



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Over a half-century ago, Martin Gouterman [1] devised his eponymous four-orbital model of porphyrin spectra, and thereby was able to classify them as belonging to three broad classes, normal, hyper and hypso [2, 3]. Briefly, hyper spectra exhibit redshifted features and/or extra features relative to normal spectra, while hypso spectra are blueshifted relative to normal spectra. Although the variety of porphyrinoid compounds is vastly greater today, Gouterman’s classification remains useful. Thus, the hyperporphyrin concept has emerged as a design principle for photosensitizers for photodynamic therapy and related applications [4]. Many hypsoporphyrins are phosphorescent and potentially useful as triplet photosensitizers, in such applications as oxygen sensing and photodynamic and related therapies [5, 6, 7]. The hypso effect had long been attributed to an increase in the HOMO–LUMO gap in certain late transition porphyrins as a result of antibonding interactions between occupied metal dπ orbitals and the porphyrin’s eg LUMOs. A recent DFT investigation, however, has indicated a different origin for the hypso effect, namely, a lowering of the a2u HOMOs in metalloporphyrins with relatively electronegative metal ions such as Pd(II) and Pt(II) [8].
Many years ago, we showed that Gouterman’s four-orbital model also applies to corroles [9, 10]. Unsurprisingly, many metallocorroles exhibit hyper spectra [11, 12, 13] and quite a few also exhibit hypso spectra [8]. Herein we show that the hypso effect also applies to chlorins [14, 15], with both the Soret and Q band maxima of chlorins blueshifting, going from Zn to Pd to Pt as the coordinated metal (Scheme 1). The finding is surprising in that the HOMO of closed-shell chlorins is invariably an a1u-type MO (in terms of D4h irreps) [3, 16, 17, 18, 19] which has no amplitude on the central nitrogen atoms and is, therefore, likely to be unaffected by the electronegativity of the central metal. Gratifyingly, the spectral data proved consistent with electrochemical cyclic voltammetry measurements and DFT/TDDFT calculations, leading to a coherent picture of hypsochlorins, as described below.
A chlorin e6 trimethyl ester metal complex with partial atom numbering.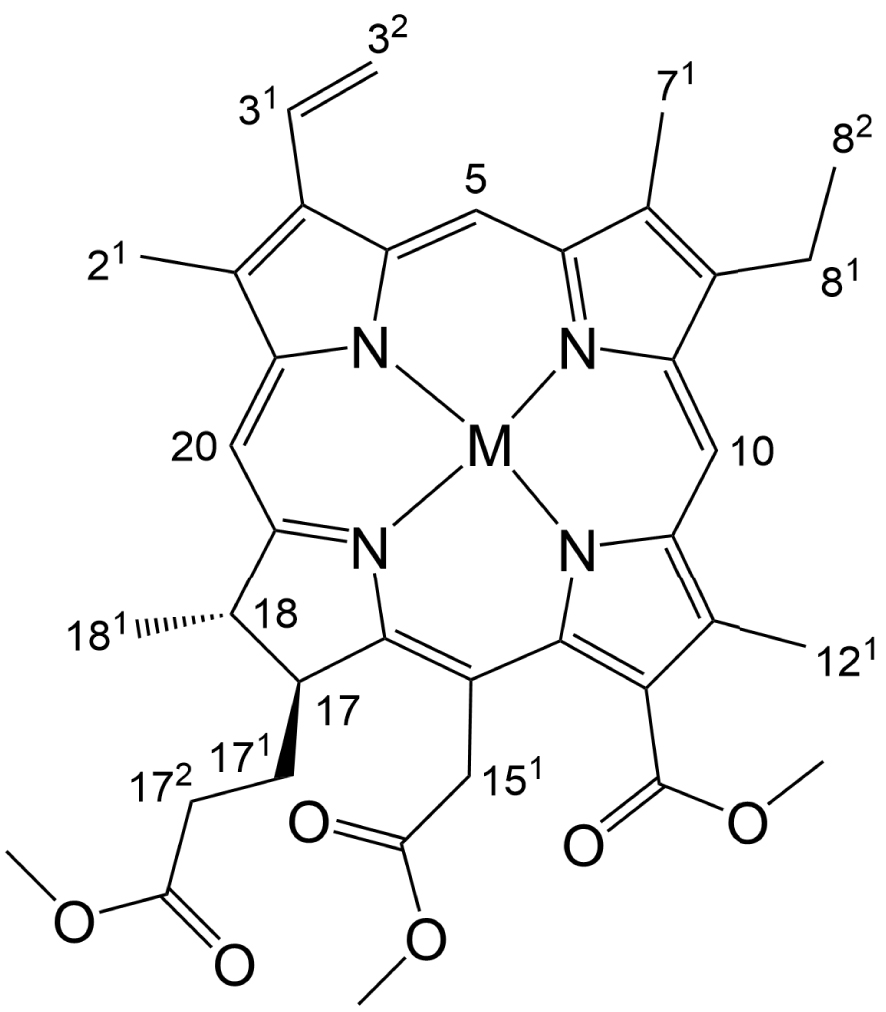
2. Results and discussion
Table 1 lists the UV–vis absorption maxima and redox potentials (V versus SCE) of the Zn, Pd, and Pt complexes of chlorin e6 trimethyl ester, M[Chl-e], where M = Zn, Pd, and Pt; the UV–vis spectra and cyclic voltammograms are depicted in Figures 1 and 2, respectively. From Zn to Pd and from Pd to Pt, the Q band blueshifts by 14 and 10 nm, respectively, and the Soret by 8 and 10 nm, respectively. The two minor features between the Soret and Q bands also blueshift in the same order. All complexes exhibit reversible 1st oxidations and 1st reductions. While there are small variations in the 1st reduction potentials there are greater variations in the 1st oxidation potentials and a clear trend of increasing oxidation potentials in the order of Zn < Pd < Pt, which in turn results in an increasing electrochemical HOMO–LUMO gap for the same series. The electrochemical data thus suggests that the hypso effect in chlorins stems primarily from a lowering of the HOMO energy in the presence of an electronegative coordinated metal.
