

1 Introduction
Phosphorous is an essential nutrient for the growth of aquatic plants and algae, but its excessive supply in water bodies causes eutrophication resulting in increased growth of algae, depletion of oxygen, and disruption in the balance of organisms. Excessive phosphate input to waters originates from anthropogenic activities such as wastewater discharges and agricultural run-off. In wastewater treatment technology, chemical and biological removal techniques are the most effective and well-established methods, but their high cost and complexity of implementation prevent their application both in rural areas and in developing countries. Recently, attention has been focused on developing alternative methods for phosphate removal from water. Among these methods, adsorption appears as an attractive option, owing to its low cost and easy application (Boyer et al., 2011; Hongbin and Ming, 2014). A large variety of materials with the availability to remove phosphate were thus described in the literature (Johansson-Westholm, 2006; Vohla et al., 2011; Wendling et al., 2013). High sorption capacity, low cost, availability in large quantity and environmentally-friendly nature are among the most important parameters in the choice of the sorbent. The present paper reports a study that aimed to develop a material satisfying all these criteria with the final objective of applying this optimized material in the treatment of wastewaters released into the Ebrié lagoon in Ivory Coast. In fact, the eutrophication of the Ebrié lagoon is dramatic in large areas, making the lagoon actually unsafe for habitation sites, for the practice of lucrative activities such as bathing and fishing (Briton et al., 2007; Koffi et al., 2009; Tuo et al., 2012; Yao et al., 2009). Interestingly, shale, sandstone and laterite are low cost and environmentally friendly geological materials easily available in large quantities in Ivory Coast. In addition to this context, these geomaterials are expected to contain separate iron oxide minerals and/or iron in structural substitution and are thus expected to be promising phosphate adsorbents.
The main objective of the present paper is to study the sorption behavior of phosphate onto three natural, available and inexpensive geomaterials from Ivory Coast to determine the best candidate for a further application in a micro-pilot unit and ultimately in a wetland system. Batch sorption experiments were carried out as a function of adsorbent dosage, pH, and contact time. The sorption kinetics and Langmuir and Freundlich isotherms were also studied. Then, the performance of the three materials for phosphate removal were evaluated in flow through column experiments. The originality of the present work lied in the understanding of the relation existing between the physicochemical properties of the adsorbents and their ability to remove phosphate from water. A particular attention was devoted to quantifying the dissolution properties of the minerals (release of FeII, AlIII, MgII, CaII in solution) in order to estimate the respective roles of dissolution and adsorption processes in the phosphate removal from water.
2 Materials and methods
2.1 Chemicals
All chemicals used were analytical grade. All experiments were conducted in polypropylene vessels that were soaked in 36% hydrochloric acid (HCl) before use and rinsed with deionized water (Milli-Q, 18.2 MΩ cm−1) and ethanol. Deionised water was used for preparing all solutions.
2.2 Adsorbent
Shale, sandstone, and laterite were collected in three regions of Ivory Coast, i.e. shale from the central region (Toumodi (6°28′ N, 4°58′ W)–Lomo North site), sandstone from the southern region (Abidjan (5°21′ N; 4°0′ W)–Akouedo site) and laterite from the eastern region (Abengourou (6°42′ N, 3°28′ W)–Sankadjo site). Sampling was performed in February 2015. Samples were prepared using the same following procedure. Large blocks (diameter of 20 to 30 cm) extracted from the surface soil without any specific tool were first coarsely screened, washed with deionised water to remove the surface-adhered particles and dried in an oven at 50 °C for 24 h. The samples were then crushed again and sieved (AFNOR sieves) to obtain granular sizes of 1–2 mm and ϕ < 400 μm. Powder samples were finally washed again and dried at 70 °C for 24 h before their use. The main physical characteristics of the three geomaterials obtained for the granular size of 1–2 mm are reported in Table S1. In addition, the chemical composition of the three substrates (Table S2) revealed that all three materials were primarily a mixture of oxides, the major one being SiO2, Al2O3, and Fe2O3. The relative amount of these oxides differs from one material to the other. Sandstone was rich in iron, while shale and laterite were Al-rich materials. All three subtrates contained large amounts of Si. The specific surface area of the three powder materials (ϕ < 400 μm) was determined by multipoint N2-BET analysis using a Belsorp max surface area analyser (Bel, Japan). The specific surface areas of shale, sandstone, and laterite were 6.3, 28.5, and 37.6 m2 g−1, respectively.
2.3 Sorption studies
Phosphate sorption onto the three substrates was studied in both batch and column experiments at room temperature. All phosphate concentrations reported in the present study refer to PO4. A stock phosphate solution (PO4 250 mg L−1) of phosphate was prepared by using sodium dihydrogen phosphate (H2NaO4P·2H2O) and diluted in subsequent experiments.
2.3.1 Batch experiments
The effect of the sorbent dose was examined in a series of experiments that used the same initial phosphate concentration (25 mg PO4 L−1) while using different adsorbent doses between 0 and 160 mg L−1. 50-mL flasks were used. The pH of the suspension was maintained at a defined value of 6.5–7.0 by manually adding 0.5 M HCl or 0.5 M NaOH at regular intervals and by using an Almemo 2690 digital pH meter (Ilmenau, Germany) and a WTW glass electrode (Weihleim, Germany). Flasks were capped and stirred magnetically at 300 rpm for 24 h to ensure approximate equilibrium. At the end of this sorption period, 10 mL were withdrawn from the reaction flasks and filtered through 0.22-μm polypropylene syringe filters for phosphorous and aqueous metal species (Ca, Mg, Al, and Fe) analyses by ICP–AES. All experiments were carried out in duplicate to check the reproducibility of the experiments. The pH effect on phosphate removal was examined with a similar procedure using a 25-mg L−1 initial phosphate concentration and 80 g L−1 (shale, sandstone and laterite) and 8 g L−1 (sandstone and laterite) adsorbent dosages while maintaining pH at different values in the range from 2 to 12. Phosphate sorption kinetics were examined in 1-L Erlenmeyer flasks, using an initial phosphate concentration of 25 mg L−1 and various adsorbent doses for ∼60 h. During this reaction period, samples were withdrawn from the flasks at regular intervals of time for phosphate and aqueous metal species determination. The pH of the suspension was maintained at a defined value (6.5–7.0). Sorption isotherm experiments were carried out at pH = 6.5–7.0 in 50-mL Erlenmeyer flasks with varying initial concentrations of phosphate ions (5–250 mg L−1) and a constant adsorbent dosage of 80 g L−1 (shale) and 8 g L−1 (sandstone and laterite). The stirring rate was 300 rpm and the equilibration time was 24 h. The phosphate removal efficiency R (%) of the three adsorbents and the phosphate uptake qt (mg g−1) at time t were calculated from Eqs. (1) and (2), respectively:
| (1) |
| (2) |
where Ci and Ct are the initial phosphate concentration and that at time t, respectively (mg L−1); V is the volume of the solution (L), and m is the mass of material (g). Note that phosphate uptake under near-equilibrium conditions (qe, mg g−1) is obtained at the equilibrium phosphate concentration Ct = Ce.
