


 CC-BY 4.0
CC-BY 4.0
1. Introduction
The incidence of cancer has been increasing over the years. It is predicted that 27.5 million new cancer cases will appear worldwide each year by 2040. This represents an increase of 61.7% from 2018 and is expected to be higher in males (67.6% increase) than in females (55.3% increase) [1]. Therefore, several classes of drugs have been developed over the years to treat cancer. For example, taxanes, monoclonal antibodies and steroids are used in clinical practice [2, 3, 4].
Steroid hormones are involved in many physiologic responses and pathologic conditions mainly by binding to their intracellular receptors, namely estrogen receptors (ERs), which are transcription factors. For example, the importance of androgens in prostatic cancer and estrogens in breast cancer led to the development of therapies that block their action in these tumors [5]. In this context, exemestane (an aromatase inhibitor) [6] and fulvestrant (a selective ERα antagonist) [7] (Figure 1) are molecules of clinical interest in the treatment of hormone-dependent breast cancers [8, 9]. Therefore, developing safer and more effective ways of preventing and treating these hormone-dependent cancers is crucial, given the significant impact that these diseases have on human health and their economic and social importance [10]. Although the use of steroid hormones and analogues has been associated with hormone-dependent cancers, evidence also suggests that they can be important in the treatment of other kinds of tumors such as lung, brain and liver cancers [9, 11].
Relevant steroids used in clinical practice as anticancer agents.
Taking into account the importance of steroids in cancer treatment, and as has been widely demonstrated in the literature, estrone (E1) and 17β-estradiol (E2) have been used as starting materials for the design and development of new and more promising anticancer drug candidates with different targets of action [8, 12, 13, 14]. For instance, the presence of a 16α-hydroxyl in E1 derivatives appeared to be associated with a high cytotoxicity and a reduced interaction with ERα [15]. In addition, the introduction of aryl groups in C-16 of the steroidal scaffold led to higher antiproliferative effects [16, 17]. Furthermore, the presence of 2-ethyl-3-O-sulfamoyl groups in estrane nucleus allowed an improvement of the antimitotic activity of E1 analogues [18]. E1 3-O-ether derivatives containing the piperazine ring also exhibited a strong cytotoxic activity against prostate cancer cell lines [19]. Interestingly, different C-13 epimeric analogues of 16β-(m-carbamoylbenzyl)-E2 showed 17β-hydroxysteroid dehydrogenase type 1 (17β-HSD1) inhibition and a weak estrogenic effect on estrogen-sensitive breast cancer cells [20]. Regarding C-ring modifications, for example, the presence of a 𝛥9,11 double bond combined with 2- and 4-substitutions in E1 nucleus was also relevant to the development of promising antiproliferative agents [21].
These findings, in addition to our continuous interest in steroidal chemistry and bioactivity [8, 22, 23, 24, 25] and the need to develop improved anticancer agents, motivated us to prepare and evaluate in in vitro conditions the cell proliferation effects of E1 derivatives. Specifically, we report herein the chemical synthesis of 𝛥9,11-E1 derivatives with A-(2,4-diiodo and 2,4-dibromo) and D-ring (16-benzylidene) modifications and their biological evaluation (cell proliferation and viability assays, E-screening assay and cell cycle distribution analysis). Docking studies on ERα, steroid sulfatase (ST) and 17β-HSD1, which are relevant potential targets of these 𝛥9,11-E1 derivatives, were also performed.
2. Experimental section
2.1. Chemistry
All chemicals received from suppliers were used without further purification. The reagents were purchased from the following suppliers: E1: Cayman Chemical (Michigan, USA); methanol (MeOH): Fisher Chemical (MA, USA); N-bromosuccinimide (NBS): Alfa Aesar (Massachusetts, USA); benzene (PhH): Merck (NJ, USA); benzaldehyde (BZ): Acros Organics (New Jersey, USA); ethanol (EtOH) 99.9%: Manuel Vieira & Ca (Torres Novas, Portugal). The reagents 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ), morpholine, E2, 5-fluorouracil (5-FU) and dimethyl sulfoxide (DMSO) as well as the remaining chemical products referred to in the text, including petroleum ether (PE) 40–60 °C, were obtained from Sigma-Aldrich (St. Louis, MO, USA). Deuterated DMSO (DMSO-d6) and deuterated chloroform (CDCl3) were purchased from Armar Chemicals (Leipzig, Germany). All reactions were monitored by thin-layer chromatography (TLC) using Al-backed aluminum/silica gel plate 0.20 mm (Macherey-Nagel 60 F254, Duren, Germany) and, after elution, the plates were visualized under ultraviolet (UV) radiation (254 nm) in a CN-15.LC UV chamber. EtOH/concentrated sulfuric acid (95:5, v:v) mixture was used to process the plates, followed by heating at 120 °C. The evaporation of solvents was achieved by using a rotary vacuum drier from Büchi (R-215). Melting points (mp) were recorded on a Büchi B-540 melting point apparatus and are uncorrected. Infrared (IR) spectra were collected on a Thermoscientific Nicolet iS10 equipped with a diamond attenuated total reflectance crystal at room temperature in the 4000–400 cm−1 range by averaging 16 scans at a spectral resolution of 2 cm−1. Nuclear magnetic resonance (NMR) spectra (1H-NMR and 13C-NMR) were acquired on a Bruker Avance 400 MHz spectrometer and were processed with the software TOPSPIN 4.07 (Bruker, Fitchburg, WI, USA). Chemical shifts are reported in parts per million (ppm) relative to tetramethylsilane (TMS) or solvent as an internal standard. Coupling constants (J values) are reported in hertz (Hz) and splitting multiplicities are described as s = singlet, brs = broad singlet, d = doublet, dd = double doublet and m = multiplet. High-resolution mass spectrometry (ESI-HRMS) was performed by the microanalysis service on a QSTAR XL instrument (Salamanca, Spain).
2.1.1. Synthesis of 3-hydroxyestra-1,3,5(10),9(11)-tetraen-17-one (2)
A stirred solution of 1 (540.8 mg, 2 mmol) in MeOH (80 mL) was heated at 45 °C. DDQ (680.9 mg) was added in one portion and the resulting solution was vigorously stirred for 5 h at 45 °C under nitrogen (N2) atmosphere. After completion of the reaction (TLC control), MeOH was evaporated and the residue was diluted in 300 mL of dichloromethane (CH2Cl2), washed with 100 mL of aqueous 10% sodium sulfite (Na2SO3), 100 mL of a saturated aqueous solution of sodium hydrogen carbonate (NaHCO3) and 100 mL of water (H2O) and dried over anhydrous sodium sulfate (Na2SO4), filtered and evaporated under reduced pressure to yield the crude product, which was recrystallized from MeOH to give compound 2 [26, 27] as beige crystals (223.5 mg, 42% yield); mp 235.2–237 °C (lit [28] 243–246 °C). IR (ʋmax, cm−1): 814, 1224, 1453, 1605, 1615, 1715, 2832-2964, 3019, 3255; 1H-NMR (400 MHz, DMSO-d6) δ: 0.82 (s, 3H, C18-CH3), 6.05 (m, 1H, C11-H), 6.46 (d, 1H, J = 2.5 Hz, C4-H), 6.55 (dd, 1H, J1 = 8.6 Hz, J2 = 2.5 Hz, C2-H), 7.43 (d, 1H, J = 8.7, C1-H), 9.28 (brs, 1H, 3-OH); 13C-NMR (100 MHz, DMSO-d6) δ: 14.21, 22.05, 27.31, 29.20, 33.56, 35.72, 37.69, 45.45, 47.02, 113.82, 114.79, 115.22, 125.03, 125.11, 135.24, 137.19, 156.23, 220.42.
2.1.2. Synthesis of 3-hydroxy-16-phenylmethylidene-estra-1,3,5(10),9(11)-tetraen-17-one (3)
To a solution of compound 2 (134.2 mg, 0.5 mmol) in MeOH (3.8 mL) was added BZ (76.4 μL) and potassium hydroxide (KOH) (192 mg). The reaction mixture was stirred for 4.5 h at room temperature. After completion (TLC control), the reaction mixture was diluted in 150 mL of CH2Cl2 and washed with 50 mL of H2O, dried over anhydrous Na2SO4 and concentrated under reduced pressure to yield the crude product, which was recrystallized from MeOH to give compound 3 as brown crystals (57.2 mg, 32% yield); mp 263.1–265.2 °C. IR (ʋmax, cm−1): 809, 1285, 1360, 1447, 1496, 1604, 1698, 2829-2958, 3021, 3060, 3324; 1H-NMR (400 MHz, DMSO-d6) δ: 0.91 (s, 3H, C18-CH3), 6.09 (m, 1H, C11-H), 6.49 (d, 1H, J = 2.3 Hz, C4-H), 6.56 (dd, 1H, J1 = 8.7 Hz, J2 = 2.4 Hz, C2-H), 7.34 (brs, 1H, H-vinyl), 7.46 (m, 4H, C1-H, , , ); 7.67 (d, 2H, J = 7.5 Hz, , ), 9.30 (s, 1H, 3-OH); 13C-NMR (100 MHz, DMSO-d6) δ: 14.89, 27.36, 29.21, 29.47, 33.60, 37.35, 45.03, 45.36, 113.86, 114.83, 115.32, 125.01, 125.11, 128.81, 129.45, 130.41, 132.07, 135.08, 135.41, 136.01, 137.19, 156.28, 208.72. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C25H24O2: 356.1776; found 356.1771.