UV–visible spectra of M[Chl-e], where M = Zn, Pd, and Pt, in CH2Cl2 at ambient temperature.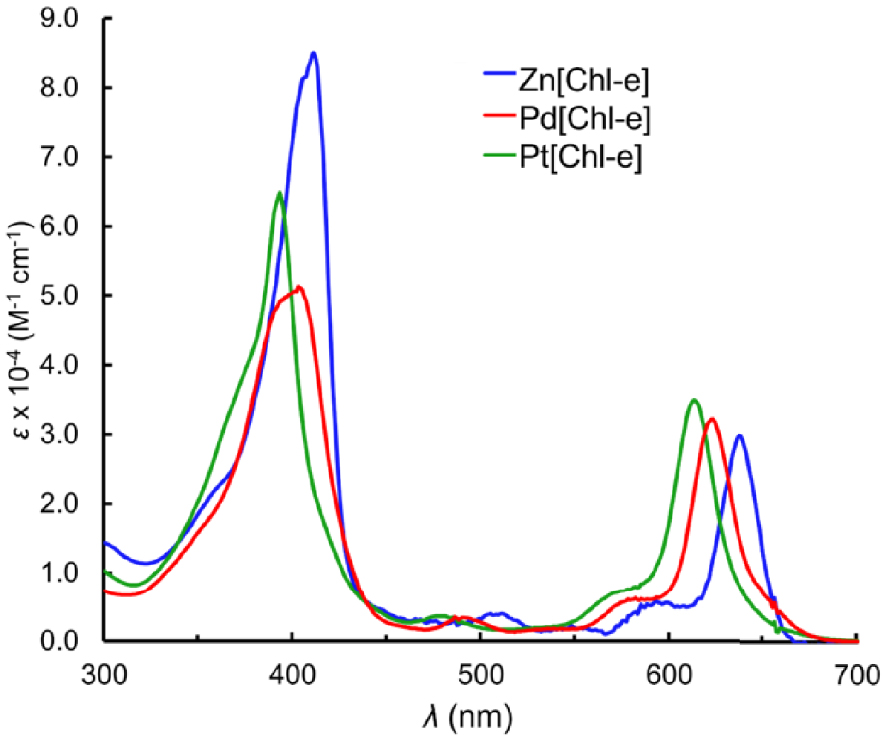
Cyclic voltammograms (V versus SCE) of M[Chl-e], where M = Zn, Pd, and Pt, in CH2Cl2 containing 0.1 M tetrabutylammonium perchlorate at ambient temperature; scan rate 100 mV⋅s−1.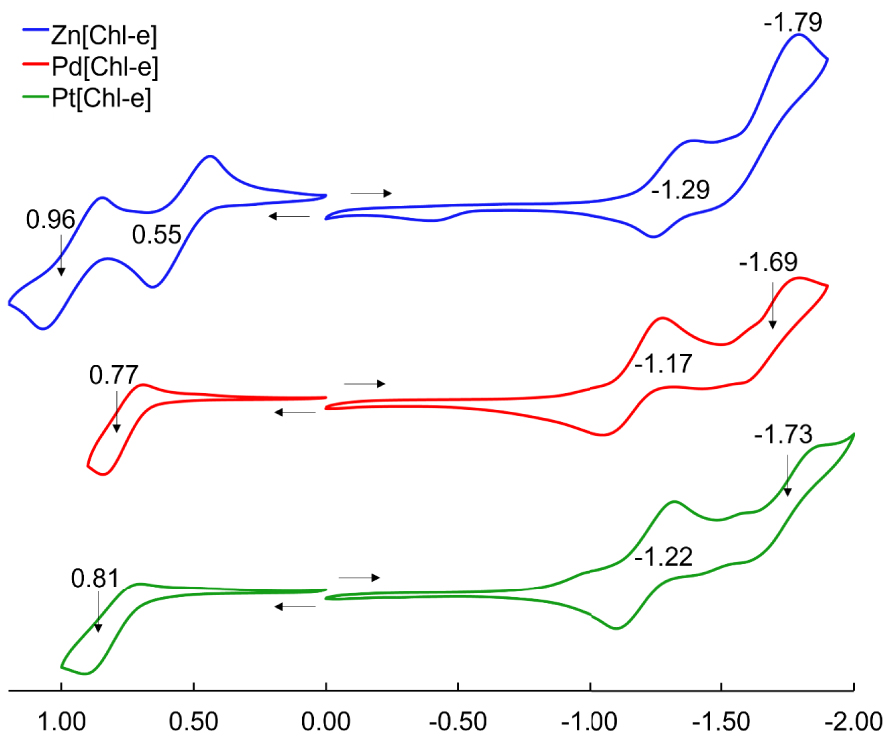
Redox potentials (V versus SCE) and UV–vis absorption maxima of the compounds studied
| Compound | E1∕2ox2 | E1∕2ox1 | E1∕2red1 | E1∕2red2 | Ered2 | 𝛥E | Soret band (nm) | Q band (nm) |
|---|---|---|---|---|---|---|---|---|
| Zn[Chl-e] | 0.96 | 0.55 | −1.29 | – | −1.79 | 1.84 | 411 | 637 |
| Pd[Chl-e] | – | 0.77 | −1.17 | −1.69 | – | 1.94 | 403 | 623 |
| Pt[Chl-e] | – | 0.81 | −1.22 | −1.73 | – | 2.03 | 393 | 613 |
To obtain a qualitative understanding of the above findings, DFT and TDDFT calculations were carried out using simple unsubstituted chlorin models of the complexes studied. The OLYP [20, 21]-D3 [22]/ZORA-STO-TZ2P optimized geometries of M[Chl] (M = Zn, Pd, Pt; Chl = unsubstituted chlorin; Figure 3) revealed essentially planar (as opposed to ruffled) geometries so differences in macrocycle conformation appear unlikely to be a significant contributor to the observed spectral differences [23]. Single-point CAMY-B3LYP [24] calculations were then carried out on the OLYP-D3 geometries. Figure 4 depicts selected CAMY-B3LYP frontier MOs along with the corresponding orbital energies, while Figure 5 presents the orbital energy levels in a graphical manner. Figure 6 depicts the simulated TD-CAMY-B3LYP-D3/Q4 STO-TZ2P-COSMO optical spectra in dichloromethane of the three M[Chl] derivatives. The key result is that, while the LUMO and LUMO+1 energies are essentially constant across all three M[Chl] derivatives, both the HOMO and HOMO-1 energies downshift going from Zn to Pd to Pt. Understandably, the downshifts for the a1u-typeHOMO are smaller than those for the a2u-type HOMO-1, given that only the latter has substantial amplitudes on the central nitrogen atoms. In spite of the simplicity of our models, these computational results appear to nicely, if qualitatively, capture the essence of both the UV–vis and electrochemical measurements.
