



 CC-BY 4.0
CC-BY 4.0
1. Introduction
α-Olefins synthesized from the diversely available and inexpensive feedstock ethylene are of high importance in chemical industry and academic research. More than 7.14 million tons of α-olefins will have been produced in 2025 from ethylene, and essentially all of them are linear in structure [1]. The most important applications of these linear α-olefins are co-feed for plastic production, and surfactant, softener, and lubricant production. Unfortunately, the synthesis of branched α-olefins employing ethylene is challenging, but recent progress in selective co-oligomerization reactions of α-olefins and ethylene or ethylene only and related catalyst development work (Figures 1 and 2) seem to enable the synthesis of numerous mono- and dibranched olefins, permitting the exploration of their advantage in comparison to their linear relatives [2, 3, 4]. Regarding advantages or applications of the polymerizable branched α-olefins, the synthesis of isotactic poly(4-ethyl-1-hexene) was reported [5] and gave rise to an ethylene-based, high-melting (220 °C) and highly transparent plastic. Furthermore, copolymerization of ethylene and 4-ethyl-1-octene via coordinative chain transfer polymerization [6] is the basis of the synthesis of a multiply and long-chain branched polyolefin with low density polyethylene (LDPE)-like properties containing two types of functional groups [7]. We report here the synthesis of 4-ethyl-1-octene, 4-ethyl-1-decene, and 4-ethyl-6-methyl-1-heptene (Figure 3). The key is the long-term stability of the catalyst employed.
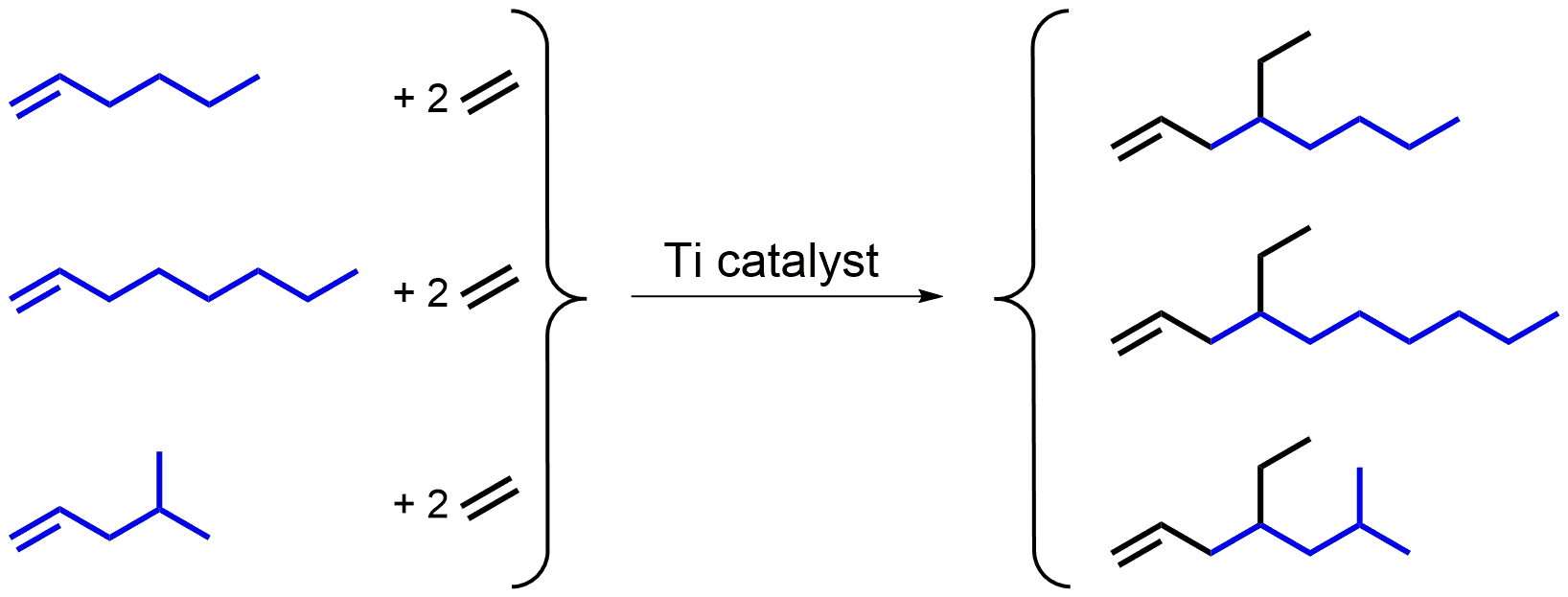
Synthesis of new mono- or multiply-branched α-olefins on a multigram scale (R = n-butyl, n-hexyl, isobutyl) [2, 3].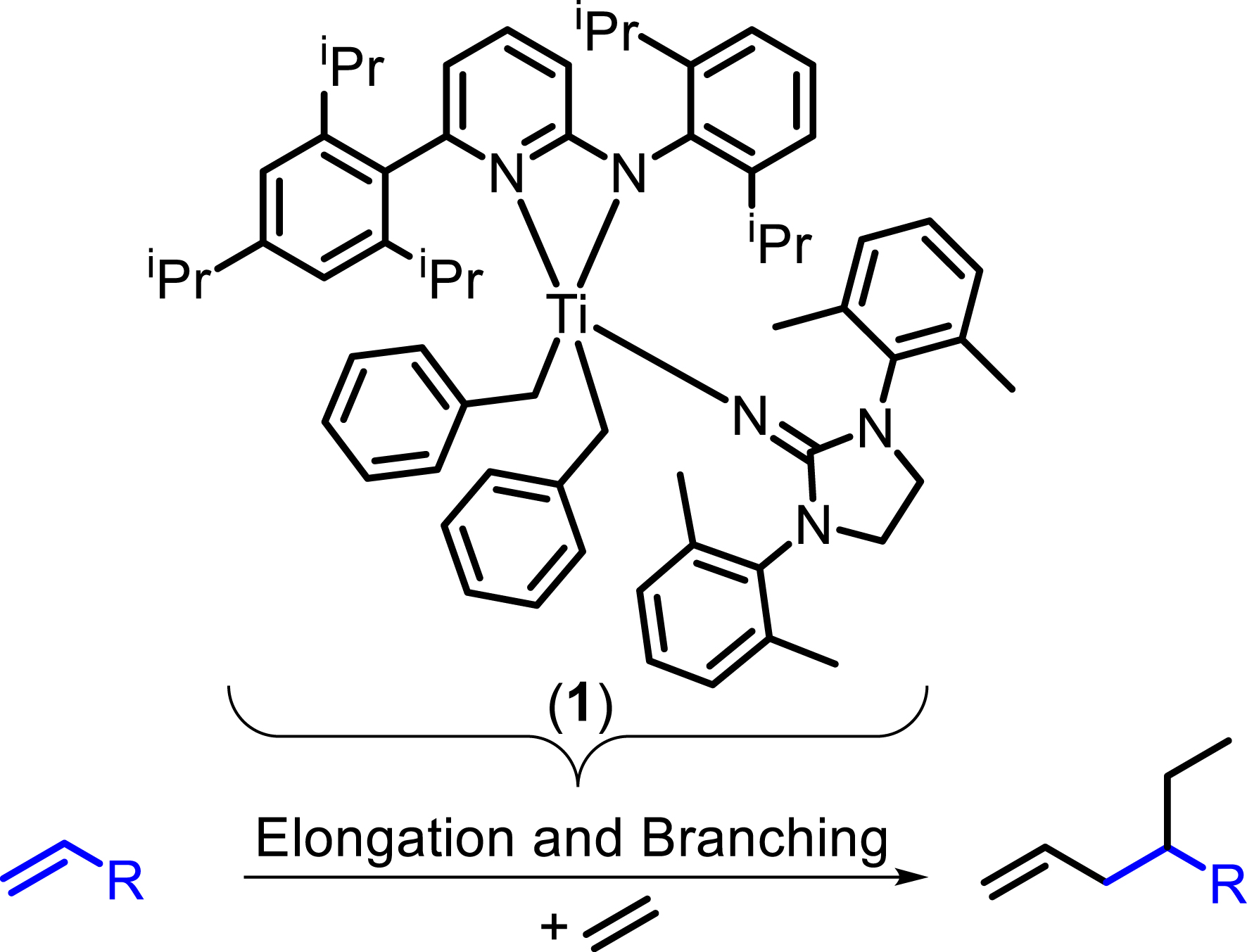
Synthesis of branched α-olefins.