2.3.2 Column experiments
Plexiglas columns 500 mm in length and with an internal diameter of 37 mm, corresponding to a volume Vtot of 473 mL, were used. The columns were packed with dried shale, sandstone or laterite (grain size of 1–2 mm) corresponding to total masses of 593, 661, and 679 g, respectively. Porosity was determined in triplicate by a volumetric method that consisted in determining the volume of water (Vw) required to saturate a known volume of shale (Vs). Porosity (ɛ) was obtained according to the following formula ɛ = Vw/Vs × 100. The porosity of all substrates was very close to 50%; indeed, the porous volume VP for the three materials was very similar, i.e. VP = 236, 246, and 241 mL for shale, sandstone, and laterite, respectively. An upward-directed water flow was used with the help of a peristaltic pump (Watson-Marlow), ensuring by this way water-saturated conditions. All the columns were first pre-equilibrated with demineralized water during 24 h before performing the phosphate removal experiments. The pH of the phosphate solution ([PO4] = 25 mg L−1) introduced into the column was close to 5, and the pH measured in the outflow was relatively constant, independently of the nature of the materials used in the columns, with mean pH values of 6.1, 6.0, and 6.4 for shale, sandstone, and laterite, respectively. For both sandstone and laterite columns, a continuous flow of 1 mL min−1, with a corresponding hydraulic retention time (HRT) of ∼ 4 h was used. The total duration of the experiments was ∼ 2.5 months. In order to mimic situations that can be encountered in constructed wetlands where water supply is often provided discontinuously, a water flow circulating discontinuously was used for the shale column, as described in our previous work (Kpannieu et al., 2018). The water flow was 0.23 mL min−1 during a first period of 104 h with a corresponding HRT ∼ 17 h followed by a shutdown of the water pump corresponding to a HRT of ∼ 64 h. The sequence (104 h, 64 h) was repeated nine times for a total duration of the experiments of ∼ 2 months. At appropriate time intervals, 10 mL of solution were sampled in the column outflow and filtered through a 0.22-μm polypropylene syringe filters before phosphate and metal species were analyzed with ICP–AES. The inlet phosphate solution was renewed every two days to limit the bacterial growth.
The phosphate uptake of the column qcol (mg g−1), which represented the total amount of adsorbed phosphate on shale, was determined using the following equation:
| (3) |
where Q (L·d−1) is the measured flow rate, m (g) is the mass of the shale in column, C0 (g·L−1) is the phosphate concentration of the incoming effluent, and C (g L−1) is the phosphate concentration of the outlet effluent.
2.4 Characterization methods
The structural properties of shale, sandstone, and laterite were investigated with X-ray diffraction (XRD) and Mössbauer spectrometry. XRD patterns were acquired with a PANalytical X’Pert Pro Multipurpose diffractometer using Co Kα radiation (λ = 1.78026 Å; 40 kV, 40 mA). Diffraction patterns were obtained over a range of 10–85o 2θ range with a step size of 0.036° and a nominal time per step of 3 s. Transmission Mössbauer spectrometry was performed with a 50-mCi source of 57Co in rhodium to identify and quantify iron phases. The Mössbauer spectra were calibrated relatively to the reference spectrum of a 25 μm thick pure iron foil recorded at room temperature. A cryostat equipped with a closed cycle helium station allows variable temperature measurement between 8 and 300 K. The spectra were fitted using Lorentzian shape lines and, in some cases, Voigt profile analysis, a convolution of a Gaussian distribution with a Lorentzian shape line, was used. Phosphate and metal species (Ca, Mg, Al and Fe) concentrations in solution after syringe filtration were determined by Inductively Coupled Plasma–Atomic Emission Spectrometry (ICP–AES) using an Ultima spectrometer (HORIBA Jobin Yvon).
3 Results and discussion
3.1 Physicochemical properties of the adsorbents
3.1.1 X-ray diffraction (XRD)
All three diffraction patterns (Fig. 1) displayed strong reflection of quartz (SiO2). Additional reflection lines of silicates minerals such as muscovite (KAl2(Si, Al)4O10(OH)2, albite (Na, Ca) (Si, Al)4O8) and clinochlore (Mg, Fe)5(Si3Al)O10(OH)8 were observed for shale, while iron oxides such as goethite (α-FeOOH) and hematite (α-Fe2O3) were observed for sandstone and laterite, as already reported (Ngon et al., 2016). Such mineral phases agreed relatively well with the chemical composition reported in Table S2, except as regards the absence of iron-bearing minerals in shale. Because iron is well known to play a key role in phosphate sorption (Altundoğan and Tümen, 2001; Mallet et al., 2013), a closer examination of iron phase minerals contained in the three substrates was carried out using Mössbauer spectrometry.