2.1.3. Synthesis of 2,4-diiodo-3-hydroxyestra-1,3,5 (10)-trien-17-one (4)
To a solution of E1 1 (270.4 mg, 1 mmol) in PhH (56 mL) were added 302.8 mg of iodine (I2) and 1536 μL of morpholine. The solution was stirred under room temperature for 17 h. After this time, 60 ml of 5% aqueous HCl solution was added and it was concentrated under reduced pressure. The result was diluted in 150 mL of CH2Cl2, washed with 50 mL of saturated aqueous solution of NaHCO3, 50 mL of H2O and dried over anhydrous Na2SO4 and evaporated under reduced pressure. Then, the residue was purified by column chromatography [ethyl acetate (EA)/PE, 1:5] to obtain compound 4 [29] as a beige solid (271 mg, 68% yield); mp 180.1–183 °C (lit [29] 200–202 °C). IR (ʋmax, cm−1): 794, 1011, 1083, 1258, 1450, 1707, 1737, 2858-2961, 3296, 3439; 1H-NMR (400 MHz, CDCl3) δ: 0.88 (s, 3H, C18-CH3), 7.60 (s, 1H, C1-H); 13C-NMR (100 MHz, CDCl3) δ: 13.96, 21.74, 26.41, 27.47, 31.61, 36.05, 37.28, 37.59, 44.09, 48.03, 50.35, 78.56, 92.19, 136.07, 136.15, 140.83, 151.70, 220.61.
2.1.4. Synthesis of 2,4-diiodo-3-hydroxyestra-1,3,5 (10),9(11)-tetraen-17-one (5)
A stirred solution of 4 (131.1 mg, 0.25 mmol) in MeOH (9.8 mL) was heated at 45 °C. DDQ (85.1 mg) was added in one portion and the resulting solution was vigorously stirred for 4 h at 45 °C under N2 atmosphere. After completion (TLC control), MeOH was evaporated and the residue was diluted in 150 mL of EA and washed with 50 mL of Na2SO3 (10%, aqueous), 50 mL of saturated aqueous solution of NaHCO3 and 50 mL of H2O, dried over anhydrous Na2SO4 and concentrated under reduced pressure. This product was purified by column chromatography (eluent: EA/PE, 1:1) to obtain compound 5 as a brown solid (72.1 mg, 55% yield); mp 225.1–227.2 °C. IR (ʋmax, cm−1): 794, 1014, 1258, 1447, 1711, 1732, 2920-2961, 3442; 1H-NMR (400 MHz, CDCl3) δ: 0.89 (s, 3H, C18-CH3), 6.12 (m, 1H, C11-H), 7.91 (s, 1H, C1-H); 13C-NMR (100 MHz, CDCl3) δ: 14.57, 22.65, 28.35, 34.22, 36.42, 36.81, 37.23, 46.38, 47.94, 79.52, 91.76, 119.27, 131.26, 134.43, 135.31, 140.06, 152.52, 221.24. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C18H18I2O2: 519.9396; found 519.9396.
2.1.5. Synthesis of 2,4-dibromo-3-hydroxyestra-1,3,5 (10)-trien-17-one (6)
To a solution of E1 1 (540.7 mg, 2 mmol) in EtOH (27.0 mL) was added 1.1 g of NBS. The solution was stirred under room temperature for 29 h. After this time, the solvent was evaporated under reduced pressure. The residue was diluted in 150 mL of CH2Cl2, washed with 50 mL of saturated aqueous solution of NaHCO3, 50 mL of H2O and dried over anhydrous Na2SO4 and concentrated under reduced pressure. Then, the product was recrystallized from MeOH to give compound 6 [30] as white crystals (353 mg, 41% yield); mp 228.2–229 °C (lit [30] 235–236 °C). IR (ʋmax, cm−1): 899, 1164, 1304, 1462, 1543, 1712, 2869-2936, 3235; 1H-NMR (400 MHz, CDCl3) δ: 0.88 (s, 3H, C18-CH3), 7.38 (s, 1H, C1-H); 13C-NMR (100 MHz, CDCl3) δ: 13.95, 21.73, 26.33, 26.69, 31.12, 31.59, 36.03, 37.54, 44.13, 48.00, 50.39, 106.68, 113.42, 128.75, 135.23, 136.66, 147.47, 220.61.
2.1.6. Synthesis of 2,4-dibromo-3-hydroxyestra-1,3,5 (10),9(11)-tetraen-17-one (7)
A stirred solution of 6 (53.5 mg, 0.125 mmol) in MeOH (4.9 mL) was heated at 45 °C. DDQ (42.6 mg) was added in one portion and the resulting solution was vigorously stirred for 5.30 h at 45 °C under N2 atmosphere. After completion (TLC control), MeOH was evaporated and then the residue was diluted in 150 mL of EA, 50 mL of Na2SO3 aqueous solution (10%), 50 mL of saturated aqueous solution of NaHCO3 and 50 mL of H2O and then dried over anhydrous Na2SO4 and concentrated under reduced pressure to obtain compound 7 as a beige solid (38 mg, 71% yield); mp 200.4–202.9 °C. IR (ʋmax, cm−1): 796, 1011, 1064, 1260, 1463, 1540, 1717, 2836-2960, 3286; 1H-NMR (400 MHz, CDCl3) δ: 0.89 (s, 3H, C18-CH3), 6.13 (m, 1H, C11-H), 7.69 (s, 1H, C1-H); 13C-NMR (100 MHz, CDCl3) δ: 14.59, 22.65, 27.60, 30.70, 34.19, 36.41, 37.25, 46.35, 47.95, 107.59, 113.14, 119.49, 127.81, 130.32, 134.47, 135.98, 148.30, 221.27. HRMS (ESI-TOF): m/z [M + H]+ calcd for C18H18Br2O2: 423.9674; found 423.9644.
2.1.7. Synthesis of 3-hydroxy-16-phenylmethylidene-estra-1,3,5(10)-tetraen-17-one (8)
To a solution of 1 (135.2 mg, 0.5 mmol) in MeOH (3.8 mL) were added BZ (76.4 μL) and KOH (192 mg). The mixture was stirred at room temperature for 4 h. After MeOH evaporation, the reaction mixture was diluted in 150 mL of CH2Cl2 and washed with 50 mL of H2O, dried over anhydrous Na2SO4 and concentrated under reduced pressure to yield the crude product, which was recrystallized from MeOH to give compound 8 [31, 32] as white crystals (162 mg, 90% yield); mp 247.5–249.7 °C (lit [32] 248–250 °C). IR (ʋmax/cm−1): 790, 1276, 1373, 1445, 1612, 1699, 2858-2920, 3019, 3053, 3350; 1H-NMR (400 MHz, CDCl3) δ: 1.00 (s, 3H, C18-CH3), 4.82 (brs, 1 H, 3-OH), 6.60 (brs, 1H, C4-H), 6.66 (d, 1H, J = 9.1 Hz, C2-H), 7.17 (d, 1H, J = 9.1 Hz, C1-H), 7.39 (m, 3H, , , ); 7.48 (brs, 1H, H-vinyl), 7.57 (d, 2H, J = 8.3 Hz, , ); 13C-NMR (100 MHz, CDCl3) δ: 14.78, 26.19, 27.01, 29.37, 29.68, 31.90, 38.20, 44.26, 48.08, 48.81, 113.10, 115.53, 126.71, 128.91, 129.49, 130.56, 132.33, 133.56, 135.82, 136.20, 138.19, 153.77, 210.04.
2.2. Bioactivity assays
2.2.1. Cell culture
Human breast (MCF-7, T47-D), prostatic (LNCaP), colon (Caco-2) and fibroblast [normal human dermal fibroblasts (NHDF)] cell lines were obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) and hepatic (HepaRG) cell line was acquired from Life Technologies—Invitrogen™ (through Alfagene, Portugal). They were cultured in 75 cm2 culture flasks at 37 °C in a humidified air incubator with 5% CO2. High-glucose Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, St Louis, MO, USA) and 1% antibiotic/antimycotic (10,000 units/mL penicillin G, 100 mg/mL streptomycin and 25 μg/mL amphotericin B) (Ab; Sigma-Aldrich, St Louis, MO, USA) was used to culture MCF-7 cells. For Caco-2 cells, high-glucose DMEM supplemented with 10% FBS and 1% of the antibiotic mixture of 10,000 units/mL penicillin G and 100 mg/mL of streptomycin (sp; Sigma-Aldrich, St Louis, MO, USA) was used. LNCaP and T47-D cells were cultured in RPMI 1640 medium with 10% FBS and 1% sp. Fibroblasts grew in RPMI 1640 medium supplemented with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate and 1% Ab. Finally, HepaRG cells were seeded in Williams’ E medium supplemented with 10% FBS, 1% sp, 5 μg/mL insulin and 5 × 10−5 M hydrocortisone hemisuccinate (Sigma-Aldrich, St Louis, MO, USA).