Synthesis of mono- or dibranched α-olefins a–c from an ethylene and α-olefin base stock of 1-hexene, 1-octene, or 4-methyl-1-pentene.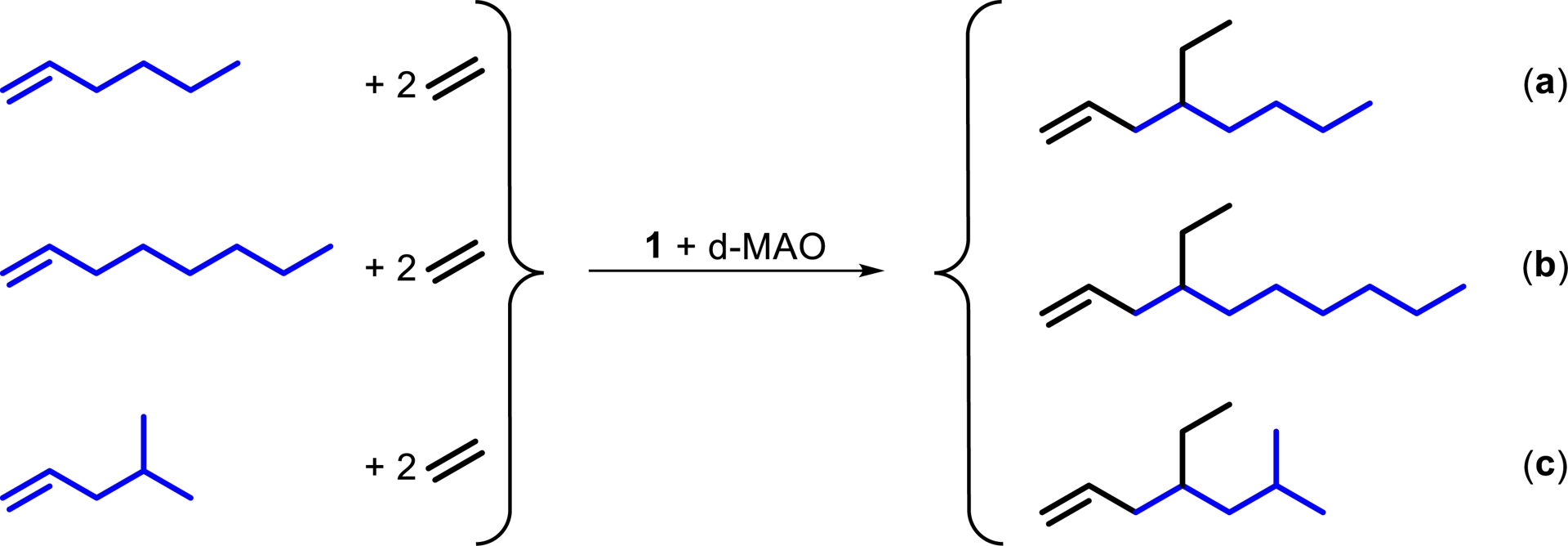
2. Experimental section
All manipulations were carried out under the rigorous exclusion of air and moisture using standard Schlenk techniques and glovebox procedures (mBraun 120-G) with high-capacity recirculation (O2 < 0.1 ppm) under an atmosphere of argon or nitrogen. The oligomerization precatalyst [1,3-bis(2,6-dimethylphenyl)-imidazolidin-2-imido]-[N-(2,6-diiso-propylphenyl)-6-(2,4,6-triisopropylphenyl)-pyridin-2-amido]-di-(phenylmethanido)titanium(IV) 1 was synthesized according to the literature [2]. Depleted methylaluminoxane (d-MAO) was prepared by removing all volatiles under reduced pressure from commercial methylaluminoxane (MAO) in toluene.
2.1. General procedure for the synthesis of branched α-olefins
In order to synthesize the branched α-olefins, 300 mL of the respective dried and degassed linear α-olefin was placed in the evacuated autoclave and the temperature adjusted to 30 °C. Subsequently, an ethylene pressure of 2.5 bar was applied in the autoclave. Stock solutions of precatalyst 1 (0.001 M), cocatalyst (0.035 M), and scavenger (0.400 M) in cumene were injected while stirring. d-MAO was employed as the cocatalyst, while triisobutylaluminum (TIBA) was used as the scavenger. Further parameters are listed in the corresponding tables. After ethylene consumptions of 5, 10, 20, 30, 50, 100, 150, and 200 Ln, samples were taken from the ongoing reaction and analyzed by gas chromatography (GC). Upon completion of the oligomerization, the product mixture was quenched using 2 mL of deionized H2O and washed using 50 mL H2O/HCl. The organic phase was distilled to separate the individual α-olefin fractions. The branched α-olefin desired was isolated and characterized by 1H and 13C NMR spectroscopy as well as GC and mass spectrometry. For the detailed methodology and equipment used for the synthesis, purification procedure, and analysis of the branched α-olefins, see the Electronic Supporting Information.
3. Results and discussion
3.1. Multigram synthesis of mono- or dibranched α-olefins
To the best of our knowledge, no synthesis of 4-ethyl-1-octene a, 4-ethyl-1-decene b, or 4-ethyl-6-methyl-1-heptene c has been reported in the literature. Compound a was observed by GC as one of nine constitutional isomers, with a selectivity of 18 mol% referring to all C10 olefin fractions [8]. Compound b was also observed by GC as one of six co-oligomerization products [9]. Compound c has no CAS number yet and seems unknown. The catalytic cycle for the synthesis of a–c and some main byproducts is depicted in Figure 4. The relevant catalytic cycles start from a cationic titanium species bearing an ethyl ligand I. Coordination of an α-olefin or an ethylene molecule to the vacant coordination site is followed by insertion into the Ti–C bond II. Subsequent ethylene insertion III and β-H elimination/transfer to a second ethylene molecule IV lead to the release of product A and regeneration of the active species. In the absence of the α-olefin insertion, 1-butene B is predominantly formed, which competes with the α-olefins added and leads to concurrent 4-ethyl-1-hexene formation. The α-value (chain propagation probability) increases with increasing temperature [2] (and pressure) and leads to more byproduct formation at higher temperature at both ends, linear byproducts and branched byproducts. We always use the bisbenzyl titanium precatalysts since we have not yet found ways of making any other suitable precatalyst despite the activation with d-MAO. d-MAO gives slightly better co-trimer selectivity than borate activators [3] and the bisbenzyl precatalysts work fine with both activators. Regarding the catalyst choice, we are very limited. We need a very low α-value to avoid formation of longer linear α-olefins and their branched derivatives. On the other hand, we need a sterically demanding catalyst to avoid β-H elimination/transfer after the co-dimer is formed. This is a very narrow gap, reducing the potential catalysts to two examples yet: the precatalysts used in the original contribution [2] and 1 used here. Both catalysts do not isomerize α-olefins under the conditions explored yet [2, 3]. Precatalyst 1 reduces co-dimer formation to about one third in comparison to precatalysts used in the original contribution.
