



 CC-BY 4.0
CC-BY 4.0
1. Mitohormesis : du stress à la persistance
L’hormèse décrit la dualité de situations biologiques où de faibles doses d’un stimulus de stress activent une réponse cellulaire adaptative pour maintenir la stabilité de l’ensemble des conditions physico-chimiques contrôlant la vie cellulaire (l’homéostasie), alors que des doses plus élevées de ce même stimulus deviennent nocives, en entraînant la faillite de l’homéostasie qui conduit ainsi à la mort cellulaire.
La mitohormèse, plus communément nommée mitohormesis [1] sous sa forme anglaise, prolonge et applique ce concept d’hormèse à la mitochondrie. Les mitochondries sont des organites essentiels à une myriade de processus cellulaires, notamment l’équilibre redox, l’homéostasie calcique, la production d’énergie, différentes voies métaboliques (anabolisme : synthèse de macromolécules ; et catabolisme : dégradation des composés organiques) et la survie et la mort cellulaire [2]. Nous retrouvons cette dualité de conséquences de l’hormèse pour différents stimuli affectant la mitochondrie. Par exemple au niveau mutationnel, un faible taux de mutations du génome mitochondrial humain de 16 kb (ADNmt) est un promoteur oncogénique plus puissant qu’un nombre trop élevé de mutations de l’ADNmt amenant l’invalidation de toutes les fonctions mitochondriales [3]. Une interprétation de ces observations est que certaines mutations modérées de l’ADNmt ne compromettent pas gravement la fonction mitochondriale, mais entraînent néanmoins une augmentation des dérivés réactifs de l’oxygène (ROS), qui agissent comme une molécule de signalisation pro-tumorigène [4]. Les ROS sont produites au niveau des mitochondries, des peroxysomes [5] et des NADPH oxydases (NOX). Les NOX sont liés à la membrane plasmique, principalement exprimés par les neutrophiles et les macrophages et produisent des ROS vers l’extérieur des cellules. Parmi toutes ces sources de ROS, nous estimons que les ROS mitochondriaux (mtROS) contribuent à hauteur de 90 % de la charge oxydative globale cellulaire (pour plus de détail sur l’origine des mtROS, voir [6]). Certains travaux ont confirmé cette hypothèse des ROS pro-tumorigènes, en montrant qu’on retrouvait dans certaines tumeurs une population homogène de mitochondries portant toutes les mêmes mutations de l’ADNmt, situation biologique appelée l’homoplasmie [4]. D’autres études au contraire ont montré qu’une hétéroplasmie mitochondriale (différents types de mitochondries portant différentes mutations au sein de la même cellule) apportait une flexibilité adaptative bénéfique à certains clones cancéreux [7].
Indépendamment des mutations de l’ADNmt, les mtROS sont le stimulus majeur de la mitohormesis qui participe à cette flexibilité adaptative des cellules cancéreuses, principalement étudiée ces deux dernières décennies au niveau du métabolisme énergétique.
2. Métabolisme énergétique des cellules cancéreuses : de Warburg à la phosphorylation oxydative
Les cellules s’appuient sur deux voies énergétiques principales alimentées par les acides aminés, les monosaccharides et les substrats dérivés de la voie des acides gras pour produire leur énergie : la glycolyse et la phosphorylation oxydative (OxPHOS) au niveau de la chaîne respiratoire mitochondriale. La glycolyse a un rendement énergétique plus faible, 2 ATP produits par la glycolyse contre 38 ATP par l’OxPHOS. En 1920, Otto Warburg a fait l’observation que les cellules cancéreuses présentent une forte consommation de glucose et produisent du lactate, privilégiant ainsi la glycolyse de type anaérobie même en présence d’oxygène. Ce mécanisme est connu sous le nom d’« effet Warburg », et sa découverte a alimenté pendant plusieurs décennies l’idée inexacte selon laquelle les cellules cancéreuses ont des défauts mitochondriaux et peuvent se passer de la chaîne de transport d’électrons (ETC) mitochondriale et de l’ATP mitochondrial pour survivre. L’hypothèse de Warburg a été depuis grandement revisitée [8]. En effet, l’activité de l’ETC est nécessaire à la génération de pyrimidines, les bases nucléiques constitutives de macromolécules d’ADN et d’ARN : la cytosine, la thymine, et l’uracile. L’enzyme mitochondriale dihydro-orotate déshydrogénase (DHODH) catalyse l’une des étapes limitantes de la biosynthèse de novo des pyrimidines. La DHODH est la seule enzyme de la biosynthèse des acides nucléiques localisée à la mitochondrie et elle requiert le coenzyme Q10 sous sa forme oxydée. Or, en conditions physiologiques d’oxygène, l’oxydation de Q10H2 en Q10 ne s’opère qu’au niveau de l’ETC. De plus, la perte d’ADNmt dans les cellules en culture, obtenue par différents protocoles expérimentaux, s’accompagne d’une auxotrophie (incapacité à synthétiser un métabolite essentiel) pour l’uridine, précurseur de la pyrimidine en aval de la DHODH. Une absence totale d’activité des ETC est donc incompatible avec la croissance inhérente au cancer [9]. En 2015, une étude rapporte que des cellules tumorales dépourvues d’ADN mitochondrial (ADNmt) ont été greffées chez des souris mais n’ont formé des tumeurs qu’après avoir acquis l’ADNmt de l’hôte via un transfert de mitochondries entières, démontrant la nécessité de mitochondries fonctionnelles dans la formation des tumeurs [10]. Depuis 2012, le nombre d’études démontrant l’existence de sous-groupes de cancers dépendants du métabolisme de l’OxPHOS n’a cessé d’augmenter (lymphome diffus à grandes cellules B, cancer du poumon, du pancréas, leucémie aigües myéloïdes, leucémie myéloïde chronique et cancer du sein …). De plus, les progrès technologiques liés aux analyses multi-omiques et à l’échelle de la cellule unique, qui démontrent l’hétérogénéité métabolique au sein d’une même entité tumorale, ont relancé le débat sur l’utilisation exclusive de la glycolyse par les cellules tumorales [11, 12, 13, 14, 15]. Les modifications dans les voies métaboliques alimentant l’OxPHOS ne constituent pas nécessairement par elle-même un événement tumorigène, mais offrent plutôt une palette de ressources moléculaires adaptatives, participant à la sélection/compétition clonale, retrouvés chez les clones cancéreux les plus agressifs [16]. Les recherches de Warburg ont posé les bases pour l’étude du métabolisme cancéreux, mettant en évidence le rôle crucial du métabolisme énergétique dans la croissance et la survie des cellules cancéreuses. Les travaux de différents groupes de recherche et de notre équipe ont plus récemment recentré l’attention scientifique sur l’OxPHOS [11, 12, 13, 14, 15, 17, 18, 19]. Nos découvertes conduisent à de nouvelles voies de recherche et ont contribué à l’émergence du domaine du métabolisme mitochondrial dans la réponse thérapeutique des cancers. Elles visent ainsi à comprendre comment les changements métaboliques peuvent être exploités pour développer de nouvelles stratégies de diagnostic et de traitement du cancer. Les leucémies aiguës myéloïdes (LAM) ont été un véritable paradigme dans cet effort de recherche. En particulier, les thérapies des LAM décrites plus bas déclenchent une signalisation mitochondriale dite rétrograde qui communique les anomalies mitochondriales (stress mitochondriaux) au noyau provoquant une réponse antérograde visant à rétablir la mitohormesis et au maintien de l’OxPHOS.
3. Les Leucémies aiguës myéloïdes, un modèle séminal de l’étude de la résistance aux traitements
Les LAM sont un groupe de tumeurs malignes agressives de la moelle osseuse les plus courantes avec un faible taux de survie après diagnostic [20, 21]. Les LAM sont caractérisées par une accumulation de cellules myéloïdes appelées blastes, bloquées à un stade plus ou moins précoce de la différenciation cellulaire et incapables de maturation terminale. Ces blastes envahissent le sang circulant et la moelle osseuse, perturbent le fonctionnement des progéniteurs normaux et entraînent une insuffisance médullaire. Le standard de soins des LAM consiste en un traitement d’induction intensif de 7 jours de cytarabine (AraC) et 3 jours d’une anthracycline (traitement communément appelé 7 + 3). Pour de nombreux patients âgés de 60 ans ou plus, ce traitement intensif a été récemment remplacé par un inhibiteur de la protéine anti-apoptotique BCL-2, le vénétoclax (VEN), associé à des agents hypométhylants comme l’azacytidine (AZA) [22]. Bien que les taux de réponse initiaux à ces traitements (7 + 3 ou VEN/AZA) soient généralement élevés, la majorité des patients atteints de LAM rechutent et leur taux de survie reste inacceptable, en particulier chez les patients plus âgés [23]. Le paradigme consensuel de la leucémogénèse est que la leucémie résulte de la transformation d’une seule cellule souche hématopoïétique normale (CSH) ou progénitrice et est entretenue par une population dite de cellules souches leucémiques (CSL) résistantes au traitement et responsables de rechutes fréquentes [24, 25]. On distingue fonctionnellement les CSL, qui ont un potentiel expérimental de xénogreffe dans des souris immunodéficientes, des non-CSL (issues de ces mêmes CSL et communément appelées blastes) qui ont perdu cette aptitude de xénogreffe [26]. Bien que l’évolution génétique et la sélection clonale des CSL soient impliquées dans la résistance aux chimiothérapies classiques et à certaines nouvelles thérapies ciblées, il est aussi établi que la résistance thérapeutique peut être médiée par des mécanismes non génétiques. Parmi les acteurs majeurs de la résistance non génétique récemment identifiés, la flexibilité métabolique des LAM a démontré la capacité des cellules cancéreuses à utiliser différents substrats pour soutenir un métabolisme oxydatif, une des caractéristiques majeures des cellules résistantes au traitement. Ces cellules constituent ce qu’on appelle la maladie résiduelle, contribuant à la rechute et à un faible taux de survie. Ces cellules sont également nommées cellules persistantes tolérantes aux traitements avec l’acquisition de mécanismes de résistances transitoires, qu’il est nécessaire de mieux comprendre afin de les cibler [15, 27].
