

1 Introduction
Les phénomènes d’oxydo-réduction jouent un rôle important dans le métier de verrier et ce, à deux titres, lors de l’élaboration, d’une part, et quant aux propriétés d’usage, d’autre part. Nous commençons par un examen général de l’état d’oxydation du verre et par l’identification des éléments du matériau susceptibles de subir des changements d’état d’oxydo-réduction. Nous examinons ensuite le rôle de l’oxydo-réduction lors de l’élaboration et enfin ce en quoi elle est importante sur le plan des propriétés d’usage du verre et, en particulier, sur la couleur.
2 L’état d’oxydo-réduction du verre
Le verre étant par nature un matériau éminemment variable, on ne peut pas parler d’un état d’oxydo-réduction, mais d’un domaine plus ou moins étendu selon l’usage.
Il convient tout d’abord de s’entendre sur le terme « verre » : nous parlerons ici essentiellement des verres d’oxydes et même, ce qui est encore plus restreint, des verres à base silicatée. À haute température, le « verre » ne présente pas une organisation différente de celle de tout liquide silicaté. Néanmoins, afin d’alléger la présentation, il nous arrivera de parler du bain de « verre » ou même, du « verre fondu ». Bien que ce soit un abus de langage, il est très commun parmi les verriers.
La teneur en oxygène de ces matériaux est évidemment grande, au point que l’expression des compositions chimiques se fait généralement sous forme de pourcentages d’oxydes, le plus souvent exprimés en pourcentages massiques dans l’industrie. Le Tableau 1 présente les compositions chimiques d’un certain nombre de verres.
Compositions chimiques approchées des principaux verres industriels en % massique (certains oxydes mineurs ne sont pas indiqués).
| Verre plat | Verre bouteille | Verre fibre isolation | Verre fibre textile | Verre Pyrex | Verre cristal | SRL 131 déchets nucléaires | |
| SiO2 | 73 | 72 | 68 | 57 | 85 | 67 | 58 |
| Al2O3 | 1 | 2 | 1 | 14 | |||
| CaO | 10 | 12 | 10 | 21 | |||
| Na2O | 15 | 13 | 15 | 1 | 2 | 10 K2O | 18 + 6 Li2O |
| B2O3 | 5 | 8 | 13 | 15 | |||
| PbO | 33 | ||||||
| Fe2O3 | 0,1 | 0,1 | 0,2 | 0,2 |
2.1 Le fer
Les dernières lignes du Tableau 1 indiquent la présence de fer en quantité variable et dans des états d’oxydation également variés.
Le verre étant fabriqué, pour une part importante, à partir de matières premières de carrière (sable et calcaire), il contient inévitablement une certaine quantité d’impuretés, parmi lesquelles la plus commune est le fer. Depuis longtemps, les verriers savent que, sauf addition de colorants particuliers, la couleur du verre dépend de sa teneur en fer, mais aussi des conditions d’élaboration, en particulier de l’addition d’éléments oxydants ou réducteurs.
Le fer étant toujours présent, on a pris l’habitude de décrire l’état d’oxydation du verre par l’intermédiaire de celui du fer. L’expression a pu évoluer, mais le principe est toujours le même : le rapport entre Fe2+ et Fe3+. Le taux de Fe2+ par rapport à la teneur totale en fer varie communément entre 10 et 90%. Des valeurs plus faibles peuvent se rencontrer pour des verres très oxydés pour des teintes spéciales ou plus élevées, dans des conditions d’élaboration particulières. Dans ce dernier cas, il est possible d’observer des particules de fer métallique.
Ces valeurs sont établies par des dosages menés sur le verre à température ambiante. On peut donc se demander si elles représentent bien son état à haute température, comme l’admettent implicitement de nombreux auteurs. Un argument parle en faveur de cette thèse : la tendance naturelle des éléments présentant plusieurs états d’oxydation est de s’oxyder quand la température baisse. Or, la diffusion des gaz, et de l’oxygène en particulier, est très lente dans le liquide. En effet, les expériences d’équilibrage du verre doivent être extrêmement longues si l’on veut vraiment atteindre l’équilibre sans brasser ou effectuer un bullage, quelques dizaines d’heures pour quelques grammes de verre. On peut conclure qu’en l’absence de ces conditions, l’état d’oxydation du verre est conservé lors du refroidissement. La question peut se poser dans le cas de la présence simultanée des deux couples redox en teneurs suffisantes : la réaction d’oxydo-réduction ne met alors pas en jeu un gaz, mais des espèces chargées, et pourrait être plus facile.
2.2 Fugacité d’oxygène
L’oxydation d’un élément Mn+ peut s’écrire de la façon suivante :
La fugacité d’oxygène influe directement sur l’état d’oxydation de l’élément M. Quant à a(O2–), c’est, pour un verre donné, une constante (cf. § 2.4).
Expérimentalement, on constate que l’état d’oxydation du verre industriel dépend en premier lieu du mélange de matières premières et, sauf opération particulière, comme un brassage mécanique, assez peu de l’atmosphère sous laquelle il a été élaboré. Cela tient à la faiblesse des vitesses de diffusion en son sein 〚1, 2〛. Cela tient par ailleurs au mode de fabrication : pour obtenir une bonne homogénéité, les verriers ont de tous temps provoqué un dégagement gazeux extrêmement intense durant l’opération d’affinage, dont nous étudierons quelques aspects plus loin. L’interaction entre le gaz et le liquide silicaté est alors beaucoup plus intime et donc incomparablement plus efficace que le simple contact avec l’atmosphère régnant au-dessus du bain de verre ; l’équilibre s’en trouve plus facilement établi.
Ce n’est donc pas l’atmosphère de combustion qui règne au-dessus du bain de verre dans un four industriel qui impose l’état d’oxydation. Partant, les verriers ont rarement ressenti le besoin de raisonner en terme de fO2.