Selected skeletal bond distances for M[Chl], where M = Zn, Pd, and Pt and Chl = unsubstituted chlorin.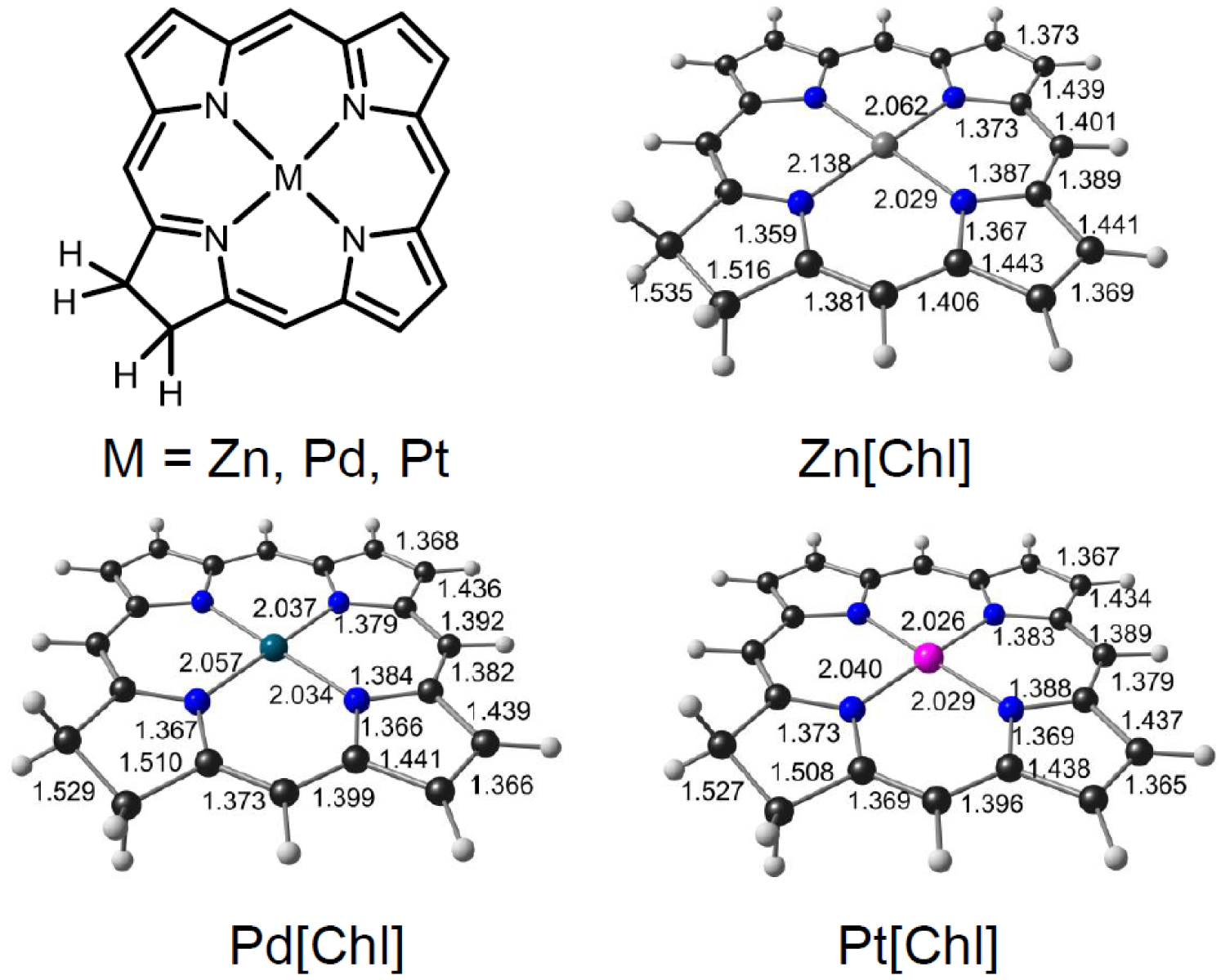
CAMY-B3LYP frontier MOs of M[Chl] (M = Zn, Pd, Pt), with C2 irreps (a and b) and orbital energies.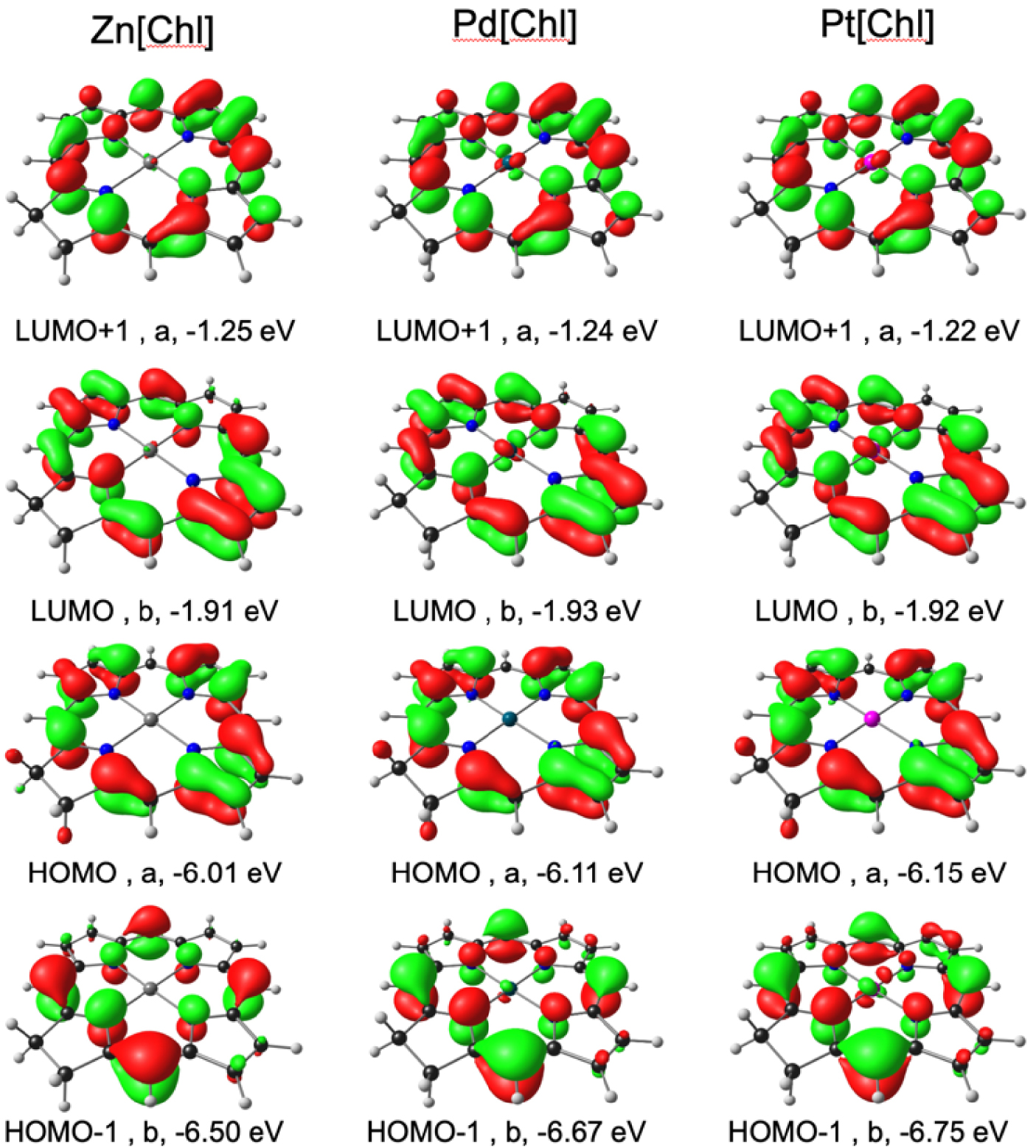
CAMY-B3LYP-D3/STO-TZ2P-COSMO frontier MO energy levels along with C2 irreps (a and b).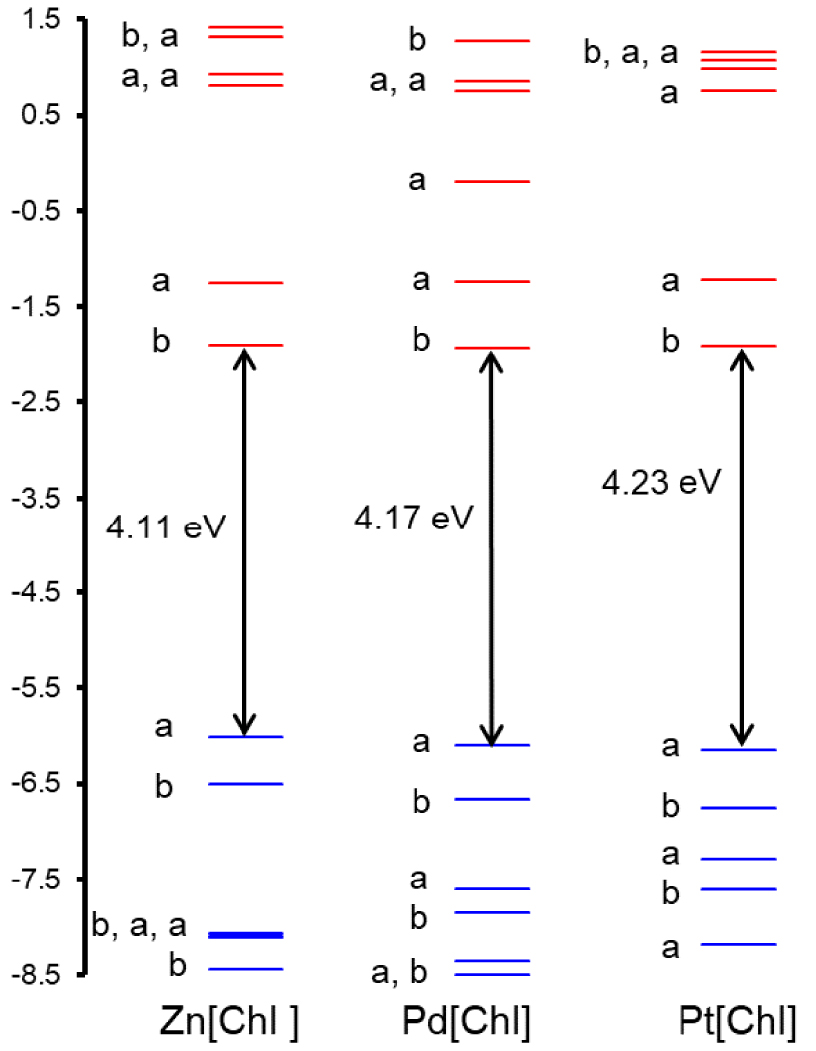
Simulated TD-CAMY-B3LYP-D3/STO-TZ2P-COSMO optical spectra in dichloromethane. The vertical lines represent calculated transitions which have then been broadened with Gaussians to generate the simulated spectra.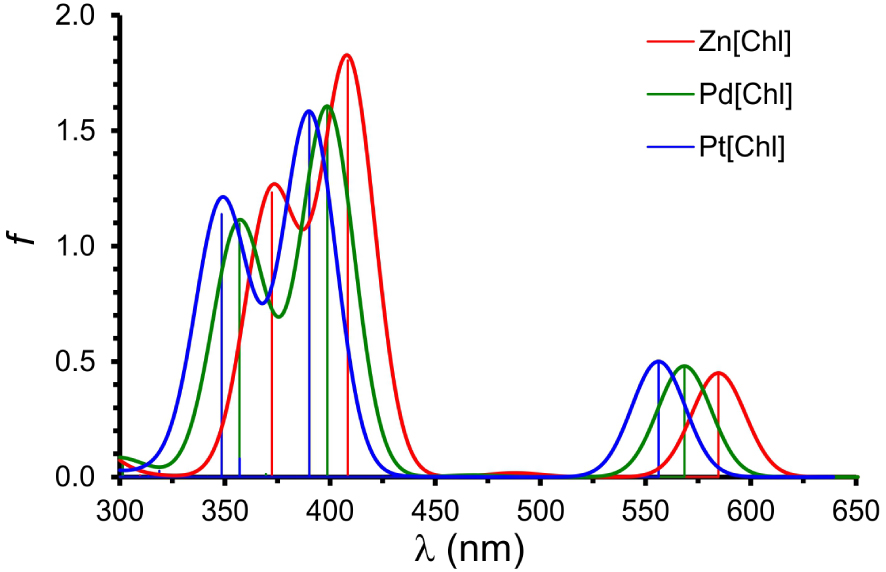
Finally, the calculated absorption maxima from CAMY-B3LYP TDDFT calculations on M[Chl] in dichloromethane (Figure 6) were found to be in surprisingly good agreement with the experimental absorption maxima for M[Chl-e]. Thus, for instance, the calculations correctly captured the larger hypsochromic shifts for the Q band relative to the Soret band. In