4. Implications physiopathologiques de l’adaptation mitochondriale au cours des chimiothérapies
Les CSH puisent leur énergie principalement par la glycolyse, tandis que les CSL dépendent principalement de la respiration mitochondriale [28, 29], notamment par le biais de la synthèse de protéines mitochondriales [30] et de la réplication de l’ADN mitochondrial [31]. Ces observations ont suscité un intérêt considérable pour l’étude du rôle du métabolisme mitochondrial dans la résistance aux traitements, en particulier suite à la cytarabine (AraC), agent chimiothérapeutique utilisé dans le traitement de première intention dit « 7 + 3 » des LAM. En effet, dans des modèles de xénogreffes de cellules issues de patients atteints de LAM, les cellules persistantes à l’AraC sont caractérisées par un métabolisme mitochondrial et oxydatif exacerbé associé à une augmentation du contenu en ROS, de la masse mitochondriale, de la consommation d’oxygène, de l’oxydation des acides gras et de la production d’ATP mitochondriale [15]. De ce fait, ces cellules sont très sensibles aux inhibiteurs mitochondriaux. Cette dépendance mitochondriale concerne une sous-population de cellules présentes lors de la maladie résiduelle avec une forte capacité de prolifération [31]. Également, des modifications post-traductionnelles sont impliquées dans le statut mitochondrial des cellules persistantes post-AraC. Entre autres, la palmitoyltransférase ZDHHC21 qui permet la palmitoylation de l’adénylate kinase 2, impliquée dans le métabolisme énergétique mitochondrial via la réaction d’interconversion de l’ADP en ATP et AMP [32]. Lors d’une rechute, les cellules issues de patients présentent une reprogrammation métabolique caractérisée par une surexpression de protéines ribosomales mitochondriales et des complexes de la chaîne de transport des électrons. Une étude réalisée en analysant des échantillons provenant de 47 patients adultes et 22 cas pédiatriques, et prélevés lors du diagnostiques, de la maladie résiduelle et de la rechute, a permis la découverte de petits peptides mitochondriaux qui pourront servir de biomarqueurs et de cibles thérapeutiques [33]. De plus, la forte expression de protéines mitochondriales permet de prédire les rechutes des patients ayant une LAM de type monocytique, M4 et M5 [34]. Enfin, une dépendance au complexe I de la chaîne de transport des électrons peut caractériser les patients qui résistent à la chimiothérapie [35].
Le métabolisme mitochondrial est également étroitement lié aux voies apoptotiques, notamment via la protéine anti-apoptotique BCL-2. Les protéines de la famille BCL-2 sont classées en trois grandes catégories : les protéines pro-apoptotiques à multi-domaines (BAX, BAK et BOK), localisées à la membrane mitochondriale, s’oligomérisent pour former des pores qui perméabilisent la membrane externe mitochondriale et permettent le relargage du cytochrome c, responsable de la mort des cellules ; les protéines pro-apoptotiques « BH3-only » (BID, BIM, BAD, NOXA et PUMA), localisées cette fois-ci au niveau du cytoplasme, permettent l’activation directe ou indirect des protéines BAX et BAK ; et enfin les protéines anti-apoptotiques (BCL-2, BCL-XL, BCL-w et MCL1) préviennent l’induction de la mort cellulaire en séquestrant les protéines pro-apoptotiques et en régulant la perméabilité de la membrane mitochondriale, empêchant la libération mitochondriale du cytochrome c dans le cytosol et de ce fait la mort cellulaire programmée (l’apoptose dépendante des caspases) et étant ainsi responsable de la survie des cellules. Il a été montré que BCL-2 joue un rôle dans la résistance à l’AraC, notamment à travers son implication dans la régulation du pore de transition de perméabilité mitochondriale (mPTP, [18]). Le mPTP est un pore de la membrane mitochondriale interne qui permet le relargage du cytochrome c dans l’espace intermembranaire des mitochondries. Le traitement par l’AraC induit une ouverture du mPTP, induisant l’apoptose des cellules. Cependant, dans certaines cellules persistantes, des mécanismes d’adaptation se mettent en place, comme la translocation de BCL-2 du cytosol à la membrane mitochondriale qui entraîne la fermeture du mPTP, empêchant le relargage du cytochrome c et protégeant ces cellules de l’apoptose induite par l’AraC [18]. Le canal 1 sélectif d’anions dépendant de la tension (VDAC1) est localisé dans la membrane mitochondriale externe, et forme un canal ionique permettant à l’ATP produit par les mitochondries de diffuser hors des mitochondries dans le cytosol. VDAC1 permet donc la communication entre la mitochondrie et le cytoplasme. Les protéines BCL-2 mitochondriales et VDAC1 sont des composantes structurelles des contacts (appelés MERCS) entre ces mitochondries et le réticulum endoplasmique, dont une des fonctions est de réguler l’homéostasie calcique mitochondriale [18]. L’augmentation des MERCS observée dans les cellules résiduelles améliore le flux de calcium du réticulum endoplasmique (RE) vers les mitochondries. Ce flux calcique régule le cycle de Krebs en stimulant les NADH déshydrogénases Ca2+-sensible et ainsi augmentant la disponibilité du NADH pour le complexe I et l’activité OxPHOS dans les cellules persistantes à l’AraC grâce à BCL-2. De plus, l’autophagie est responsable de la dégradation des acides gras présents dans les gouttelettes lipidiques (lipophagie) au niveau des MERCS, permettant ainsi l’alimentation de l’OxPHOS via l’oxydation des acides gras [36]. Collectivement, ces travaux soulignent le rôle majeur de ces protéines anti-apoptotiques et de VDAC1 dans le contrôle non seulement de l’induction de la mort cellulaire via la mitochondrie mais également dans la régulation des fonctions métaboliques des mitochondries.
