

1 Introduction
Fullerenes, C60 and C70, linked to multiple redox- and/or photoactive entities are useful candidates to build molecular/supramolecular electronic devices and artificial light energy harvesting systems. Toward this aim, electron donors such as porphyrin, ferrocene, N,N-dimethylaminophenyl, ruthenium(II) trisbipyridine, and tetrathiafulvarene have been employed to form fullerene-electron donor type dyads, whereas electron acceptors such as benzoquinone derivatives, spiroannelated methano groups, cyano groups, fluorine atoms, TCNQ and DCNQI derivatives, ammonium cations, and nitroaromatic substituents have been employed to form covalently linked fullerene-electron acceptor type dyads [1–12]. Molecular and supramolecular systems are made by combining two or more of these systems to form dyads and hybrid triads bearing three or more redox- or photo-active entities. Assembly is accomplished by covalently linking subunits and/or by self-assembly by coordinate metal–ligand bonds, H-bonding, electrostatics, or by π–π porphyrin–fullerene interactions [2]. Determination of the geometric and electronic structure of these newly developed dyads and triads pose a formidable task, but have been elucidated from optical absorption, electrochemical, and computational methods.
Computational calculations often provide the much needed information on the geometry, and in some instances, the correct electronic structure of molecular/supramolecular systems. Choice of a computational procedure, however, depends upon the level of the needed accuracy, size of the molecular system, and the available computational facility. Often, semi-empirical calculations at the AM1 and PM3 level provide the geometry; however, they may or may not predict the electronic structure accurately. Higher level ab initio calculations could predict both geometry and the electronic structures accurately, however, such calculations are very time consuming for molecules and supramolecules bearing large donor and acceptor entities. We have been trying to identify a low-to-moderate level ab initio or density functional method that could predict the geometry and electronic structure of large size molecular and supramolecular dyads with some certainty, while utilizing reasonable computational time and commercially available computational software. Among the different procedures tested on the molecular/supramolecular systems developed in our laboratory, B3LYP/3-21G(*) seems to work best. We hereby summarize examples of successful application of the DFT B3LYP/3-21G(*) model, on a wide range of molecular/supramolecular systems, to the rationalization of the geometric and electronic properties of these very large systems and compare to other models where available for these large systems.
1.1 Computational strategy
The procedures and examples shown here utilize a computational strategy popularized by Pople:
- ● select a computational ‘Model’. Here we consider only computational quantum chemistry models, specifically the surprisingly successful B3LYP/3-21G(*) method, but other methods such as PBE, PW91 or BP86, combined with 3-21G(*) or other basis sets, have promise for properties of systems of these sizes;
- ● explore ‘Chemistry’ in the ‘Model Universe’ (here the B3LYP/3-21G(*) universe);
- ● when results from the computational ‘model’ adequately overlap experimental results one can ‘predict’ (interpolate) results for similar, but experimentally unknown systems;
- ● with greater risk, one can extend (extrapolate) the ‘model’ to systems less similar to the systems used to justify the ‘model’;
- ● if ‘model’ results are in error but have a consistent trend (e.g., 10% high) for a certain property or subset of systems, one can apply ‘standard’ empirical corrections to the ‘model’ results for these situations.
1.2 Computational method—B3LYP/3-21G(*) model optimized geometries
The B3LYP/3-21G(*) method has been successfully used to predict the geometry and electronic structure of molecular and self-assembled supramolecular dyads and triads. Generally in this method, the dyads or triads were ‘built’ utilizing the GAUSSVIEW program, and then optimized to a stationary point on the Born–Oppenheimer potential-energy surface, usually to the defaults in the GAUSSIAN-03 [13] program. For some of the large systems treated here, semi-empirical Hamiltonians, AM1 or PM3, were used for a few geometry optimization cycles to remove large geometry distortions. For systems with SCF convergence problems, a smaller minimal basis set, such as STO-2G or STO-3G, is used to provide a stable electronic wavefunction to ‘start’ the calculation.
For association energies of supramolecules or bond energies of covalently bound systems, the energy of the complex is compared to the sum of the optimized energies of the separated porphyrin, fullerene, ferrocene, etc. parts, or in some cases, by calculating a single point energy (non-relaxed) or a few cycles of geometry optimization (relaxed) of a separated system created by simply ‘pulling’ the supramolecule apart to a nearest atom distance of greater than an 8 Å. It may be mentioned that some of the dyads and triads are very flexible and multiple minima are possible. Under these conditions, the optimization process requires much experience and considerable computational time to confidently achieve an optimized structure at the B3LYP/3-21G(*) level. Insight is gained by using a variety of starting geometries, then evaluating the resulting local minima properties with respect to their consistency. Semi-empirical and molecular mechanics conformational searches may help to identify potential minima, but often the energy surfaces are not similar, leading to false semi-empirical minima or missed B3LYP/3-21G(*) minima.
1.3 Special advantages of the B3LYP/3-21G(*) model
- ● Calculations are possible on large systems containing one or more C60 and porphyrin entities.
- ● The shape and location of the B3LYP/3-21G(*) Kohn–Sham (KS) orbitals agree with chemical intuition.
- ● The orbital energies track oxidation and reduction potentials and maintain functional group integrity.
- ● The highest occupied molecular orbital–lowest unoccupied molecular orbital (HOMO–LUMO) energy gap correlates well with the S0–S1 optical transition and the sum of the oxidation and reduction potentials from electrochemical measurements. The agreement with optical transitions (and by inference with the electrochemical redox gap) is improved by explicit calculation using time dependent density functional theory (TD-DFT).
- ● The total calculated energy is group additive for dyad and triad supra molecular donor–acceptor assemblies with weakly interacting groups.
- ● B3LYP/3-21G(*) calculations demonstrate an interaction potential for weakly bound systems that appears like a dispersion interaction with a minimum that is close to experimental distances.
- ● HOMO and LUMO localization on different regions of a supramolecular assembly may be associated with charge transfer on excitation.
- ● Delocalized KS orbitals may be associated with through–space interactions leading to rapid energy transfer.
1.4 Disadvantages of the B3LYP/3-21G(*) model
- ● The small basis set leads to a large Basis Set Superposition Error (BSSE) such that, in some cases, normal correction by the counterpoise method completely removes dispersion bonding. A half-correction [14,15], however, may yield usable results, while no correction is preferable in most cases.
- ● B3LYP/3-21G(*) bond energies and association energies for loosely bound systems (such as systems self assembled by coordinate metal–ligand bonds, electrostatics, or by π–π porphyrin–fullerene interactions, van der Waals, etc.) are consistently much larger than experiment, probably due to the large BSSE, or Basis Set Incompleteness Error (BSIE) inherent in the 3-21G(*) basis. This may be an advantage, however, for the initial location of shallow minima on the potential energy surface (PES) of loosely bound entities, since the resulting geometries are, in general, quite good. Since the association values are consistently high, except for the most loosely bound complexes, an empirical correction can be applied for similar systems. The bond energy can be substantially improved by a single point B3LYP/6-31G* energy calculation of the joined and separated systems, or for small systems, optimization (relaxation) of the systems at the B3LYP/6-31G* or higher level. This reduction of BSSE and BSIE by using larger basis sets is preferable to applying counterpoise corrections. Other DFT methods have been suggested for improvement of bond dissociation energies; e.g. BP86 for cobalt–chlorin interactions [15] and X3LYP for van der Waals complexes [16,17].
2 Studied examples: fulleropyrrolidine derivatives
2.1 Acid–base properties of fulleropyrrolidines
The acid–base equilibria of 2-(n-alkyl)fulleropyrrolidines, 1 and N-methyl-2-(n-alkyl)fulleropyrrolidines, 2 in aqueous micellar media of a sodium dodecyl sulfate surfactant were examined experimentally and modeled theoretically by using Hartree–Fock 3-21G(*) methods [18]. The pKa values of these compounds determined both by HCl and NaOH titration with potentiometric detection of the end point, are equal to 6.3 and 7.5, respectively, some 5 pKa units lower than unsubstituted pyrrolidine. The calculations were performed on four conformations of the pyrrolidine substituents (i.e. N–R1 anti or syn and C–R2 cis or trans) of the fulleropyrrolidine and with small model compounds designed to quantitate the electronic and structural effects causing the large pKa shift, namely pyrrolidine alone, pyrrolidine with fused saturated dicyclopentapentalene ring, and pyrrolidine with fused unsaturated cyclopenta-acenaphthylene ring. The results indicate that the fullerene cage fused to a pyrrolidine ring increases the acidity of the protonated pyrrolidine nitrogen mainly due to the induced electronic effects and, to a much smaller extent, by structural effects. The HF/3-21G(*) estimated free energy difference, ΔE (with solvation corrections incorporated based on the Cramer–Trular SM5.4 solvation model), between the protonated and unprotonated fulleropyrrolidines, and the experimental pKa values have been correlated for 37 different nitrogenous bases. A 0 < pKa < 14 range was covered with these bases which include fulleropyrrolidines (Fig. 1). Correlation between the calculated ΔE and experimental pKa values was quite good for such a large variety of bases (pKa = [280.49 – ΔE3-21G(*)(sol)]/2.3126, correlation coefficient = 0.95, standard deviation pKa = 0.87) and indicates that the effect on pKa of the micellar environment, utilized in the study for solubilization of the fulleropyrrolidines, is negligible. Comparison of the calculated HOMO and LUMO energy levels for the protonated and free-base forms of the investigated fulleropyrrolidines indicates that electron deficiency of the C60 cage and the HOMO–LUMO energy gap caused by N-protonation are increased. Later studies [19] utilizing the B3LYP/3-21G(*) model gave similar but slightly better correlations for a larger range of bases (59 bases covering the pKa range from –5 to +14, correlation coefficient = 0.968, mean absolute deviation, MAD = 0.78), and reasonable results for acids were achieved [19] with the B3LYP/3-21+G(*) model (used in order to adequately treat the anions involved with acid dissociation).
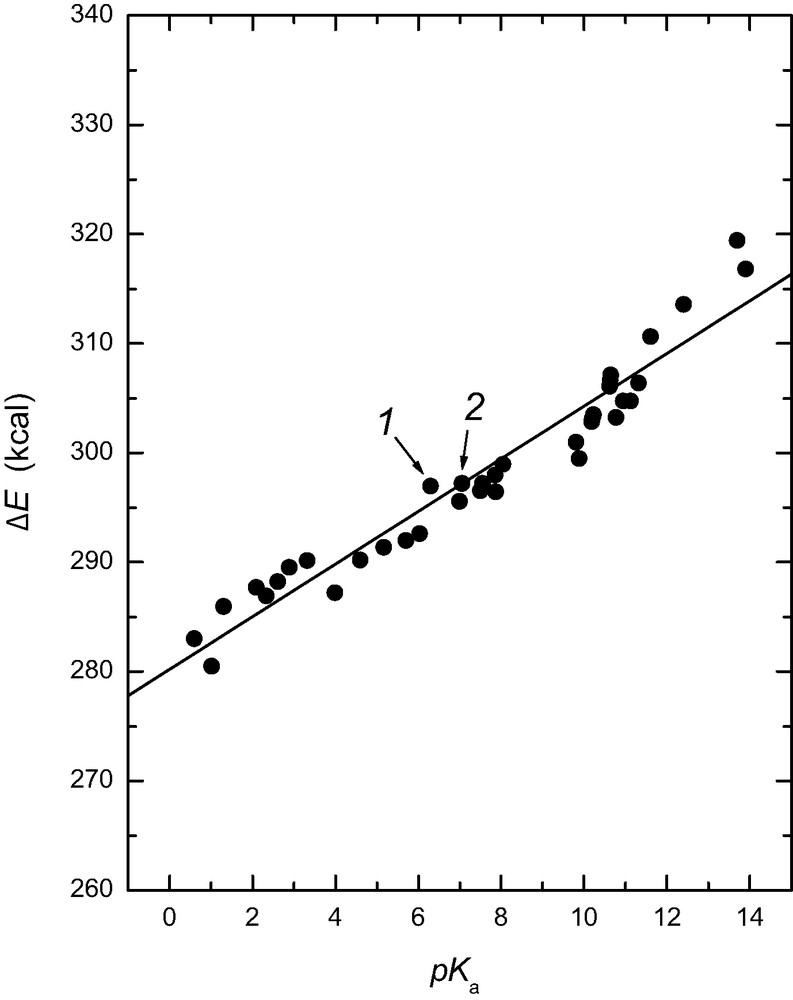
Plot of the HF/3-21G(*) calculated energy difference, ΔE, between the protonated and unprotonated nitrogenous bases corrected for solvation as a function of the experimental pKa values. The datum point for 2-(n-alkyl)fulleropyrrolidine, 1 and datum point for N-methyl-2-(n-alkyl)fulleropyrrolidine, 2 are indicated with arrows (taken from Ref. [18]).