all three cases, the Q band consists of approximately 95% HOMO → LUMO character, with the remainder made up of HOMO-1 → LUMO+1 character (Table 2). The Soret band, by comparison, consists of a mixture of HOMO-1 → LUMO and HOMO → LUMO+1 character. Since both the HOMO and HOMO-1 downshift from Zn through Pd to Pt, it makes sense that both the Q and Soret absorption maxima blueshift in the same order.
CAMY-B3LYP-D3/STO-TZ2P TDDFT results, including transition energies (E) and wavelengths (𝜆), oscillator strengths (f), MO compositions, and symmetries
| Molecule | Peak | E (eV) | 𝜆 (nm) | f | MO composition | State symmetry | ||
|---|---|---|---|---|---|---|---|---|
| From | To | Weight (%) | ||||||
| Zn[Chl] | Q | 2.12 | 584.6 | 0.45 | HOMO | LUMO | 92.6 | B |
| HOMO-1 | LUMO+1 | 6.3 | B | |||||
| Soret | 3.04 | 408.4 | 1.80 | HOMO | LUMO+1 | 62.3 | A | |
| HOMO-1 | LUMO | 35.8 | A | |||||
| 3.33 | 372.3 | 1.23 | HOMO-1 | LUMO+1 | 91.4 | B | ||
| HOMO | LUMO | 6.4 | B | |||||