Malgré tout, afin d’être capable d’évaluer l’impact de l’atmosphère sur le verre ou, plus exactement, sur sa surface, puisque toute l’épaisseur (plus d’un mètre dans les fours industriels) est rarement concernée, il s’est révélé utile de disposer de la valeur de la constante d’équilibre du fer, au moins pour les verres sodocalciques, les plus courants, proches des verres plats et verres à bouteille. L’équation chimique est la suivante :
La valeur de K’ varie en fonction de la température selon la loi habituelle ΔG = –R T ln K’.
Expérimentalement, le verre est équilibré par bullage à pression partielle d’oxygène connue jusqu’à l’obtention de deux dosages de Fe2+ consécutifs identiques. On a pu ainsi déterminer la relation suivante :
Cette loi peut être traduite sous forme d’abaque commode à utiliser (Fig. 1). On peut ainsi constater que le verre à 1500 °C est stable à l’air si son état d’oxydation correspond à Fe2+/Fetotal = 0,2.

Relation entre l‘état d’oxydo-réduction du verre traduit par Fe2+/Fetotal et la pression partielle de O2 en fonction de la température.
Un verre élaboré dans des conditions telles que Fe2+/Fetotal = 0,8, ce qui correspond à une grande partie des verres « ambre », est équilibré « sous » une pression partielle d’oxygène de 10–5–10–6 atm à 1500 °C. Ce verre n’est pas stable si une pression partielle de 10–2 atm à 1200 °C lui est imposée durant la suite du procédé. La surface en est immanquablement oxydée et la couleur disparaît.
La seule possibilité de fabriquer ce type de verre dans un four à flamme dans la chambre duquel la pression partielle d’oxygène est imposée par la combustion (∼10–3–10–2 atm) suffit d’ailleurs à démontrer la faible influence de l’atmosphère sur la masse du verre. La modification de l’extrême surface peut cependant générer des problèmes de coloration, mais aussi de moussage, si une espèce susceptible de passer à l’état gazeux est présente.
Des essais systématiques sur de nombreux couples redox menés au cours de ces dernières années ont permis d’établir pour le verre une échelle d’oxydo-réduction plus « normalisée ».
Ce sont en particulier les travaux de Schreiber et al. sur le verre de stockage de déchets nucléaires SRL131 (borosilicate de sodium) à 1150 °C 〚1〛. La technique utilisée par ces auteurs consiste à placer une très petite quantité de verre (0,2–0,4 g) dans une atmosphère contrôlée et à attendre l’équilibre (entre 3 et 15 h). Les résultats sont présentés sur le graphique de la Fig. 2, extrait de la référence 〚1〛.
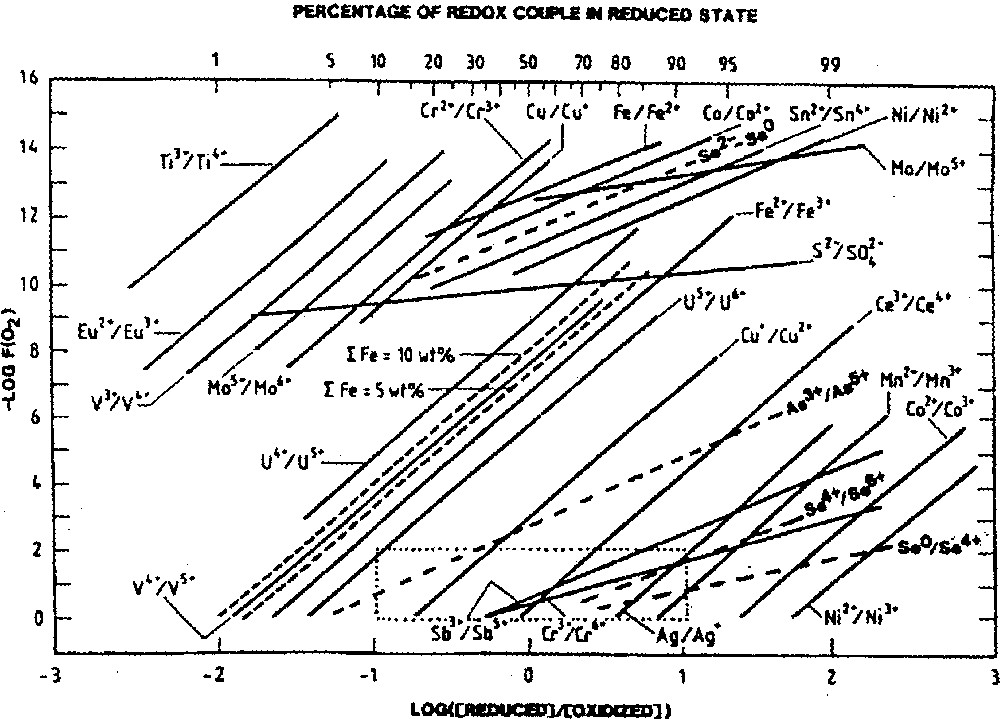
Couples redox en fonction de la pression partielle d’oxygène à 1150 °C selon 〚1〛.
La comparaison entre nos résultats et ceux de Schreiber est intéressante (Tableau 2). Les valeurs trouvées sont extrêmement proches l’une de l’autre, bien que les verres soient très différents. Sauf coïncidence, cela permet de penser que le terme est très similaire dans les deux types de verre. Cela autorise à utiliser les données de Schreiber pour d’autres couples redox. Avec ce travail très complet, on dispose d’une échelle d’oxydo-réduction permettant le classement des éléments et la détermination des espèces possibles dans une très large gamme de pressions partielles d’oxygène.