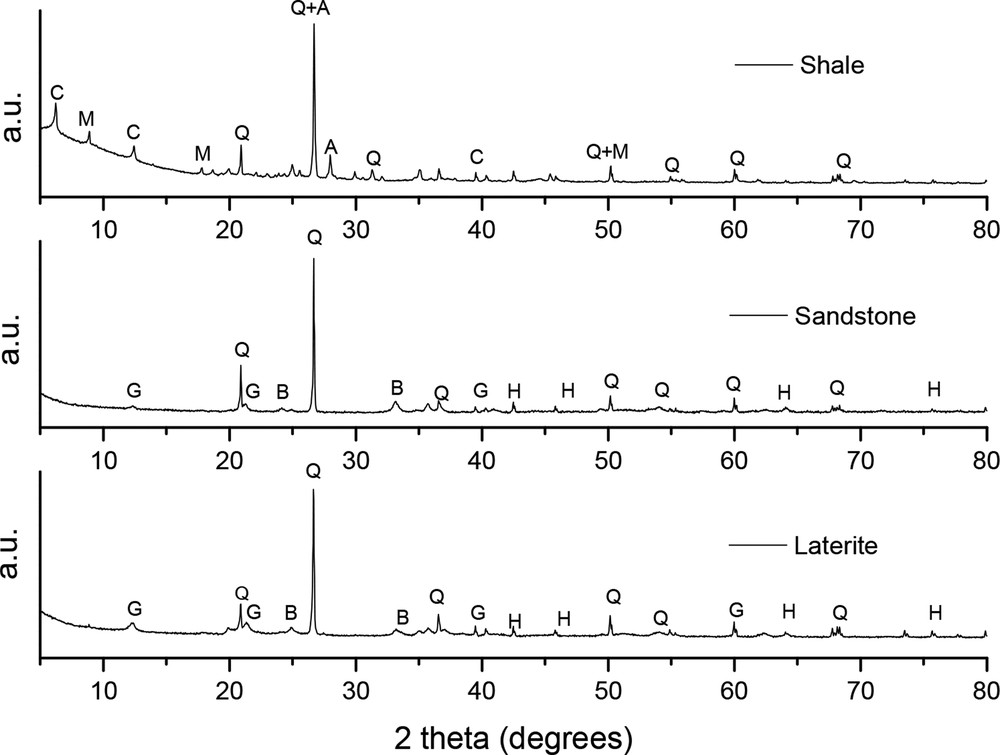
XRD patterns of naturally occurring shale, sandstone, and laterite from Ivory Coast. The following abbreviations are used: A: albite; B: birnessite; C: clinochlore; G: goethite; H: hematite; M: muscovite, and Q: quartz.
3.1.2 Mössbauer spectrometry
The Mössbauer spectra of sandstone and laterite recorded at room temperature (Fig. 2) exhibit both two magnetic sextets attributed to hematite α-Fe2O3 and goethite α-FeOOH (Fritsch et al., 2011) and a ferric paramagnetic doublet D1 (Table S3), in agreement with XRD analyses. Mössbauer analysis at lower temperature was needed to distinguish whether the paramagnetic doublet D1 recorded at room temperature corresponded to iron oxides or to FeIII species embedded in the structure of clays. The Mössbauer spectra of sandstone and laterite recorded at 9 K (Fig. 3) clearly showed that the doublet observed at 300 K has been transformed into a magnetic sextet attributed to α-FeOOH (Kone et al., 2009). The corresponding Mössbauer hyperfine parameters (Table S4) revealed that only a very low intensity (RA of ∼ 3%) paramagnetic doublet remained on the 9 K Mössbauer spectrum of laterite and could be attributed to structural FeIII species into clays. Therefore, for both sandstone and laterite, FeIII species were essentially included into ferric oxides (α-Fe2O3 and α-FeOOH), and the ferric doublet recorded at room temperature (Fig. 2) was attributed to superparamagnetic goethite (Janot et al., 1973). This means that the size of some of the goethite particles in sandstone and laterite is quite small. This must be particularly true for laterite, since the ferric doublet recorded at room temperature is relatively intense (RA of ∼ 55%) and narrow and does not exhibit any relaxation effect (Table S3). This last result may be cautiously correlated to the BET analysis showing that laterite and sandstone had the highest surface specific area, although the materials were composed of a mixture of minerals. In fact, it can be hypothesized that the presence of small iron particles in laterite and sandstone contribute to a global increase of the specific surface area. The status of iron in shale was very different, and the Mössbauer spectrum recorded at 12 K (Fig. 3) exhibits a ferric (D1) and a ferrous (D2) paramagnetic doublet. The total relative areas of both doublets are relatively high, representing ∼ 2/3 of the total spectra. This means that, for the shale, most of the iron species was embedded in the clay minerals (muscovite, clinochlore), contrary to sandstone and laterite, where iron was present in iron oxides. However, the shale did also contain some iron in goethite, as shown by the magnetic sextet of α-FeOOH observed at 12 K (Fig. 3).

Mössbauer spectra of naturally occurring sandstone (a) and laterite (b) from Ivory Coast at room temperature.