2.2 C60–ferrocene–dinitrobenzoyl triad–orbital energies
One of the first successful applications of B3LYP/3-21G(*) to the interpretation of electrochemical and optical properties of a donor–acceptor system involving fullerene and ferrocene entities was the (a) 2-(ferrocenyl)fulleropyrrolidine dyad, 3 and the (b) N-(nitroaromatic functionalized)-2-(ferrocenyl)fulleropyrrolidine triads, 4–5 and similar systems [20,21] (Scheme 2).
Electrochemical studies on 4 revealed multiple redox processes involving all three redox active, ferrocene, C60, and dinitrobenzoyl entities. A total of eight reversible redox couples within the accessible potential window of o-dichlorobenzene containing 0.1 M (TBA)ClO4 are observed. A comparison between the measured redox potentials with those of the starting compounds revealed absence of any significant electronic interactions between the different redox entities, and the reduction potentials alternated between the C60 and dinitrobenzoyl groups.
Our attempts by using semi-empirical PM3 and Hartree–Fock ab initio methods with a 3-21G(*) basis resulted either in incorrect geometry or electronic structure for the studied dyad and triads, i.e. the HOMO orbital was found to lie on the C60 entity. However, the energy optimized B3LYP/3-21G(*) structure was in agreement with the electrochemical studies [20]. The first HOMO was found to be completely localized on the ferrocene entity, whereas the first LUMO was localized mainly on the C60 entity with small orbital contributions on the dinitrobenzoyl entity (Fig. 2). The HOMO–LUMO gap of 2.12 eV is larger than the electrochemical 1.24 eV, which, from later work, is presumably due to the ferrocene HOMO energies being calculated too low (note that the TD-DFT calculated singlet–singlet transition is somewhat smaller, thus closer to experiment). Surprisingly (at the time), the subsequent LUMOs were found to track the observed site of electrochemical reductions of the triad (Fig. 3), alternating from C60 to dinitrobenzoyl groups. Note that for the compound 5, N-(4′-nitrobenzoyl)-2-(ferrocenyl)-fulleropyrrolidine, that where the electrochemical reduction potentials overlap (Fig. 4), the corresponding B3LYP/3-21G(*) orbitals are near degenerate with considerable delocalization over both the C60 and nitrobenzoyl acceptors [21].
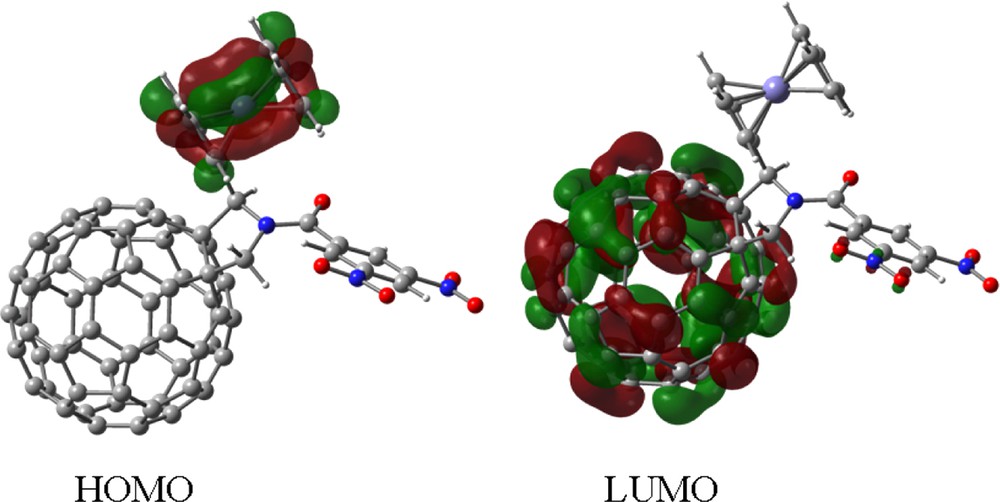
Frontier HOMO and LUMO, of the investigated N-(3′,5′-dinitrobenzoyl)-2-(ferrocenyl)-fulleropyrrolidine triad, 4.
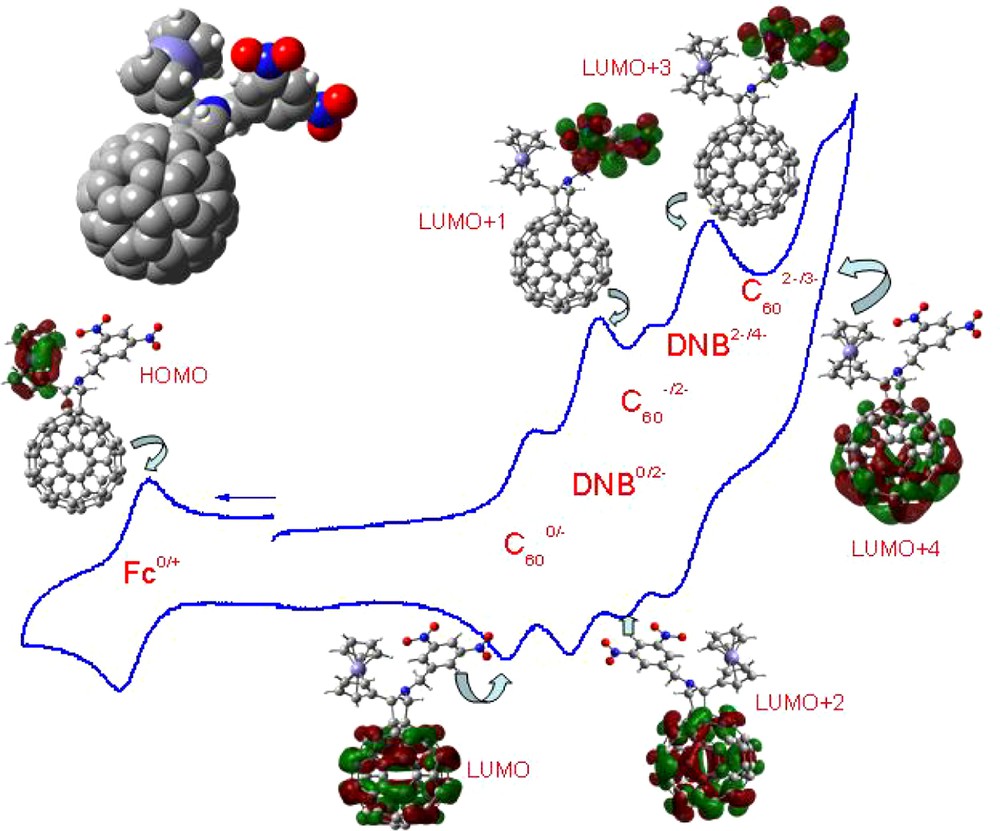
Top left: Space filling model of the B3LYP/3-21G(*) optimized structure of N-(3,5-dinitrobenzyl)-2-ferrocenyl fulleropyrrolidine, 4. The cyclic voltammogram of the title compound in 0.1 M tetrabutylammonium perchloride in o-dichlorobenzene (scan rate = 100 mV s–1) along with the frontier HOMO and LUMO, LUMO + 1, LUMO + 2, LUMO + 3, and LUMO + 4 orbitals corresponding to each of the indicated redox couple showing the occurrence of the sequence of the site of electron transfer (see Refs. [20,21] for additional details).
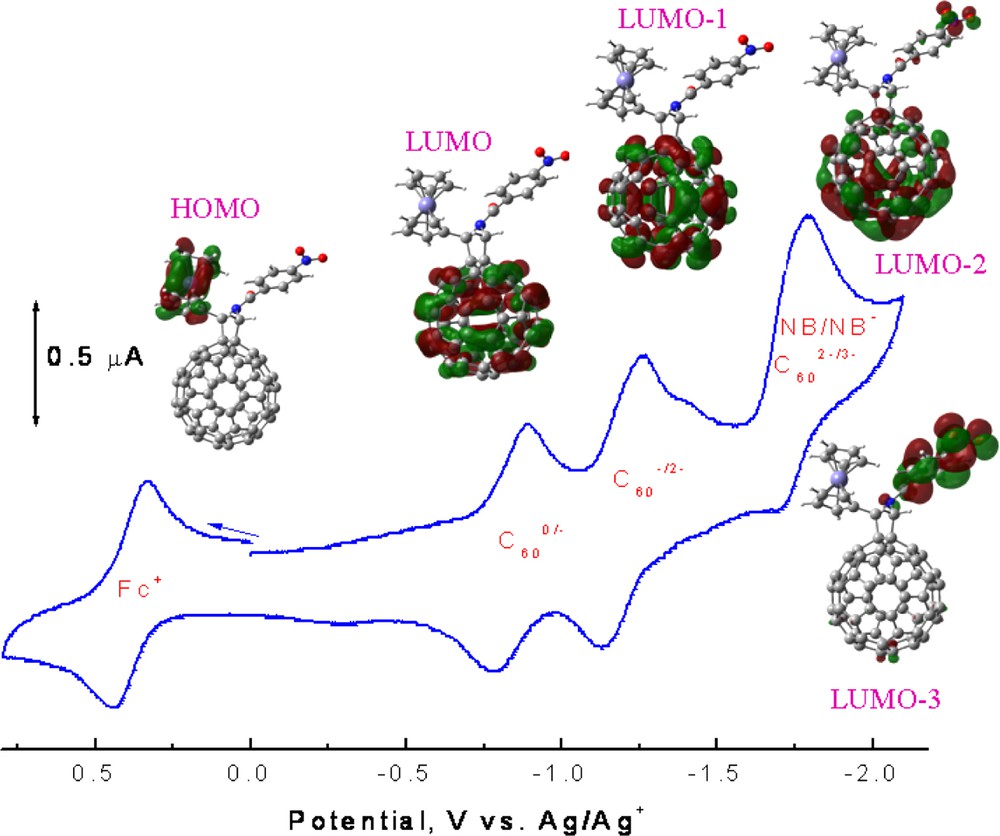
The cyclic voltammogram of 5 in 0.1 M tetrabutylammonium perchloride in o-dichlorobenzene (scan rate = 100 mV s–1) along with the frontier HOMO and LUMO, LUMO – 1, LUMO – 2, and LUMO – 3, orbitals corresponding to each of the indicated redox couple showing the occurrence of the sequence of the site of electron transfer.