5. Flexibilité métabolique en réponse aux thérapies ciblées
Des travaux précédents [69] ont défini un sous-type de CSL par leur faible contenu en ROS (ROSlow) dont il sera question dans cette partie. De façon intéressante, cette sous-populations de CSL montre également une dépendance à BCL-2 et l’OxPHOS [29]. L’utilisation de vénétoclax (VEN) pour inhiber BCL-2 permet donc la sensibilisation de ces cellules à la chimiothérapie en altérant l’homéostasie calcique et en facilitant le déclenchement d’une réponse apoptotique. Cependant, il a été constaté que lorsque l’AraC et VEN sont utilisés conjointement, une nouvelle dépendance pour le complexe I de la chaîne respiratoire apparaît. Le ciblage du complexe I en consolidation de la combinaison VEN/AraC permet alors de sensibiliser ces cellules et de retarder la rechute [18]. D’autre part, la combinaison VEN/AZA, évoquée précédemment, a également montré son efficacité anti-leucémique dans de nombreuses études précliniques [37, 38]. En effet, les CSL ROSlow présentes lors du diagnostic initial dépendent également du catabolisme des acides aminés pour l’OxPHOS et leur survie, et l’utilisation de la combinaison VEN/AZA permet d’éradiquer ces cellules en ciblant le transport et la biosynthèse des acides aminés, notamment la cystéine [39, 40]. En revanche, lors d’une rechute, les CSL ROSlow présentes ne dépendent plus du métabolisme des acides aminés en raison de leur plasticité métabolique qui leur permet d’utiliser, cette fois, l’oxydation des acides gras pour produire de l’énergie (paragraphe ci-dessous) [40]. Néanmoins, les cellules résistantes à VEN montrent également une dépendance pour la voie de biosynthèse de la purine. L’utilisation de ME-344, un isoflavone qui inhibe l’OxPHOS dans les cancers solides, diminue la synthèse des purines et améliore l’apoptose induite par VEN dans les lignées cellulaires de LAM, y compris dans les cellules résistantes à l’AraC. Son effet synergique avec VEN a été montré dans les cellules de patients mais également in vivo [41]. Ces travaux mettent en exergue l’hétérogénéité métabolique des CSL ROSlow et leur flexibilité catabolique sous la pression de sélection des thérapies.
La β-oxydation des acides gras (FAO en anglais), aussi appelée spirale de Lynen, est la voie métabolique de dégradation des molécules d’acides gras qui génère les co-enzymes réduits NADH, FADH2, et NADPH, ainsi que l’acétyl-CoA qui alimente le cycle de Krebs aussi appelé cycle des acides tricarboxyliques (TCA en anglais). L’essentiel de l’énergie chimique potentielle issue de la FAO est produite sous forme de pouvoir réducteur constituant un vaste apport de proton pour l’OxPHOS qui permet une production d’ATP par le complexe V de la chaîne respiratoire mitochondriale. La FAO, qui se déroule dans les mitochondries, ou dans les peroxysome dans le cas des acides gras très longs, est mobilisée dans des conditions de stress métabolique [42]. La surexpression du transporteur mitochondrial d’acides gras CPT1a est un facteur de mauvais pronostic dans les LAM, et le ciblage de CPT1a augmente l’effet anti-leucémique de VEN [43]. Le traitement VEN/AZA induit une diminution du métabolisme des acides aminés, mais n’entraîne pas de baisse de l’OxPHOS dans les cellules résistantes, qui utilisent un métabolisme alternatif pour conduire leur métabolisme énergétique. En effet, les cellules persistantes augmentent les métabolites précurseurs impliqués dans la biosynthèse des acides gras au niveau de la mitochondrie, comme le citrate et le malate observés spécifiquement chez les patients mutés pour la voie de signalisation RAS [44]. En cohérence avec ces observations, l’utilisation du 8-chloro-adénosine qui serait un inhibiteur de la FAO et de l’OxPHOS, permettrait d’éradiquer les CSL en combinaison avec VEN [45]. L’oxydation des acides gras est également régulée par la protéine anti-apoptotique MCL1 qui peut se lier directement à l’acyl-CoA déshydrogénase à très longue chaîne (VLCAD), une enzyme clé de cette voie oxydative [46]. Ainsi, l’utilisation d’un inhibiteur de CPT1a ou de MCL1 permet de diminuer le métabolisme des acides gras et éliminer les cellules résistantes à VEN/AZA [44]. Les désaturases d’acides gras 1 et 2 (FADS1 et FADS2) sont également enrichies à la suite du traitement par VEN/AZA, ces désaturases fonctionnent comme un mécanisme de recyclage du NAD+ pour assurer la survie des CSL. Ainsi, leur inhibition génétique ou pharmacologique entraîne une diminution de la viabilité des cellules lors de la rechute suite à un traitement VEN/AZA [47]. Enfin, d’autres gènes impliqués dans le transport des acides gras sont surexprimés suite au traitement par chimiothérapie intensive, notamment le récepteur CD36 (Figure 1) [15, 48].