2.2.2. Preparation of compound solutions
Stock solutions of compounds were prepared in DMSO at 10 mM and stored at 4–8 °C. The maximum DMSO concentration in cell studies was 1% and previous experiments revealed that this solvent level has no significant effects on cell proliferation (data not shown).
2.2.3. Antiproliferative assays
Cytotoxicity of compounds 1–8 was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich, St Louis, MO, USA) assay against MCF-7, T47-D, LNCaP, HepaRG, Caco-2 and NHDF cells. After reaching near confluence, cells were trypsinized and counted with a hemocytometer by means of the trypan-blue exclusion of dead cells. Then, 100 μL of cell suspension (2 × 104 cells/mL) were seeded in 96-well culture plates and left to adhere for 48 h. After adherence, the medium was replaced by several solutions of the compounds in the study (30 μM for screening assays and 0.1, 1, 10, 25, 50 and 100 μM for concentration–response studies) in the appropriate culture medium for approximately 72 h. After this period, cells were washed with 100 μL of phosphate buffer saline (PBS; NaCl 137 mM, KCl 2.7 mM, Na2HPO4 10 mM and KH2PO4 1.8 mM, pH 7.4), and then 100 μL of the MTT solution (5 mg/mL), prepared in the appropriate serum-free medium, was added to each well, followed by incubation for approximately 4 h at 37 °C. Then, the MTT containing medium was removed and formazan crystals were dissolved in DMSO. Absorbance was measured at 570 nm using a microplate reader Bio-rad xMark spectrophotometer. After background subtraction, cell proliferation values were expressed as percentage relative to the absorbance determined in negative control cells. Untreated cells were used as the negative control and the clinical drug 5-FU was used as the positive control. Each experiment was performed in quadruplicate and independently repeated.
2.2.4. E-screening assay
T47-D cells (2 × 104 cells/mL) were seeded in 96-well culture plates in 100 μM of RPMI 1640 medium supplemented with 10% FBS and allowed to attach. After overnight incubation, the medium was replaced every 3 days with fresh phenol red free RPMI 1640 medium supplemented with 5% of dextran-coated charcoal-treated fetal calf serum (DCC-FCS) and containing the compounds under study. After 6 days of exposure, the proliferation of T47-D cells was estimated by the MTT assay described in the previous section. 0.1, 0.01 and 0.001 μM were the concentrations tested for E2 and for the synthesized selected compounds. Each experiment was performed in quadruplicate and independently repeated. After background subtraction, cell proliferation values were expressed as percentage relative to the absorbance determined in negative control cells.
2.2.5. Flow cytometric analysis of cell viability
The analysis of cell viability on HepaRG cells was performed by flow cytometry after staining dead cells with propidium iodide (PI) (solution of PI 1 mg/ml in 0.1% of sodium azide and water; Sigma-Aldrich, St Louis, MO, USA). Briefly, 3 mL of cell suspension was seeded in 6-well plates (5 × 104 cells/mL) in a complete culture medium. After 48 h, they were treated with 50 μM of compound 2. Untreated cells were used as the negative control and 5-FU was used as the positive control. Each experiment was performed in duplicate and independently repeated. At the end of 24 h of incubation, the supernatant of each well was collected; cells were harvested by trypsinization and pooled with the supernatants. The resulting cell suspension was kept on ice, pelleted by centrifugation and resuspended in 400 μL of complete medium. Afterward, 395 μL of the cell suspension was transferred to a FACS tube and 5 μL of PI with EDTA (0.5 μL at 0.123 M) was added. A minimum of 20000 events was acquired using a BD Accuri C6 (San Jose, USA) flow cytometer in the channels forward scatter (FSC), side scatter (SSC) and fluorescence channel-3 (FL3, for PI). Acquisition and analysis were performed with BD Accuri Software. In the FSC/FL3 contour plot, three regions were created, one corresponding to viable cells (R1), another to dead cells (R2) and a third to an indeterminate cell population between the other two regions (R3) excluding debris that were not considered in the analysis (data not shown). The percentage of viability is the percentage of cells in R1 as compared to the total number of events in R1, R2 and R3.
2.2.6. Flow cytometric analysis of cell cycle
After 24 h of treatment with 50 μM of compound 2 (6-well plates, 5 × 104 cells/mL), HepaRG cells were collected and washed with PBS and resuspended in 450 μL of a cold solution of 0.5% bovine serum albumin (BSA; Amresco, USA) in PBS with EDTA (204 μM in 25 mL), followed by fixation with 70% of EtOH and incubation at − 20 °C. After, at least, 2 days at − 20 °C, fixed cells were washed twice with PBS and resuspended in a solution of PI (50 μg/mL) prepared in 0.5% BSA in PBS with EDTA and then incubated with Ribonuclease A from bovine pancreas at a final concentration of 0.5 μg/μL (solution in 50% glycerol, 10 mM Tris-HCl, pH 8; Sigma-Aldrich, St Louis, MO, USA) for 15 min in the dark. For comparison, untreated cells were used as the negative control and cells treated with 5-FU at 50 μM were used as the positive control. Each experiment was performed in duplicate and independently repeated. A minimum of 10000 events was acquired using BD Accuri Software and analysis was performed by Modfit software (Becton Dickinson, San Jose, CA, USA).
2.2.7. Flow cytometry carboxyfluorescein succinimidyl ester assay
HepaRG cells were trypsinized, counted and seeded in two 12-well culture plates (1 mL/well; 8 × 104 cells/mL) and left to adhere for 48 h. After this period, cells were rinsed twice with PBS and then carboxyfluorescein succinimidyl ester (CFSE; BD Horizon, San Jose, USA) was added at 10 μM and incubated for 15 or 30 min. After incubation, the wells were rinsed with PBS and the medium with compound 2 (50 μM) was added, followed by incubation for approximately 48 and 72 h. Untreated cells were used as the negative control in each plate. For 15 min of CFSE incubation, each experiment was performed in duplicate. For 30 min of incubation, one experiment was performed for 48 h and another for 72 h. At the end of the incubation period, cells were trypsinized, centrifuged and resuspended in 300 μL of medium with 5 μL of EDTA. A minimum of 20000 events was acquired using a BD Accuri C6 flow cytometer in the channels forward scatter (FSC), side scatter (SSC) and fluorescence channel-1 (FL1, for CFSE). Acquisition and analysis were performed with BD Accuri Software.
2.2.8. Statistical analysis
Data were expressed as mean ± standard deviation (SD). Comparison among groups was performed by using the t-Student test (two groups) and one-way ANOVA (three groups) followed by Bonferroni post hoc tests to determine statistically significant differences among the means. Difference between groups was considered statistically significant for a p-value lower than 0.05 (p < 0.05). The determination of IC50 was carried out by sigmoidal fitting analysis considering a confidence level of 95%.
2.3. Molecular docking studies
2.3.1. Preparation of proteins for molecular docking
The crystal structures of ERα, ST and 17β-HSD1 were obtained from the Protein Data Bank (PDB code: 1A52, 1P49 and 3KLM, respectively) [33, 34, 35]. The coordinates of all non-standard residues were deleted using the software Chimera (v. 1.10.1). Then, non-polar hydrogens were merged in AutoDockTools (v. 1.5.6) and Kollman and Gasteiger partial charges were added. Finally, the prepared structure was converted from the PDB format to PDBQT for posterior use in the docking study.
2.3.2. Preparation of ligands
All ligands were constructed using the Chem3D (v. 12.0) software. Energy minimization and geometry optimization were performed by the same software and the final structures were saved as a PDB file format. The process of energy minimization was applied in a range from −20 to −40 kcal⋅mol−1. Then, the ligands were completely prepared choosing torsions and the structures were converted from the PDB to the PDBQT format in the software AutoDockTools.
2.3.3. Grid parameters
The grid parameters were calculated using AutoDock Vina and AutoDockTools based on the coordinates of the ligand crystallized for each case: E2, N-acetyl-D-glucosamine and 5α-dihydrotestosterone (DHT), with the respective macromolecule. The grid box was centered on the ligand with the following coordinates: for ERα, the coordinates were x = 107.27, y = 13.94, z = 96.38; for ST, x = 62.033, y = −12.215, z = 52.512; and for 17β-HSD1, x = 11.643, y = 9.297, z = −11.887. The size of the grid box was 20 × 20 × 20 with a spacing of 1.0 Å.