Comparaison entre les états d’oxydo-réduction exprimés par Fe2+/Fetotal selon les données de H.D.Schreiber et les nôtres. Les pentes sont déterminées par le nombre d’électrons mis en jeu : une valeur suffit donc à la comparaison.
| PO2 | Schreiber | Saint-Gobain Recherche |
| 10–5 | 0,28 | 0,30 |
On constate ainsi que certains ions peuvent exister dans deux états, comme le fer, voire trois différents, comme le cuivre. En revanche, pour Co ou Ni, par exemple, seul l’état +II est favorisé, sauf dans des verres réduits où l’on peut rencontrer du cobalt ou, plus facilement encore, du nickel métallique.
On observe qu’à 80% de Fe2+, il ne peut exister plus de 0,000 001% de Cr6+ par rapport au chrome total. Cette particularité est utilisée pour garantir l’absence de cet ion toxique dans le verre à bouteille réduit.
2.3 Potentiel d’oxydo-réduction
La méthode la plus classique pour exprimer les potentiels d’oxydo-réduction est l’utilisation des méthodes électrochimiques 〚3〛. La potentiométrie à courant nul est la mesure de la différence de concentration en oxygène entre le verre et une électrode de référence via celle de la différence de potentiel entre les deux électrodes. En l’occurrence, la concentration mesurée est exprimée par la pression partielle d’oxygène dans le verre correspondant à l’équilibre (1) ci-dessus.
On utilise deux électrodes de platine : l’une est plongée dans le verre et l’autre balayée par un courant du gaz de référence, avec une pression partielle d’oxygène connue. Ces deux électrodes sont séparées par un matériau conducteur des O2–, de l’oxyde de zirconium dopé à l’oxyde d’yttrium en général. S’il existe une différence de PO2 entre le verre et le gaz de référence, une différence de potentiel est créée entre les électrodes.
Si on met les électrodes en court-circuit, des réactions redox aux électrodes tendent à neutraliser la différence de potentiel. À l’électrode sur laquelle la pression partielle d’oxygène est la plus forte aura lieu la réaction suivante :
Dans des essais de potentiométrie, les électrodes sont connectées avec un voltmètre à impédance élevée. Un courant très faible traverse le système. On mesure alors la différence de potentiel Em entre les deux électrodes comme :
La voltamétrie 〚3〛 permet de déterminer E0. Le Tableau 3 donne quelques résultats issus des travaux de Russel 〚3〛 sur des couples redox très courants, à commencer par (FeII/FeIII) dans un verre silico-sodocalcique.
Potentiels normaux obtenus par voltamétrie à 1250 °C et calculs des rapports forme oxydée/forme réduite à l’air à 1100 °C.
| Couples redox | E0 à 1250°C | log (Cox/Cred) à PO2 = 0,21 atm à 1100 °C |
| ZnII/Zn0 | –0,63 | 5,36 |
| FeIII/FeII | –0,50 | 2,06 |
| NiII/Ni0 | –0,49 | 4,33 |
| CuI/Cu0 | –0,40 | 1,69 |
| AsV/AsIII | –0,06 | 1,25 |
Ces résultats concernant le couple FeIII/FeII peuvent être comparés aux nôtres. À 1100°C, à l’air, nous trouvons 〚Fe3+〛/〚Fe2+〛 = 40, Russel, 〚Fe3+〛/〚Fe2+〛 = 115. Nos données sont obtenues à partir d’expériences réalisées à des températures beaucoup plus élevées. On peut penser que, compte tenu de cette incertitude, l’accord est assez bon et, là encore, utiliser les résultats de cette étude pour évaluer les interactions possibles entre oxydes.
2.4 Influence de la composition chimique du verre
Revenons à l’équation d’oxydo-réduction :
Le terme a(O2–) traduit la contribution de l’environnement chimique de l’ion oxygène, qui varie en fonction de la composition chimique du verre. La concentration de cette espèce étant très grande dans le matériau, on ne peut en aucun cas assimiler concentration et activité. À même fugacité d’oxygène, le terme , donc l’état d’oxydo-réduction du verre, varie avec la composition.
L’établissement de lois générales de variation des constantes d’équilibre d’oxydo-réduction en fonction de la composition chimique du verre est pratiquement impossible à faire a priori, car les activités restent très mal évaluées dans les verres, malgré les progrès réalisés ces dernières années 〚4〛. Un certain nombre de travaux sont en cours de réalisation dans ce domaine, en utilisant en particulier les données de cristallisation ou la température de liquidus, indiquant la température à laquelle la phase qui précipite a une activité égale à 1. Malheureusement, le verre n’a rien d’un corps simple et l’étendue du domaine de compositions à explorer est telle que le calcul parfait des activités reste du domaine du futur.
Parmi les travaux expérimentaux les plus pertinents sur la relation entre constante d’équilibre et composition chimique du verre, on trouve ceux de Paul et Douglas 〚2〛 et de Budd 〚5〛. La Fig. 3 représente l’évolution de K′Fe en fonction de la teneur en oxydes alcalins dans les trois silicates binaires Li2O, Na2O et K2O·SiO2. On constate que la valeur de K′ varie dans le même sens que la teneur en oxyde alcalin, autrement dit que a(O2–). D’autres essais ont permis de montrer que l’addition de CaO aux dépens de SiO2, donc l’augmentation du taux d’oxygènes non pontants, exerce une influence similaire (Fig. 4).