| Pd[Chl] | Q | 2.18 | 568.4 | 0.48 | HOMO | LUMO | 94.3 | B |
| HOMO-1 | LUMO+1 | 4.5 | B | |||||
| Soret | 3.11 | 398.7 | 1.60 | HOMO-1 | LUMO | 59.8 | A | |
| HOMO | LUMO+1 | 38.2 | A | |||||
| 3.47 | 356.9 | 1.10 | HOMO-1 | LUMO+1 | 92.5 | B | ||
| HOMO | LUMO | 4.5 | B | |||||
| Pt[Chl] | Q | 2.23 | 556.1 | 0.50 | HOMO | LUMO | 94.8 | B |
| HOMO-1 | LUMO+1 | 3.9 | B | |||||
| Soret | 3.18 | 390.1 | 1.58 | HOMO | LUMO+1 | 56.5 | A | |
| HOMO-1 | LUMO | 40.9 | A | |||||
| 3.56 | 348.4 | 1.14 | HOMO-1 | LUMO+1 | 92.6 | B | ||
| HOMO | LUMO | 3.8 | B | |||||
In summary, a combined experimental and theoretical study indicates the plausible occurrence of a hypso effect in chlorins. Thus, both the Soret and Q bands blueshift going from Zn to Pd to Pt as the coordinated metal. As in the case of porphyrins, the blueshifts appear to be largely attributable to the presence of an electronegative coordinated atom, which lowers the orbital energies of the HOMO and HOMO-1, while leaving the LUMOs relatively unaffected.