2.3.4. Docking simulations
After ligands and protein preparation, molecular docking was performed by AutoDock Vina executable, which uses an iterated local search global optimizer. The parameter exhaustiveness of the performed experiments was defined as 8 (default). The results of molecular docking were visualized in Discovery Studio Visualizer program from BIOVIA and in PyMOL software.
2.3.5. Validation of the molecular docking performance
Scoring functions are essential for molecular docking performance. In order to verify those functions, it is necessary to validate the docking performance of AutoDock Vina. This step is required to verify the performance by analysis of the difference between the real and best-scored conformations. For the docking process to be considered successful, the root-mean-square distance (RMSD) value between those two conformations must be less than 2.0 Å. In this case, the validation method was performed by re-docking ERα with E2, ST with N-acetyl-D-glucosamine and 17β-HSD1 with DHT. Low RMSD values were obtained for all cases, which means that the docking process was reliable and validated.
3. Results and discussion
3.1. Chemistry
Four 𝛥9,11-E1 derivatives were synthesized by the general synthetic procedure described in Scheme 1. To the best of our knowledge, three of these derivatives have been synthesized for the first time (compounds 3, 5 and 7). All compounds were characterized by spectral analysis (IR, 1H- and 13C-NMR) and HRMS was also obtained for the new steroids prepared. All spectral data are in agreement with the presented structures. For example, the presence of a 𝛥9,11 double bond was observed from the signal of the C-11 proton that appeared between 6.05 and 6.13 ppm in the 1H-NMR spectra, in accordance with the results described in the literature [27].
Synthetic route to prepare 𝛥9,11-estrone derivatives. Reagents and conditions: (a) DDQ, MeOH, reflux; (b) benzaldehyde, KOH, MeOH, room temperature; (c) I2, morpholine, PhH, room temperature; (d) NBS, EtOH, room temperature.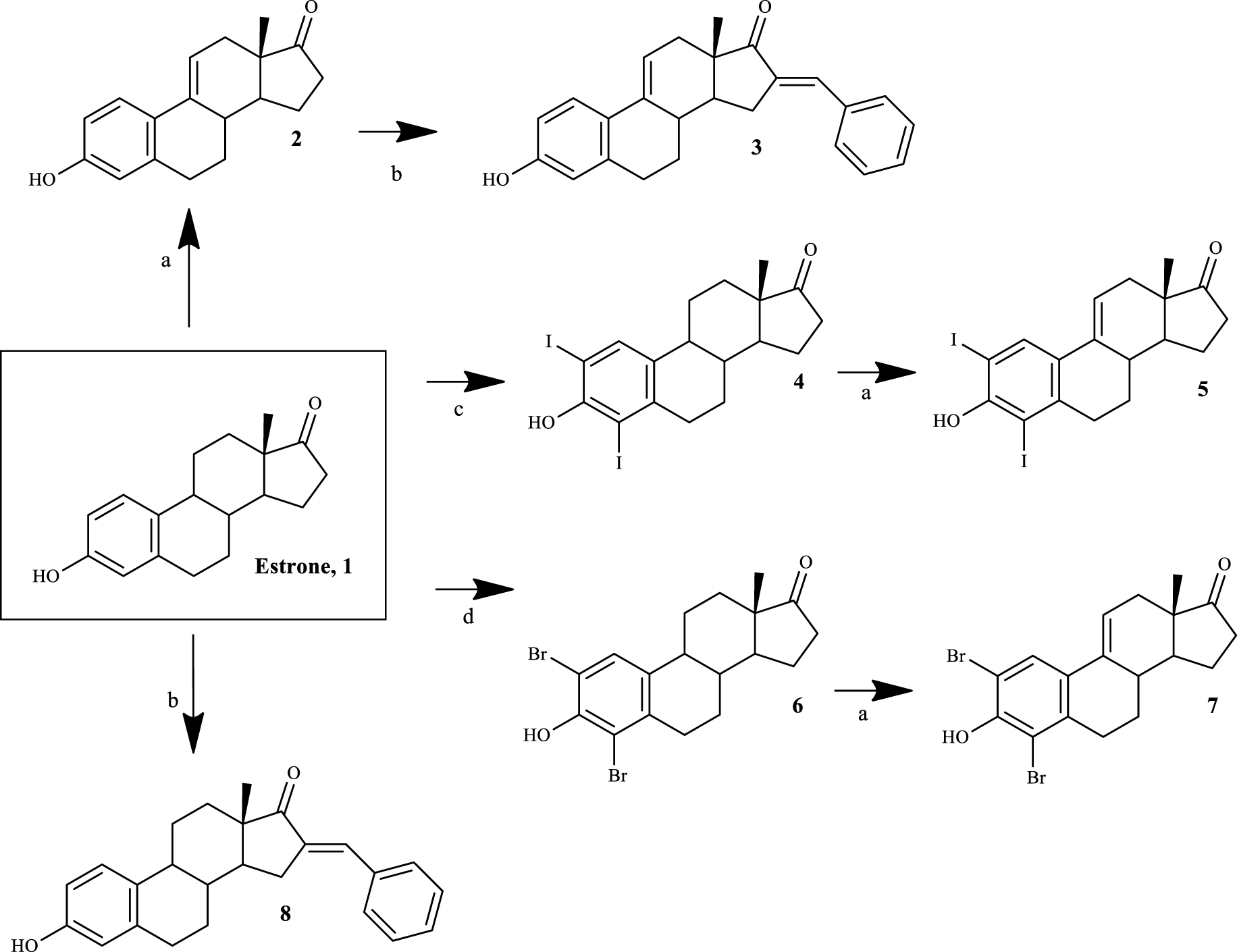
The synthesis of 𝛥9,11-E1 derivatives can be performed using adamantyl carbonium ion as the dehydrogenating agent [36]. In addition, Brown et al. [37] described a simpler route using DDQ to obtain these compounds from E1 in high yield. Later, this last procedure was improved by other research groups [27]. Although the preparation of 𝛥9,11-estrane derivatives has been known since the 1960s, the biological activities of this group of compounds, specifically their potential anticancer activity, continue to be relatively unexplored. In this context, Milic et al. [21] described promising cytotoxicity results of 2- and 4-substituted 𝛥9,11-E1 derivatives in different cancer cell lines, evidencing the interest for this modification in the C-ring of E1. Based on this information, in order to obtain compounds with promising cytotoxic effects, modifications in the A-ring (2- and 4-positions) of E1 were combined with the 𝛥9,11 double bond. In addition, due to the fact that the presence of a 16-arylidene group in the steroid skeleton is also associated with notable cytotoxic properties in several cell lines [16, 35], this modification was likewise explored by us.