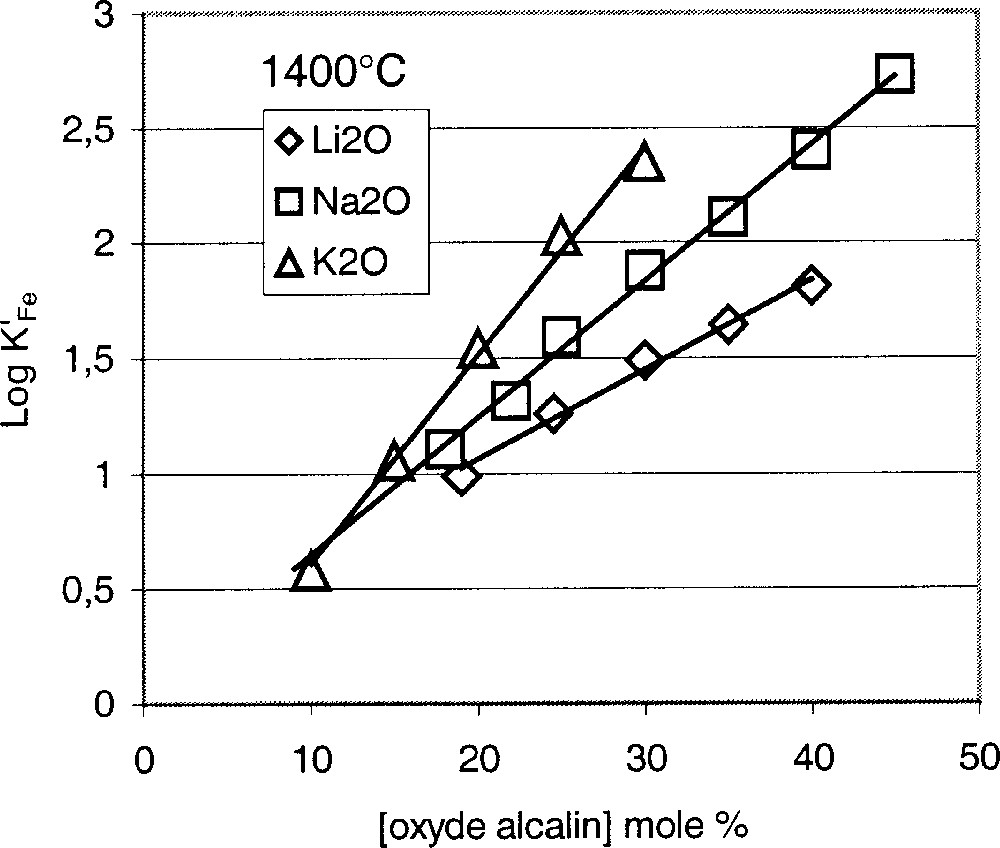
Constante d’équilibre du fer en fonction de la teneur en oxyde alcalin à 1400 °C.
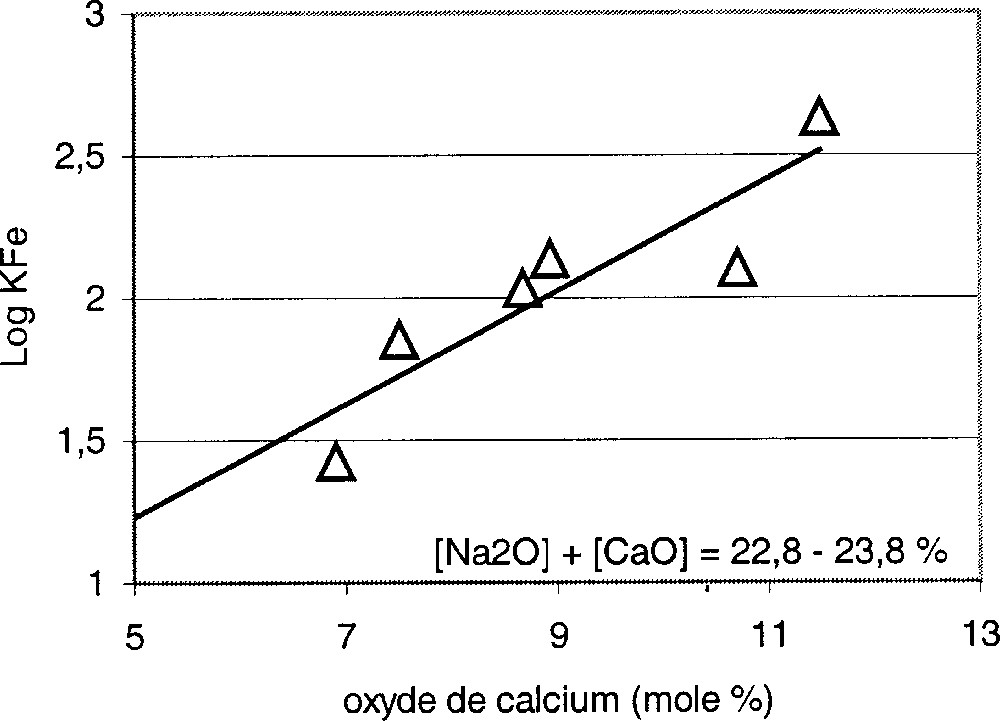
Constante d’équilibre du fer en fonction de l’addition de CaO.
On considère alors que M est sous forme « complexée», ce qui est d’ailleurs vrai, dans le silicate. Un certain nombre de couples redox ont été étudiés de cette manière, le cérium 〚6, 7〛, le chrome 〚7, 8〛, pour lesquels le résultat a été le même : plus le verre contient de cations modificateurs, plus il est oxydé à même pression partielle d’oxygène.