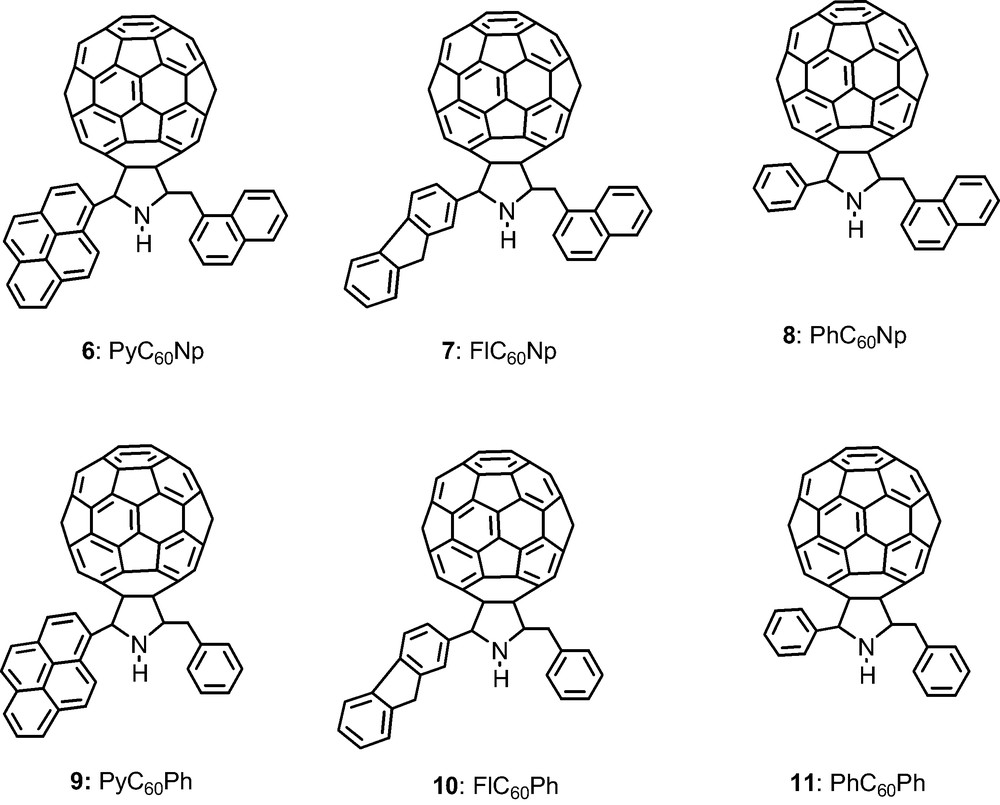
2.3 Group additive properties: fluorophore(s) appended fullerene dyads and triads
Fullerene, C60, was functionalized to possess one or two fluorophore entities, 6–11 (Scheme 3), such that the dyads contained either a naphthalene, pyrene, or fluorene entities while the triads contain either a pyrene or fluorene entity in addition to a naphthalene entity [22]. While photoinduced energy transfer is expected in the case of the dyads upon excitation of the fluorophore, at least two photochemical events were envisioned for the triads, that is, energy transfer from one of the excited fluorophores to the fullerene, or stepwise energy transfer from one fluorophore to the other followed by energy transfer from the excited fluorophore to the fullerene. Additionally, electron transfer from the excited fluorophore to the fullerene is also possible. Hence, there might be an ‘antenna effect’ for light induced energy transfer in the molecular triads. Electrochemical measurements using cyclic voltammetry showed that only C60 reductions were observed whereas the oxidations were dependent on the fluorophore and were irreversible. Fig. 5 shows the B3LYP/3-21(*) optimized structures of the six compounds with the respective total energies in atomic units. It is remarkable that differences in energy are constant; implying a Benson like group additivity for the non-interacting groups, i.e. the energy increment from phenyl to naphthyl is a constant 152.7936 a.u. (good to 0.00003 a.u. or about 0.02 kcal mol–1). Similarly, the energy increment from phenyl to fluorenyl is 267.6900 a.u. and from fluorenyl to pyrenyl is 113.7096 a.u. with equal accuracy.
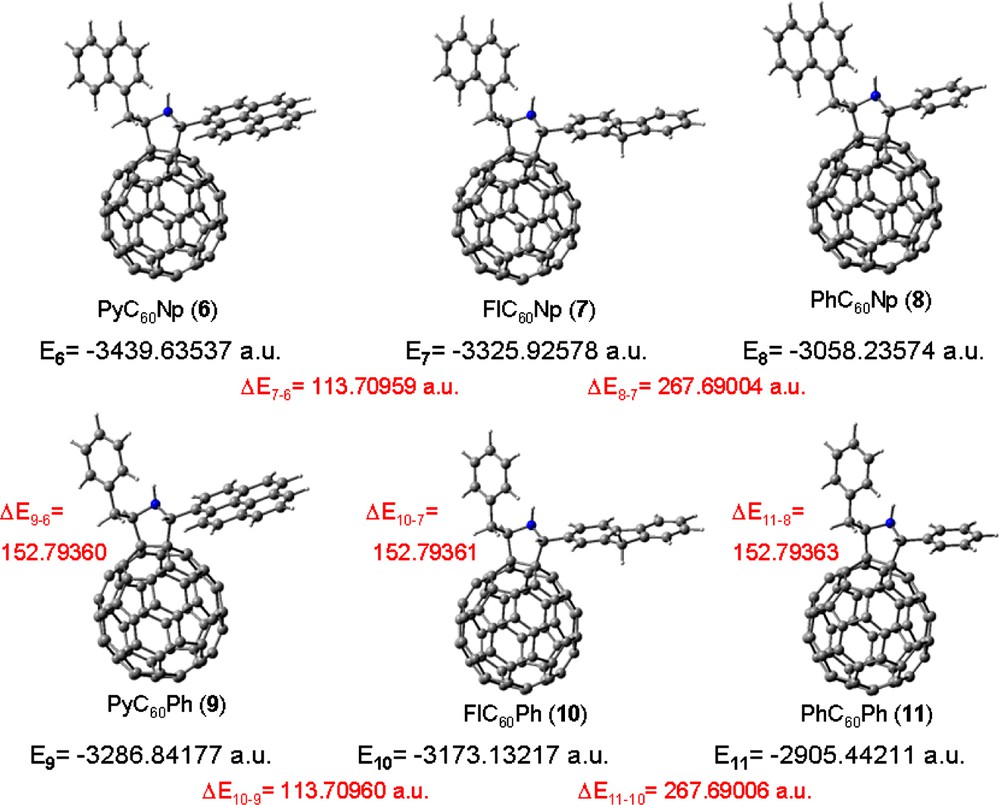
B3LYP/3-21G(*) optimized structures for compounds 6–11 with their respective total energies in atomic units. The ΔE refers to the energy difference between the different compounds.
It is also remarkable that the orbital profiles (location) and associated energies are astoundingly constant across the series of compounds (Fig. 6). That is, the HOMO located on pyrene is constant at –5.55 eV in compounds 6 and 9, while the HOMO located on fluorene is constant at –5.94 eV in compounds 7 and 10, i.e. irrespective of phenyl or naphthyl ‘antenna’. More remarkable is that the HOMO–1 in compounds 6 and 7 and the HOMO of compound 8 have the same profile (delocalized over the fullerene and naphthyl ‘antenna’) all with energy –5.99 eV. Similarly, the HOMO–2 in compounds 6 and 7 and the HOMO – 1 in compound 8, all have similar profile (localized mostly on C60) with energy –6.12 eV. It is even more insightful that the HOMO–1 in compounds 9 and 10 and the HOMO of compound 11 all are localized on the fullerene with energy –6.01 eV, illustrating that compound 11 without a fluorophore will be oxidized at the fullerene. Also, HOMO–2 in compounds 8–10 and HOMO–1 in compound 11 all are fullerene localized with energy –6.18 eV. Thus, B3LYP/3-21G(*) orbitals in these compounds with non-interacting groups have remarkably constant profiles and energies, suggesting that this model has the ability to predict relative oxidation potentials and site of electron removal, i.e. for relatively isolated groups dominating an orbital, one can assign orbital energies to these groups. It might be expected that the model has sufficient capability to adjust or perturb these group assignments for interacting groups, but this has not yet been adequately tested. Similar relations should apply to LUMOs, i.e. reductions, but for these compounds all reductions are nearly identically localized on the fullerene.
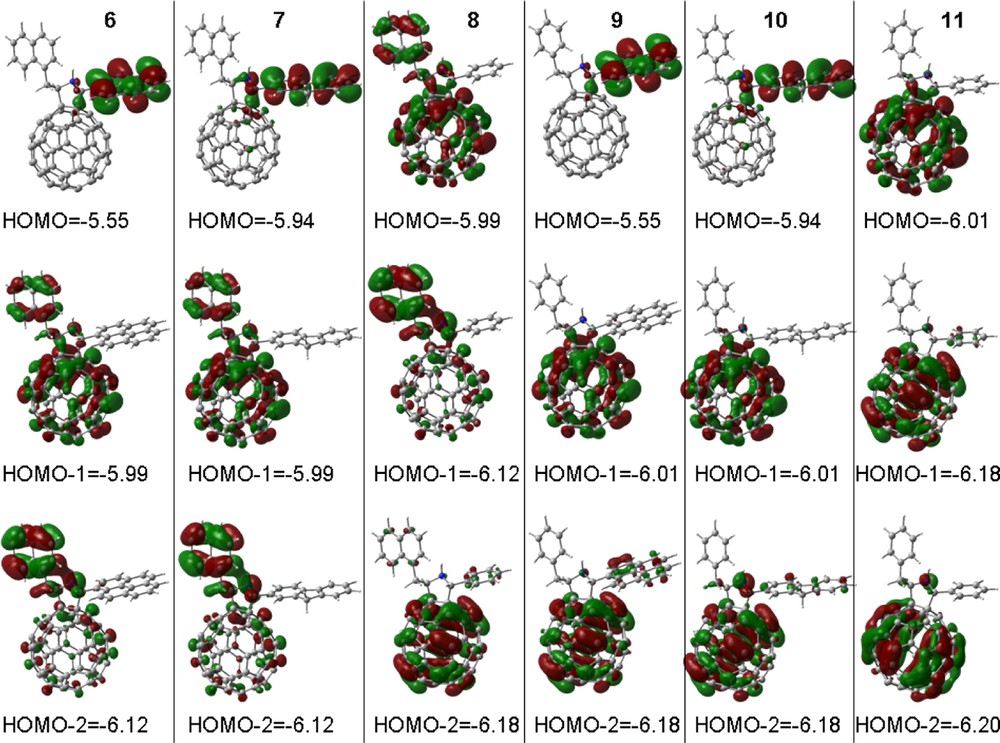
B3LYP/3-21G(*) calculated HOMO, HOMO – 1 and HOMO – 2 for compounds 6–11. Orbital energies in eV.