Relative cell proliferation of MCF-7, T47-D, LNCaP, HepaRG, Caco-2 and NHDF cells incubated with the synthesized compounds, for 72 h at a 30 μM concentration, determined by the MTT assay, spectrophotometrically quantifying formazan at 570 nm. Data are expressed as a percentage of cell proliferation relative to the negative control and are indicated as means ± SD and are representative of at least two independent experiments. ∗ p < 0.05 vs control.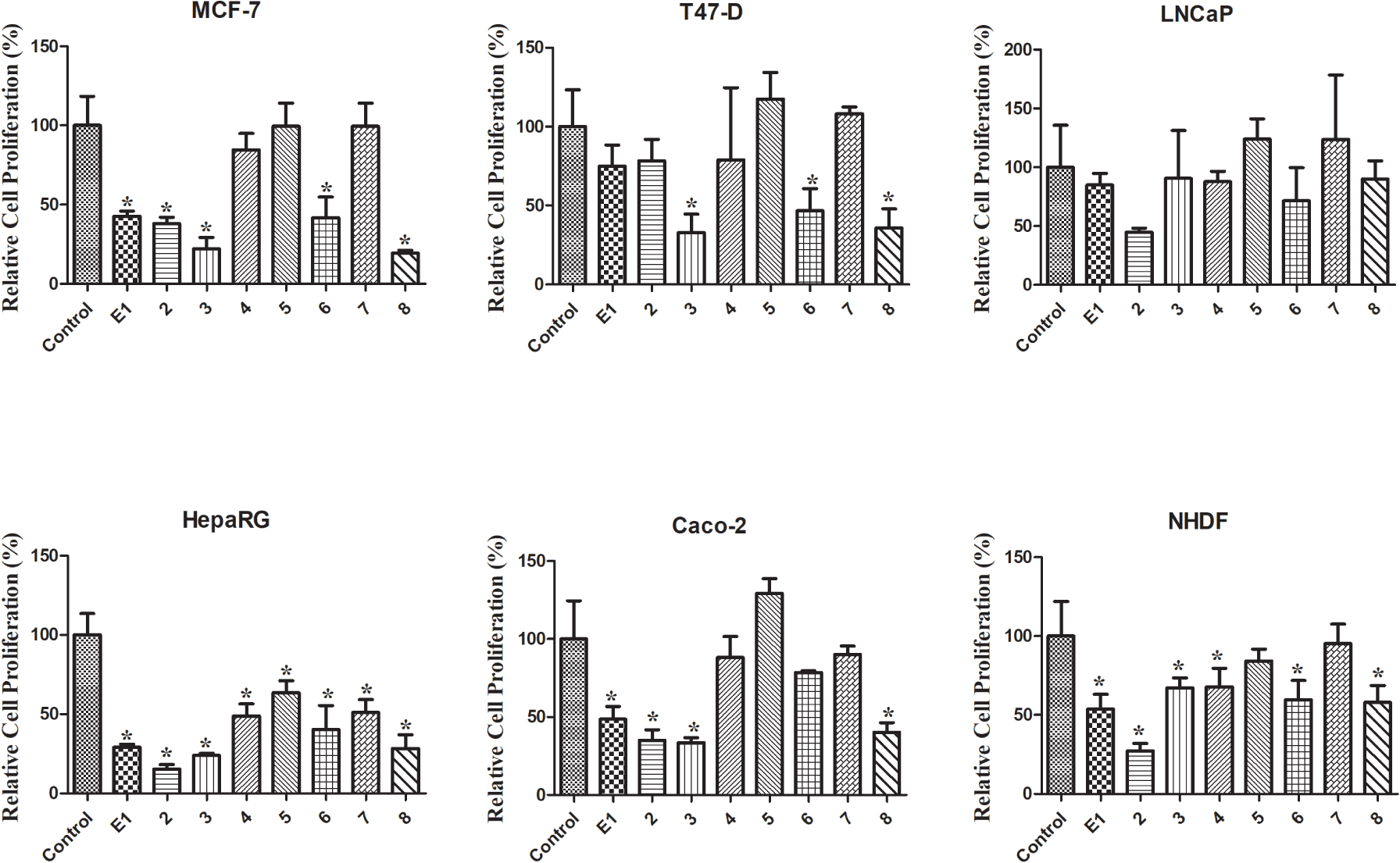
Thus, the introduction of the 𝛥9,11 double bond in E1 yielded compound 2. This process was successfully carried out using DDQ, as described in the literature [27]. Then, using BZ and KOH, compound 3 was easily synthesized through a base-mediated aldol reaction [38], where the corresponding 16E-benzylidene steroid was obtained [25]. In this context, the signal of the methine-bridged proton at C-16 appeared at 7.34 ppm in the 1H-NMR spectra [39], and an E-configuration was assigned to this double bond based on previous reports [25]. E1 A-ring iodination (using I2) and bromination (using NBS) were performed to obtain 2,4-diiodoestrone (compound 4) [40] and 2,4-dibromoestrone (compound 6) [30], respectively. Among these two types of aromatic halogenation, bromination was more simple to perform than iodination. In fact, two other greener strategies were tried before the successful use of I2/morpholine/PhH for the iodination [40]. This last procedure was preferable instead of the combination of sodium iodide and sodium chlorite [41], which only allowed the synthesis of 2-iodoestrone in low yields. In addition, on using I2 and copper (II) chloride dihydrate (CuCl2.2H2O) [42], after the reaction, it was very difficult to separate the isomers 2- and 4-iodoestrone by column chromatography. This iodination also needs a non-oxidant atmosphere, which is more time-consuming. Then, the intermediates 4 and 6 were used to prepare the two new 𝛥9,11 derivatives 5 and 7 by DDQ, as described above. Interestingly, under similar reaction conditions, a higher yield of the product was observed in the dehydrogenation of 2,4-dibromoestrone. Finally, compound 8 was also prepared from E1 by aldol condensation, aiming to improve structure-relationship data by comparing its bioactivity with that observed for compounds 3 and even E1.
Cytotoxicity (IC50 in μM) of the synthesized compounds (1–8) as well as 5-FU against breast (MCF-7 and T47-D), prostatic (LNCaP), hepatic (HepaRG) and colon (Caco-2) cancer cell lines and normal human dermal fibroblasts (NHDF)a
| Compound | MCF-7 | T47-D | LNCaP | HepaRG | Caco-2 | NHDF |
|---|---|---|---|---|---|---|
| IC50 | IC50 | IC50 | IC50 | IC50 | IC50 | |
| 1 | 41.93 | ND | ND | 29.53 | 42.69 | 61.82 |
| 2 | 40.87 | ND | 32.30 | 6.67 | 39.17 | 20.83 |
| 3 | 25.14 | 25.06 | ND | 27.07 | 46.31 | ND |
| 4 | ND | ND | ND | 29.67 | ND | ND |
| 5 | ND | ND | ND | ND | ND | ND |
| 6 | 47.89 | 51.41 | ND | 18.46 | ND | ND |
| 7 | ND | ND | ND | ND | ND | ND |
| 8 | 26.70 | 34.27 | ND | 23.15 | 35.54 | ND |
| 5-FU | 1.71 | 0.54 | 7.79 | 1.78 | 1.31 | 3.61 |
aCells were treated with different concentrations (0.1, 1, 10, 25, 50 and 100 μM) for 72 h. The cell proliferation effects were determined by the MTT assay. The data shown are representative of at least two independent experiments. ND: not determined.
3.2. Biological testing
3.2.1. Cell growth effect
The MTT colorimetric assay was performed to evaluate the cytotoxicity of compounds 1–8 on hormone-dependent (MCF-7, T47-D and LNCaP) and hormone-independent (HepaRG and Caco-2) cancer cells and on NHDF. First, a screening at 30 μM was performed for all compounds in all cell lines (Figure 2). When the reduction of cell proliferation was higher than 50%, the IC50 was determined. As shown in Table 1, the most relevant reduction of cell proliferation was observed with compound 2 in HepaRG cells (IC50 = 6.67 μM). Interestingly, the introduction of the 𝛥9,11 double bond in E1 increased the cytotoxic effects for all cell lines studied, except for T47-D cells. The presence of the 16E-benzylidene group (compound 3) augmented the cytotoxic effects on MCF-7 (IC50 = 25.14 μM) and T47-D cells (IC50 = 25.06 μM) when compared with compound 2. When comparing the bioactivity of compounds 3 and 8, it was interesting to note that the presence of the 𝛥9,11 double bond in these 16E-benzylidenes also led to an increase in the cytotoxicity in breast cell lines but not in HepaRG and Caco-2 cells. In addition, the introduction of iodine in positions 2 and 4 of E1 (compound 4) allowed selective cytotoxicity against HepaRG cells (IC50 = 29.67 μM). The dibrominated steroid 6 generally had higher cytotoxicity than the corresponding iodinated analogue 4. On the other hand, no pronounced reduction of cell proliferation was observed for compounds 5 and 7 in all the cell lines tested. Therefore, contrary to what was observed for E1 and compound 2, the presence of iodine and bromine in positions 2 and 4 of 𝛥9,11-E1 was not a favorable structural change for the development of potential antiproliferative agents.
Regarding the values of selectivity index (SI) (Table 2), it is known that a value of 2 or greater indicates high selectivity for cancer cells [43]. According to this information, the selectivity of compound 2 against the HepaRG cell line is very interesting (SI > 3).