Le cas d’un autre élément s’écrit plus naturellement en suivant ce modèle d’équation :
Holmquist 〚9〛 a étudié cet équilibre à 1200 °C en équilibrant des verres binaires SiO2·Na2O avec un mélange de SO2 + O2. La saturation en sulfate est facilement détectée, car elle provoque la formation de sulfate de sodium, qui se sépare du verre fondu et « flotte » à sa surface. L’activité de Na2SO4 est alors égale à 1. La valeur de la constante d’équilibre peut, par exemple, être calculée à partir des tables de Janaf 〚10〛. On obtient K = 1010,76 à 1200 °C. De l’expression , on déduit la valeur de a〚Na2O〛 si l’on connaît les pressions partielles (en Pa). La Fig. 5 présente les résultats obtenus : la valeur calculée à basse teneur en oxyde de sodium (en fraction molaire) est remarquablement en accord avec cette teneur, mais ce n’est plus le cas au-delà. Cela reste néanmoins potentiellement une solution élégante pour évaluer les activités des espèces dans les verres fondus.

Relation entre teneur en alcalin et activité calculée.
2.5 Influence de la température
Le diagramme d’Ellingham, bien connu, montre que, plus la température s’élève, plus les éléments sont réduits. La règle s’applique complètement au verre. KM décroît quand la température monte, ΔG étant positif (Fig. 1).
3 Les phénomènes liés à des réactions d’oxydo-réduction au cours de l’élaboration du verre
Nous avons dit que l’équilibre du verre final était généralement obtenu lors de l’élaboration. En effet, le procédé de fabrication à haute température comporte un certain nombre de réactions génératrices de gaz : le dégagement gazeux intense a comme but l’homogénéisation du liquide. Il est donc nécessaire que l’ensemble du volume liquide soit concerné. La génération de gaz est le plus souvent réalisée par l’intermédiaire de réactions d’oxydo-réduction. Du même coup, c’est ce processus d’affinage qui impose l’équilibre d’oxydo-réduction final au système, tout comme pourrait le faire un bullage généré de manière beaucoup plus classique.
D’autres aspects de l’élaboration reposent sur des réactions d’oxydo-réduction : par exemple, les interactions verre–matériaux réfractaires et en particulier verre–molybdène, métal très utilisé pour fabriquer les électrodes de chauffage des fours industriels. Nous verrons une illustration de cette réaction.
3.1 Affinage du verre
3.1.1 Réactifs
Deux processus assez différents, quoique reposant tous deux sur des réactions d’oxydo-réduction, sont utilisés : l’affinage par le couple (sulfate/réducteur) et l’affinage par le couple (oxyde d’arsenic ou d’antimoine/nitrate). Ni le cation venant avec l’espèce sulfate ni celui venant avec l’espèce nitrate n’ont véritablement d’importance, attendu qu’ils se trouvent à l’état ionique dans le mélange de carbonates et de silicates fondus après la première étape de montée en température du mélange vitrifiable. Ce sont néanmoins, le plus souvent, des sels alcalins.
Dans les deux cas, on a affaire à un processus en deux étapes : la première à basse température, assurant la formation d’espèces susceptibles de réagir dans une seconde étape, à la température voulue, suffisamment élevée pour obtenir une efficacité maximum, le liquide étant le plus fluide possible.
3.1.2 Réactions
3.1.2.1 L’affinage par le couple (oxyde d’arsenic ou d’antimoine/nitrate)
Les deux étapes du processus sont les suivantes :
Ce processus a été parfaitement mis en évidence par des auteurs comme Cable et al. 〚11〛. Le graphique suivant (Fig. 6), établi à partir des données de Cable 〚11〛, montre l’évolution rapide des bulles contenant initialement CO2, le gaz provenant de la décomposition des carbonates en bulles d’O2 majoritaires par suite de la réaction (2). On observe alors une augmentation de la taille des bulles, ce qui leur permet de monter plus rapidement vers la surface du bain de silicate fondu et de s’échapper. L’addition de nitrate de sodium accentue nettement le phénomène : au-delà de 0,5% en As2O3, plus aucune bulle ne reste dans le verre au bout de 30 min, d’où l’absence des points correspondants.

Contenu en CO2 et O2 des bulles après 30 min d’élaboration en présence d’oxyde d’arsenic : en blanc sans NaNO3, en gris avec 1% de Na2O en NaNO3.
En parallèle à l’homogénéisation, ce dégagement gazeux intense équilibre le verre, avec une atmosphère constituée à plus de 90% de O2.
Remarque. Il est bien entendu que Na2O est écrit symboliquement en considérant que l'ion sodium s'intègre au silicate liquide avec la quantité d'ion oxygène nécessaire et non pas comme une entité indépendante. Il en va d'ailleurs de même pour l'écriture en oxyde que nous avons choisie. Elle est habituelle aux verriers, sinon parfaitement normalisée !
3.1.2.2 L’affinage par le couple (sulfate/réducteur)
Les étapes du processus sont les suivantes :
On s’aperçoit que la seconde réaction ne peut se produire que s’il reste du sulfate après la première. Il est d’ailleurs possible que deux autres réactions se produisent :
En réalité, l’ensemble de ces réactions génère un mélange gazeux en fonction de l’équilibre :
Le mélange gazeux est donc constitué de SO2, O2 et S2 dans des proportions variables, sachant que le maximum en pression partielle de O2, donné par la réaction (6), est de 1/3.
3.1.3 États d’oxydo-réduction obtenus
L’état d’oxydo-réduction des verres peut donc varier comme fO2 de 1 pour les verres affinés par le système arsenic/nitrate à des valeurs très basses pour le système sulfate/réducteur.
3.1.4 Équilibre entre (S4+/S6+) et (Fe2+/Fe3+)
L’état d’oxydation du fer dépend de la fugacité d’oxygène comme celui du soufre, selon les équations précédentes. Il en découle une relation entre les deux couples redox :
Si l’on se réfère à la réaction (7), le mélange de gaz créé lors de l’élaboration en présence de sulfate est SO2 + O2. On peut donc écrire :
Or, PO2 peut être extrait de la constante d’équilibre de (8) (équivalent à (1) pour le couple Fe2+/Fe3+) :
La Fig. 7 présente 〚SO42–〛 en fonction de . 〚SO42–〛 est exprimé en % SO3 pour 100 g de verre et représente la teneur totale en soufre dans le verre, toutes formes confondues. Les données ont été obtenues par élaboration de verre silico-sodo-calcique (Tableau 1) en présence de sulfate de sodium et de coke comme réducteur. Sauf au tout début, la première partie de la courbe est quasiment linéaire, ce qui indique que le terme entre crochets est très voisin de 1, donc que PO2 est négligeable devant PSO2. On en conclut que la plupart des verres élaborés avec du sulfate de sodium sont équilibrés par une atmosphère de SO2.