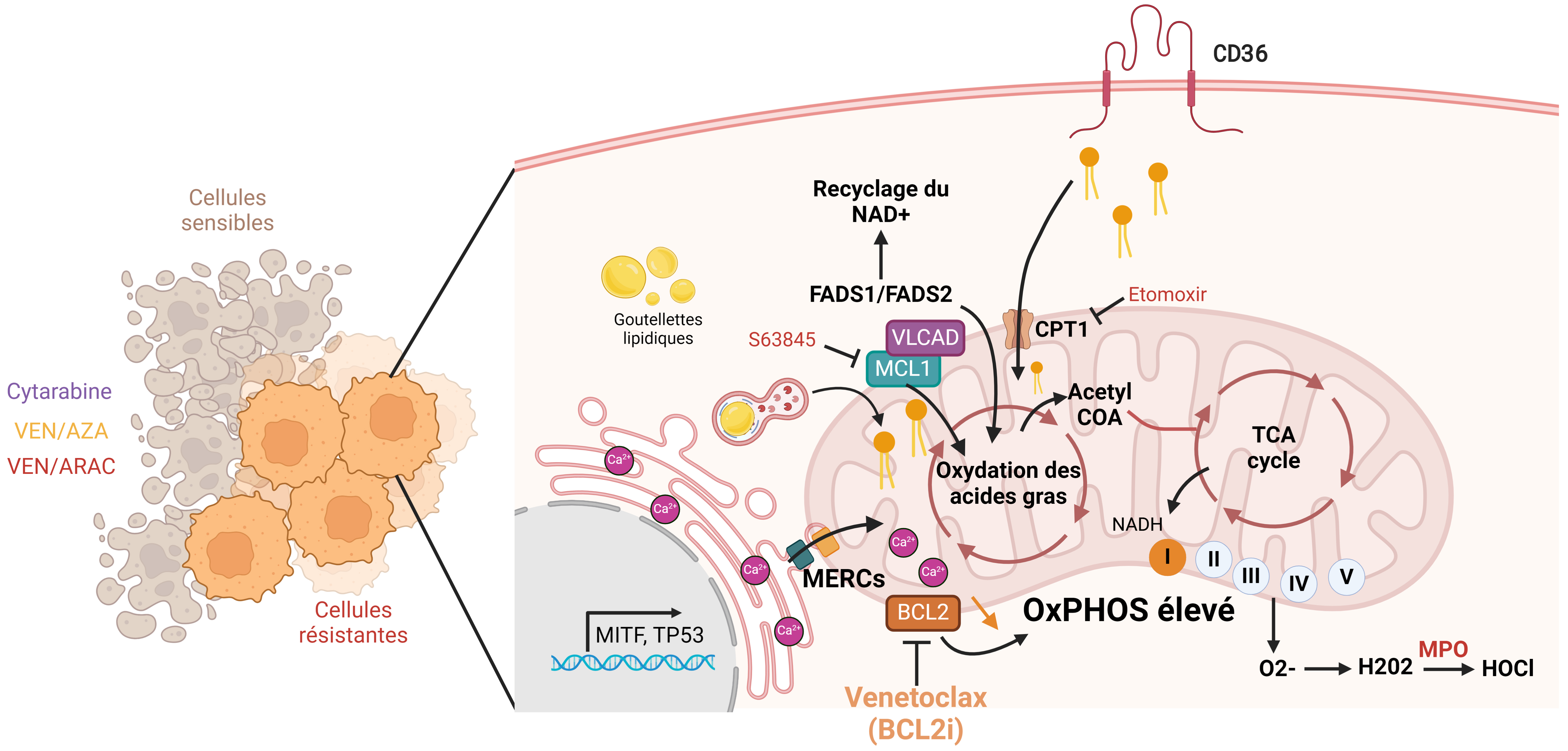
Rôle du métabolisme des lipides dans la réponse thérapeutique dans les LAM.