Catalytic cycle for the synthesis of branched α-olefins A and byproducts 1-butene B and 4-ethyl-1-hexene C. The coordination site is shown as a square (R = butyl, hexyl, isobutyl).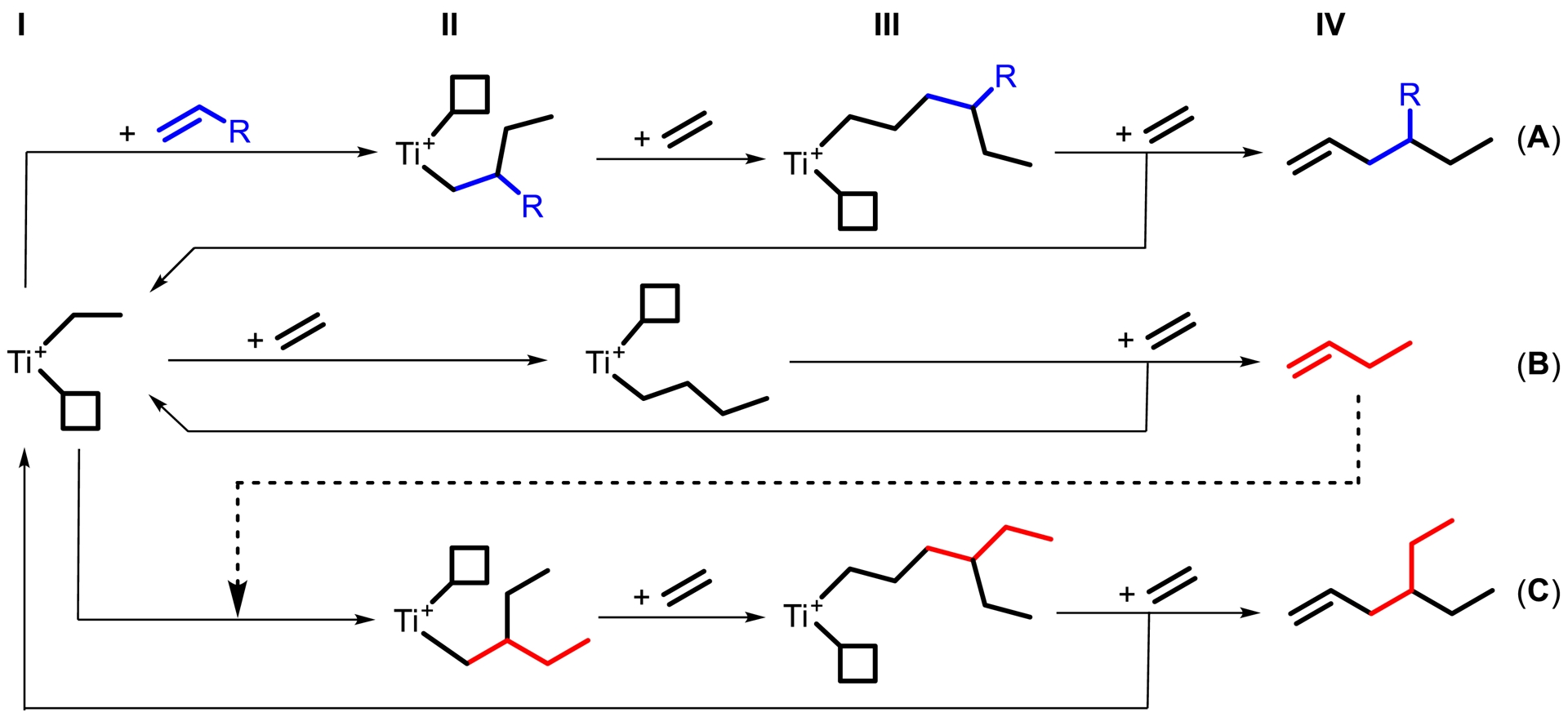
3.1.1. Synthesis of 4-ethyl-1-octene a
Compound a was synthesized on a multigram scale according to the reaction scheme shown in Figure 5. 1-Hexene was used as the starting material. The corresponding reaction parameters are summarized in Table 1. The amount and mass of a formed after consumption of a specific ethylene volume are given in Table 2. A plot of the amount of a synthesized depending on the consumption of ethylene is depicted in Figure 6. The synthesis of the branched olefins desired on a multigram scale requires a long-term stable catalyst system. Figure 7 shows the ethylene consumption during co-oligomerization of 1-hexene and ethylene plotted against the reaction time required for the corresponding consumption.
Synthesis of 4-ethyl-1-octene a from ethylene and 1-hexene.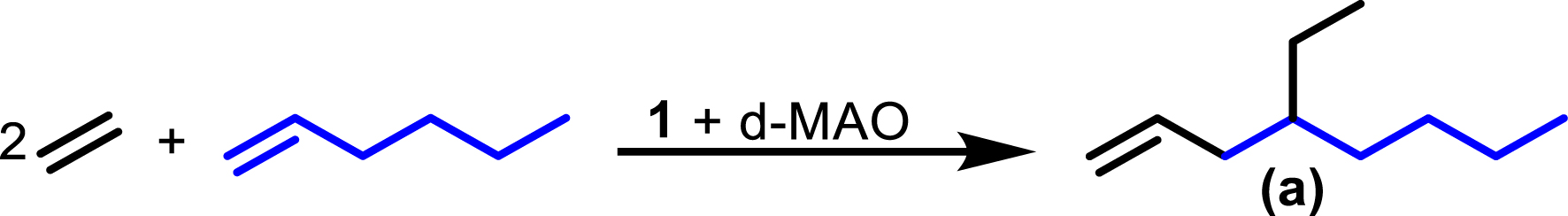
Amount of 4-ethyl-1-octene a synthesized as a function of ethylene consumption.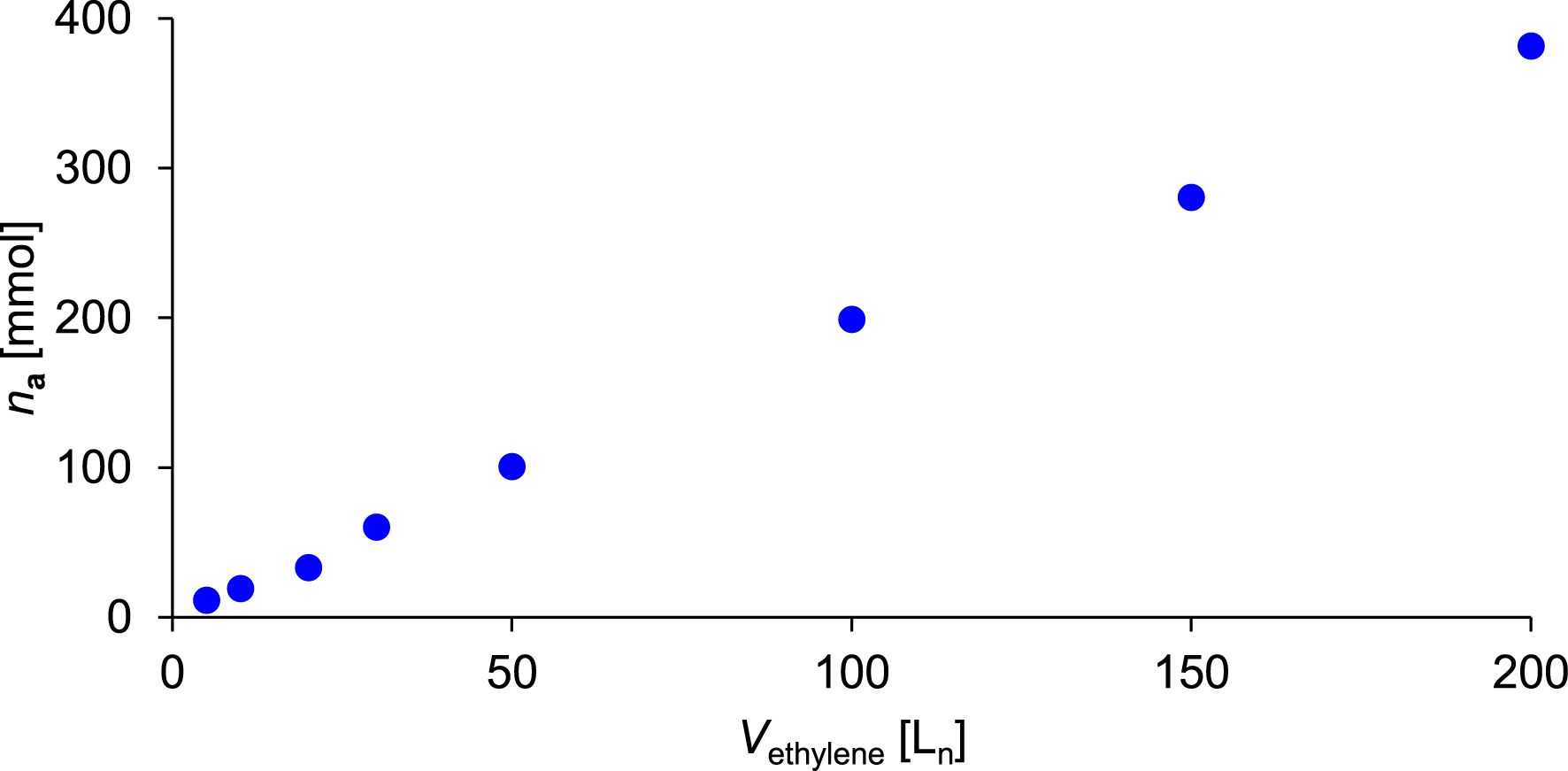
Ethylene consumption over time during the synthesis of 4-ethyl-1-octene a from ethylene and 1-hexene.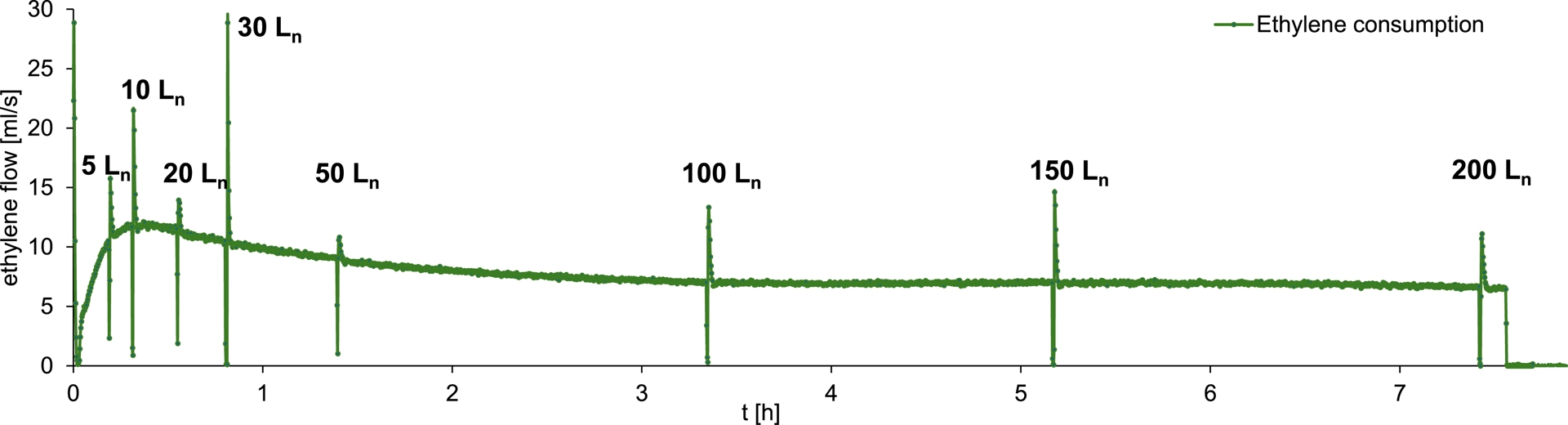
Parameters for 4-ethyl-1-octene a synthesis*
| n1 (μmol) | md‐MAO (mg) | nTIBA (μmol) | V1‐hexene (mL) | pethylene (bar) | T ( °C) | Vethylene (Ln) | t (h) |
|---|---|---|---|---|---|---|---|
| 1.0 | 2.0 | 400 | 300 | 2.5 | 30 | 200 | 7.5 |
*n(d-MAO)/n(Ti) = 34.5.
Amount and mass of 4-ethyl-1-octene a synthesized by consumption of the indicated ethylene volumes
| Vethylene (Ln) | 5 | 10 | 20 | 30 | 50 | 100 | 150 | 200 |
| na (mmol) | 11.3 | 19.2 | 33.0 | 60.3 | 100.8 | 198.9 | 280.5 | 381.4 |
| ma (g) | 1.6 | 2.7 | 4.6 | 8.5 | 14.1 | 27.9 | 39.4 | 53.5 |