Relation entre la teneur en SO3 résiduelle dans le verre et l’état d’oxydo-réduction.
Le maximum de ces courbes correspond à , décomposition stœchiométrique du sulfate, soit PO2=1/3, et peut donc être facilement calculé à toutes les températures via la constante d’équilibre du fer (cf. § 2.2).
Connaissant K′Fe et , il est facile d’extraire K′S, correspondant à l’équilibre d’oxydation du soufre :
L’ordonnée à l’origine non nulle observable sur le graphique correspond à des verres très réduits. Dans ces verres, l’élément S est présent sous plusieurs formes : SVI, mais aussi SIV et SII–, ce qui complique beaucoup les équilibres à prendre en compte. Outre la présence probable de sulfite et de sulfure, qui se retrouvent dosés dans les échantillons comme élément soufre, on devrait probablement écrire :
Étant donné que les espèces concernées sont gazeuses, il est possible d’appliquer les données de la thermodynamique 〚10〛 pour calculer les pressions partielles des trois gaz (à 1400 °C sur la Fig. 8). On constate effectivement la présence de S2 quand est supérieur à 0,8. Ces calculs confirment d’ailleurs que SO2 est le gaz majoritaire si , sa pression partielle étant très proche de 1 pour compris entre 0,26 et 0,8, mais que PS2 monte et ne devrait plus être négligé quand est supérieur à 0,84.

Équilibre S2–SO2–O2 en fonction de l’état d’oxydo-réduction indiqué par le taux de fer(II) dans le verre.
Le problème des verres réduits est donc beaucoup plus complexe à traiter et nous ne l’aborderons pas davantage.
3.2 Interaction verre–matériau réfractaire : exemple du molybdène
Le molybdène est utilisé comme matériau d’électrodes pour la fusion électrique du verre ou, dans les four à flammes, comme matériau d’électrodes d’appoint électrique.
Sa vitesse de corrosion est influencée par un certain nombre de paramètres imposés par le procédé, plus ou moins liés entre eux, comme, par exemple, la température d’interface molybdène–verre, la densité de courant alternatif, la puissance électrique...
Elle est également influencée par la composition du verre et, en particulier, par sa teneur en éléments oxydants, qui sont les moteurs de la corrosion du molybdène.
En considérant un verre ne contenant que les oxydes de fer en tant que couple oxydo-réducteur potentiellement actif dans le domaine de stabilité du verre, les réactions de corrosion du molybdène peuvent s’écrire :
- • réactions anodiques, éventuellement combinées :
- • réactions cathodiques, éventuellement combinées :
En général, ces réactions sont sous contrôle cinétique mixte, c’est-à-dire qu’elles sont à la fois contrôlée par la cinétique des réactions d’interface et par le transfert de matière (c’est-à-dire les phénomènes de diffusion–convection).
La Fig. 9 montre l’influence de la teneur en oxydes de fer ferrique Fe3+ sur la vitesse de corrosion du molybdène.
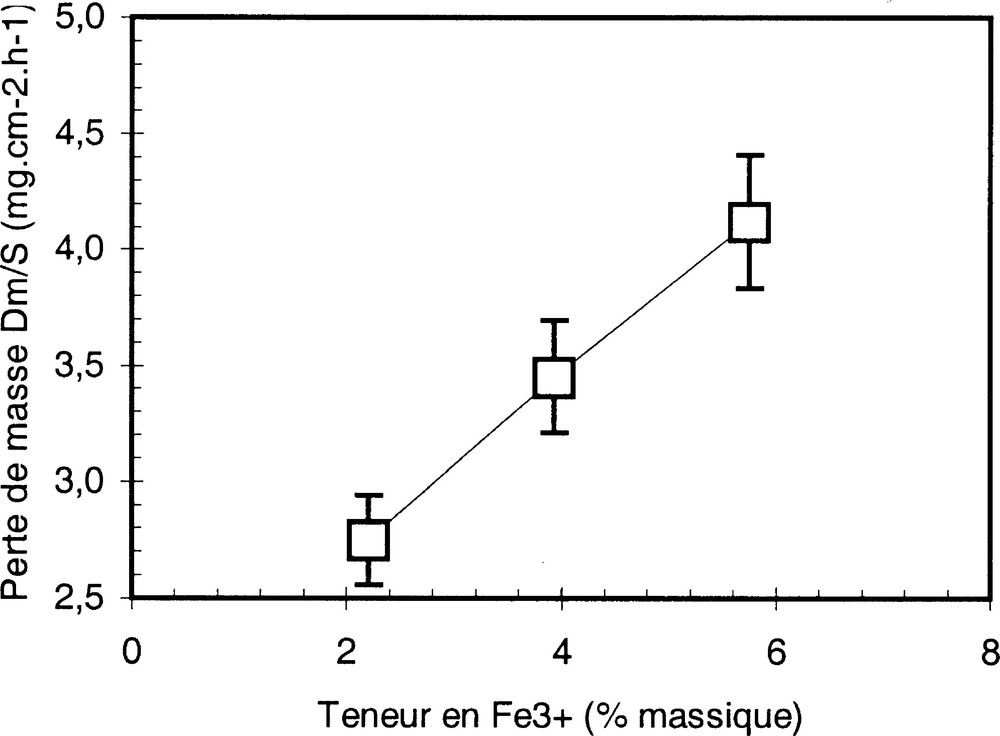
Perte de masse d’un échantillon de molybdène en fonction de la teneur en fer(III) dans le verre.
