C–Cl) cm−1; HMRS-ESI: calcd. for C10H10Cl3N2 [M+H]+: 262.9904. Found: 262.9899.
4.7.8. 5,15-Bis(trifluoromethyl)-10,20-bis-(4-methoxyphenyl)porphyrin (6a)
Under argon, to a solution of 5-(trifluoromethyl)dipyrromethane 3a (643 mg, 3 mmol, 1 equiv.) and 4-methoxybenzaldehyde (0.36 mL, 3 mmol, 1 equiv.) in CH2Cl2 (300 mL) was added BF3⋅OEt2 (2.5 M, 0.4 mL, 1 mmol, 0.33 equiv.). The mixture was stirred at room temperature for 4 h before DDQ (1.01 g, 4.5 mmol, 1.5 equiv.) was added and the stirring maintained for 10 min. The mixture was passed over a short silica gel plug (CH2Cl2/petroleum ether 1:2). The following purification by flash chromatography (CH2Cl2/petroleum ether 1:4) afforded 6a as a dark purple solid (88 mg, 0.13 mmol, 9%).
RF = 0.15 (silica, CH2Cl2/petroleum ether 1:4). 1H NMR (CDCl3) 𝛿 = 9.59 (m, 4H, β-H), 8.93 (d, 3 JH,H = 5.0, 4H, β-H), 8.06 (d, 3 JH,H = 8.4, 4H, Ar–H), 7.31 (d, 3 JH,H = 8.4, 4H, Ar–H), 4.13 (s, 6H, CH3), − 2.64 (br s, 2H, NH) ppm; 19F NMR (CDCl3) 𝛿 = −37.0, −45.5 ppm; 13C {1H} NMR (CDCl3) 𝛿 = 160.0 (C), 135.6 (CH), 134.3 (C), 133.8 (CH), 130.0 (CH), 128.1 (d, 1 JC,F = 275, C), 122.0 (C), 112.5 (CH), 105.5 (d, 2 JC,F = 30.5, C), 55.8 (CH), 53.6 (CH), 29.9 (CH3) ppm; IR:v∼ = 3299.2 (w, N–H), 2918.7, 2843.1 (m, C𝛽-H) cm−1; UV–Vis (CH2Cl2): 𝜆max(𝜀) = 416.0 (180 530), 514.1 (10 730), 550.0 (8 490), 594.9 (4 300), 648.0 nm (7220 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 656, 722 nm; HRMS-ESI: calcd. for C36H25F6N4O2 [M+H]+: 659.1876. Found: 659.1877.
4.7.9. 5,15-Bis(perfluoropropyl)-10,20-bis-(4-methoxyphenyl)porphyrin (6b)
A solution of 5-(perfluoropropyl)dipyrromethane 3b (471 mg, 1.5 mmol, 1 equiv.) and 4-methoxybenzaldehyde (0.18 mL, 1.5 mmol, 1 equiv.) in CH2Cl2 (300 mL) was stirred under argon before the addition of TFA (1.16 mL, 15.1 mmol, 10 equiv.). The mixture was stirred at room temperature for 4 h before DDQ (570 mg, 2.25 mmol, 1.5 equiv.) was added. The resulting mixture was stirred for 16 h before being passed over a short silica gel plug (CH2Cl2/petroleum ether 1:2). The following purification by flash chromatography (CH2Cl2/petroleum ether 1:4) afforded 6b as a dark purple solid (29 mg, 0.03 mmol, 4.5%).
RF = 0.25 (silica, CH2Cl2/petroleum ether 1:4). 1H NMR (CDCl3) 𝛿 = 9.45 (br s, 4H, β-H), 8.94 (d, 3 JH,H = 5.1, 4H, β-H), 8.07 (d, 3 JH,H = 7.1, 4H, Ar–H), 7.31 (d, 3 JH,H = 8.6, 4H, Ar–H), 4.13 (s, 6H, CH3), − 2.53 (br s, 2H, NH) ppm; 19F NMR (CDCl3) 𝛿 = − 76.1 (CF3), − 81.8, − 119.9 ppm; 13C {1H} NMR spectra could not be obtained due to the poor solubility in deuterated solvents; IR:v∼ = 3271.6 (w, N–H) cm−1; UV–Vis (CH2Cl2): 𝜆max(𝜀) = 414 (189,000), 514 (9910), 551 (11,430), 595 (4640), 646 nm (10,655 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 656, 719 nm; HRMS-ESI: calcd. for C48H23F30N4O2 [M–H]−: 857.1603. Found: 857.1602.