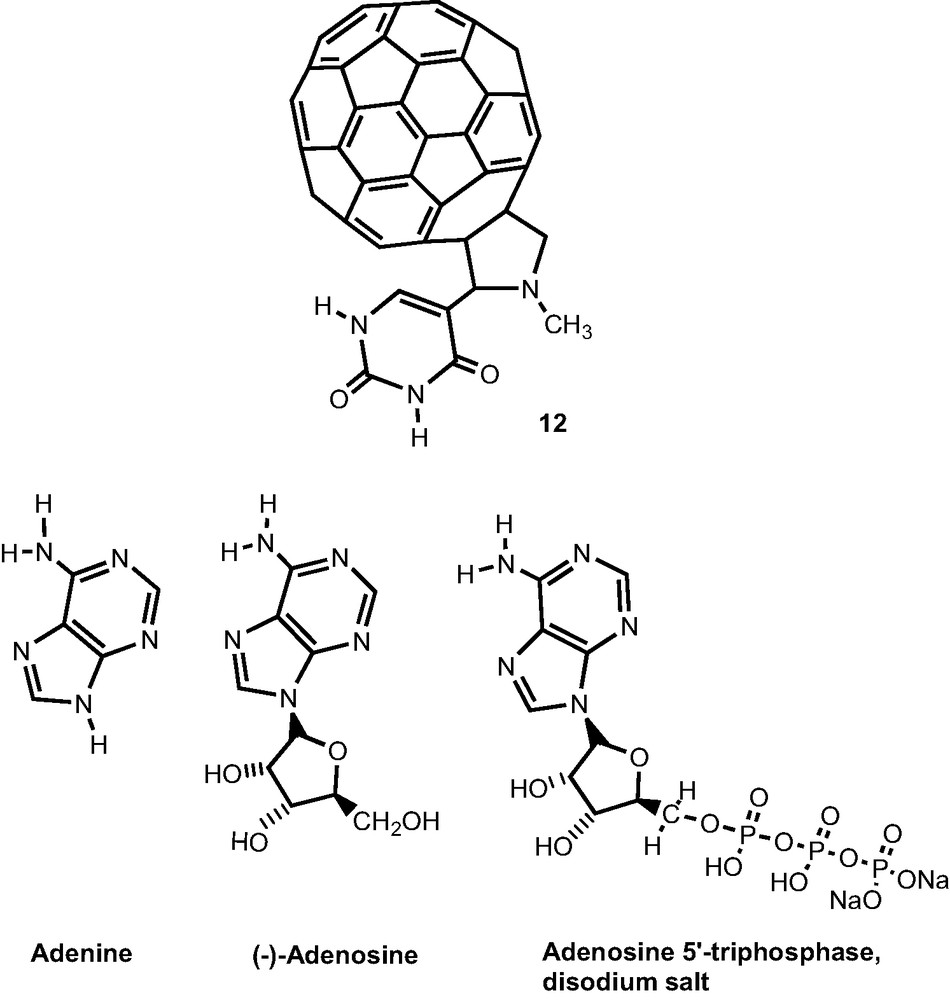
2.4 Hydrogen bonded complexes: uracil appended fullerene–adenine (adenosine or ATP) conjugates
A C60–uracil adduct, 12 capable of hydrogen bonding, via complimentary base pairing, of adenine, adenosine, and adenosine 5′-triphosphate (ATP) was synthesized and characterized by UV–visible and 1H NMR spectroscopy, cyclic voltammetry and differential pulse voltammetry, as well as ESI-mass spectrometry [23,24]. Stable ‘expanded liquid’ Langmuir films of the C60–uracil/adenine, C60–uracil/adenosine and C60–uracil/ATP complexes were prepared and characterized by isotherms of surface pressure versus area per molecule as well as the Brewster angle microscopy imaging (Scheme 4).
Molecular modeling by B3LYP/3-21G(*) calculations revealed the Watson–Crick type base pairing [23]. In the base-paired C60–uracil/adenine complex, the uracil and adenine rings were positioned in a plane and did not reveal any steric constraints due to the presence of the bulky fullerene spheroidal entity. The H-bond distances for the base pairs, i.e. N–H–···–O and N–H–···–N, were calculated as 1.85 and 1.58 Å, respectively, suggesting formation of a stable Watson–Crick type base paired complex.
The complex formation energy for the base pair formation, i.e. the energy difference between that of the C60–uracil/adenine complex and the sum of the energies of separated C60–uracil and adenine was calculated by B3LYP/3-21G(*) to be –29.1 kcal mol–1 suggesting formation of a stable complex of similar geometry and bond strength as a smaller model adenine/uracil hydrogen bonded complex. The corresponding B3LYP/3-21G(*) complexation energies for other small model complexes; the formamide dimer, the cytosine dimer, the uracil/adenine complex, and the pyrrolidine–uracil/adenine complex are –28.9, –31.4, –28.6 and –28.4 kcal mol–1, respectively, compared to the high level BSSE corrected results at the MP2/6-311++G (3df, pd) level of Sponer and Hobza [25] of –13.0, –18.8 and –13.9 kcal mol–1 for the first three complexes.
The B3LYP/3-21G(*) complexation energies are known to be systematically high for several reasons, mainly due to effects of the small basis set, the neglecting of BSSE, and the reported inability of B3LYP to “properly capture the intersystem electron correlation effects.” The magnitude of these effects was explored by comparing computations of the formamide and cytosine dimers, and the uracil/adenine complex at various levels with different bases sets with the high level results of Sponer and Hobza [25]. The correlation effects are, fortuitously, nearly correct at the B3LYP/3-21G(*) level; that is, for the formamide and cytosine dimers and the uracil/adenine complex the B3LYP-HF difference is about +6.0 kcal mol–1 at the 3-21G(*) basis and +4.0 kcal mol–1 at the 6-31G* and 6-31G** bases compared to the MP2-HF difference of +2.5 kcal mol–1 at the 6-31G** basis and +5.4 kcal mol–1 at the 6-311++G(3df, pd) level [25]. However, the 3-21G(*) basis overestimates the deformation (relaxation) energy by about 1 kcal mol–1 and is subject to a high BSSE of about +11.0 kcal mol–1 compared a BSSE of +6.3 kcal mol–1 at the 6-31G* level, +2.6 kcal mol–1 at the 6.31G** level, +2.1 kcal mol–1 at the MP2/6-31+G(2d,p) [22] level and +1.9 kcal mol–1 at the cc-pVTZ basis. The corrected B3LYP/3-21G(*) results of 14.6 kcal mol–1 (i.e. addition of about 2.0 + 1.0 + 11.0 = 14.0 to the calculated value of –28.6 kcal mol–1), therefore, agrees well with the high-level calculations of –13.1 kcal mol–1, for uracil/adenine, while predicting that the complexation energy for C60–uracil/adenine is slightly higher.
For the C60–uracil/adenine complex, the majority of the HOMO is located on the pyrrolidine ring with a small portion on the C60 and uracil ring atoms. However, the LUMO is entirely located on the C60 spheroid. The HOMO energy of C60–uracil alone being equal to –5.93 eV, reveals a slight change, that is, it is –5.80 eV after base pairing of the adduct with adenine, thus it is predicted to be slightly easier to oxidize after base pairing. The edge-to-edge distance, that is the overall size of the C60–uracil/adenine complex, was estimated to be around 1.6 nm, agreeing with the value used to estimate area per molecule in the Langmuir film studies.
3 Self-assembled fullerene–porphyrin complexes
3.1 Supramolecular metalloporphyrin–fullerene dyads via metal ligand axial coordination
Self-assembled donor–acceptor dyads were constructed by axial coordination of pyridine or imidazole functionalized fulleropyrrolidines, 12–14 to metalloporphyrins, 15–16 (metal = zinc or magnesium) or metallonaphthalocyanines, 17–18 [26–29]. The B3LYP/3-21G(*) optimized structures predicted that the Zn–N bond length of the newly formed coordinate bond is very close to the Zn–N or Mg–N bond lengths of the porphyrin nitrogen atoms, with the metal atom being pulled some 0.4 Å out of the porphyrin plane, a feature that subsequently agreed well with the X-ray structures [30,31]. As predicted from electrochemical studies, the HOMO and LUMO orbitals were well localized on the porphyrin and fullerene entities, respectively, as shown for a representative dyad, 15:14 (: represents coordinate bond) in Fig. 7. The HOMO–LUMO gap was slightly smaller than the electrochemical and optical values, suggesting that B3LYP/3-21G(*) porphyrin HOMOs are computed too high. Similar results were obtained for a zinc naphthalocyanine appended imidazole–fulleropyrrolidine dyad, 17:14 [28] (Scheme 5).
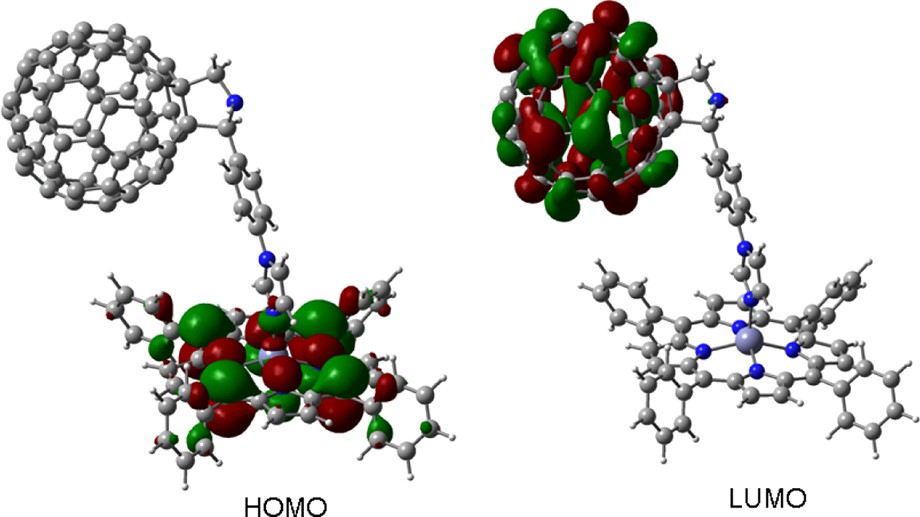
B3LYP/3-21G(*) calculated HOMO and LUMO for dyad, 15:14.
The B3LYP/3-21G(*) was able to predict the intermolecular association of the metalloporphyrin and C60 entities of the dyads in essential agreement with the subsequent X-ray structures [30,31] (see Fig. 8 for the representative 16:14 dyad), even though several studies, using large basis sets on small molecules, have indicated that there are no dispersive forces associated with B3LYP [32–35]. The 16:14 dyad agrees with the experiment as being twice as tightly bound as the 16:12 dyad. Tsuzuki and Luthi [32] show that another high level DFT method, PW91/aug-cc-pV5Z, does predict that the argon dimer, Ar2, is bound by 0.33 kcal mol–1 at a bond distance of 4.0 Å compared to the experimental 0.28 kcal mol–1 at 3.78 Å, while argon dimer is unbound at the high B3LYP/aug-cc-pV5Z level and indeed with B3LYP using bases as small as aug-cc-pVDZ. Unreported, however, is the fact that argon dimer IS bound using the B3LYP/3-21G(*) model by 0.10 kcal mol–1 at 3.8 Å (Fig. 9). This anomaly is probably due to basis set incompleteness and/or BSSE of the small 3-21G(*) basis set. This apparent pseudo-dispersion force in B3LYP/3-21G(*) yields excellent association distances for the dyads and triads treated here, and does not disappear with basis sets as large as 6-31G* for these large systems. In fact, it appears that this pseudo-dispersion force causes the association energy of the self assembled complex (the imido–Zn bond) to be consistently greatly overestimated by B3LYP/3-21G(*) but nearly correctly estimated by B3LYP/6-31G*. Due to its deeper and shorter ‘dispersion force’, Wang and Lin [33] recommend the PBE/DZP method for calculations of the porphyrin–C60 interaction potential with no BSSE corrections.
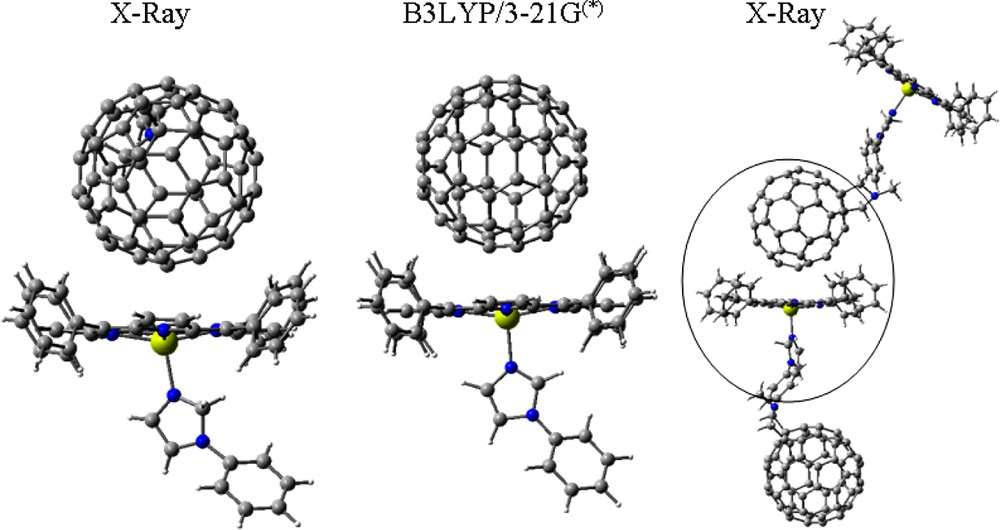
Intermolecularly interacting porphyrin and fullerene segments from the X-ray packing diagram of dyad, 16:14 (first and last) and that obtained from B3LYP/3-21(*) modeling (middle).