3. Experimental section
3.1. Materials
All reagents were purchased from Sigma-Aldrich and used as received except chlorin e6 trimethyl ester (CAS 35038-32-5) which was purchased from PorphyChem. Silica gel 60 (0.04–0.063 mm particle size, 230–400 mesh, Merck) was employed for flash chromatography. Silica gel 60 preparative thin-layer chromatographic plates (20 cm × 20 cm × 0.5 mm, Merck) were used for final purification of all compounds.
3.2. General instrumental methods
UV–visible spectra were recorded on an HP 8454 spectrophotometer. 1H NMR spectra were recorded on a 400 MHz Bruker Avance III HD spectrometer equipped with a 5 mm BB/1H SmartProbe and referenced to either residual C6H6 at 7.16 ppm or residual CHCl3 at 7.26 ppm. High-resolution electrospray-ionization mass spectra were recorded on an Orbitrap Exploris 120 spectrometer using methanolic solutions. Cyclic voltammetry was carried out at ambient temperature with a Gamry Reference 620 potentiostat equipped with a three-electrode system: a 3 mm disc glassy carbon working electrode, a platinum wire counter-electrode, and a saturated calomel reference electrode (SCE). Tetra(n-butyl)ammonium perchlorate (CAUTION!) was used as the supporting electrolyte. Anhydrous CH2Cl2 (Sigma-Aldrich) was used as solvent. The electrolyte solution was purged with argon for at least 2 min prior to all measurements, which were carried out under an argon blanket. The glassy carbon working electrode was polished using a polishing pad and 0.05 micrometer polishing alumina from ALS Japan. All potentials were referenced to the SCE.
3.3. Synthesis
The Zn complex was made by adding a solution of Zn(OAc)2∙2H2O (29.5 mg, 4 eq.) in 1 mL MeOH to a solution of chlorin e6 trimethyl ester (21.9 mg) in 5 mL chloroform. The resulting solution was refluxed for 1 h. The Pd complex was made by dissolving chlorin e6 trimethyl ester (17.3 mg) and Pd(OAc)2 (19.7 mg, 3 eq.) in 5 mL pyridine and refluxing the resulting solution for 4 h. The Pt complex was made by dissolving chlorin e6 trimethyl ester (18.0 mg) and Pt(OAc)2 (26.3 mg, 3 eq.) in 5 mL benzonitrile and refluxing the resulting solution for 10 h. Following reflux, each mixture was rotary-evaporated to dryness and the residue dissolved in DCM and placed on a silica column. Dichloromethane/ethyl acetate 20:1 eluted a blue fraction which was evaporated to dryness and further purified by preparative TLC using n-hexane/ethyl acetate 1:1 as solvent. Yields and analytical details are as follows:
3.3.1. Zn[Chl-e]
Yield 16.8 mg, 70%. UV–Vis (CH2Cl2) 𝜆max (nm) [𝜖 × 10−4 (M−1⋅cm−1)]: 411 (8.50), 505 (0.40), 593 (0.58), 637 (2.98). 1H NMR (400 MHz, C6D6, 𝛿 in ppm): 9.54 (s, 1H, 5), 9.41 (s, 1H, 10), 8.50 (s, 1H, 20), 8.05 (dd, J = 17.8, 11.5 Hz, 1H, 31), 6.18 (d, J = 18.0 Hz, 1H, 32 trans), 5.91 (d, J = 1.8 Hz, 1H, 32 cis), 5.45 (d, J = 19.0 Hz, 1H, 151), 5.28 (d, J = 19.0 Hz, 1H, 151), 4.28 (d, J = 10.2 Hz, 1H, 17), 4.01 (t, J = 7.1 Hz, 1H, 18), 3.95 (s, 3H, OCH3), 3.59 (s, 3H, OCH3), 3.47 (q, J = 7.5 Hz, 2H, 81), 3.38 (s, 3H, OCH3), 3.26 (s, 3H, 121), 3.25 (s, 3H, 21), 3.01 (s, 3H, 71), 2.30 (dt, J = 15.9, 7.9 Hz, 1H, 171), 2.07 (d, J = 10.0 Hz, 1H, 171), 2.00–1.91 (m, 1H, 172), 1.67 (d, J = 7.2 Hz, 3H, 181), 1.60 (t, J = 7.6 Hz, 3H, 82), ∼1.55 (1H, 172 concealed by overlapping peaks). MS (ESI, positive mode): m/z calcd for C37H40N4O5ZnH 701.2312; [M + H+] found 701.2307.