Outre les combinaisons thérapeutiques décrites précédemment, de nombreuses autres thérapies ciblées ont vu le jour, visant des mutations spécifiques des LAM également impliquées dans la reprogrammation métabolique des cellules mutées. C’est notamment le cas des inhibiteurs ciblant les mutations des isocitrate déshydrogénases 1 et 2 (IDH1/2), qui rendent les cellules persistantes dépendantes du métabolisme mitochondrial. Par l’inhibition de la production de l’oncométabolite 2-hydroxyglutarate (2-HG), ces cellules rétablissent l’activité de la méthylcytosine dioxygénase TET2 et l’expression du gène suppresseur de tumeur, la phosphatase and tensin homologue (PTEN), entraînant l’activation du facteur de transcription clé dans la biogenèse mitochondriale, le peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1a) [19]. L’utilisation d’inhibiteurs d’OxPHOS en association avec des inhibiteurs d’IDH1 permet la sensibilisation des cellules mutées [19].
Par ailleurs, après thérapie ciblée giltéritinib (un inhibiteur spécifique de la Fms-like tyrosine kinase 3 (FLT3), le récepteur à tyrosine kinase le plus fréquemment muté dans les LAM), il a été montré que les cellules résistantes au giltéritinib sont soutenues par un métabolisme utilisant la glutamine pour alimenter l’OxPHOS et maintenir l’homéostasie redox. L’association du giltéritinib avec un inhibiteur de la glutaminolyse permet de sensibiliser les cellules leucémiques et de prolonger ainsi la survie des souris xénogreffées [49]. Ces cellules montrent également une dépendance pour la voie de biosynthèse des purines après thérapie ciblée giltéritinib. Le blocage de l’enzyme DHODH permet d’inhiber cette voie de biosynthèse et d’éradiquer les cellules leucémiques résistantes au giltéritinib [50]. Toujours dans le cadre des patients mutés pour FLT3, le facteur de transcription CCAAT/enhancer-binding protein-alpha (C/EBPα) est impliqué dans la régulation de la biosynthèse des acides gras et la ferroptose. La ferroptose est un processus de mort cellulaire programmée dû à l’accumulation de ROS lipidiques, appelés « lipides péroxydés », présents notamment à la membrane plasmique. L’enzyme stéaroyl-CoA désaturase (SCD) est nécessaire à la conversion d’acides gras saturés en acides gras monoinsaturés, protégeant ainsi les cellules de la ferroptose. L’inhibition de FLT3 concomitante à celle de C/EBPα réduit l’incorporation d’acides gras mono-insaturés dans les membranes en diminuant l’expression de la SCD, conduisant à la ferroptose. Ainsi, cibler l’homéostasie des lipides permet de sensibiliser à la thérapie ciblée giltéritinib [51]. Au-delà des mutations, des modifications post-transcriptionnelles peuvent également être impliquées dans la progression tumorale, notamment par l’intermédiaire de modifications épigénétiques. Une étude récente a démontré que le lactate peut également être utilisé par les cellules persistantes suite à l’inhibition des protéines à Bromodomain et Extra-Terminal Domain (BET), qui sont des régulateurs épigénétiques, impliquées dans la progression tumorale. Les cellules traitées par les inhibiteurs de BET freinent la glycolyse et par compensation utilisent le lactate extracellulaire comme substrat pour alimenter le cycle de Krebs [52]. L’entrée du lactate extracellulaire est induite dans ces cellules via le transporteur MCT1, et produit le pyruvate par la lactate déshydrogénase (LDH) associée à la membrane mitochondriale soutenant le cycle de Krebs et maintient l’OxPHOS [52], illustrant une nouvelle fois la flexibilité dans les substrats respiratoires des cellules leucémiques en réponse aux stress thérapeutiques [53].