The amount of a increases approximately linearly with the progressing ethylene consumption, as shown in Figure 6. Since 1-hexene, which was initially loaded, is also formed during the catalytic process, its consumption appears to have had no observable influence on the formation of a. After the consumption of 200 Ln ethylene, 53.5 g (381.4 mmol) of a was synthesized, as determined by GC. Based on ethylene consumption, catalyst activity is 9500 kgethylene⋅mol−1⋅h−1⋅bar−1. Regarding the branched products synthesized, the catalyst system based on 1 exhibited a selectivity of 58.0 wt% (55.2 mol%) toward a. Additionally 136.9 g 1-butene, 41.0 g 1-hexene, and 24.5 g 4-ethyl-1-hexene were formed. A mass balance of all α-olefins synthesized during this catalytic process is provided in the Electronic Supporting Information. As illustrated in Figure 7, precatalyst 1 enables a continuous and long-term stable oligomerization of the substrates. Moreover, a short interruption of the ethylene feed for sampling appears to have had no significant impact on the further progression of the reaction. After the consumption of 200 Ln of ethylene, approximately 460 mL of the liquid product and reactant fraction was obtained. The resulting organic phase was subjected to fractional distillation to isolate a. The compound exhibited a boiling point of 110 °C at a pressure of 200 mbar. After purification, 38.5 g (274.5 mmol) of a was isolated with a purity of 97.2 mol%. Figure 8 displays the 1H NMR spectrum of compound a. All resonances observed can be assigned to the protons of 4-ethyl-1-octene. The integrals were referenced to the two vinylic protons of a. As expected, the integral corresponding to the two methyl groups accounts for approximately six protons. Additionally, trace amounts of cumene are visible in the spectrum. Cumene was used as the solvent for the stock solutions employed during synthesis. Its boiling point of 152.4 °C at ambient pressure is close to that of a, preventing complete removal by distillation. A 13C NMR spectrum and a mass spectrum were recorded for further characterization. Both are shown in the Electronic Supporting Information.