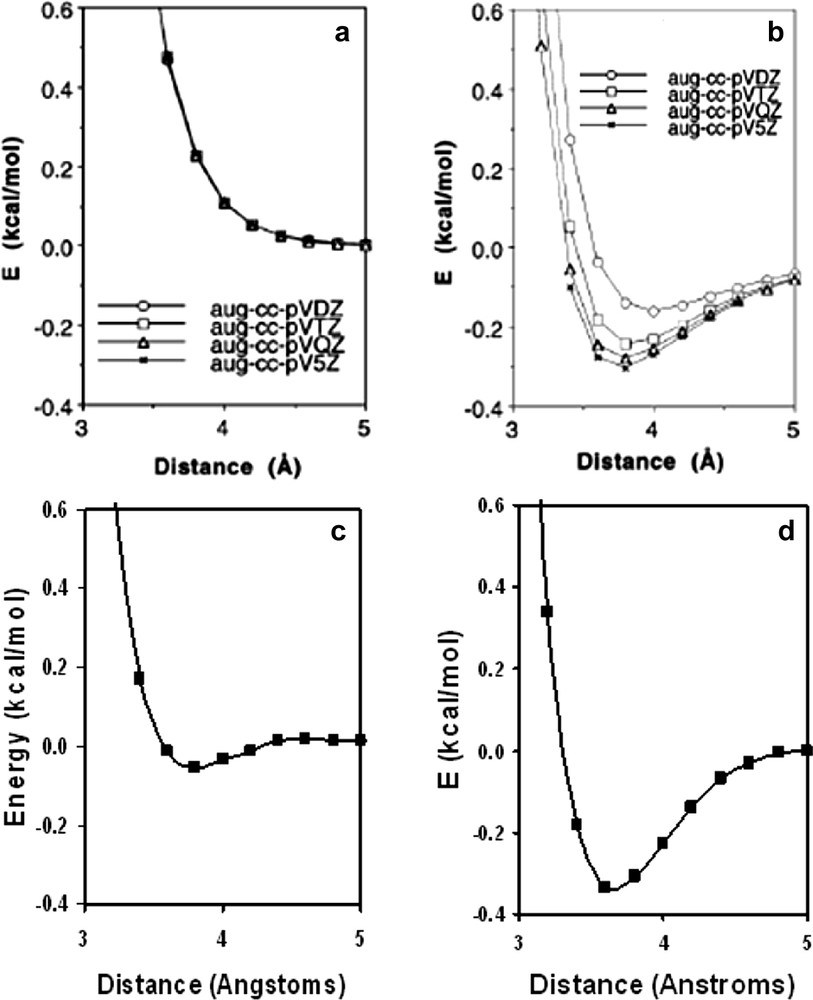
The (a) B3LYP/aug-cc-PVXZ, (b) PW91/aug-cc-PVXZ, (c) B3LYP/3-21G(*), and (d) PBE/3-21G(*) potentials for argon dimer. Figs a and b are from Ref. [32].
3.2 Bis porphyrin–fullerene and bis naphthalocyanine–fullerene triads
A supramolecular complex with a large association constant was assembled from a fulleropyrrolidine bearing two axially coordinating pyridine entities, 19 and a covalently linked zinc porphyrin dimer, 20 [36]. Efficient intramolecular photoinduced electron transfer from the singlet excited state of zinc porphyrin to the fullerene was observed. The rate and quantum yield of charge separation were found to be better than that observed in the triad formed by self-assembling two equivalents of 15–19 [37] (Scheme 6).
The geometry and the electronic structure of the supramolecular complex was visualized by performing DFT calculations at the B3LYP/3-21G(*) level. In the optimized structure (Fig. 10), the two ZnP rings were aligned in a V-shape with an angle of 70°. No steric hindrance was observed. The distance of the center of the C60 to the nearest zinc atom was found to be 10.3 Å while the Zn–Zn distance was found to be 13.8 Å. As predicted, the HOMOs formed two sets of degenerate orbitals and were alternately located exclusively on the ZnP entities while the LUMO was located on the C60 entity [36]. The lack of delocalization of the HOMOs on both the ZnP entities is suggestive of little of or an absence of intramolecular interactions between the two ZnP entities of the dimer.
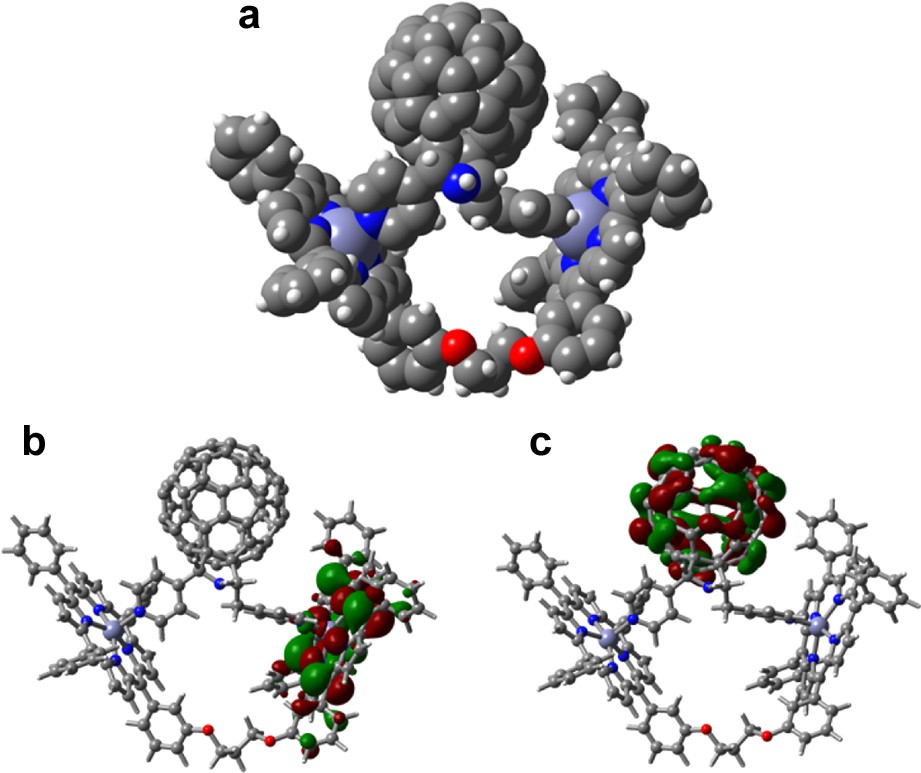
(a) The B3LYP/3-21G(*) optimized structure, (b) HOMO, and (c) LUMO of the supramolecular complex, 19:20 (from Ref. [36]).
Another supramolecular triad was constructed by self-assembling via metal–ligand axial coordination, two zinc naphthalocyanines, 17, and a functionalized fullerene bearing two pyridine entities, 19 [38]. The B3LYP/3-21G(*) optimized structure of the triad revealed a shape resembling the Star Wars Tiefighter Ship (Fig. 11), with the two naphthalocyanine wings almost coplanar with a small tilt angle and separation of about 16 Å, and the fullerene occupying the center position via axial coordination. The large planar–rectangular shape of the zinc naphthalocyanine (as opposed to their smaller zinc porphyrin or zinc phthalocyanine analogs) acted as the two wings leaving the central fullerene spheroid as the cockpit (body) of the vehicle! This serves as a nice example of utilizing self-assembly approach to build exotic supramolecular structures capable of undergoing light induced processes. Little or no π–π interactions were observed both experimentally or via computation. The main quenching pathway involved charge-separation from the singlet excited 17–19, with a rate constant, kCS, of 5.7 × 109 s–1, indicating an efficient photochemical electron transfer process.
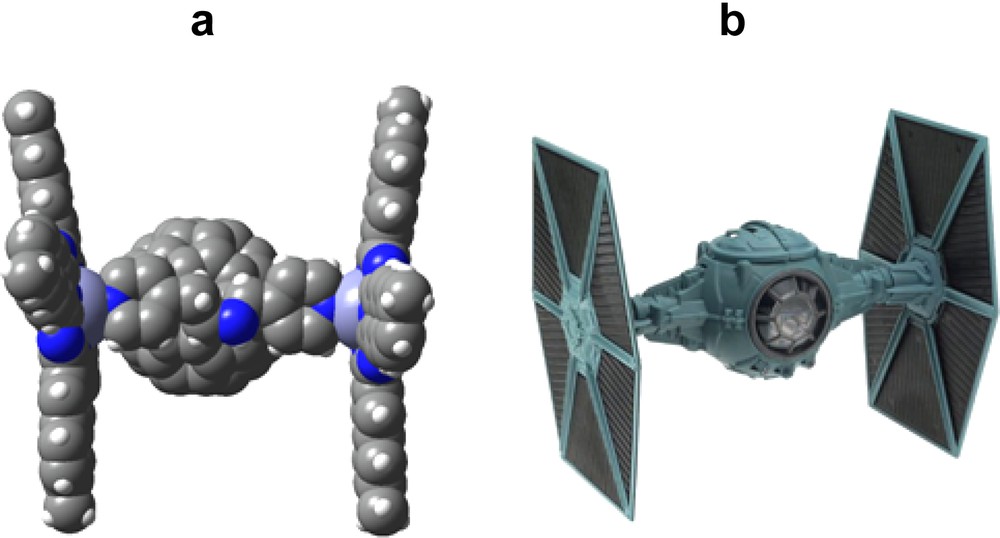
(a) Space-filling model of the B3LYP/3-21G(*) optimized structure of two zinc naphthalocyanines, 17, interacting with bis pyridine functionalized fulleropyrrolidine, 19 (b) Model of Star Wars Trefighter Ship.
3.3 Axial coordination of fullerene to covalently linked zinc porphyrin–ferrocene and zinc porphyrin–boron dipyrrin dyads
Supramolecular triads have been constructed by using covalently linked zinc porphyrin–ferrocene dyad, 21 self-assembled via axial coordination to pyridine- or imidazole-appended fulleropyrrolidines, 12, 14 [39]. These triads were characterized by optical absorption, computational, and electrochemical methods. The calculated binding constants revealed stable complexation and suggested the existence of intermolecular interactions between the ferrocene and fullerene entities. Accordingly, the optimized geometry obtained by the B3LYP/3-21G(*) method revealed closely spaced ferrocene and fullerene entities in the studied triads (Scheme 7).
The structure of one of the fully optimized triads 21:14 is shown in Fig. 12. It may be mentioned here that the triads were very flexible and multiple minima were possible, therefore, the optimization process required much experience and considerable computational time to confidently achieve an optimized structure at the B3LYP/3-21G(*) level. Geometric parameters that are helpful to understand the spectral and photochemical properties (kinetics of charge separation and charge-recombination), were obtained from the B3LYP/3-21G(*) optimized structures. The metal–metal distances between Zn and Fe were found to be about 10.7 Å for the directly linked zinc porphyrin–ferrocene dyads in the closed conformation. Importantly, intermolecular-type interactions between the C60 and ferrocene units were observed corresponding to the close edge-to-edge distances between the ferrocene and C60 entities computed to lie around 3 Å, well within the van der Waals interacting distances. Such interactions between the ferrocene and C60 entities are expected to modulate the photochemical properties; that is, because of the close proximity of these entities, efficient charge recombination of the ferrocene cation and C60 anion radical during photochemical electron transfer is expected to occur. The ordering of the HOMOs were found to be inverted so that the porphyrin-like orbitals were higher than the ferrocene-like orbitals—presumably due to B3LYP/3-21G(*) predicting both ferrocene HOMO energies too low and porphyrin HOMO energies too high.