3.3.2. Pd[Chl-e]
Yield 1.9 mg, 9.4%. UV–Vis (CH2Cl2) 𝜆max (nm) [𝜖 × 10−4 (M−1⋅cm−1)]: 403 (5.14), 486 (0.35), 582 (0.62), 623 (3.22). 1H NMR (400 MHz, CDCl3, 𝛿 in ppm): 9.57 (s, 1H, 5/10), 9.56 (s, 1H, 5/10), 8.62 (s, 1H, 20), 7.98 (dd, J = 17.8, 11.5 Hz, 1H, 31), 6.16 (dd, J = 17.8, 1.7 Hz, 1H, 32 trans), 6.03 (dd, J = 11.5, 1.7 Hz, 1H, 32 cis), 5.14 (d, J = 19.1 Hz, 1H, 151), 4.93 (d, J = 19.0 Hz, 1H, 151), 4.48–4.35 (m, 2H, overlapping 17 and 18), 4.19 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.75 (q, J = 7.6 Hz, 2H, 81), 3.62 (s, 3H, OCH3), 3.44 (s, 3H, 121), 3.33 (s, 3H, 21), 3.29 (s, 3H, 71), 2.58–2.46 (m, 1H, 171), 2.24–2.11 (m, 1H, 171), 2.07–1.95 (m, 1H, 172), 1.75 (d, J = 7.1 Hz, 3H, 181), 1.69 (t, J = 7.6 Hz, 3H, 82) ∼1.69 (1H, 172 concealed by overlapping peaks). MS (APCI, positive mode): m/z calcd for C37H40N4O5PdH 743.2055; [M + H+] found 743.2065.
3.3.3. Pt[Chl-e]
Yield 5.8 mg, 24.7%. UV–Vis (CH2Cl2) 𝜆max (nm) [𝜖 × 10−4 (M−1⋅cm−1)]: 393 (6.49), 478 (0.36), 613 (3.49). 1H NMR (400 MHz, CDCl3, 𝛿 in ppm): 9.56 (s, 1H, 5), 9.52 (s, 1H, 10), 8.70 (s, 1H, 20), 7.98 (dd, J = 17.8, 11.5 Hz, 1H, 31), 6.14 (dd, J = 17.8, 1.7 Hz, 1H, 32 trans), 6.02 (dd, J = 11.4, 1.6 Hz, 1H, 32 cis), 5.13 (d, J = 19.1 Hz, 1H, 151), 4.93 (d, J = 19.1 Hz, 1H, 151), 4.46–4.34 (m, 2H, overlapping 17 and 18), 4.20 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.71 (q, J = 7.6 Hz, 2H, 81), 3.63 (s, 3H, OCH3), 3.50 (s, 3H, 121), 3.31 (s, 3H, 21), 3.24 (s, 3H, 71), 2.54 (ddd, J = 16.1, 9.0, 7.0 Hz, 1H, 171), 2.20 (ddd, J = 16.2, 7.2, 4.8 Hz, 1H, 171), 2.09–1.97 (m, 1H, 172), 1.76 (d, J = 7.1 Hz, 3H, 181), 1.68 (t, J = 7.6 Hz, 3H, 82), ∼1.71 (1H, 172 concealed by overlapping peaks). MS (APCI, positive mode): m/z calcd for C37H40N4O5PtH 832.2668; [M + H+] found 832.2670.
3.4. Computational method
The three M[Chl] (M = Zn, Pd, Pt] complexes were optimized with scalar-relativistic DFT calculations, with the OLYP [20, 21] functional augmented with Grimme’s D3 [22] dispersion correction, all-electron ZORA STO-TZ2P basis sets, fine integration grids, and tight criteria for both SCF cycles and geometry optimizations, as implemented in the ADF 2019 program system [25]. All the optimizations were carried out with a C2 symmetry constraint and the COSMO (Conductor like Screening Model) [26] solvation model with dichloromethane as solvent. TDDFT calculations were performed on the OLYP-D3 optimized geometries using the CAMY-B3LYP [24] range-separated functional (which has been extensively calibrated for calculations of porphyrin spectra [4, 7, 27]), with basis set and other parameters the same as in the ground-state calculations).
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
This research was supported by the Research Council of Norway (grant no. 262229 to AG), the CNRS (grant UMR UBCNRS 6302), and the South African National Research Foundation (grant nos. 129270 and 132504 to JC).
Acknowledgment
The authors also warmly thank Mrs. Sandrine Pacquelet for technical assistance.