1H NMR spectrum of 4-ethyl-1-octene a (300 MHz, 293 K, C6D6).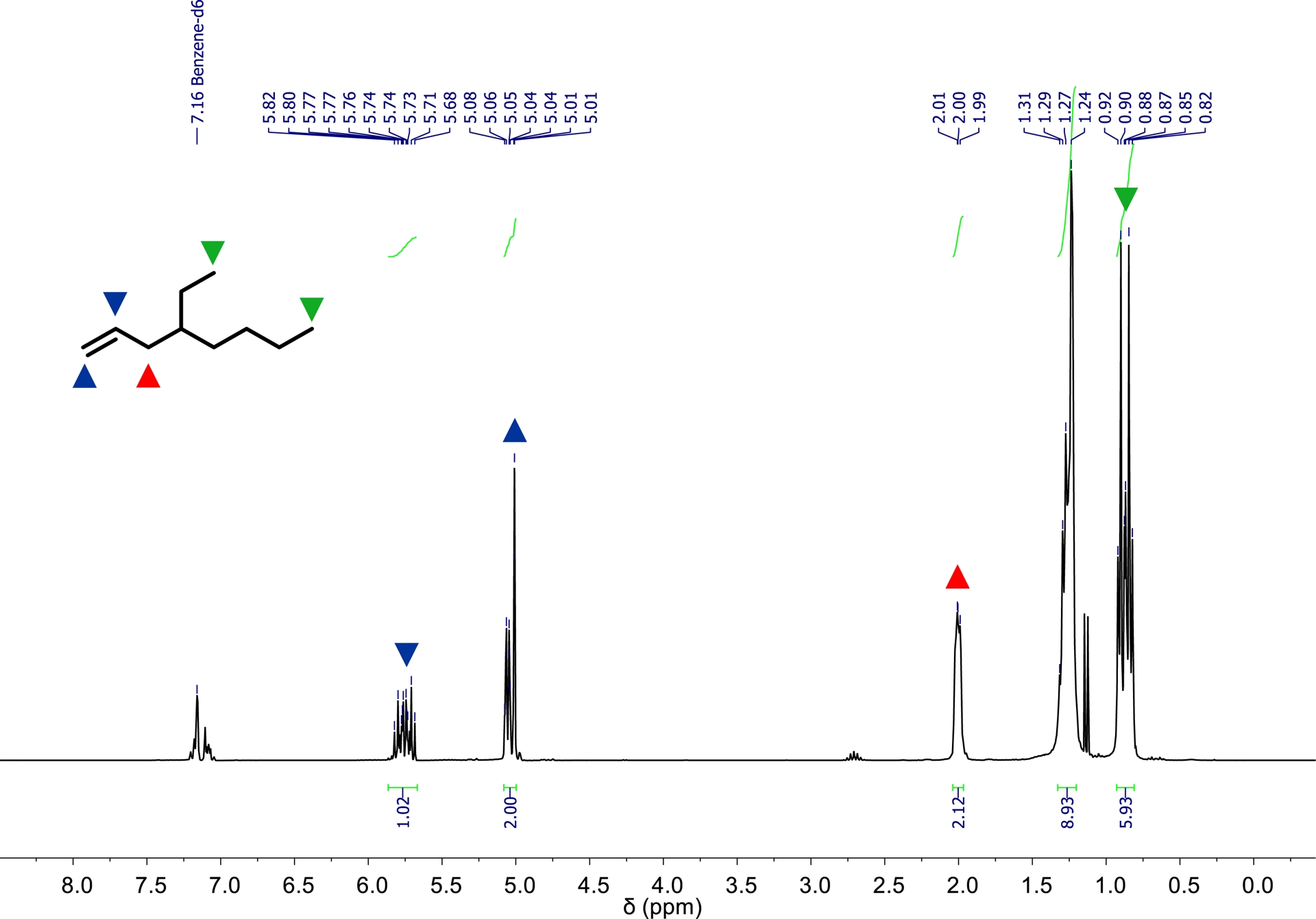
3.1.2. Synthesis of 4-ethyl-1-decene b
The methodology applied for the synthesis of 4-ethyl-1-decene b (Figure 9) is analogous to that used for the preparation of a. The corresponding reaction parameters are summarized in Table 3. The amount and mass of b obtained after a defined ethylene consumption are listed in Table 4. Figure 10 displays the plot of the synthesized amount of b versus the corresponding ethylene consumption.