Proliferation of estrogen-sensitive T47-D cells after treatment with 17β-estradiol and compounds 2 and 3 for 24 h. Each bar represents the mean ± SD (originated from two independent experiments). ∗ p < 0.05 vs control; ∗∗ p < 0.01 vs control; ∗∗∗ p < 0.001 vs control.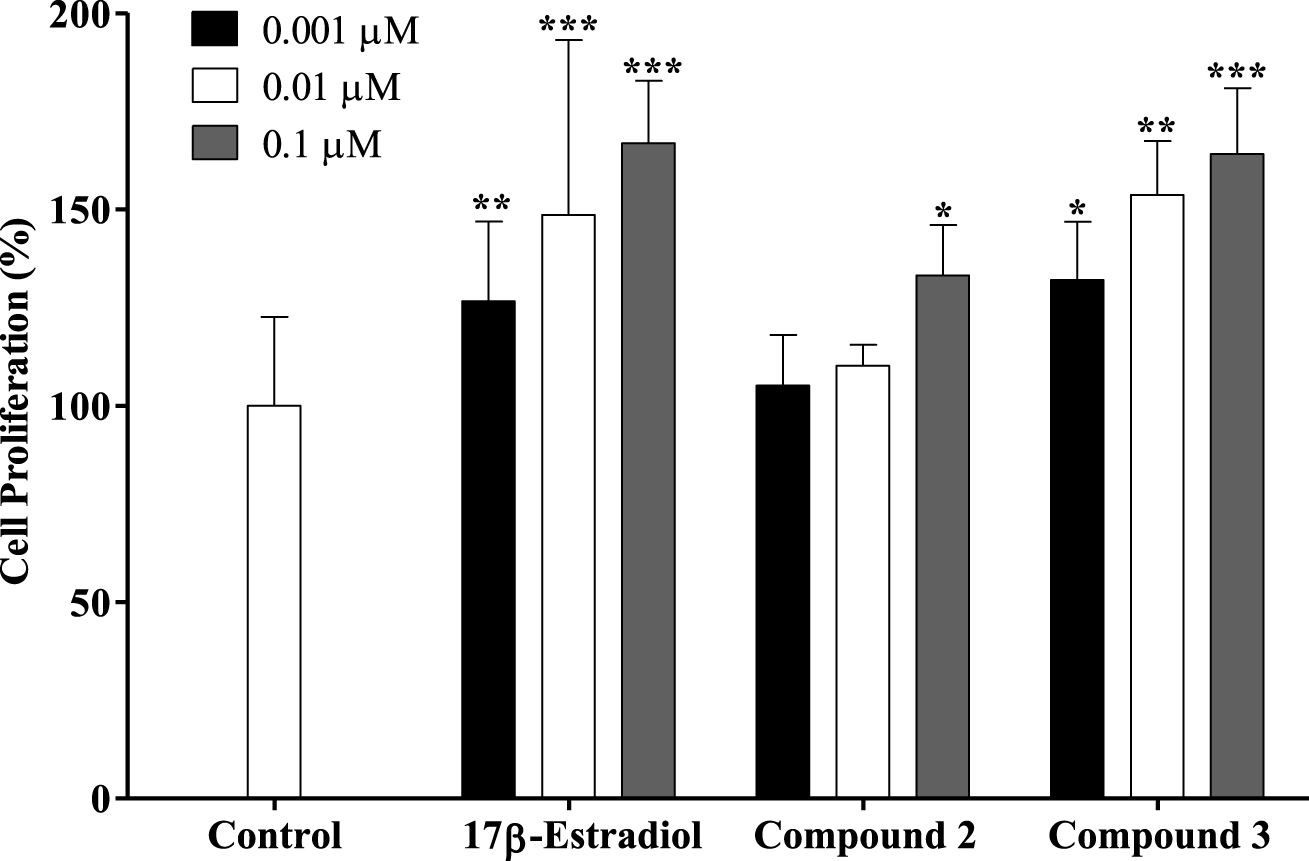
Selectivity indexa of compounds 1, 2 and 5-FU
| Compound | MCF-7 | T47-D | LNCaP | HepaRG | Caco-2 |
|---|---|---|---|---|---|
| 1 | 1.47 | ND | ND | 2.09 | 1.45 |
| 2 | 0.51 | 0.53 | 0.64 | 3.12 | 0.53 |
| 5-FU | 2.11 | 0.49 | 0.46 | 2.03 | 2.76 |
aSelectivity index is the ratio of the IC50 values of the treatments of non-tumor cells (NHDF) and tumor cells (MCF-7, T47-D, LNCaP, HepaRG and Caco-2). ND: not determined.
A new drug candidate should be devoid of estrogenic activity as a pre-requisite for use in cancer therapy. In order to investigate the potential estrogenic profile of the synthesized compounds with the most relevant antiproliferative activities (steroids 2 and 3), their effect on cell growth was measured on the estrogen-sensitive breast cancer T47-D cells (ER+) in a serum-free culture medium. This proliferative/estrogenic activity was expressed as the difference between the cell proliferation (in percentage) caused by a given compound and the basal cell proliferation fixed at 100% (Figure 3) [44, 45]. The natural estrogen E2 was also tested as a reference compound. As expected, E2 had a proliferative effect on T47-D cells in all concentrations tested. Unfortunately, compound 2 also stimulated the cell proliferation at 0.1 μM (133%) when compared with the negative control. Compound 3 also favored cell proliferation in all concentrations tested. In this context, Palomino et al. [46], using X-ray crystallography and molecular modeling studies, showed that the 𝛥9,11 unsaturation in E2 (receptor binding affinity, RBA = 1000) caused a flattening of B-, C- and D-rings and consequently reduced the binding to the ER by one-fifth (RBA = 196). Although the presence of the 𝛥9,11 double bond can change the spatial conformation and reduce the interaction with the ER, it did not eliminate the estrogenic effect characteristic of these compounds as evidenced by our results. In addition, Sakac et al. [47] confirmed the estrogenic effect of compound 3,6β-dihydroxyestra-1,3,5(10),9(11)-tetraene-17β-yl propionate using an immature rat uterine weight assay (approximately 73% of uterus proliferation compared with control). The antiestrogenic activity of this compound was also assessed using an antiuterotrophic method that showed a weak effect (3.22% of antagonism effect versus 62.80% for reference drug tamoxifen) [15]. Novel C-16 and C-17 modified E1 derivatives were synthesized by the Alsayari group [15], showing potent inhibition of cell growth stimulated by E2 and high selective affinity to ERα. In addition, 2-methoxyestra-1,3,5(10),9(11)-tetraen-17-one, which has a 𝛥9,11 double bond, showed estrogenic activity and displayed good binding affinities to ERα (4.09 μM) and ERβ (19.19 μM). It was also demonstrated that a 2-bromoethyl side chain at C-3 and that a carbamoylbenzyl chain at C-16 removed the residual estrogenic activity associated with the estrogen nucleus [44, 48, 49]. However, our data showed that the introduction of the benzylidene group at C-16 was not sufficient to reduce the estrogenic effect of this E1 derivative on T47-D cells.
3.2.2. Cell survival and cell cycle distribution evaluation
Compound 2 was further tested to evaluate its possible mechanism of action by flow cytometry after PI staining. This assay was performed in HepaRG cells, and 5-FU was used as the positive control. In this cell line, it was observed that compound 2 led to a 34% reduction in cell viability after 24 h of treatment (Figure 4). This effect was higher than that caused by 5-FU. In addition to this flow cytometry study, cells were also observed with an optic microscope (Figure 5) and, after 24 h of treatment with compound 2, it was possible to see small modifications in HepaRG cells. The cells lost their shape, becoming more rounded.