6. Les réponses cellulaires liées aux stress mitochondriaux
Les traitements anticancéreux induisent plusieurs voies de signalisations dont la réponse aux stress mitochondriaux (en anglais MSR). La MSR est un concept général qui englobe différents mécanismes tels que l’Unfolded Protein Response (UPR) mitochondriale (UPRmt), la réponse intégrative au stress mitochondrial (ISRmt), la mitophagie, la fusion et fission mitochondriales. Les mécanismes de la mitophagie et de la fusion–fission mitochondriales ne seront pas détaillés ici [54, 55]. Les traitements de premières lignes des patients atteints de LAM induisent des ROS en particulier des mtROS qui entraînent une mauvaise conformation des protéines du RE, des protéines mitochondriales et des protéines du cytosol. Cette perturbation de l’homéostasie des protéines (protéostase) active l’UPR du RE (UPRER) et l’UPRmt. UPRER et UPRmt se distinguent par leurs acteurs moléculaires déclenchant différentes vagues transcriptionnelles ayant pour même mission de rétablir la protéostase, de combattre le stress oxydatif, et de rétablir la mitohormesis. L’UPRER utilise la signalisation de la protéine kinase RNA-like endoplasmic reticulum kinase (PERK). L’UPRmt quant à elle est une voie de signalisation multi-branchée qui garantit la mitohormesis en augmentant le contrôle de qualité de la protéostase, la machinerie antioxydante, la biogenèse mitochondriale et les autres fonctions mitochondriales. La réponse UPRmt comporte plusieurs axes. Le premier axe de l’UPRmt conduit à l’activation de C/EBP Homologous Protein (CHOP), l’activating transcription factor 4 (ATF4) et ATF5. L’axe sirtuine de l’UPRmt est une réponse antioxydante conduisant à la désacétylation et à la relocalisation du facteur de transcription Forkhead box O3 (FOXO3A) vers le noyau où il induit l’expression de l’enzyme anti-oxydante superoxide dismutase 2 mitochondriale (SOD2). Notons que chez Caenorhabditis elegans, l’UPRmt est principalement activée lors de la réduction du potentiel mitochondrial (𝛥ψm) par l’arrêt de l’importation de protéines mitochondriales portant une séquence d’adressage chargée positivement et attirées par la charge négative de la membrane interne mitochondriale au travers du transporteur TIM23. Les liens possibles entre 𝛥ψm, thérapies anticancéreuses et mitohormesis ne seront pas développés ici. Les stress mitochondriaux activent également l’ISR. L’ISR est indifféremment initiée par l’une des quatre kinases eucaryotes du facteur d’initiation 2 alpha (famille EIF2A) qui intègrent divers stress cellulaires tels que la privation d’acides aminés (EIF2AK4-GCN2), le déficit en hème (EIF2AK1-HRI), le mauvais repliement de protéines ER (PERK) ou l’infection virale (EIF2AK2-PKR) [56]. Au cœur de l’activation de l’ISR se trouve la phosphorylation de eIF2α en aval de ces quatre kinases. La phosphorylation de eif2a résulte en la traduction d’ATF4, facteur de transcription clé dans la MSR. Il permet également d’établir un lien direct entre ISR et UPR, de par son interaction avec le facteur de transcription ATF5, impliqué dans l’UPR [56]. Le lien entre l’ISR et la mitochondrie a été démontré par l’intermédiaire de l’activation de la protéase OPA1, située dans la mitochondrie, qui suite à un stress mitochondrial, induit le clivage de la protéine DELE1 (death-associated protein-3 binding cell death enhancer-1), située dans un premier temps au niveau de la membrane interne mitochondriale, puis déplacée vers le cytosol, afin d’activer la réponse HRI-eif2a-ATF4 et permettre la restauration de l’activité mitochondriale [57]. L’activation de la voie PERK-eiF2a-ATF4 participe à l’assemblage des supra-complexes respiratoires au niveau des mitochondries [58]. Ces voies cellulaires peuvent ainsi s’inscrire comme activateur de l’ISR mitochondriale et faire le lien entre réponse de stress du RE et de la mitochondrie. L’augmentation des MERCS observée dans les cellules persistantes suggère un lien étroit avec la signalisation du RE. Enfin, la monosomie 5 et les délétions du chromosome 5q (−5/del(5q)), anomalies chromosomiques présentes dans 40 % des LAM secondaires de mauvais pronostic, induisent une haploinsuffisance pour DELE1, bloquant ainsi l’activation de la voie OMA1-DELE1-HRI, responsable de la résistance aux traitements [59]. En effet, comme décrit précédemment, ATF4 est responsable de l’adaptation mitochondriale, mais également de la transcription de CHOP, inducteur de la mort cellulaire. De ce fait, inhiber cette voie peut également avoir un effet protecteur sur les cellules. D’où l’intérêt de mesurer la balance entre activités pro- et anti-apoptotique selon les situations.
7. Rôle de la mitohormesis dans la résistance thérapeutique des LAM
Le facteur de transcription ATF4 activé par l’ISR joue un rôle essentiel dans la réponse de stress après traitement par l’AraC [17, 60]. ATF4 contrôle un programme transcriptionnel comprenant des régulateurs du métabolisme des acides aminés, de l’équilibre redox, de l’autophagie et de la synthèse des protéines [61, 62] conduisant une restauration de l’activité mitochondriale et une adaptation métabolique responsable de la chimiorésistance [17]. Une expression élevée des gènes cibles d’ATF4 a été mesurée à l’aide d’un rapporteur ATF4-GFP dans les CSH normales et les CSL dans les LAM [63]. Cette étude de van Galen et al. a néanmoins identifié un paradoxe qu’il faudra élucider : les cellules CSH et CSL ont de faibles niveaux d’eIF2 par rapport à des progéniteurs hématopoïétiques normaux ou des blastes, malgré une forte signature ATF4. La mise en place dans les cellules leucémiques de mécanismes de régulation du stress via l’ISR dépendant d’ATF4 a également été montrée dans les CSL présentes dans l’échantillon au diagnostic. La privation en acides aminés entraîne une augmentation d’ATF4 dépendante de la phosphorylation de eIF2α, favorisant la survie des CSL [63]. Parmi les quatre kinases activant l’ISR par phosphorylation de eIF2α (cf. partie précédente), les données de spectrométrie de masse de cette étude suggèrent l’implication de EIF2AK4-GCN2 pour les CSH normales bien que ces cellules n’aient pas été privées d’acides aminés. Il est possible que cette privation résulte néanmoins du processus de tri cellulaire. La kinase phosphorylant eIF2α dans les CSL reste à être identifiée.