Synthesis of 4-ethyl-1-decene b from ethylene and 1-octene.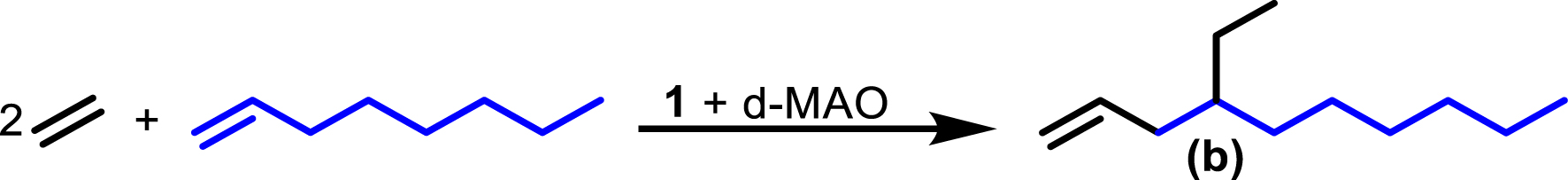
Amount of 4-ethyl-1-decene b synthesized as a function of ethylene consumption.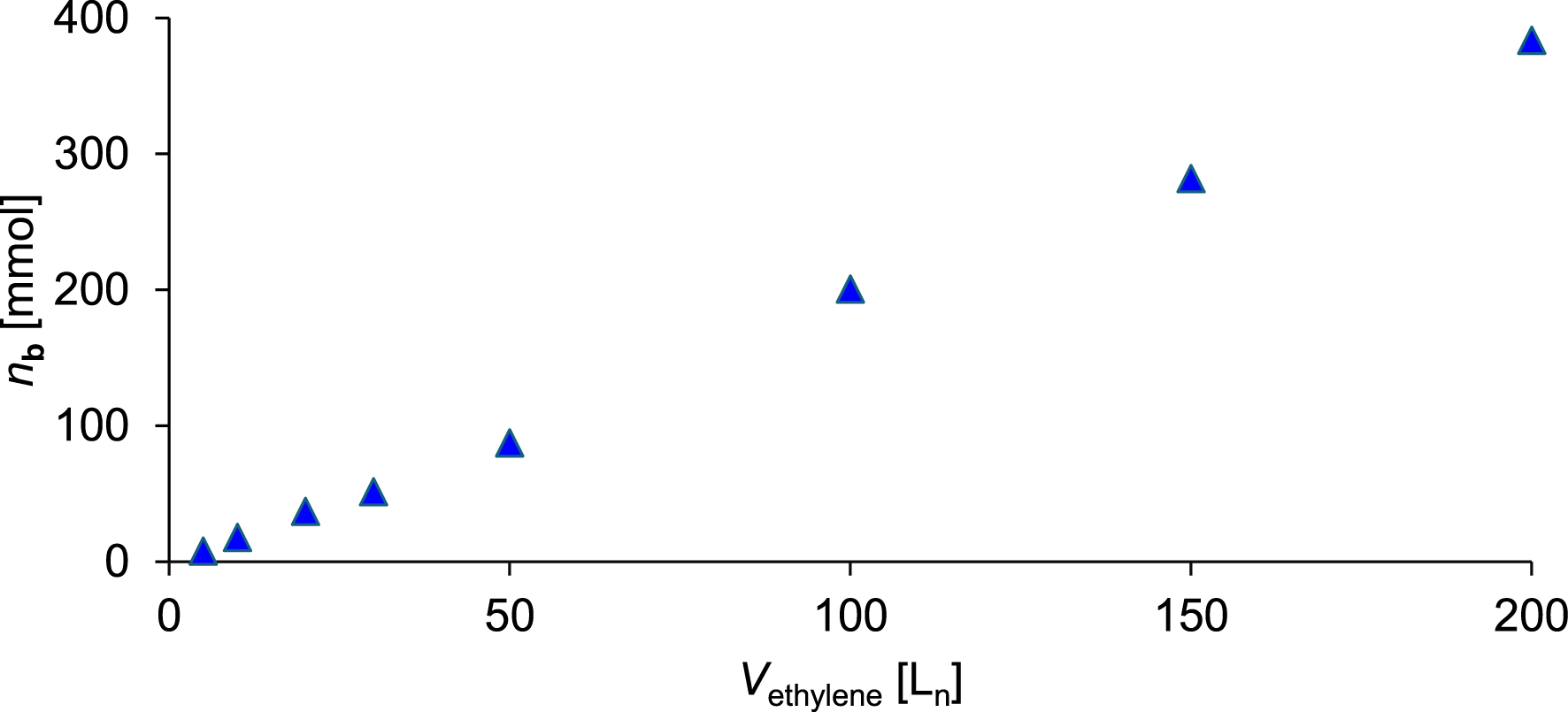
Parameters for 4-ethyl-1-decene b synthesis*
| n1 (μmol) | md‐MAO (mg) | nTIBA (μmol) | V1‐octene (mL) | pethylene (bar) | T (°C) | Vethylene (Ln) | t (h) |
|---|---|---|---|---|---|---|---|
| 1.0 | 2.0 | 400 | 300 | 2.5 | 30 | 200 | 7.2 |
*n(d-MAO)/n(Ti) = 34.5.
Amount and mass of 4-ethyl-1-decene b synthesized by consumption of the indicated ethylene volumes
| Vethylene (Ln) | 5 | 10 | 20 | 30 | 50 | 100 | 150 | 200 |
| nb (mmol) | 8.0 | 17.9 | 37.3 | 51.6 | 87.6 | 200.9 | 282.1 | 383.8 |
| mb (g) | 1.3 | 3.0 | 6.3 | 8.7 | 14.7 | 33.8 | 47.5 | 64.6 |
The parameters of the synthesis affording 4-ethyl-1-decene b (Table 3) are nearly identical to those used for the synthesis of a (Table 1). As shown in Table 4 and Figure 10, the amount of b increases approximately linearly with increasing ethylene consumption. After the consumption of 200 Ln of ethylene, 64.6 g (383.8 mmol) of 4-ethyl-1-decene b was obtained, as determined by GC analysis. Based on ethylene consumption, catalyst activity is 9900 kgethylene⋅mol−1⋅h−1⋅bar−1. Regarding the branched products synthesized, 1 exhibits a selectivity of 58.9 wt% (53.0 mol%) toward b. Additionally 126.9 g 1-butene, 43.7 g 1-hexene, 13.4 g 1-octene, and 22.3 g 4-ethyl-1-hexene were formed. Upon completion of the synthesis, approximately 490 mL of a liquid product and reactant mixture were recovered. The workup was carried out analogously to a. The organic phase was subjected to fractional distillation to isolate b. 4-Ethyl-1-decene b exhibits a boiling point of 115 °C at 15 mbar. After purification, 44.7 g (265.6 mmol) of b was isolated with a purity of 94.1 mol%. 1H and 13C NMR spectra as well as a mass spectrometry spectrum were recorded for the characterization of b. The referring 1H NMR spectrum is shown in Figure 11. All signals observed in the spectrum can be assigned to the resonances of 4-ethyl-1-decene. The integrals were referenced to the two vinylic protons of b. The high-field region contains the signals of the protons in the two terminal methyl groups of the branched 4-ethyl-1-decene, showing an integral value corresponding to approximately six protons.
1H NMR spectrum of 4-ethyl-1-decene b (300 MHz, 293 K, C6D6).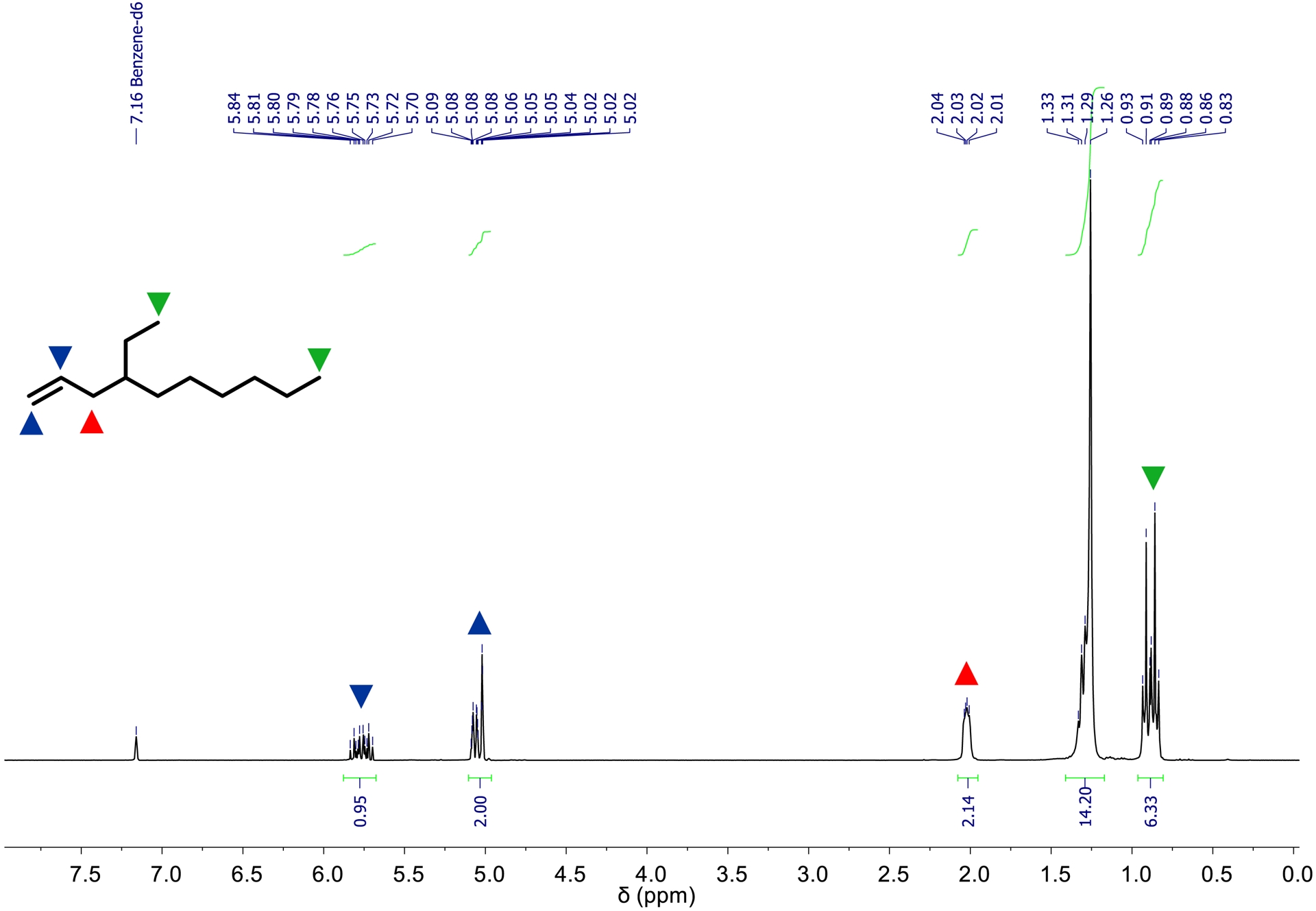
3.1.3. Synthesis of 4-ethyl-6-methyl-1-heptene c