L’étude a été conduite au laboratoire, à 1300 °C. Les vitesses de corrosion ont été évaluées par essais d’électrochimie (méthode de Stern et Gary), en conditions statiques.
Au-delà d’un certain seuil, la vitesse de corrosion du molybdène suit linéairement l’augmentation de la concentration en ions ferriques. Dans les conditions étudiées, on a donc l’expression suivante pour la vitesse v de corrosion du molybdène en fonction de la teneur en ions ferrique du verre :
Dans ce domaine de composition et pour une température de 1300 °C, les courants de corrosion et pertes de masse croissent linéairement avec la teneur en oxydants Fe3+ selon les lois calculées suivantes :
4 Relation entre couleur du verre et oxydo-réduction
En dehors des aspects liés à son élaboration, la principale propriété du verre concerné par l’oxydo-réduction est sa couleur. En effet, le verre est essentiellement coloré par des espèces absorbant dans le visible et non par des pigments, au contraire des émaux. Les espèces colorantes sont tout à fait similaires à celles qui absorbent dans l’eau, par exemple. Le cas le plus courant de coloration est d’ailleurs lié aux ions de métaux de transition, le mécanisme en étant des transitions électroniques entre deux niveaux d’énergie déterminés par le champ cristallin imposé par l’environnement chimique des cations, de l’oxygène le plus souvent. Le second type de coloration est lié au phénomène de transfert de charge entre le cation et l’anion, l’anion le plus courant étant là encore l’oxygène, mais aussi parfois le soufre ou le sélénium. Enfin, le troisième type de coloration est lié à l’apparition d’agrégats métalliques, comme Ag, Au ou Cu, ou bien semi-conducteurs, comme CdS ou CsSe.
Ces mécanismes de coloration dépendent évidemment pour une part essentielle de l’état d’oxydation des espèces colorantes.
4.1 Coloration par les métaux de transition
Compte tenu de ce que nous avons vu au § 1, il est clair que tous les états d’oxydation ne sont pas possibles pour tous les métaux de transition. Par exemple, Co et Ni ne se trouvent qu’à l’état +II dans les verres ordinairement élaborés. La coloration qu’ils apportent au verre ne varie donc pas avec fO2, à environnement chimique identique. En revanche, d’autres éléments de transition peuvent adopter des états d’oxydation beaucoup plus changeants (vanadium, chrome, manganèse, fer et cuivre), donc conférer au verre une couleur variable en fonction de son état d’oxydation (Tableau 4).
Métaux de transition : formes oxydées rencontrées dans les verres de silicates et coloration attribuée à l’absorption par une transition électronique due au champ cristallin (selon C.R. Bamford 〚12〛).
| Élément | État d’oxydation | Positions des bandes d’absorption | Couleur conférée au verre |
| Chrome | Cr3+ (octaédrique) | 450 et 650 nm | Vert |
| Manganèse | Mn3+ (octaédrique) | 490 nm | Mauve |
| Fer | Fe2+ (octaédrique) | 1050 nm | Bleu |
| Fe3+ (tétraédrique) | 380, 420, 435 nm | Jaune | |
| Cobalt | Co2+ (octaédrique) | 530, 590, 645 nm | Bleu |
| Nickel | Ni2+ (octaédrique) | 450, 930, 1800 nm | Brun |
| Ni2+ (tétraédrique) | 560, 630, 1200 nm | ||
| Cuivre | Cu2+ (octaédrique) | 780 nm | Bleu |
Les coefficients d’absorption des différents ions sont extrêmement variables. Nous avons représenté ceux des ions Fe2+, Fe3+, Cr3+ sur la Fig. 10 〚12〛.
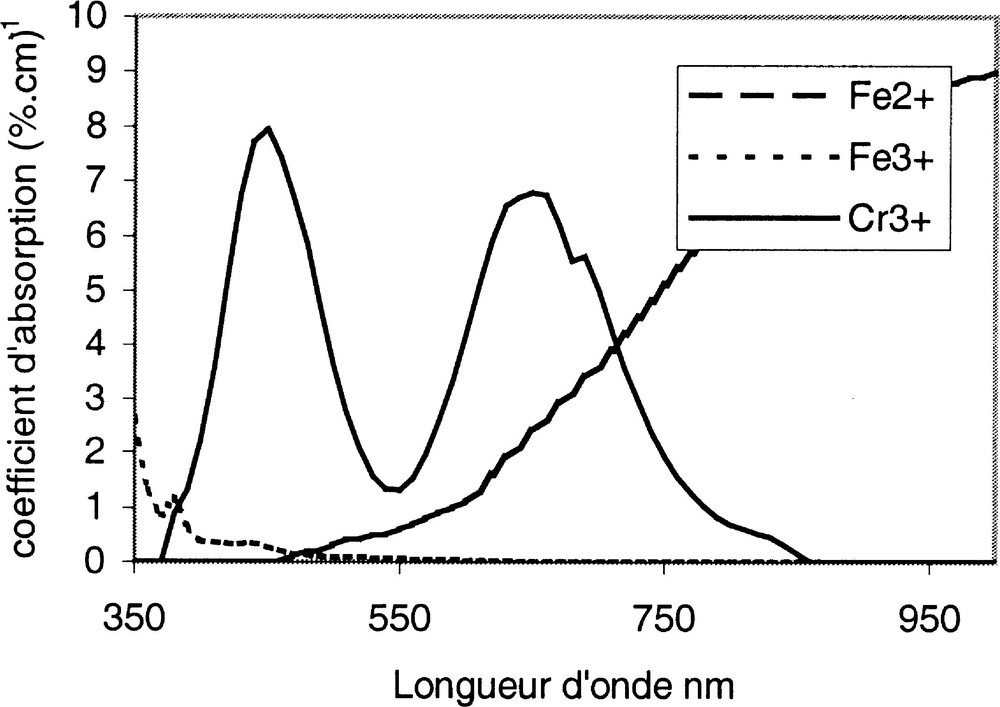
Coefficients d’absorption des ions Fe2+, Fe3+, Cr3+ dans un verre silico-sodo-calcique de formulation analogue aux verres plat et à bouteille du Tableau 1.