4.7.10. 5,15-Bis(perfluoroheptyl)-10,20-bis-(4-methoxyphenyl)porphyrin (6c)
A solution of 5-(perfluoroheptyl)dipyrromethane 3c (771 mg, 1.5 mmol, 1 equiv.) and 4-methoxybenzaldehyde (0.18 mL, 1.5 mmol, 1 equiv.) in CH2Cl2 (300 mL) was stirred under argon before the addition of TFA (1.16 mL, 15.1 mmol, 10 equiv.). The mixture was stirred at room temperature for 4 h before DDQ (570 mg, 2.25 mmol, 1.5 equiv.) was added and the stirring maintained for 16 h. The mixture was passed over a short silica gel plug (CH2Cl2/petroleum ether 1:2). The following purification by flash chromatography (CH2Cl2/petroleum ether 1:4) afforded 6c as a dark purple solid (37 mg, 0.03 mmol, 4%).
RF = 0.4 (silica, CH2Cl2/petroleum ether 1:4); 1H NMR (CDCl3) 𝛿 = 9.45 (br s, 4H, β-H), 8.94 (d, 3 JH,H = 5.1, 4H, β-H), 8.07 (br s, 4H, Ar–H), 7.31 (d, 3 JH,H = 8.4, 4H, Ar–H), 4.13 (s, 6H, CH3), − 2.52 (br s, 2H, NH) ppm; 19F NMR (CDCl3) 𝛿 = − 80.5 (CF3), − 81.1, − 115.1, − 120.7, − 121.4, − 122.3, − 125.9 ppm; 13C {1H} NMR spectra could not be obtained due to the poor solubility in deuterated solvents; IR:v∼ = 2958, 2922, 2854 (s, C𝛽-H) cm−1; UV–Vis (CH2Cl2): 𝜆max(𝜀) = 415 (198,140), 514 (10,420), 552 (12,680), 596 (5070), 647 nm (10,610 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 655, 720 nm; HRMS-ESI: calcd for C40H23F14N4O2 [M–H]−: 1257.1347. Found: 1257.1349.
4.7.11. 5,15-Bis(fluoroacyl)-10,20-bis-(4-methoxyphenyl)porphyrin (6d)
A solution of 5-(chlorodifluoromethyl)dipyrromethane 3d (345 mg, 1.5 mmol, 1 equiv.) and 4-methoxybenzaldehyde (0.18 mL, 1.5 mmol, 1 equiv.) in CH2Cl2 (150 mL) was stirred under argon before the addition of BF3⋅O(Et)2 (2.5 M, 0.2 mL, 0.5 mmol, 0.33 equiv.). The mixture was stirred at room temperature for 4 h before DDQ (510 mg, 4.5 mmol, 1.5 equiv.) was added and the stirring maintained for 10 min. The crude product was passed over a short silica gel plug (CH2Cl2). The following purification by flash chromatography (CH2Cl2/petroleum ether 1:1 then 2:1) afforded 6d as a dark purple solid (7 mg, 15 μmol, < 2%).
RF = 0.6 (silica, CH2Cl2/petroleum ether 2:1). 1H NMR (CDCl3) 𝛿 = 9.61 (m, 4H, β-H), 9.01 (d, 3 JH,H = 4.9, 4H, β-H), 8.09 (d, 3 JH,H = 8.5, 4H, Ar–H), 7.34 (d, 3 JH,H = 8.5, 4H, Ar–H), 4.13 (s, 6H, CH3), − 2.68 (br s, 2H, NH) ppm; 19F NMR (CDCl3) 𝛿 = − 67.62 ppm; 13C {1H} NMR spectra could not be obtained due to little quantity; IR: v∼ = 2921, 2853 (vs, C𝛽-H) cm−1; UV–Vis (CH2Cl2): 𝜆max(𝜀) = 419 (204,600), 521 (9650), 564 (11,780), 663 nm (11,290 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 690 nm; HRMS-ESI: calcd. for C36H25N4O4F2 [M+H]+: 615.1838. Found: 615.1838.