The synthesis of c was carried out according to the reaction sequence depicted in Figure 12 and proceeded analogously to the synthesis of a and b. The corresponding reaction parameters are summarized in Table 5. The amount and mass of c formed after the consumption of a specific volume of ethylene are given in Table 6. A plot of the amount of c synthesized depending on the consumption of ethylene is depicted in Figure 13. The amount of precatalyst 1 required for the synthesis of c had to be increased to 3.0 μmol and the mass of d-MAO had to be increased to 6.0 mg to achieve the consumption of 200 Ln of ethylene within the desired reaction time of eight hours. This corresponds to a threefold increase in the amounts of both precatalyst and activator compared to the synthesis of a and b. Based on ethylene consumption, catalyst activity is 3000 kgethylene⋅mol−1⋅h−1⋅bar−1. As evident from Table 6 and Figure 13, the reactivity ratio of 1 toward branched α-olefins is significantly lower compared to linear α-olefins. Under otherwise identical conditions, the synthesis of 4-ethyl-6-methyl-1-heptene c from ethylene and 4-methyl-1-pentene only afforded 122.6 mmol of c. This corresponds to approximately one-third of the amount of product obtained in the synthesis of a from ethylene and 1-hexene (Table 2) or b from ethylene and 1-octene (Table 4). During the synthesis of c, an exponential increase in product formation was observed between 5 and 30 Ln of ethylene consumption (Figure 13), which transitions into a linear increase beyond 30 Ln. After the consumption of 200 Ln of ethylene, 17.2 g (122.6 mmol) of 4-ethyl-6-methyl-1-heptene c was obtained, as determined by GC analysis. Regarding the branched products synthesized, 1 exhibits a selectivity of 25.3 wt% (24.1 mol%) toward c. Additionally 136.7 g 1-butene, 32.8 g 1-hexene, and 24.4 g 4-ethyl-1-hexene were formed. Upon completion of the co-oligomerization, approximately 430 mL of a liquid product and reactant mixture was recovered. After workup, the organic phase was subjected to fractionated distillation. 4-Ethyl-6-methyl-1-heptene c exhibits a boiling point of 95 °C at 300 mbar. After purification, 15.3 g (109.1 mmol) of c was isolated with a purity of 95.8 mol%. The 1H NMR spectrum of c is shown in Figure 14. All resonances observed in the spectrum can be assigned to the protons of c. The integrals were referenced to the two vinyl protons in the low-field region of the spectrum. The proton signal of the three terminal methyl groups appears in the high-field region, with an integral corresponding to approximately nine protons. Additionally, two signals assigned to the protons at the two saturated tertiary carbon centers of c were observed in the high-field region of the spectrum.