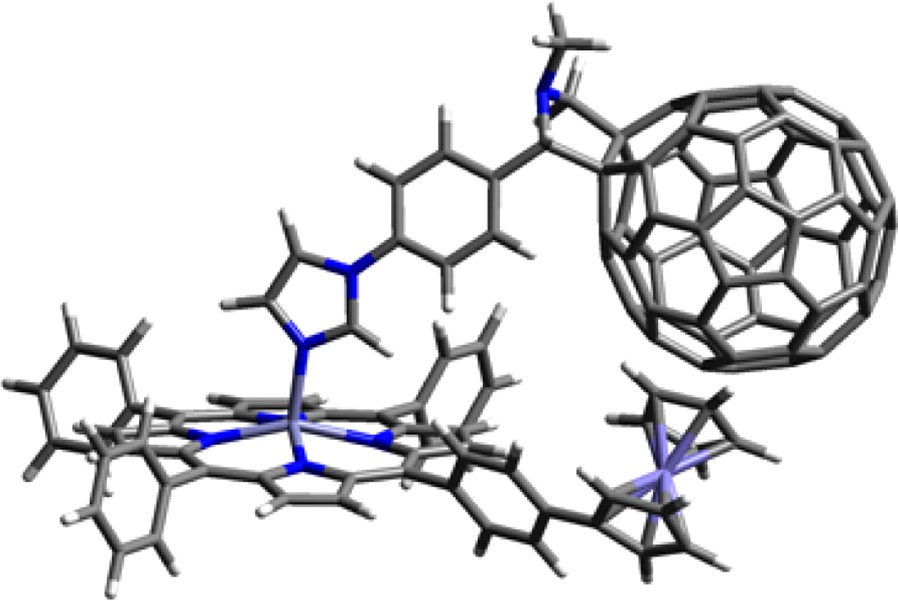
B3LYP/3-21G(*) optimized structure of supramolecular triad, 21:14. (from Ref. [39]).
A similar complex was prepared using porphyrin linked by a boron dipyrrin (BDP) entity, 22 dyad [40]. Here, BDP acts as energy absorbing and transferring antenna, and zinc porphyrin (ZnP) acts as an energy acceptor from the antenna which promotes electron transfer to the fullerene moiety by using the excitation energy, with the fullerene, 14 being the electron acceptor. The electrochemical studies indicated that reduction of BDP at –1.33 V was close to reduction of ZnP at –1.35 V, relative to Fc. The B3LYP/3-21G(*) optimized structure of 22 (not shown) revealed an open structure with HOMO and LUMO π-type orbitals on the porphyrin and BDP entities, respectively. The location of the LUMO on the BDP moiety is consistent with the electrochemical results where the possibility of BDP acting as an electron acceptor from the singlet excited ZnP is possible in addition to being an excited-state energy donor to the ZnP was revealed. This dyad is especially suitable to probe such events because of wavelength selectivity where ZnP and BDP can be excited at 550 and 388 (or 500) nm, respectively. The self-assembled triad, 22:14 was optimized with B3LYP/3-21G(*) at two reasonable ‘extreme’ geometries, an ‘open’ arrangement with little steric interaction and a “closed” form with close BDP–C60Im interaction (other conformations might be possible). As expected from the electrochemical studies, for both forms the HOMO was found to be located on the ZnP moiety while the LUMO was located on the fullerene, that is, the LUMO moved to the fullerene from its location on the BDP moiety in the dyad, suggesting fullerene being the final electron acceptor in this novel supramolecular triad (Fig. 13).
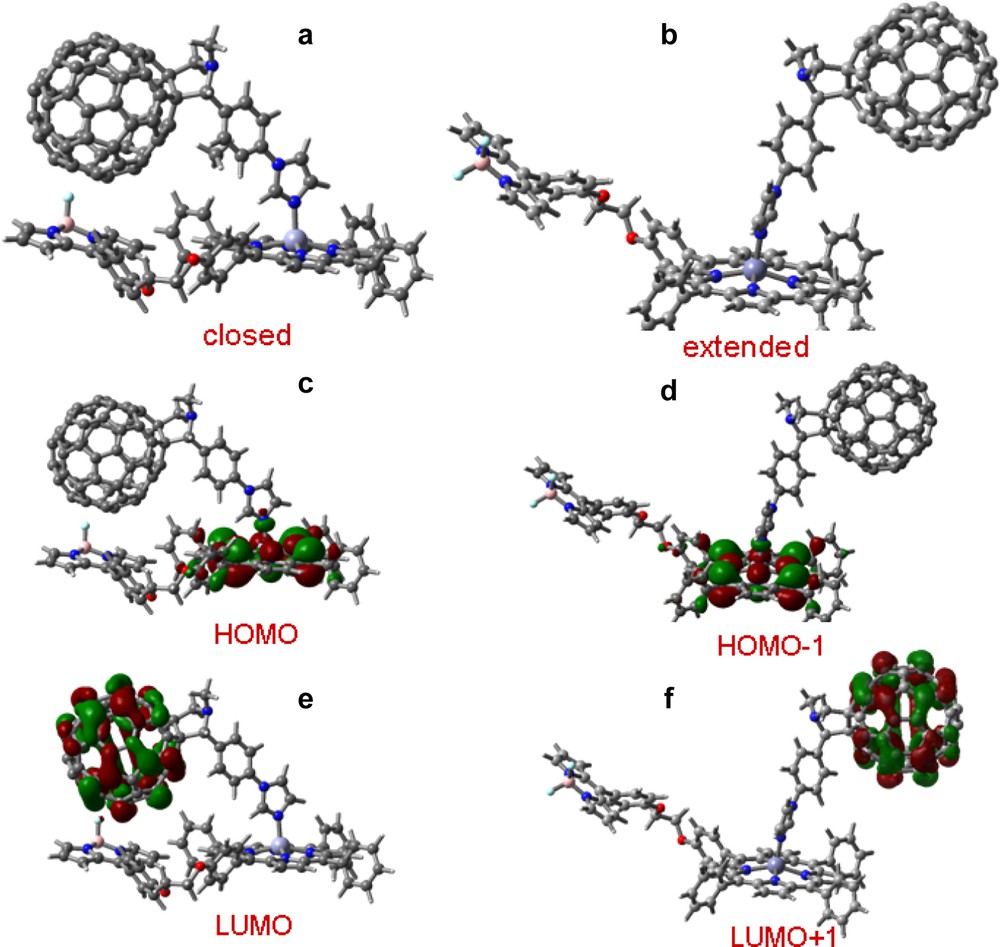
B3LYP/3-21G(*) optimized structure at local minima for the supramolecular triad formed by coordinating 14–22 in (a) closed and (b) expanded forms. The corresponding HOMO and LUMO are shown in figures c–f (from Ref. [40]).
3.4 Two-point bound porphyrin–fullerene conjugates—axial-coordination and carboxyl pyrrolidine hydrogen bonded complexation
A newly developed ‘two-point’ binding strategy was used to produce stable self-assembled supramolecular triads with defined distance and orientation, composed of either –COOH or –CONH2 functionalized zinc porphyrins, 23–24 binding to either ferrocene, N,N-dimethylaminophenyl or N,N-diphenylaminophenyl functionalized fullerene–pyridine conjugates, 25–27 [41,42]. For this, zinc porphyrin was derivatized to bear ‘hydrogen-bonding’ functionalities, carboxylic acid or amide groups, and C60 was functionalized to bear a ligating group, pyridine, and a second electron donor group. The supramolecular triads were characterized by spectroscopic and electrochemical techniques and were modeled using B3LYP/3-21G(*). Evidence was obtained for axial coordination of the pyridine entity to the zinc metal center, with Zn–N distance as short as in the four porphyrin bonds, and H-bonding between the pyrrolidine ring nitrogen and the pendant carboxylic acid or amide groups, with N–O distance comparable, but slightly longer, than a smaller comparison system, pyrrolidine/acetic acid.
For all systems, B3LYP/3-21G(*) predicts a pseudo six-member ring involving two H-bonds between the pyrrolidine N–H and the carboxyl O–H or amide N–H. In the supramolecular triads, the second electron donors promote efficient charge separation upon excitation of the zinc porphyrin to yield the radical ion pairs. The radical ion pairs thus generated undergo slow charge recombination to yield relatively long-lived (30–40 ns) charge-separated states (Scheme 8).
The HOMO and HOMO–1 profiles of the representative triad 23:26 (Fig. 14) indicate porphyrin π (Gouterman) orbitals, while the HOMO–2 is mostly located on the dimethylaminophenyl group with small portion on the pyrrolidine ring. The close spacing between the first oxidation of zinc porphyrin and the dimethylaminophenyl entities suggests the possibility of the occurrence of a hole migration or hole exchange between ZnP+● and the dimethylaminophenyl entity formed during the photoinduced electron-transfer process. The LUMO, LUMO+1, and LUMO+2 were all found, as expected, on the C60. The electrochemical studies verified that the B3LYP/3-21G(*) orbital energies and profiles, track the magnitude of the potentials of the different redox entities, that is, the sequence of the site of electron–transfer [41,42].
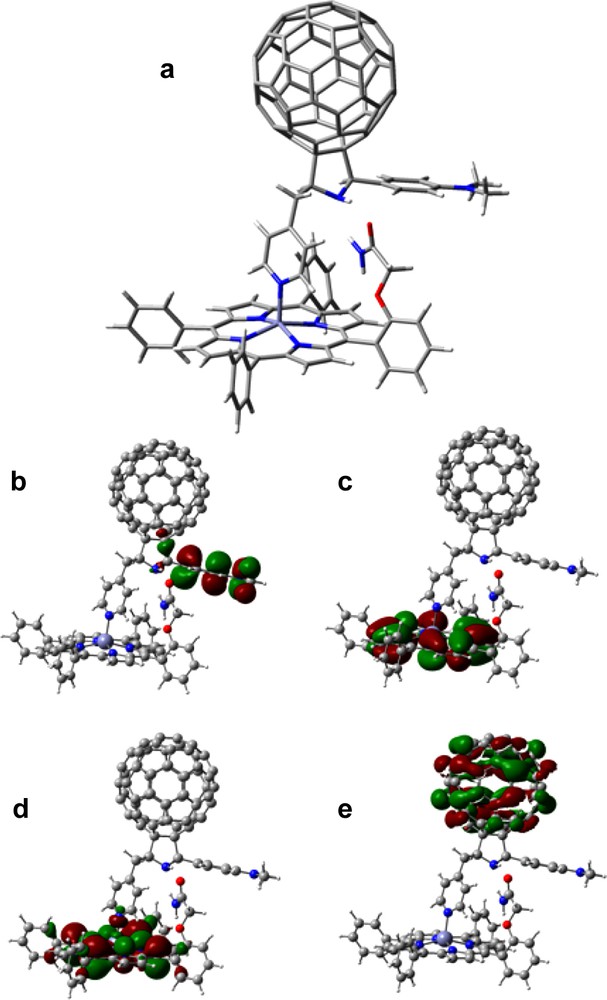
B3LYP/3-21G(*) calculated (a) optimized structure, (b) HOMO – 2, (c) HOMO – 1, (d) HOMO and (e) LUMO for the self-assembled triad, 23:26 (from Ref. [41]).
3.5 Two-point bound porphyrin–fullerene conjugates—axial-coordination and cation–crown ether complexation or cation–crown ether complexation and π–π type interactions
Highly stable porphyrin–fullerene conjugates with defined distance and orientation were formed using a newly developed ‘two-point’ binding strategy involving axial-coordination and cation–crown ether complexation [43,44]. Towards this, zinc porphyrin was functionalized to bear an 18-crown-6 entity at different positions of one of the phenyl rings, 28–29 while fullerene was functionalized to bear an alkyl ammonium cation, 30 or both pyridine and alkyl ammonium entities, 31. Crown-ether binding to ammonium cation, and axial coordination of pyridine to zinc was observed in dyads formed by porphyrins 28–29 with fullerene, 31. When porphyrins 28–29 were complexed with 30, π−π type interactions between porphyrin and fullerene along with crown ether–cation complexation was observed thus fulfilling the two-point binding strategy [44]. Photochemical studies performed in benzonitrile revealed efficient charge separation and slow charge-recombination in the supramolecular complex (Scheme 9).