Percentage of HepaRG viable cells after 24 h treatment with 50 μM of compound 2 evaluated through propidium iodide (PI) flow cytometry assay. Control corresponds to untreated cells and 5-FU (50 μM) was used for comparison. The percentage of survival is the percentage of cells in R1 (live cells) as compared to the total number of events in R1, R2 (dead cells) and R3 (undetermined cells). Each bar represents the mean ± SD (originated from two independent experiments). ∗∗ p < 0.01 vs control; ∗∗∗ p < 0.001 vs control.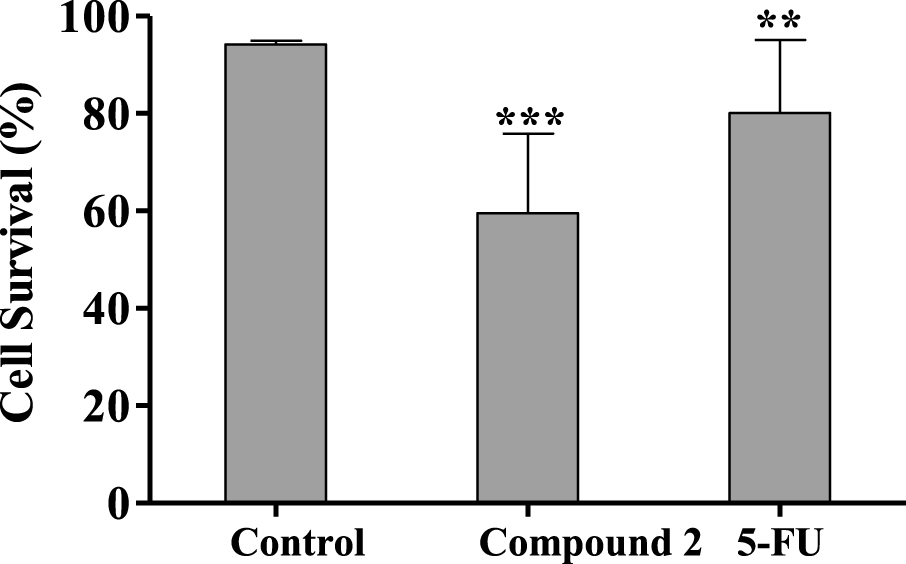
Photographs of HepaRG cells (A) treated with 50 μM of compound 2 (B) and 5-FU (C) for 24 h. Amplification of 100×.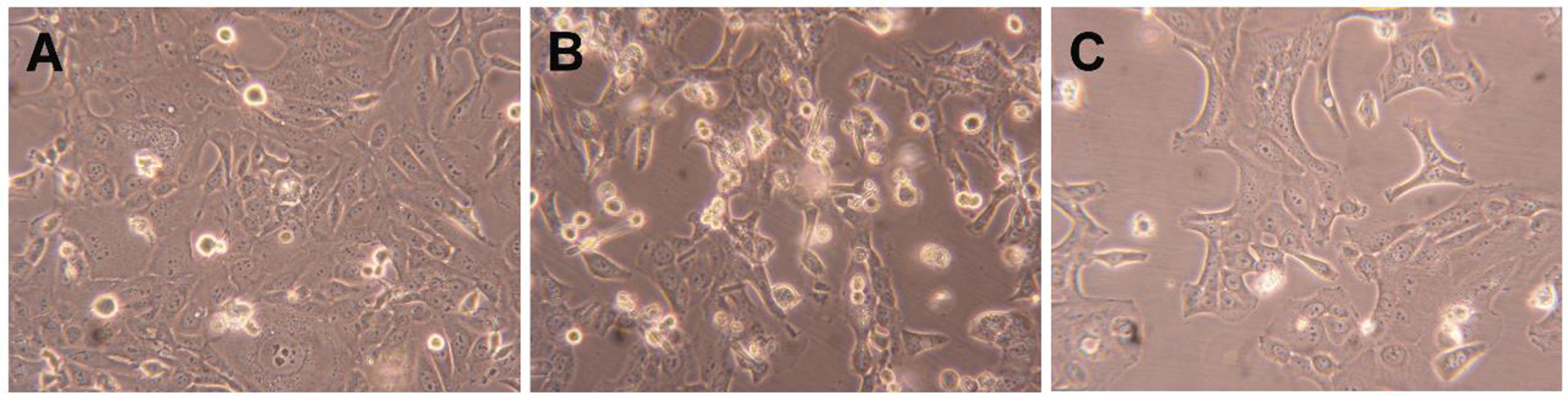
Some studies showed that different steroids led to cell cycle blockage and inhibited some enzymes important for cell cycle regulation. For instance, 16β-triazolyl-17α-estradiol 3-benzyl ethers of the 13α-E2 series showed G2/M cell cycle arrest and caspase inhibition [50]. In addition, new 3-benzyloxy-16-hydroxymethylene-estradiol derivatives led to a G1 phase accumulation and to a proapoptotic effect through the elevation of the apoptotic sub-G1 phase on MDA-MB-231 cells after 24 h treatment (0.1–30 μM). In addition, these compounds were observed to have an antimetastatic activity by inhibition of kinase phosphorylation in a concentration-dependent manner [51]. Taking into account this information, the interference of compound 2 in cell cycle distribution was evaluated by flow cytometry. Interestingly, it was found that the treatment with compound 2 (50 μM, 24 h) induced an apparent G0/G1 cell cycle arrest (Figure 6), reducing the percentage of cells in the S phase (DNA replication). The observed cell cycle arrest in the G0/G1 phase can be related to the interference with one or more of the many proteins that participate in the highly regulated cellular mechanisms which delay or initiate DNA replication [52]. Further studies will be necessary to elucidate which are the signaling pathways that are affected and to ascertain whether other mechanisms are involved in the cytotoxicity of these compounds.
Cell cycle distribution analysis of HepaRG cancer cells after treatment with compound 2 (at 50 μM) for 24 h. A negative control (untreated cells) and a positive control [5-fluorouracil (5-FU), 50 μM] were included. The analysis of cell cycle distribution was performed after propidium iodide (PI) staining and then by flow cytometry. Each bar represents the mean ± SD (originating from two independent experiments). ∗∗ p < 0.01 vs control; ∗∗∗ p < 0.001 vs control.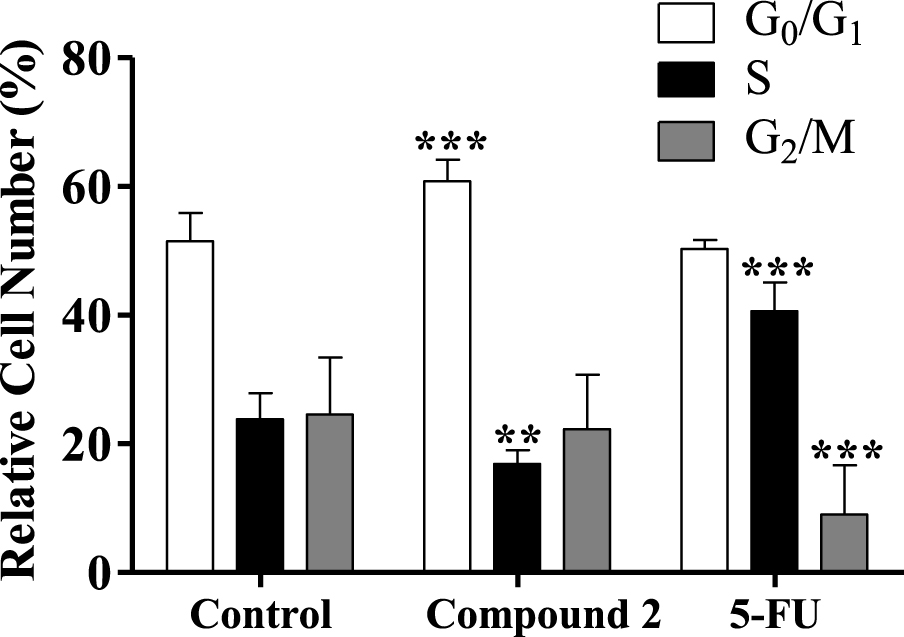
Regarding the effect of compound 2 on the HepaRG cell cycle, we also decided to study HepaRG cell proliferation after 72 h using an adapted protocol with carboxyfluorescein succinimidyl ester, a dye that labels cell cytoplasm and is diluted on cell division [53]. HepaRG cells treated with compound 2 had a higher intensity signal than control cells (Figure 7), meaning that they accumulated a lower number of cell divisions and therefore are less proliferative. Decreased proliferation confirms that cell accumulation in G0/G1 is due to arrest in the cell cycle rather than faster cell cycle progression through S and G2/M phases.
3.3. Molecular docking studies
Molecular docking studies are a determinant in structure-based drug design as it is possible to predict the binding conformation of small molecule ligands to appropriate target binding sites, binding energies and binding mode in the target. In this context, characterization of the binding behavior plays an important role in rational drug design and helps us to elucidate fundamental biochemical processes [54]. This study aimed to evaluate the existence of potential interactions between these 𝛥9,11-estrone derivatives and proteins that are known to interact with these types of steroids.