Synthesis of 4-ethyl-6-methyl-1-heptene c from ethylene and 4-methyl-1-pentene.
Amount of 4-ethyl-6-methyl-1-heptene c synthesized as a function of ethylene consumption.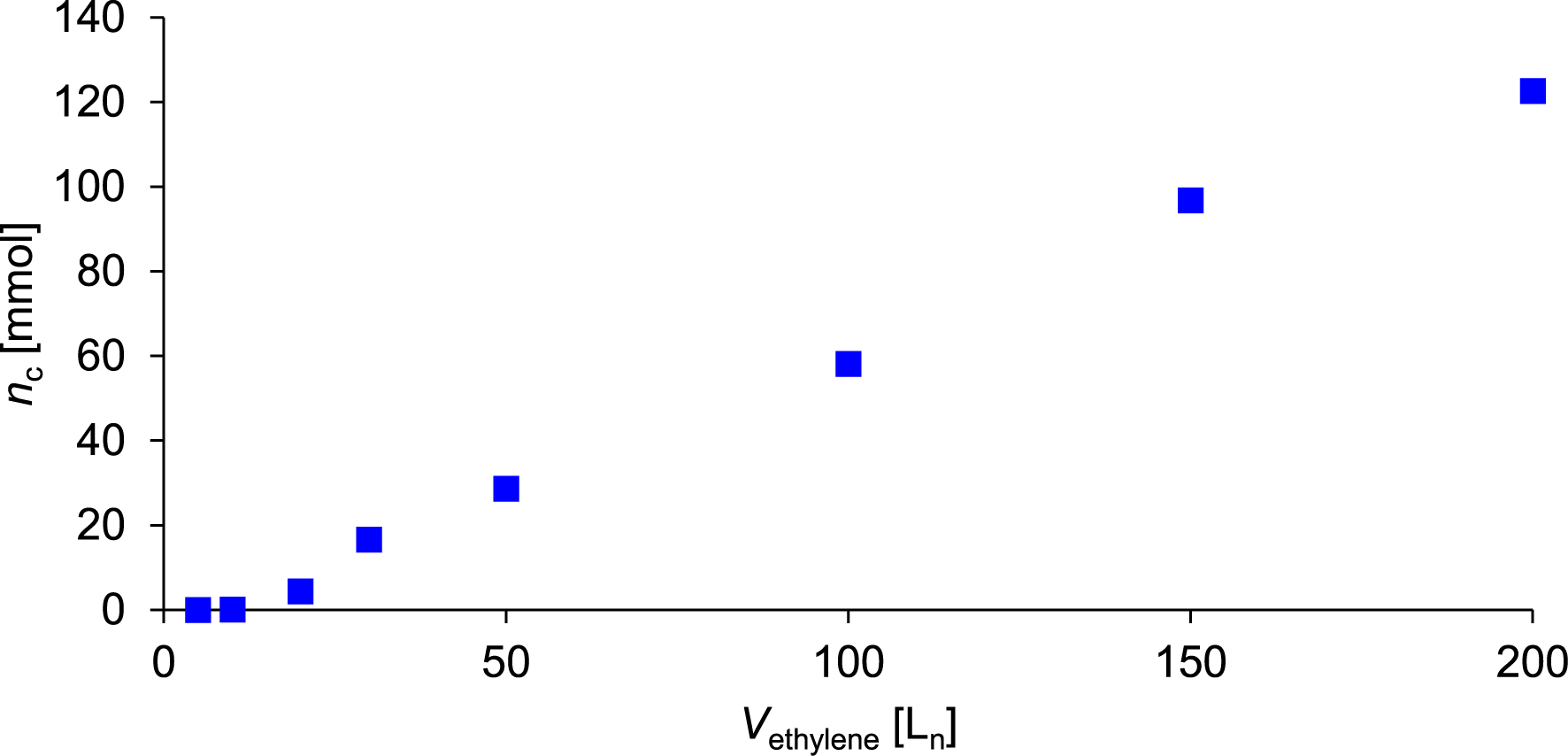
1H NMR spectrum of 4-ethyl-6-methyl-1-heptene c (300 MHz, 293 K, C6D6).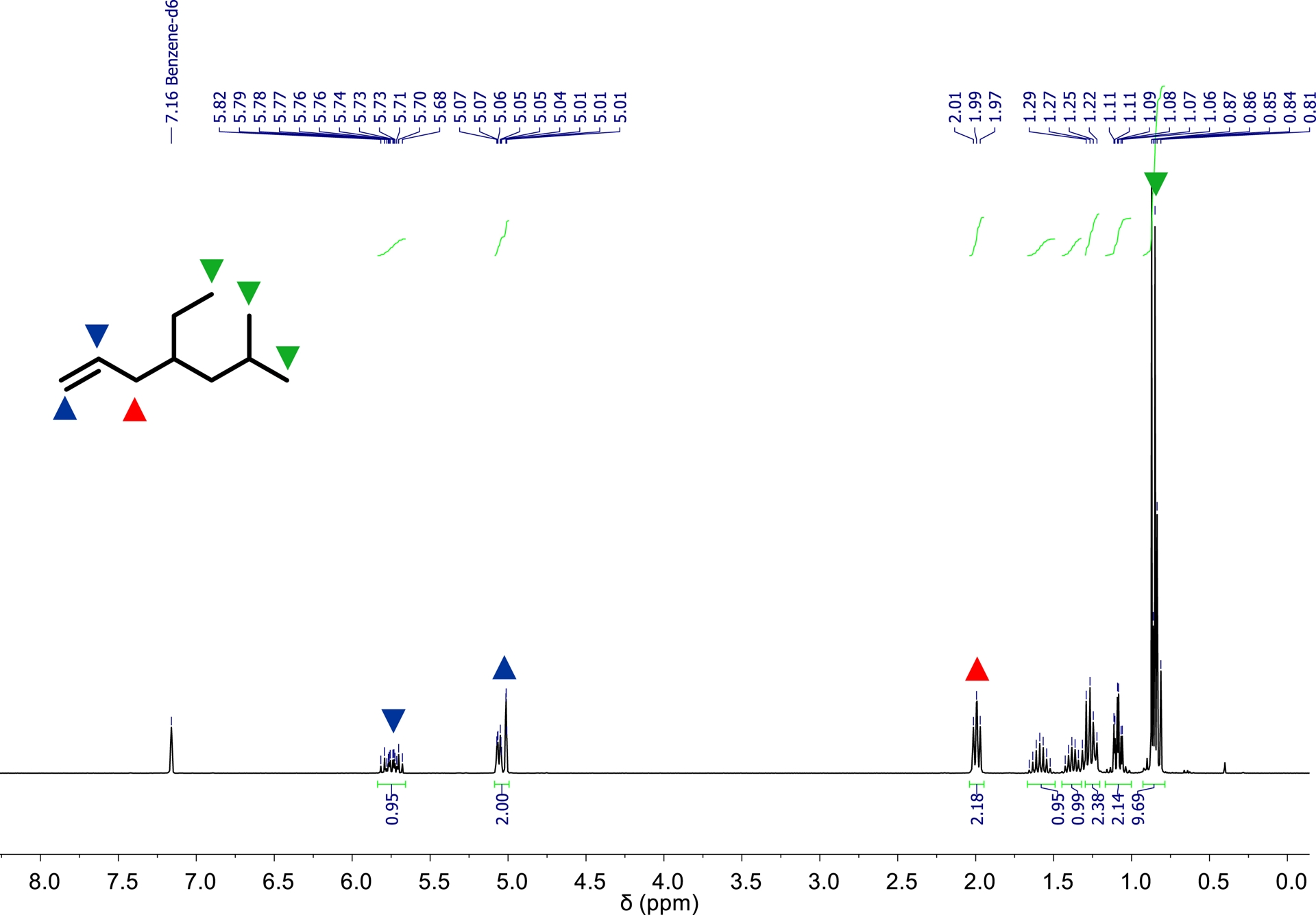
Parameters for the synthesis of 4-ethyl-6-methyl-1-heptene c*
| n1 (μmol) | md‐MAO (mg) | nTIBA (μmol) | V4‐methyl‐1‐pentene (mL) | pethylene (bar) | T (°C) | Vethylene (Ln) | t (h) |
|---|---|---|---|---|---|---|---|
| 3.0 | 6.0 | 400 | 300 | 2.5 | 30 | 200 | 8.0 |
*n(d-MAO) / n(Ti) = 34.5.
Amount and mass of 4-ethyl-6-methyl-1-heptene c synthesized by consumption of the indicated ethylene volumes
| Vethylene (Ln) | 5 | 10 | 20 | 30 | 50 | 100 | 150 | 200 |
| nc (mmol) | 0 | 0.1 | 4.3 | 16.6 | 28.6 | 58.1 | 96.8 | 122.6 |
| mc (g) | 0 | 0.01 | 0.6 | 2.3 | 4.0 | 8.1 | 13.6 | 17.2 |
4. Conclusion
In conclusion, we report the multigram synthesis of 4-ethyl-1-octene, 4-ethyl-1-decene, 4-ethyl-6-methyl-1-heptene from ethylene and commercially and abundantly available, inexpensive starting materials. The catalyst employed is stable in long-time runs of more than seven hours and the change in product distribution over time results mostly from 1-butene byproduct formation and its elongation/branching reaction forming 4-ethyl-1-hexene. The same phenomenon is relevant for the synthesis of 4-ethyl-1-hexene [3] but in a beneficial way since 1-butene concentration is kept high via byproduct formation. We work with a well-defined precatalyst under relatively low ethylene pressure and at low temperature. All three conditions are helpful to avoid polymer byproduct formation. In addition, we form a lot of branched products with a better solubility than purely linear products. We do not see polymer formation/precipitation even in an over-seven-hour run. Conventional oligomerization catalysts that operate via a Cossee–Arlman mechanism type eliminate as soon as the co-dimer is formed and cannot insert another ethylene molecule according to the formal ethylene–α-olefin–ethylene insertion sequence [3] needed to form 4-ethyl-1-alkenes.
Acknowledgements
We thank the Chair of Macromolecular Chemistry II of the University of Bayreuth for MS measurements.
Declaration of interests
The authors do not work for, advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliation other than their research organizations.
Funding
The research was supported by the Deutsche Forschungsgemeinschaft (DFG KE756 / 35-1).
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.434 or from the author.