4.7.12. 5,15-Bis(formyl)-10,20-bis-(4-methoxyphenyl) porphyrin (6e)
A solution of 5-(dichloromethyl)dipyrromethane 3e (230 mg, 1 mmol, 1 equiv.) and 4-methoxybenzaldehyde (0.12 mL, 1 mmol, 1 equiv.) in CH2Cl2 (200 mL) was stirred under argon before the addition of TFA (0.77 mL, 10 mmol, 10 equiv.). The mixture was stirred at room temperature for 4 h before DDQ (340 mg, 1.5 mmol, 1.5 equiv.) was added and the stirring maintained for 16 h. The mixture was passed over a short silica gel plug (CH2Cl2/AcOEt 9:1). The following purification by flash chromatography (CH2Cl2, then CH2Cl2/AcOEt 2.5%) afforded 6e as a dark purple solid (31 mg, 0.05 mmol, 11%).
RF = 0.6 (silica, CH2Cl2/AcOEt 97.5:2.5). 1H NMR (CDCl3) 𝛿 = 12.53 (s, 2H, CHO), 10.01 (d, 3 JH,H = 4.8, 4H, β-H), 9.01 (d, 3 JH,H = 4.8, 4H, β-H), 8.08 (d, 3 JH,H = 8.5, 4H, Ar–H), 7.33 (d,3 JH,H = 8.5, 4H, Ar–H), 4.13 (s, 6H, CH3), − 2.30 (br s, 2H, NH) ppm; 13C {1H} NMR spectra could not be obtained due to the poor solubility in deuterated solvents; IR: v∼ = 3318 (w, N–H), 2957, 2921, 2852 (m, C𝛽-H), 1672 (vs, C =O) cm−1; UV–Vis (CH2Cl2): 𝜆max(𝜀) = 430 (193,270), 510 (4040), 538 (6900), 587 (17,130), 686 nm (14,880 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 707, 771 nm; HRMS-ESI: calcd. for C36H27N4O4 [M+H]+: 579.2027. Found: 579.2031.
4.7.13. 5,15-Bis(formyl)-10,20-bis-(4-cyanophenyl) porphyrin (7)
A solution of 5-(dichloromethyl)dipyrromethane 3e (345 mg, 1.5 mmol, 1 equiv.) and 4-cyanobenzaldehyde (197 mg, 1.5 mmol, 1 equiv.) in CH2Cl2 (300 mL) was stirred under argon before the addition of TFA (1.15 mL, 15 mmol, 10 equiv.). The mixture was stirred at room temperature for 2 h before DDQ (570 mg, 2.25 mmol, 1.5 equiv.) was added and the stirring maintained for 10 min. The mixture was passed over a short silica gel plug (CH2Cl2/AcOEt 9:1). The following purification by flash chromatography (CH2Cl2, then CH2Cl2/AcOEt 2.5%) afforded 7 as a dark purple solid (39 mg, 0.07 mmol, 9%).
RF = 0.5 (silica, CH2Cl2/AcOEt 97.5:2.5). 1H NMR (DMSO-d6, 85 °C) 𝛿 = 12.56 (s, 2H, CHO), 10.19 (d, 3 JH,H = 5.1, 4H, β-H), 8.95 (d, 3 JH,H = 5.1, 4H, β-H), 8.43 (d, 3 JH,H = 8.0, 4H, Ar–H), 8.31 (d, 3 JH,H = 8.0, 4H, Ar–H), − 2.40 (br s, 2H, NH) ppm; 13C {1H} NMR spectra could not be obtained due to the poor solubility in deuterated solvents; IR:v∼ = 3316 (w, N–H), 1671 (vs, C =O) cm−1; UV–Vis (CH2Cl2): 𝜆max (𝜀) = 427 (102,970), 508 (2250), 535 (3840), 579 (8460), 680 nm (7470 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 688, 757 nm; HRMS-ESI: calcd. for C36H21N6O2 [M+H]+: 569.1721. Found: 569.1716.