DFT studies using B3LYP/3-21G(*) were performed to visualize the geometry and electronic structure of these strongly bound complexes. The geometry optimized structure revealed the presence of both the zinc–pyridine coordinate bond and a remarkably stable (computationally) ammonium cation–crown ether complex for 28 or 29 interacting with 31 (Fig. 15). The apparently highly strained requirements for the two-point binding was, however, accomplished in case of 29:31 such that the B3LYP/3-21G(*) dissociation energy was within 2 kcal mol–1 of the expected more favorable arrangement of the 28:31 complex. The resulting geometry of 28:31 showed the crown ether conformation bowed to maintain both the strong cation–ether bond and the axial coordinate bond. Two sets of N–O bond distances were found for the ammonium cation–crown ether complex, 2.80 and 3.00 Å, the former representing N–H–···–O type H-bonding. The ‘gas phase’ dissociation energy for the complex, calculated as the energy difference between the dyad and the sum of the energies of the individual porphyrin and fullerene moieties, was about 137 kcal mol–1 unrelaxed and 117 kcal mol–1 relaxed, an energy nearly four times that calculated by B3LYP/3-21G(*) for a similar conjugate bound only by the coordinate bond, indicating substantial bonding through the ammonium ion–crown ether, as expected, and substantial reorganization upon relaxation. Both calculations are undoubtedly very high because of substantial BSSE and BSIE errors associated with the small basis but might be expected to give reasonable relative energies.
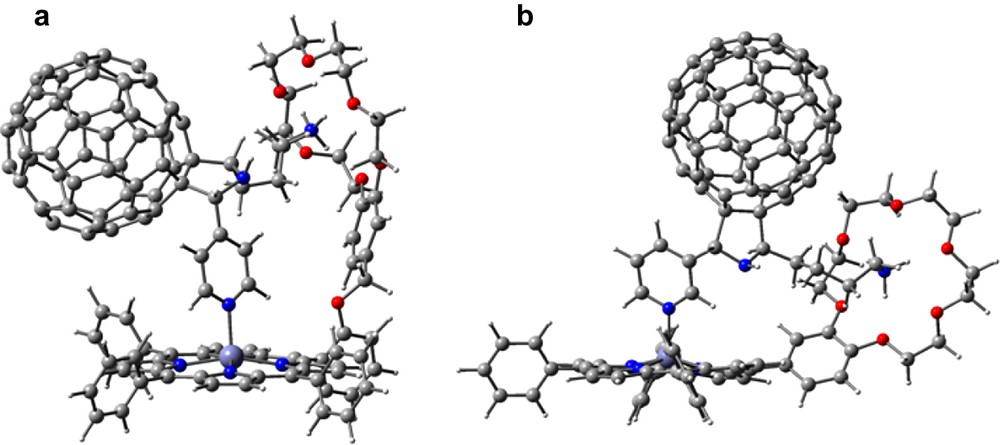
B3LYP/3-21G(*) calculated structures of the self-assembled dyads (a) 28:31, and (b) 29:31.
As predicted, the HOMO and LUMO were found to be located entirely on the porphyrin and fullerene moieties, respectively, indicating that the ammonium cation–crown ether complex did not perturb the electronic structure of the donor and acceptor entities of the conjugate. It should be noted that the complex modeled, was the singlet supramolecular cation with no explicit anion present in the calculations (although adding a chloride ion pseudo bonded to the ammonium ion through the crown ether resulted in little change in geometry or complex strength of the supramolecule).
Self assembled ‘two-point’ bound supramolecular conjugates utilizing cation–crown ether bonding and π–π type interactions were prepared and characterized by using porphyrins 28 or 29 interacting with 30 (Fig. 16a) [44]. The B3LYP/3-21G(*) optimized geometries show that the fullerene hovers nearly centered over the zinc porphyrin with a spacing of about 2.6 Å (note that the zinc atom is pulled toward the C60) by some 0.3 Å (Fig. 16a). The association energy is –107.3 kcal mol–1 (unrelaxed), some 30 kcal mol–1 less than the axial coordinated pyridine compound discussed above. The B3LYP/6-31G* association energy is –72.8 kcal mol–1 (unrelaxed), presumably much closer to the correct result. Importantly, in support of the optical absorbance results, evidence for the π–π interaction was obtained from computational studies. That is, the computed HOMO revealed partial delocalization of the frontier orbitals on the fullerene while the LUMO revealed partial delocalization on the porphyrin entity, as shown in Fig. 16b, c. Addition of pyridine to these supramolecular dyads broke the π−π interactions and gave an opportunity to explore their effect on charge separation and charge recombination [44].
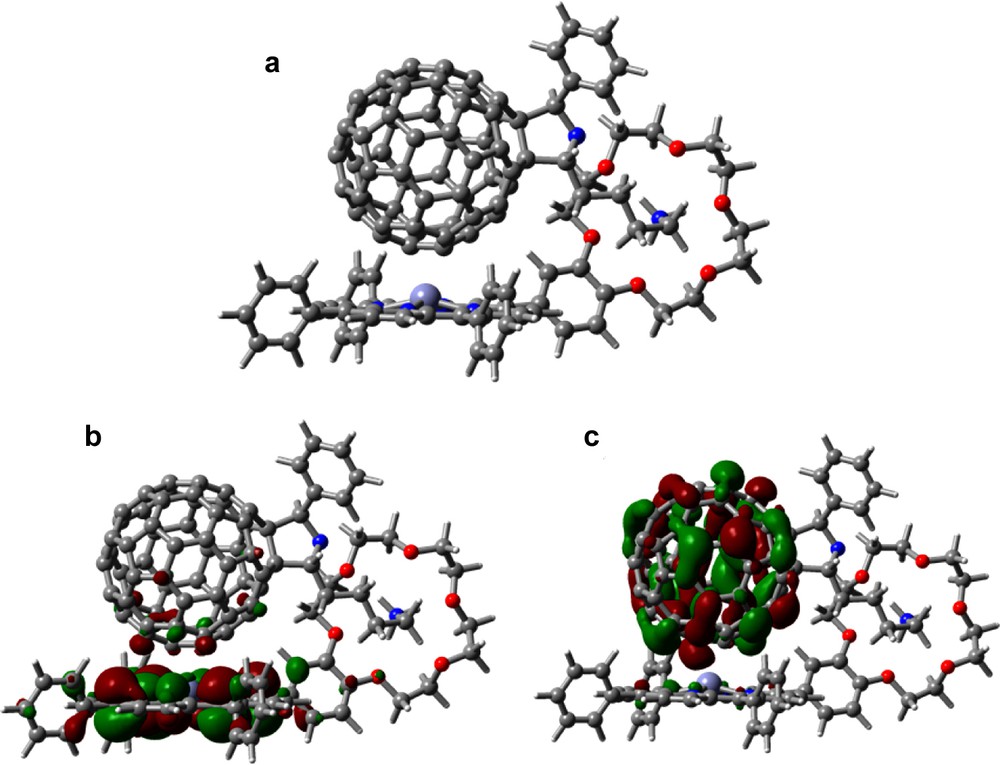
B3LYP/3-21G(*) calculated (a) optimized structure, (b) HOMO, and (c) LUMO of the self-assembled 29:30 dyad.
Unpublished B3LYP/3-21G(*) studies on intermolecularly interacting pristine C60/TPP and C60/ZnTPP model complexes, show that the fullerene interacts with the free-base porphyrin and zinc porphyrin in a similar way. In both complexes, the optimized geometries have the C60 oriented with the 6–6 bond nearest the porphyrin with about 2.6 Å spacing aligned along an N–N direction in near agreement with close contacts observed in crystal structures (2.7–3.0 Å). The zinc atom of C60/ZnTPP is displaced about 0.4 Å toward the C60, contrary to observations reported by Boyd and Reed [2]. The B3LYP/3-21G(*) association energy for C60/ZnTPP is about 26 kcal mol–1 (unrelaxed) and 17 kcal mol–1 (relaxed) agreeing well with the 16.25 kcal mol–1 reported by Wang and Lin [33] using PBE/DZP without BSSE correction. They report that BLYP/DZP gives a much smaller value of 5.8 kcal mol–1. The B3LYP/3-21G(*) association energy for C60/TPP is much smaller, whereas Wang and Lin [33] report a larger value of 17.33 kcal mol–1 agreeing with a best estimate of 16.5 kcal mol–1 from an exhaustive MO and DFT study by Shephard and Paddon-Row [45]. They report that the computationally demanding MP2 is among the methods that give values much too high while many methods are too low, especially if counterpoise corrected.
4 Covalently linked porphyrin–fullerene conjugates
To probe the effect of axial ligation on the photoinduced charge separation and charge recombination, a series of covalently linked porphyrin–C60 dyads was investigated by functionalizing at the ortho or para positions of one of the aryl groups of tetraphenylporphyrin to bear a fulleropyrrolidine entity through a flexible ethylene dioxide bridge, 32–37 [46,47]. Electrochemical studies revealed redox processes corresponding to both porphyrin and fullerene entities while the oxidation potential of the donor porphyrin followed the order: MgP < ZnP < H2P. UV–visible absorption spectra revealed charge–transfer type interactions between the electron donor (porphyrin) and the electron acceptor (fullerene) entities in these dyads [46,47] (Scheme 10).
The geometric and electronic structures of the dyads probed by B3LYP/3-21G(*) methods also revealed the existence of charge–transfer interactions. All of the dyads were fully optimized to a bonafide minimum such that the C60 entity hovered over the porphyrin entity with a spacing of 3.2–3.9 Å for the various forms, although the optimization of these flexible molecules proceeded to a minimum with great difficulty (modredundant restraints were necessary to start the optimization since a very small change in bond length or angle of the linker caused huge spacing changes) (see Fig. 17 for representative dyads, 32, 33). Energetically, the para-derivatized dyads are more stable by ca. 3 kcal mol–1 than the ortho-derivatized dyads and the intra-entity spacing is larger for the free base forms. Little or no distortion of the porphyrin ring is observed for the para-substituted derivatives, while substantial distortion of the porphyrin ring atoms near the substituted meso-phenyl ring is observed in the case of the ortho-substituted derivatives.
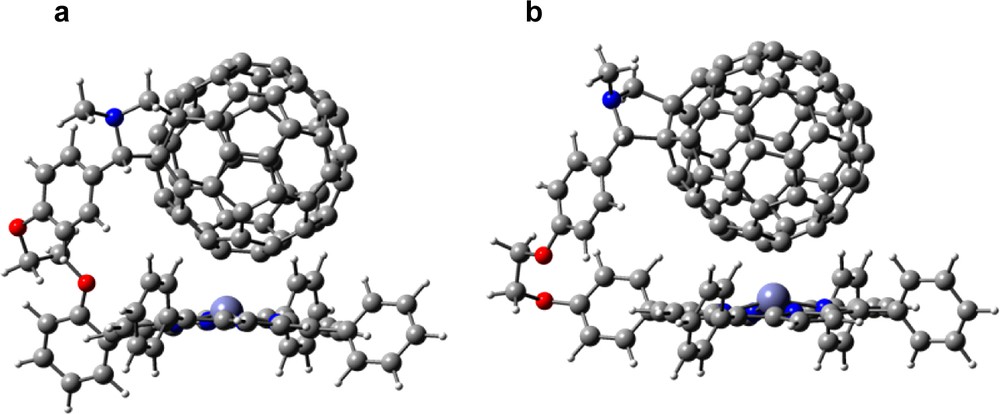
B3LYP/3-21G(*) calculated geometric structures of (a) ZnPo~C60, 32 and (b) ZnPp~C60, 33.