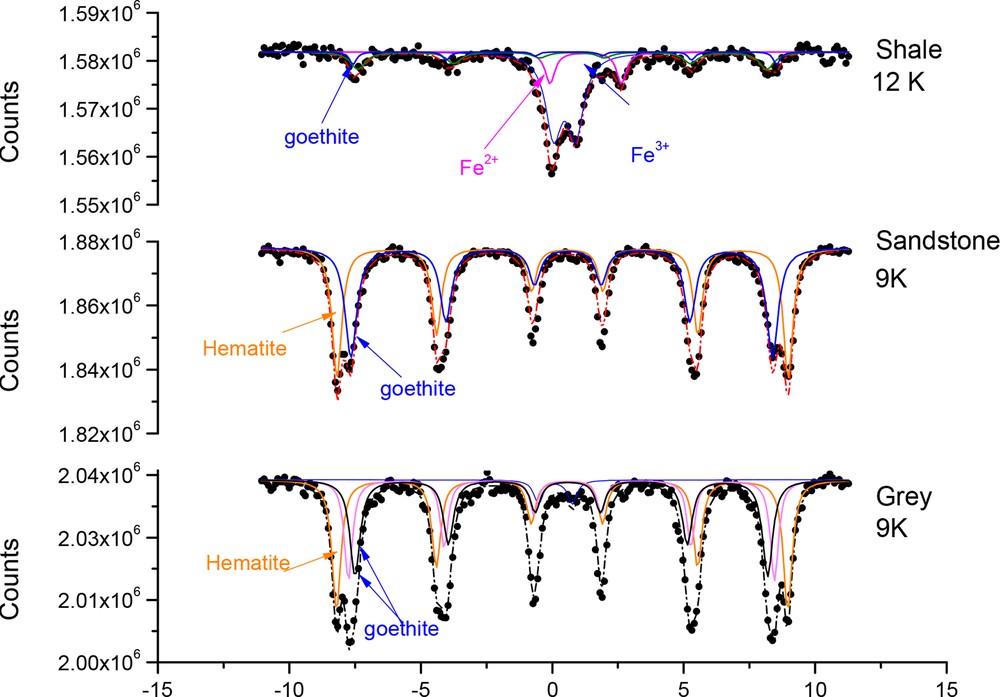
Mössbauer spectra of naturally occurring shale, sandstone and laterite from Ivory Coast at 12 K (shale) and 9 K (sandstone and laterite).
3.2 Phosphate sorption in batch experiments
3.2.1 Effect of adsorbent dosage
All three substrates displayed not surprisingly an increase in the removal efficiency with increasing adsorbent dosage (Fig. 4). A high removal efficiency of 100% was achieved with laterite and sandstone at 20 and 40 g L−1, respectively, while the removal efficiency remained less than 100% in the whole sorbent dose studied for shale (Fig. 4a). In addition, the phosphate uptake strongly decreased as the dosage increased and reached a saturation level at high sorbent doses (Fig. 4b). Such results reflected that adsorbed phosphate species were distributed among more available sorption sites at high sorbent doses, thus decreasing the sorption capacity (Ádám et al., 2007; Babatunde et al., 2009).
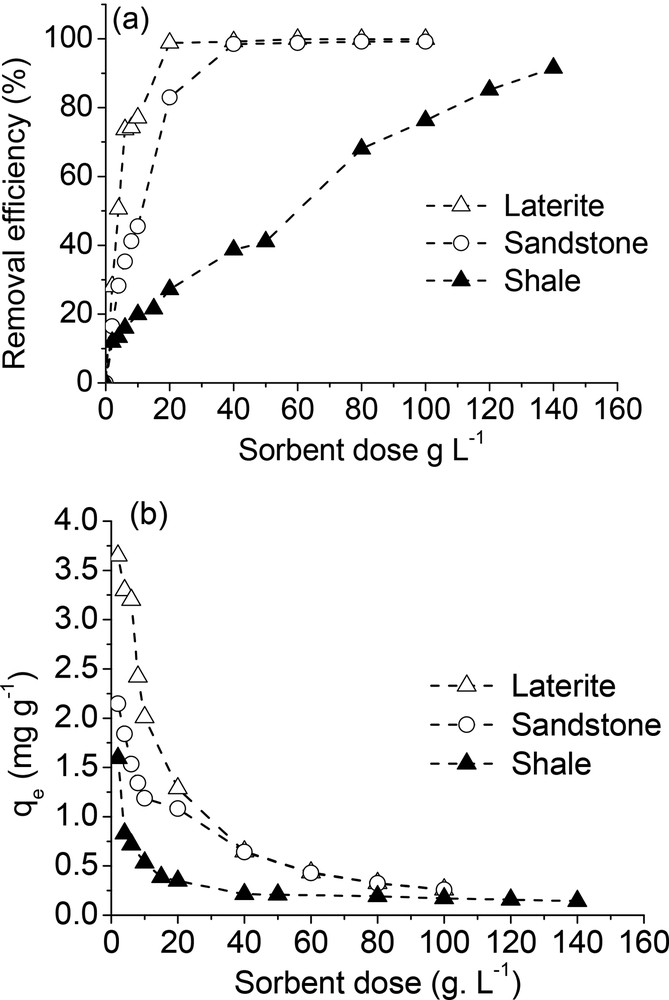
Phosphate removal (a) and phosphate uptake (b) for shale, sandstone, and laterite as a function of the dose of adsorbent ([PO4]initial = 25 mg L−1; time contact: 24 h).
3.2.2 Effect of pH on phosphate removal sorption
The pH is an important factor that strongly influences the phosphate sorption mechanisms at the solid–solution interfaces. Small amounts of phosphate are determined at pH values lower than 8 and 10 for sandstone and laterite, respectively, which indicated a phosphate removal efficiency close to 100% (Fig. 5). Beyond these two pH values, phosphate concentration strongly increased. A different behavior was observed for shale. In fact, an increase in phosphate concentration was observed in the pH range of 2–11, followed by a strong decrease beyond pH 11. The inset in Fig. 5 displays the phosphate concentration determined in solution for a laterite and sandstone dosage of 8 g L−1. The phosphate concentration increased over the whole pH range studied. It can thus be concluded that increasing the pH resulted in a decrease of the phosphate removal efficiency (at phosphate removal efficiencies lower than 100%), except for shale, for which a stronger removal efficiency was observed at pH 12. With the exception of this high pH value, the phosphate removal efficiency of shale was much smaller than those for sandstone and laterite.