Relative fluorescence intensity of carboxyfluorescein succinimidyl ester (CFSE) of HepaRG cells, evaluated by flow cytometry after treatment with compound 2 (50 μM) for 48 and 72 h. Each bar represents the median with range of two samples. ∗∗ p < 0.01 vs control; ∗∗∗ p < 0.001 vs control.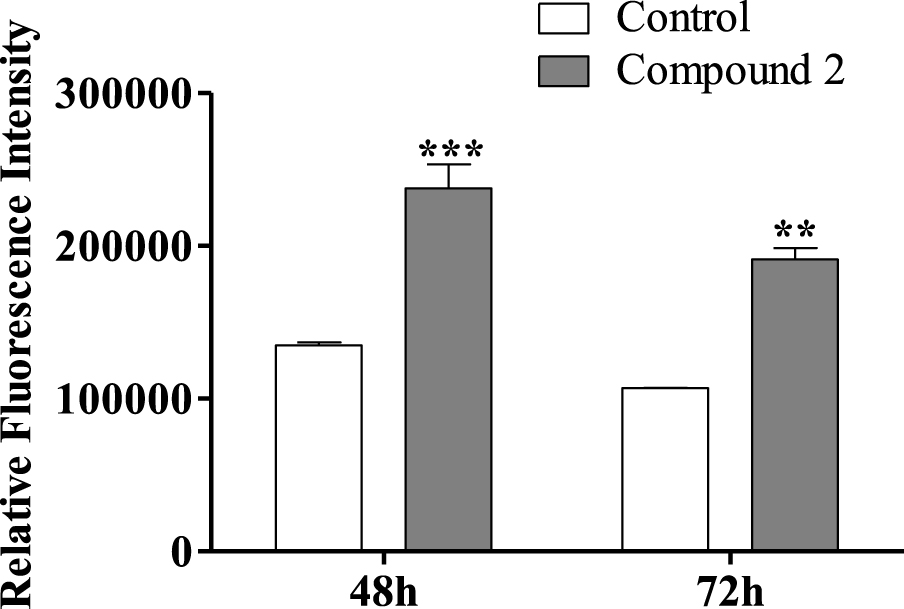
Analysis of predicted ERα binding orientations for the best ranked compound, 2 (binding energies lower than re-docking energies). (A) 3D and (B) 2D docking results showing the main interactions: H bonds with His 524 and Glu 353.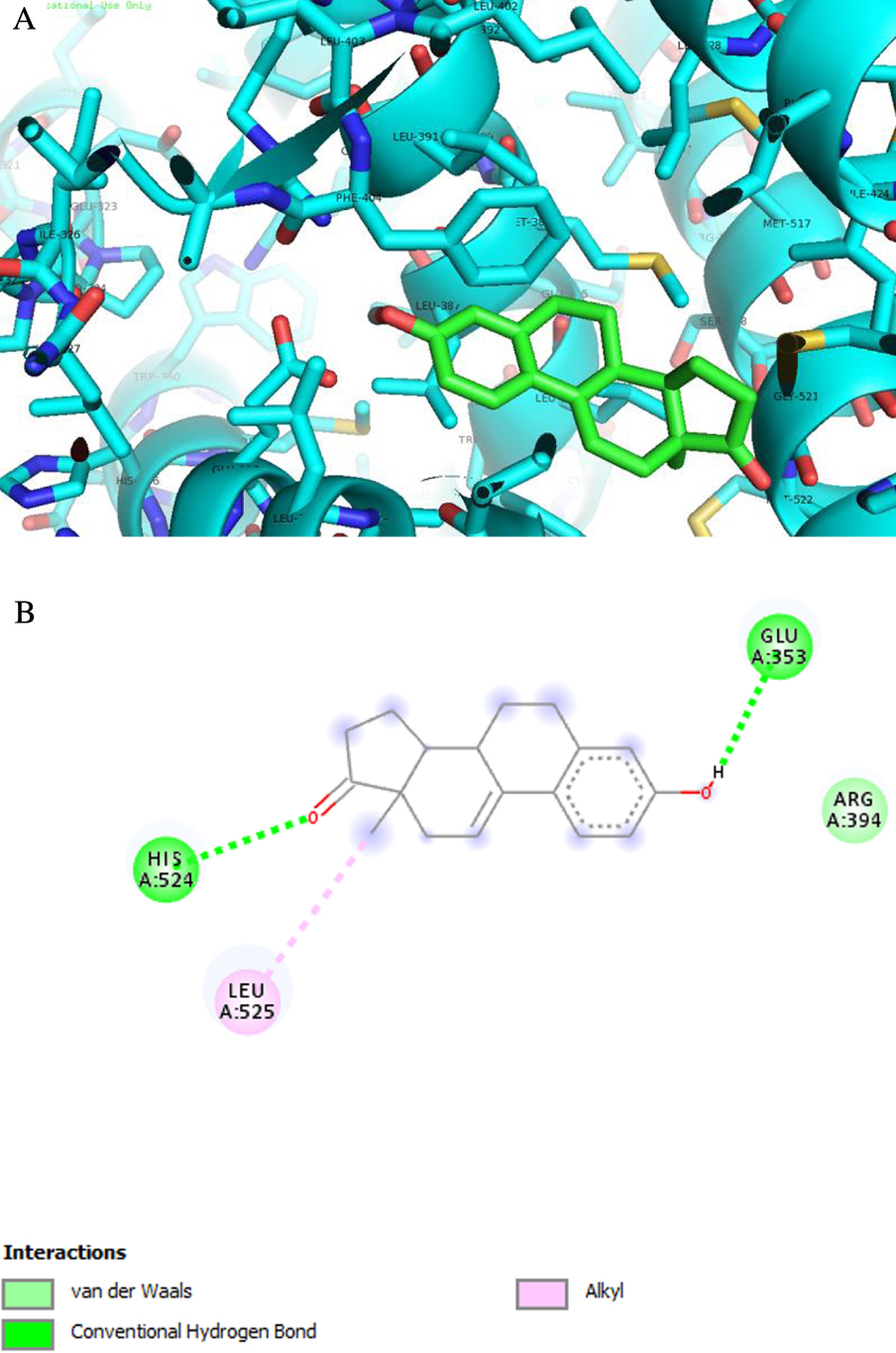
Predicted binding energies of compounds 1–8 calculated against ERα, ST and 17β-HSD1 by AutoDockTools with Vina executable. Binding energies of ligand present in the X-ray crystal structures were calculated by re-docking
| Compounds | Lowest energy (kcal⋅mol−1) | ||
|---|---|---|---|
| ERα | ST | 17β-HSD1 | |
| 1 | −10.3 | −6.2 | −8.1 |
| 2 | −10.9 | −5.9 | −8.2 |
| 3 | −6.9 | −6.3 | −9.8 |
| 4 | −4.3 | −6.0 | −7.7 |
| 5 | −4.1 | −6.5 | −8.0 |
| 6 | −6.8 | −6.3 | −8.1 |
| 7 | −6.3 | −6.3 | −8.3 |
| 8 | −8.3 | −6.9 | −8.5 |
| 17β-estradiol | −9.9a | - | - |
| N-acetyl-D-glucosamine | - | −7.2a | - |
| DHT | - | - | −8.3a |
aThe RMSD between re-docked ligands and the corresponding X-ray crystal structure coordinates was ⩽2.
ERα is a transcription factor that is involved in the regulation of many complex physiological processes in humans. The association between ERα activity and the cell cycle reveals that this receptor also regulates cell proliferation as well as therapeutic resistance [55, 56, 57]. ST and 17β-HSD type 1 are enzymes also involved in cell proliferation [58]. In fact, ST converts estrone sulfate into E1 and 17β-HSD type 1 reduces the 17-ketone of androstane and estrane steroids to the corresponding 17β-hydroxylated derivatives, leading to estrogenic activity [59]. Therefore, their deregulation can contribute to the progression of several hormone-dependent cancers. Three-dimensional structural coordinates of these three protein receptors were obtained from the PDB, and molecular docking was performed using the program AutoDock Vina. To validate the docking method, simulations were carried out and compared to crystallized ligands/drugs complexed with the respective proteins: all control re-docking simulations were able to reproduce the ligand–protein interaction geometries present in the respective crystal structures with an RMSD ⩽2.0 Å. All compounds were docked for ERα, ST and 17β-HSD1 as observed in Table 3. Interestingly, the results revealed that compound 2 can bind ERα at a lower energy than the control 17β-estradiol. From Figure 8 and regarding the ERα target, compound 2 exhibits two hydrogen bonds between the ketone group at C-17 and Hist 524 and between the hydroxyl group at C-3 and Glu 353. These interactions are similar to those observed for E2. As was expected, regarding 17β-HSD1, the lowest energy compared to the control DHT was obtained with compound 3, followed by compound 8. In fact, many studies were published involving modifications at C-16 of E1 and C-2 of E2 to develop 17β-HSD 1 inhibitors [12, 31, 48, 49]. Compound 3 has a 16E-benzylidene group at C-16, which contributes to the interaction with the 17β-HSD 1 target. In this context, it was demonstrated that a flexible linker at the C-16 position gave better 17β-HSD1 inhibition than those with a rigid alkene linker [60]. In addition, Bacsa et al. [29] showed that 2- and/or 4-halogenated 13𝛽- or 13𝛼-estrone derivatives led to a competitive reversible inhibition of 17β-HSD1 and ST enzymes. Regarding the ST enzyme, none of the studied compounds showed relevant interaction with this target. It is known that the presence of a free or N-unsubstituted sulfamate group (H2NSO2O–) is a pre-requisite for potent and irreversible ST inhibition as shown by inhibitors like EMATE [61].
4. Conclusion
In summary, several estranes with A-, C- and D-ring modifications, including three new 𝛥9,11-E1 derivatives, were prepared under mild reaction conditions. The introduction of a 𝛥9,11 double bond and a 16E-benzylidene group in E1 increased the cytotoxic activity on hormone-dependent breast (MCF-7 and T47-D) cancer cells when compared with E1. The introduction of 2,4-diiodo groups in E1 seemed to favor a selectivity for HepaRG cells. However, the presence of 2,4-diiodo and 2,4-dibromo groups in 𝛥9,11-E1 seemed to have no benefit for antiproliferative activities in all the cell lines studied. The most promising result was observed with 𝛥9,11-E1, which exhibited relevant antiproliferative activity against HepaRG cancer cells and presented moderate cytotoxicity on normal human cells. Nevertheless, this compound also showed an estrogenic effect on T47-D cells at 0.1 μM, and flow cytometry analysis revealed a cell cycle arrest at the G0/G1 phase of HepaRG cells. Molecular docking studies estimated a strong interaction between this compound and ERα. In conclusion, the presence of a 𝛥9,11 double bond in estrane derivatives can be of interest in the development of new and interesting antitumor agents.
Acknowledgments
The authors acknowledge the support provided by FEDER funds through the POCI—COMPETE 2020—Operational Programme Competitiveness and Internationalisation in Axis I—Strengthening Research, Technological Development and Innovation (Project POCI-01-0145-FEDER-007491) and National Funds by FCT—Foundation for Science and Technology (Project UID/Multi/00709/2013). CC would also like to thank Professor Silvia Socorro’s group (CICS-UBI) for providing DCC-FCS.
Conflict of interest
The authors confirm that this article content has no conflict of interest.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.17 or from the author. The experimental details and randomization protocols are provided.