Le fer constitue la pollution principale des verres blancs : c’est l’espèce Fe2+ qui est la plus colorante. Les efforts des verriers ont longtemps porté sur l’oxydation de cette espèce : on utilisait pour cela le manganèse, le « savon des verrier » qui, étant plus oxydant que le fer, en transformait un maximum en Fe3+, éliminant du même coup la coloration bleue générée par le fer réduit. L’ajout d’oxyde d’arsenic au mélange vitrifiable remplissait à peu près le même rôle : nous avons vu qu’elle imposait au verre une pression partielle d’oxygène très élevée.
Les métaux de transition possèdent tous des électrons dans leur couche électronique 3d. Dans un verre, ils se trouvent soumis au champ électrostatique crée par les anions du réseau, le plus souvent de l’oxygène, les ligands. Le champ de ligand ou champ cristallin génère l’éclatement des niveaux énergétiques et donc la possibilité de transitions électroniques normalement interdites 〚12〛. Les intensités d’absorption sont généralement assez faibles, contrairement aux suivantes (Fig. 11).

Coefficients d’absorption de Fe3+–S2– et Fe3+–Se2–.
4.2 Absorption dans le spectre visible et ultraviolet par des transitions électroniques de type transfert de charge entre cation métallique et anion du réseau
La présence d’ions métalliques dans le verre génère une absorption intense dans l’ultraviolet par transfert d’électron des ligands (le réseau vitreux oxygéné) vers l’ion métallique central. C’est le cas par exemple des ions de métaux de transition Cu2+, de Cr6+ ou de Fe3+. Étant donné le désordre caractéristique du matériau, les bandes dont le maximum se situe dans l’ultraviolet s’étalent jusqu’au domaine visible. Le verre devient jaunâtre.
Cette transition électronique étant autorisée contrairement aux transitions d, son intensité est généralement très grande. Cette absorption, recherchée comme protection pour le conditionnement des liquides ou les vitrages anti-fanage des tissus, devient gênante si l’on veut faire des verres très transparents dans l’ultraviolet, pour des applications laser par exemple.
Le remplacement de l’anion oxygène par d’autres tels que S ou Se provoque un glissement de la longueur d’onde de l’absorption maximale vers le visible. Les verres deviennent tout à fait jaunes (Fig. 11).
Dans le cas des sulfures, les verres sont réduits (Fe2+/Fetotal ≈ 0,8–0,85), afin d’obtenir l’éléments S à l’état –II, tout en conservant suffisamment de fer à l’état ferrique. La pression partielle équivalente est de l’ordre de 10–5 à 10–6 atm (Fig. 1). La coloration jaune due au sélénium est obtenue dans des conditions assez différentes, beaucoup plus oxydantes, d’autant plus que l’on recherche aussi le sélénium à l’état élémentaire, qui colore en rouge par formation d’agrégats.
4.3 Coloration par formation d’agrégats
Parmi les colorations avec formation d’agrégats 〚12, 13, 14〛, la plus connue est peut-être le « rouge à l’or » , obtenu par précipitation d’or métallique au sein du verre, sous forme de sphères diffusant la lumière, sans pour autant atteindre une taille telle que la transparence du matériau en soit affectée (1 à 50 nm). On n’obtient de couleur intéressante que si les caractéristiques optiques (n et k) du matériau changent en fonction de la longueur d’onde dans le visible. C’est le cas pour Au, Cu et Ag.
Le métal est introduit dans le mélange vitrifiable sous forme de sel, donc du métal sous forme oxydée, même dans le cas de l’or 〚14〛, pour lequel il est nécessaire d’ajouter un oxydant. Pour obtenir une couleur vive, il convient que la précipitation des particules métalliques s’opère seulement lors de la recuisson du verre car, à plus haute température, on forme des agrégats de grande taille et la couleur s’opacifie. Dans ce but, on introduit un élément multivalent, comme l’oxyde d’étain, qui est sous forme réduite à haute température et s’oxyde à température de recuisson, en provoquant la réduction du métal intéressant, l’or ou le cuivre. L’addition d’un nucléant, comme CeO2, permet d’obtenir une précipitation homogène.
On voit que la fabrication de ce type de verre requiert un dosage très précis des réactifs, afin que les phénomènes d’oxydo-réduction mutuelle entre éléments conduisent au bon équilibre à la température idéale. Les « recettes » concernant ces verres ont toujours été un secret bien gardé !
5 Conclusion
Les phénomènes provoqués par des réactions d’oxydo-réduction sont nombreux dans le processus de fabrication du verre ainsi que dans ses propriétés. Nous n’avons pas tenté d’en faire une description exhaustive, mais uniquement de donner un certain nombre d’exemples, témoignant à la fois de leur importance, mais aussi, souvent, du fait que nous sommes très loin de tout en savoir. Dans le même ordre d’idée, la littérature sur ce sujet est extrêmement abondante et nous n’avons retenu que les articles illustrant au mieux, selon nous, notre propos. Nous espérons que ce travail suscitera des vocations dans la communauté scientifique et que les domaines encore insuffisamment connus seront mieux explorés dans l’avenir.