4.7.14. 5,15-Bis(formyl)-10,20-bis-(4-chlorophenyl) porphyrin (8)
A solution of 5-(dichloromethyl)dipyrromethane 3e (345 mg, 1.5 mmol, 1 equiv.) and 4-chlorobenzaldehyde (0.21 mL, 1.5 mmol, 1 equiv.) in CH2Cl2 (300 mL) was stirred under argon before the addition of TFA (1.15 mL, 15 mmol, 10 equiv.). The mixture was stirred at room temperature for 2 h before DDQ (570 mg, 2.25 mmol, 1.5 equiv.) was added and the stirring maintained for 10 min. The mixture was passed over a short silica gel plug (CH2Cl2/AcOEt 9:1). The following purification by flash chromatography (CH2Cl2) afforded 8 as a dark purple solid (8 mg, 13 μmol, < 2%).
RF = 0.7 (silica, CH2Cl2). 1H NMR (CDCl3) 𝛿 = 12.54 (s, 2H, CHO), 10.04 (4H, β-H), 8.97 (d, 3 JH,H = 4.9, 4H, β-H), 8.11 (d, 3 JH,H = 7.9, 4H, Ar–H), 7.79 (d, 3 JH,H = 7.9, 4H, Ar–H), − 2.38 (br s, 2H, NH) ppm; 13C {1H} NMR spectra could not be obtained due to the poor solubility in deuterated solvents; IR:v∼ = 1671 (vs, C =O) cm−1; UV–Vis (CH2Cl2): 𝜆max (𝜀) = 427 (116,900), 535 (3630), 583 (10,310), 682 nm (9140 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 692, 763 nm; HRMS-ESI: calcd. for C34H21Cl2N4O2 [M+H]+: 587.1036. Found: 587.1038.
4.7.15. 5,15-Bis(formyl)-10,20-bis-(4-tert-butylphenyl)porphyrin (9)
A solution of 5-(dichloromethyl)dipyrromethane 3e (173 mg, 0.75 mmol, 1 equiv.) and 4-tert-butylbenzaldehyde (0.13 mL, 0.75 mmol, 1 equiv.) in CH2Cl2 (150 mL) was stirred under argon before the addition of TFA (0.58 mL, 7.5 mmol, 10 equiv.). The mixture was stirred at room temperature for 2 h before DDQ (285 mg, 1.13 mmol, 1.5 equiv.) was added and the stirring maintained for 10 min. The mixture was passed over a short silica gel plug (CH2Cl2/AcOEt 9:1). The following purification by flash chromatography (CH2Cl2, then CH2Cl2/AcOEt 2.5%) afforded 9 as a dark purple solid (29 mg, 0.045 mmol, 12%).
RF = 0.8 (silica, CH2Cl2). 1H NMR (CDCl3) 𝛿 = 12.51 (s, 2H, CHO), 9.98 (d, 3 JH,H = 4.8, 4H, β-H), 9.00 (d, 3 JH,H = 4.8, 4H, β-H), 8.09 (d, 3 JH,H = 8.1, 4H, Ar–H), 7.81 (d, 3 JH,H = 8.1, 4H, Ar–H), 1.64 (s, 18H, tertbutyl-H), − 2.37 (br s, 2H, NH) ppm; 13C {1H} NMR spectra could not be obtained due to the poor solubility in deuterated solvents; IR:v∼ = 3302 (vw, N–H), 2962 (w, C𝛽-H), 1671 (vs, C =O) cm−1; UV–Vis (CH2Cl2): 𝜆max (𝜀) = 427 (206,460), 539 (6160), 585 (17,490), 684 nm (15,410 mol−1⋅L⋅cm−1); Fluorescence(CH2Cl2): 𝜆ex = 590 nm, 𝜆em = 696, 767 nm; HRMS-ESI: calcd. for C42H39N4O2 [M+H]+: 631.3068. Found: 631.3068.
1No other porphyrin was detected by TLC or during the chromatographic purification.