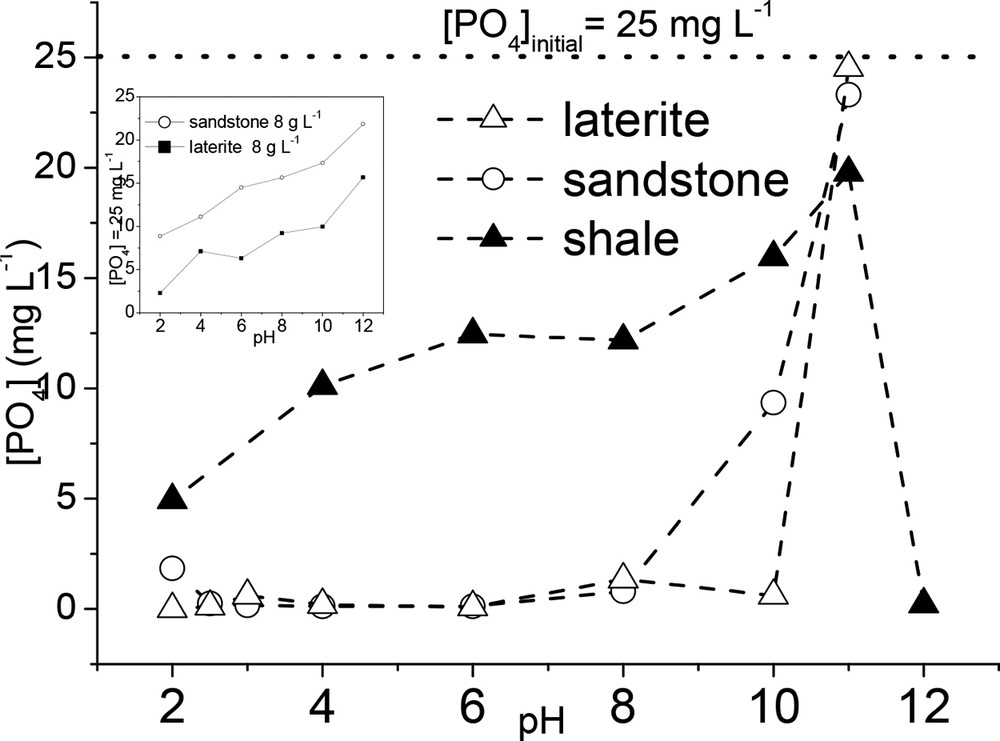
Aqueous phosphate concentration as a function of pH after sandstone, laterite and shale reaction with a 25mg L−1 initial phosphate solution ([sorbent] = 80 g L−1 and 8 g L−1 (inset); time contact: 24 h).
3.2.3 Ions concentration as a function of pH
To further investigate the phosphate removal mechanism, aqueous ions release was studied as a function of pH in blank (without phosphate) and after adsorbents reaction with a 25mg L−1 phosphate solution (Fig. 6 and supplementary Figure S1). Blank experiments revealed, in the low pH range, a strong release of Ca, Mg and Al, and to a lesser extent Fe species for shale, compared to sandstone and laterite. Dissolution processes without added phosphate are thus much more pronounced for shale in acidic conditions. Moreover, sandstone and laterite clearly exhibited a behavior towards phosphate sorption different from that of shale. In fact, low amounts of Ca, Mg, Al, and Fe were determined in solution after shale reaction with phosphate ions and whatever the pH was (Fig. 6). Additionally, calcium and magnesium concentrations did not significantly differ in blank experiments and after phosphate reaction with sandstone and laterite, even at a lower dosage of 8 g L−1 (data not shown). Some differences in ion concentrations were observed at low pH, where the presence of phosphate ions induced an increase in Fe and Al dissolution processes in laterite and sandstone, respectively. From Figs. 5 and 6, it clearly appears that shale displayed a different sorption behavior from those of sandstone and laterite. The weak dissolution processes on phosphate removal in the case of sandstone and laterite combined to a much more efficient phosphate removal clearly suggested that phosphate sorption was dominated by adsorption processes. Adsorption processes controlled phosphate sorption in a pH range of 4–11 for shale, but dissolution of shale followed by precipitation of the released aqueous species with phosphate competes with adsorption under low pH conditions (Kpannieu et al., 2018).
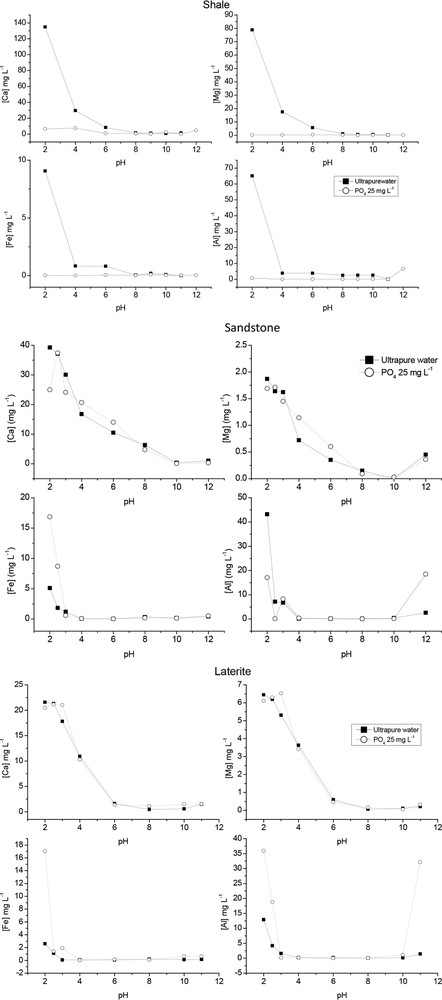
Ion concentrations in solution in blank experiments (without PO4) and after reaction with a 25mg L−1 initial phosphate solution ([sorbent] = 80 g L−1; pH ∼ 7; time contact: 24 h).