The existence of intramolecular interactions between the porphyrin ring and the C60 moiety was evidenced by the frontier HOMO and LUMO. Fig. 18a shows the HOMO for the ZnPp–C60 dyad, 33. The majority of the HOMO is located on the porphyrin ring while small amounts of the HOMO are also found on the C60 carbons located within the interacting distances, suggesting through space interactions. Similarly, the majority of the LUMO is located on the C60 spheroid, with small amounts located on the porphyrin ring atoms. The delocalization of HOMO and LUMO over the zinc porphyrin and C60 entities suggests though–space charge transfer interactions both in the ground state and in the excited state of the zinc porphyrin–C60 dyads. Similar charge–transfer interactions for the ZnPo-C60, 32 and Mg porphyrin derivatives, 36–37 were observed [47]. However, for free-base porphyrin derivatives, 34–35, because of the larger separation between the porphyrin and the C60 entities, such effects are not observed; that is, the HOMO was found entirely located on the porphyrin ring while the LUMO is on the C60 spheroid. The calculated gas phase HOMO–LUMO energy gap was found to follow the nature of the metal ion in the porphyrin cavity: MgP < ZnP < H2P. This trend is in full agreement with the electrochemical ‘HOMO–LUMO’ gap values (that is, the difference between the first oxidation potential and first reduction potential). Again, the orbital energies were found to track the electrochemical redox potentials with a majority of the HOMO and HOMO – 1 orbitals located on the porphyrin entity while the majority of the LUMO and LUMO + 1 located on the C60 entities. However, a specific trend for higher LUMO orbitals was difficult to establish.
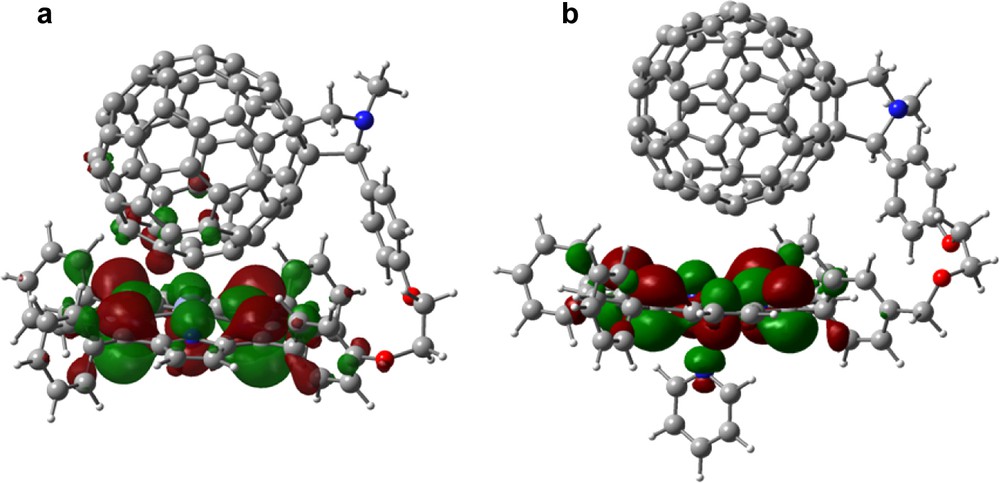
B3LYP/3-21G(*) calculated HOMO for 33 (a) in and absence and (b) in the presence of axially bound pyridine.
When pyridine was coordinated to MP–C60 to form a penta-coordinated species in case of zinc porphyrin [46] and hexa-coordinated species in the case of magnesium porphyrin [47], the first and second oxidation peaks corresponding to the metalloporphyrin oxidation revealed a cathodic shifts. This potential shift was about 15–20 mV for zinc porphyrins and was about 110 mV for magnesium porphyrins, suggesting that the pyridine bound metalloporphyrins are better electron donors. The HOMO and LUMO orbitals generated for the pyridine bound dyads revealed delocalization of HOMO orbitals over the pyridine ring (Fig. 18b). The delocalization of the frontier HOMO over the axially bound pyridine ligand would dissipate the positive charge formed during initial charge separation and slow the charge recombination process. This effect is in addition to the increased distance between the MP and the C60 entities of the pyridine bound dyads.
The remarkable ability of B3LYP/3-21G(*) to provide optimized geometries of loosely bound complexes in agreement with the experimental observations again implies that the ‘dispersion forces’ are well represented with this small basis set model. The orbital shapes, locations, and energies adequately describe the electronic properties of these molecules.
5 Other computational studies
Following is a brief summary of computational work reported by others on porphyrin–fullerene conjugates. Exhaustive ab initio MO and DFT studies of the porphyrin–C60 non-bonded (π–π) interactions for several metal porphyrins are reported by Basiuk [48], and Shephard and Paddon-Row [45], and the interaction energy of C60 and C70 with zinc and free-base tetraphenylporphyrins was decomposed into the electrostatic, orbital, and Pauli-repulsion terms by Wang and Lin [33]. Yamaguchi [49] made a theoretical prediction of electronic structures of fully π-conjugated zinc oligoporphyrins with curved surface structures. The excitation energies and oscillator strengths for the ring and ball structures were calculated with TD-DFT.
Tappura et al. [50] used molecular mechanics simulations to explore the energetically accessible conformational space of a covalently tethered C60–porphyrin dyad, followed by semi-empirical and DFT optimization of the favorable conformations. The results in vacuum and in two different explicitly simulated solvents (over 500 solvent molecules) were analyzed as to the temperature dependence of folded and open structures.
The electron transfer characteristics and geometries of porphyrin–fullerene dyads was explored computationally by several groups, including Schuster et al. [51–53], using semi-empirical and molecular dynamics treatments, by Parusel [54] treating excited-states and spectra using semi-empirical methods, by Armaroli et al. [55], treating differing spectroscopic features of various self-assembled complexes, by Sun et al. [56], treating semi-empirically the spectral characteristics of fulleride anions and pyrrolidine-functionalized C60 derivatives, and by Liu et al. [57] using INDO/CI methods for a comparative study of the spectra and second-order non-linear polarizabilities for donor–acceptor ensembles between Zn porphyrin–fullerene and Zn porphyrin–naphthalenediimide.
Characterization of the experimental properties (crystal structures) of fullerene–porphyrin associations and complexes is excellently summarized by Boyd and Reed [2]. Fullerene–porphyrin architectures important for devising model systems to transmit and process solar energy were treated by Guldi [58]. The π–π interactions of supramolecular complexes of fullerene joined to jaw-like clefts in metalloporphyrin dimers was described by Sun et al. [59]. The photochemical and electrochemical properties of zinc and free-base chlorin–C60 dyads compared to porphyrin analogs was reported by Fukuzumi et al. [60], and the excited-state interchromophore interactions in porphyrin–fullerene π-stacks were treated by Chukharev et al. [61].
6 Summary and outlook
Described here are a large number of applications of a single method, B3LYP/3-21G(*), to a wide variety of fullerene–porphyrin molecular and supramolecular complexes. By studying these results, one can come to a consensus view of the adequacy of the method to explain and correlate structural, electronic, electrochemical, and spectral properties of the covalently and self-assembled complexes treated here. No one would expect this low level procedure to be reasonably exact, especially for the extremely large systems treated here, but one could hope that trends and correlations could allow reasonable predictions to be made for systems similar to those analyzed here. Other DFT methods and higher level treatments may be warranted in cases of poor agreement with experiment, but there is no guarantee that more lengthy methods will necessarily yield better results, i.e. convergence is slow and solvent and cooperative effects may be more important than higher level theoretical methods. It appears that in the B3LYP/3-21G(*) method, there is a fortuitous trade-off or cancellation of errors (reminiscent of HMO theory) in the combination of the hybrid DFT treatment of correlation effects, mixed with Hartree–Fock components of the 3-parameter empirically adjusted functional, combined with the quite small basis set (BSSE and BSIE errors). Although not emphasized here, it would be necessary to consider the solvent environment surrounding the complexes with perhaps an empirical solvation method (e.g. Cramer–Trular methods), polarized continuum methods, or molecular dynamics simulation with explicit solvent molecules.
Historically, we continue to be amazed at the agreement between (a) experimental optical S0 → S1 transition energies and (b) the difference between the reduction and oxidation potentials—both commonly referred to as a measure of the ‘HOMO–LUMO’ gap (see Ref. [62] for insight into this relationship). The computed HOMO and lower levels have often been correlated with experimental oxidation potentials as a direct generalization of Koopman's theorem, and occupied orbital energies correlate nicely with photoelectron spectroscopy. Associating the LUMO with reduction is problematic with Hartree–Fock, leading to the extremely large predicted gap with HF calculations. DFT calculations considerably lower the calculated gap, however, the interpretation of DFT orbital profiles and energies have changed rapidly over the past 5 years. Prior to 2000, it was widely believed that the ‘orbitals’ of DFT implementations were purely mathematical constructs useful in the construction of the density with little physical meaning.
When we noticed that the DFT calculated HOMOs and LUMOs of large donor–acceptor complexes tracked the electrochemical reduction and oxidation potentials as outlined here, we were at a loss for theoretical explanation, but were happy to see argument for DFT orbital meaning in Stowasser and Hoffmann's [63] paper, and use by others to interpret redox and spectral behavior. The orbital profiles are similar to HF orbitals, but are located in better agreement with chemical intuition, and changes in the surprisingly localized HOMO and LUMO regions graphically illustrate charge transfer characteristics of optical transitions. In this regard, Cramer points out that DFT treats both HOMO and LUMO orbitals the same (i.e. from the density) in contrast to HF, which treats virtual and occupied orbitals differently. “All KS orbitals, occupied and virtual, are subject to the same external potential. HF orbitals, on the other hand, experience varying potentials, and in particular, HF orbitals experience the potential that would be felt by an extra electron being added to the molecule. As a result, HF virtual orbitals tend to be too high in energy and anomalously diffuse compared to KS virtual orbitals” [64]. This could be the main reason for the better agreement between DFT calculated orbital energies (HOMO–LUMO gap) and the experimentally measured optical excitation energies and electrochemical redox energies. The agreement with optical transition energies (and by inference with the redox energies) could be improved by TD-DFT calculations of the excitation energy.
As discussed in the Stowasser and Hoffmann [63] paper, there is no real Koopman's theorem for DFT (except that in ‘exact’ DFT there is correspondence with the first ionization potential). However, there is much empirical and some theoretical justification for the success of functionals including self-interaction or statistical average of orbitals potential (SOAP) [65]. Recently, it has been proposed that for commonly used DFT functionals, especially B3LYP, there is an offset or shift that allows extremely useful correlations of HOMO and LUMO energies with oxidation and reduction potentials [66]. Additionally, there is evidence that the correlation does not depend highly on basis set size as long as the LUMO energies are negative—as is true for every calculation reported here.
Lastly, we should point out that common consistent errors occur with B3LYP/3-21G(*) calculations that can be corrected by empirical adjustment; (a) orbital energies are mostly correct for the donor acceptors treated here but porphyrin HOMOs are too high and ferrocene orbitals are too low, (b) association energies are too high but can be corrected by a single point energy evaluation with B3LYP/6-31G* calculation, (c) HOMO–LUMO gaps are normally too large (except when porphyrin HOMOs are involved) but can be ‘corrected’ by TD-DFT calculations (larger basis sets do not seem to help here), (d) supramolecular assemblies involving more than four fullerene and/or TPP groups challenge current computer resources but might be overcome by treating most of the system with lower level methods (semi-empirical or molecular mechanics). We eagerly await a usable theoretical treatment or method with demonstrated greater success than the B3LYP/3-21G(*) model, when consistently applied across the wide range of systems and properties treated here.
Acknowledgments
The authors are thankful to the National Science Foundation (Grant 0453464 to F.D.), Petroleum Research Funds administered by the American Chemical Society, and the Dryfus Foundation for support of this work.