3.2.4 Phosphate sorption kinetics
Sorption kinetics were obtained with 80 g L−1 (shale) and 8 g L−1 (laterite and sandstone) sorbent doses (Fig. 7). The results obtained at additional sorbent doses for laterite and sandstone are displayed as supporting information (Supplementary Figures S2 and S3). All curves displayed almost the same shape. The sorption rate was rapid at the beginning of the experiments, and then decreased until a pseudo-plateau was reached. The first step may be due to phosphate sorption at the external surface of the sorbents, while the second step probably reflected some slow sorption process due to phosphate diffusion into the pores of the sorbent (Sanyal and De Datta, 1991; Yang et al., 2006). The phosphate uptake decreased in the following order: laterite > sandstone > shale. In particular, the observed maximal phosphate uptakes determined from Fig. 7 were 3.2, 2.8, and 0.23 mg g−1 for laterite, sandstone, and shale, respectively. The sorption capacity of shale was thus smaller than those of laterite and sandstone by more than a factor of 10. Common pseudo-first-order and pseudo-second-order equations were used to model the experimental data using a linear fit approach (Eqs. (4) and (5)):
- • pseudo-first-order equation:
| (4) |
- • pseudo-second-order equation:
| (5) |
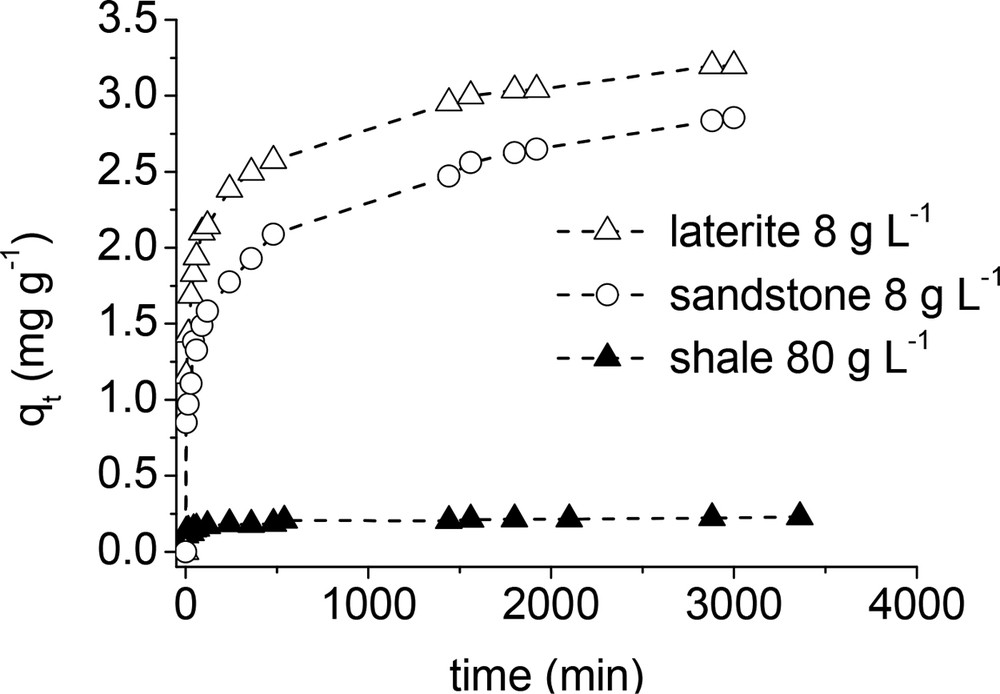
Effect of contact time on phosphate uptake capacity by sandstone, laterite, and shale ([PO4]initial = 25 mg L−1; pH ∼ 7; contact time: 24 h).
where qe (mg g−1) is the equilibrium sorption amount, qt (mg g−1) is the amount of phosphate adsorbed at any time t and k1 (min−1) and k2 (g mg−1 min−1) are the pseudo-first-order and pseudo-second-order rate constants, respectively.
The linear plots of phosphate sorption kinetics obtained and the corresponding kinetic parameters are given in Supplementary Figures S4 and S5, and Table S5 respectively. The kinetic data could only be well described using the pseudo-second-order kinetic model. High correlation coefficients were in fact obtained and the equilibrium sorption amounts (qcal) closely match the experimental data, while the pseudo first-order-model did not fit the data.
Similar kinetics results were reported in the literature for various phosphate adsorbents (Cai et al., 2012; Namasivayam and Prathap, 2005; Zhu et al., 2009). Such results indicated that the sorption process of phosphate on these three materials may be based on chemisorption. As the sorbent dose increased and the other experimental conditions remained unchanged, the value of k2 increased by a factor higher than 10.
3.2.5 Phosphate sorption isotherms
Isotherms are commonly used to describe the sorption behavior of a given adsorbent (Liu and Zhang, 2015; Yang et al., 2014). The sorption behavior of laterite and sandstone was clearly different from that of shale (Fig. 8). As it is the case in most studies, the shale curve displayed a saturation point at which increasing the initial phosphate concentration did not cause a significant increase in the phosphate sorption capacity (Zhu et al., 2009). The presence of this saturation level allowed us to fit the data by using the common Langmuir and Freundlich models (S). The obtained parameters for both Langmuir and Freundlich models are reported in Supplementary Table S6. The data were better fitted by using the Langmuir model. The Langmuir linear plot of shale is displayed in Supplementary Figure S6. Isotherms obtained for laterite and sandstone did not display any saturation level, which made the prediction of the sorption capacity unreliable. A careful examination of the data suggested that sandstone and laterite isotherms are close to C-type isotherms (Limousin et al., 2007). In fact, coefficient correlations of 0.989 and 0.988 were obtained for sandstone and laterite, respectively, by using a linear data fitting approach (data not shown). Such a behavior has already been reported in the literature for phosphate sorption by substrates used in constructed wetlands and phosphate retention in filter materials such as shellsand and filtralite P® (Boeykens et al., 2017; Drizo et al., 1999). In any way, the maximum phosphate uptake varied between 0.48 for shale and 8.3 mg g−1 for laterite. Note that the shale dose was ten times higher than those of sandstone and laterite. The uptake capacities of laterite were higher than those reported for other natural minerals such as bauxite (0.6 mg g−1) (Drizo et al., 1999), limestone (0.55 mg g−1) (Drizo et al., 1999), zeolite (0.25 mg g−1) (Drizo et al., 1999), dolomite (0.3 mg g−1) (Pant et al., 2001), and apatite (0.41 mg g−1) (Bellier et al., 2006). Additionally, all these literature data were obtained at higher initial phosphate concentrations (40–150 mg L−1). The ranking order of the maximum sorption capacity was therefore again: laterite > sandstone > shale. Remind that the specific surface areas of shale, sandstone, and laterite were 6.3, 28.5, and 37.6 m2 g−1, respectively. It thus appeared that the material with the lowest specific surface area had the lowest phosphate uptake capacity. One reason for this may be that phosphate sorption was decreased by limited available surface sorption sites in the case of shale. Another reason explaining the lowest capacity of shale for phosphate removal was the status of iron. Indeed, the shale contained the lowest quantity of iron (Table S2) and the lowest proportion of Fe in iron oxides, i.e. 31% compared with 97% and 100% observed for laterite and sandstone, respectively. Iron oxide particles, in particular those with low crystal size, are well known to develop strong bonding sites for surface complexation by phosphate species. The superiority of laterite for phosphate removal in comparison with shale and sandstone may be correlated to the existence of goethite particles with very low crystal size, as testified by the superparamagnetic behaviour observed for this mineral at room temperature by Mössbauer spectroscopy. Interestingly, sorption capacities were recalculated by taking into account the specific surface area of laterite and sandstone; similar values close to 0.2 mg m−2 were obtained, thus suggesting that the difference in reactivity was due to a particle size effect, in agreement with Mössbauer analysis.
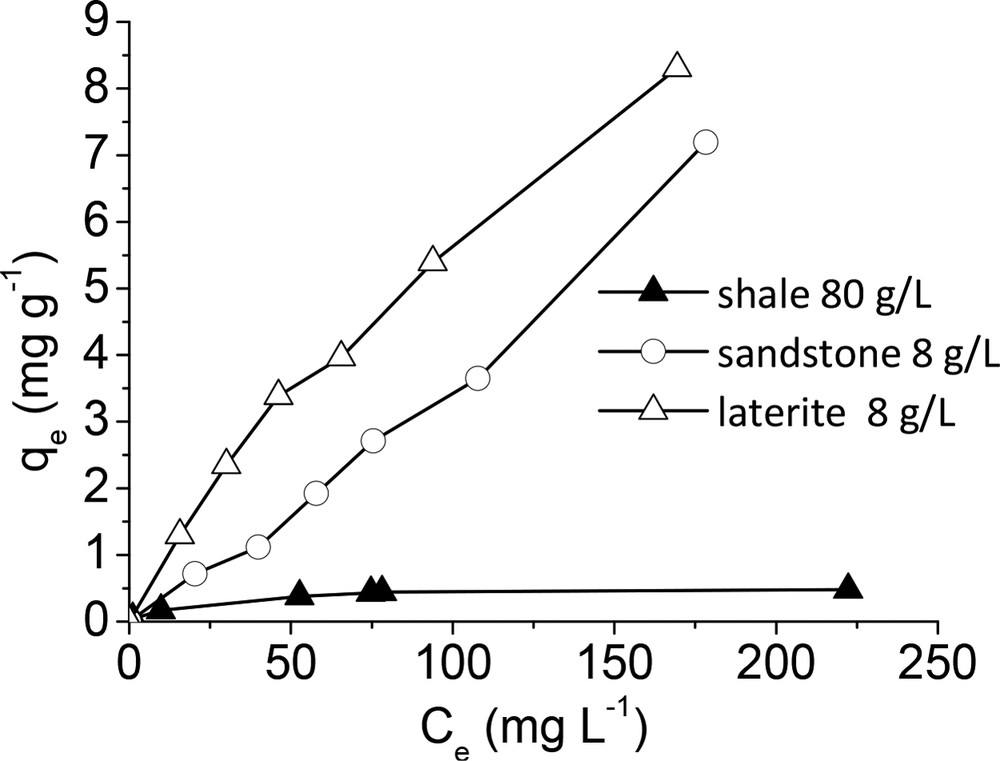
Adsorption isotherms of phosphate for the three different sorbents (pH ∼ 7; time contact: 24 h). The dashed line is only drawn as a visual help.
3.3 Phosphate sorption in hydrodynamic conditions: column tests
The breakthrough curves of sandstone and laterite obtained at a flow rate of 1 mL min−1 and a corresponding HRT of ∼ 4 h are displayed in Fig. 9. We noticed that the breakthrough curve of laterite presented an unsual shape, with two plateaus. The origin of this behaviour was outside the scope of the present study. As expected from the batch experiments, the breakthrough observed for laterite occurred later than the one observed for sandstone. Indeed, if a threshold of [PO4] = 1 mg L−1 was chosen, i.e. a C/C0 of 0.04 with C0 = 25 mg L−1, the breakthrough occurred at a value of V/VP of ∼ 147 and 132 for laterite and sandstone, respectively. These values were about 8 times higher than the breakthrough observed previously for shale, i.e. V/VP ∼ 17, even though the mean HRT of ∼ 35 h used for shale was much higher than the one used for sandstone and laterite. Therefore, the column experiments confirmed that the capacity of shale to remove phosphate was the lowest, as already observed in batch experiments. The removal capacity at the breakthrough qbreak can be computed as qbreak = 0.17, 1.23, and 1.30 mg PO4 g−1 for shale, sandstone, and laterite, respectively. Calcium, magnesium, and phosphate ions concentrations are displayed as a function of time (Fig. 10). The initial calcium (4.9 and 9.2 mg L−1 for laterite and sandstone, respectively), magnesium (1.8 and 1 mg L−1), aluminum (not detected) and iron (not detected) concentrations in the outflow were in good agreement with the equilibrium data recorded at a pH of 5 in batch experiments. The soluble calcium and magnesium concentrations in the outflow decreased continuously and reached a value lower than 0.5 mg L−1 at the breakthrough. Experiments carried out in hydrodynamic mode required a preliminary step in which the column was pre-equilibrated with ultra-pure water during 24 h before the phosphate solution flowed through the column. The aqueous species concentrations determined at t = 0 thus reflected the equilibria solubility that occurred during this period (Fig. 10). In this experimental situation, phosphate removal partially occurred through reaction with the soluble species released in solution during the column equilibration. In fact, a correlation between the decrease in Ca and Mg concentrations and the increase in phosphate concentration at the outflow of the column was evidenced. Iron concentration as a function of time remained below the detection limit (data not shown). Such behavior had already been reported in the literature for shellsand and filtralite P® filter materials (Ádám et al., 2007). According to this study, phosphate removal occurred through the accumulation of large amounts of Ca (45–260 mg L−1) and Mg ions (5–65 mg L−1) at the beginning of the column experiment. The removal capacity at the breakthrough measured for filtralite P® and shellsand can be calculated from the data provided in this previous work (Ádám et al., 2007) and was evaluated to be respectively ∼ 0.6 and 0.9 mg PO4 g−1 for a contact time of ∼ 4 days. Therefore, the removal capacity of laterite and sandstone from Ivory Coast was at least of the same order of magnitude than the one reported for the shellsand from Hordaland Tørkeri in Norway, which was recognized as a very efficient granular natural material for phosphate removal in hydrodynamic conditions.
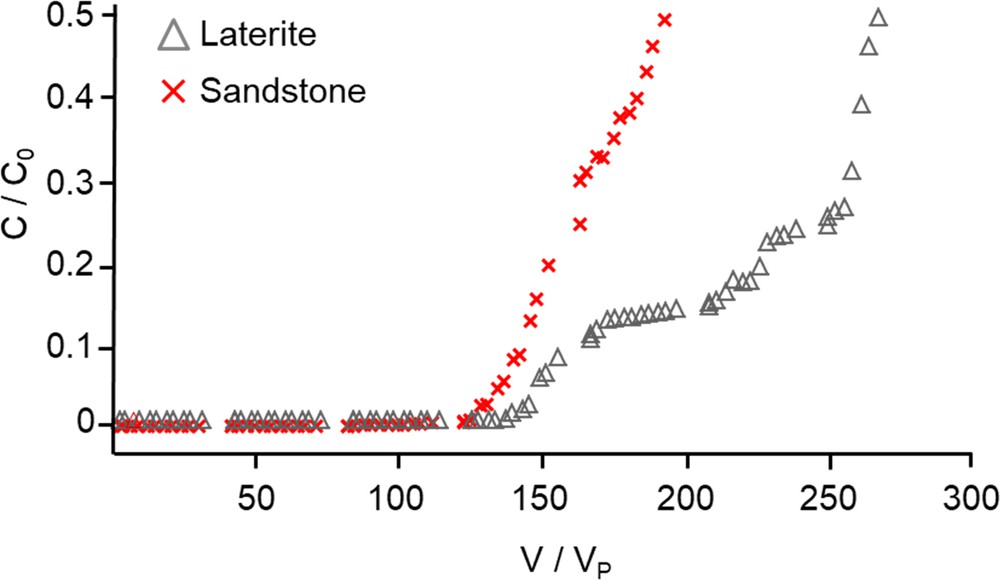
Breakthrough curves obtained for the sandstone and laterite column experiments in hydrodynamic conditions (flow of 1 mL min−1, C0 = 25 mg PO4 L−1).
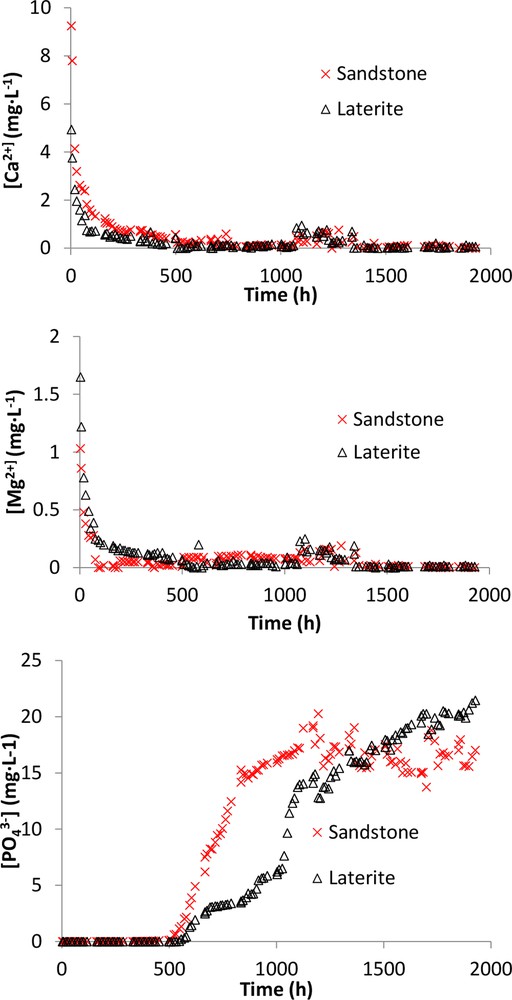
Ca2+, Mg2+, and PO43– concentrations as a function of time in sandstone and laterite column experiments in hydrodynamic conditions (ϕ 1–2 mm).
4 Conclusion
The removal of phosphate by three natural materials from Ivory Coast was studied in homogeneous batch suspensions and in static/hydrodynamic conditions in column experiments. Interestingly, the three geomaterials exhibit different sorption behaviors: phosphate removal is dominated by adsorption for laterite and sandstone, while dissolution processes may compete for adsorption at low pH for shale. The investigations revealed that all three materials retained phosphate to various extents. Batch experiments showed that the most efficient phosphate adsorbent was laterite, followed by sandstone and shale, the sorption efficiency of shale being strongly smaller than those of laterite and sandstone. The removal of phosphate by adsorption on iron oxides was certainly very limited in shale studied here, since most of the iron species were situated in structural substitution into the bulk of the solid clay minerals. In contrast most of the iron in laterite and sandstone occurred as separate iron oxides minerals (goethite and hematite). Laterite was also the most efficient mineral for removing phosphate in flow through conditions with a phosphate removal capacity at the breakthrough higher than 1 mg PO4 g−1. Therefore, laterite is a very promising mineral and may be considered for phosphate removal at a larger scale, i.e. in a micro-pilot unit before ultimately being tested in wetlands in Ivory Coast.
Acknowledgements
The authors thank G. Medjahdi (Centre of X-gamma Competence; UMR 7198 CNRS–Université de Lorraine, Nancy, France) for XRD analyses, and C. Genois from the LCPME is gratefully acknowledged for ICP analyses.