L’utilisation de l’antiparasitaire atovaquone, analogue de l’ubiquinone et inhibiteur du complexe III de la chaîne respiratoire, permet de sensibiliser les cellules leucémiques en induisant l’activation d’ATF4 par la phosphorylation d’eIF2α [64]. Cela induit la transcription des gènes cibles d’ATF4, les protéines pro-apoptotiques CHOP et CHAC1 et permet la transcription de REDD1, qui inhibe la voie mTORC1, mimant ainsi l’activité de la rapamycin. En plus d’induire la mort cellulaire, l’atovaquone affecte la consommation d’oxygène et l’OxPHOS, impliquée dans la résistance aux chimiothérapies [64]. L’atovaquone est actuellement en essai clinique dans le traitement de consolidation des LAM pédiatriques après chimiothérapie intensive (NCT03568994) [65]. De même, suite aux traitements par l’AraC et doxorubicine, une réponse de stress de type UPRmt s’active en partie par l’intermédiaire du facteur de transcription GATA1 qui permet la transcription de l’enzyme hydroxy-3-méthylglutaryl-CoA synthase 1 (HMGCS1). HMGCS1 est une enzyme clé en amont de la voie du mévalonate pour la synthèse du cholestérol et de l’ubiquinone cofacteur de la chaîne respiratoire mitochondriale surexprimée dans les LAM [66]. Cette réponse entraîne une augmentation du nombre de mitochondries et une baisse des dommages du réticulum endoplasmique, permettant une résistance à l’apoptose cellulaire. L’HMGCS1 est donc une nouvelle cible thérapeutique potentielle pour le traitement des patients leucémiques réfractaires ou en rechute post-chimiothérapie [66].
De nombreuses études ont utilisé la technologie CRISPR-Cas9 afin de découvrir quels sont les gènes impliqués dans la sensibilité ou la résistance à VEN. La protéine chaperonne CLPB, surexprimée dans les LAM, est impliquée dans le maintien de la structure mitochondriale et des crêtes mitochondriales, grâce à son interaction avec la protéine OPA1. La perte de la protéine CLPB entraîne des défauts structurels et fonctionnels des mitochondries, à l’origine d’une réponse induisant un stress mitochondrial élevé dépendant d’ATF4 et conduisant à l’apoptose des cellules [67]. Ce même processus a été observé suite à l’inhibition de la traduction mitochondriale [68]. Le traitement par VEN affecte la structure des mitochondries, avec une augmentation de la taille et de l’aire des mitochondries, suggérant une augmentation de la fusion mitochondriale en réponse au stress mitochondrial induit [68]. Ce stress mitochondrial conduit à l’activation de la synthèse protéique mitochondriale et participe au maintien de leur activité, favorisant la survie des cellules résistantes. Cibler la traduction mitochondriale permet de sensibiliser les cellules résistantes pour augmenter l’efficacité du traitement à VEN par l’activation d’une ISR élevée favorisant l’apoptose cellulaire [68]. L’équipe de J. Tyner a également montré que la résistance à VEN est due à une protection des cellules face au stress mitochondrial et à une augmentation de l’OxPHOS, due à la protéine TP53 [69]. De même, dans des blastes leucémiques traités soit par VEN, soit par un inhibiteur de MCL-1, soit pas la combinaison VEN/AZA, la perte de MFN2 et MARCH5, deux gènes responsables de la mitophagie, permet d’améliorer la réponse à ces traitements. Du reste, les cellules résistantes augmentent l’élimination des mitochondries par la mitophagie suite à l’induction d’un stress mitochondrial [70]. La mitophagie agit donc comme un mécanisme pro-survie pour éliminer les mitochondries endommagées et empêcher l’apoptose des cellules. L’utilisation d’inhibiteurs de MFN2 permet de potentialiser l’effet des différents mimétiques de BH3 [70]. Enfin, l’actinomycine D, actuellement utilisée en clinique pour traiter des patients dont les cellules résistantes présentent la mutation NPM1c+, entraîne une diminution très précoce de l’activité mitochondriale en ciblant en partie le complexe II de la chaîne respiratoire, ce qui conduit à l’activation d’une réponse de stress mitochondrial et de la voie cGAS. Ceci active une réponse inflammatoire avec l’activation de NF-kB et des interférons mais également une forte production de ROS, ce qui entraîne la formation des corps nucléaires PML, l’activation de TP53 et la mort cellulaire par sénescence et apoptose. Ce mécanisme est amplifié par l’utilisation de VEN (Figure 2) [71].
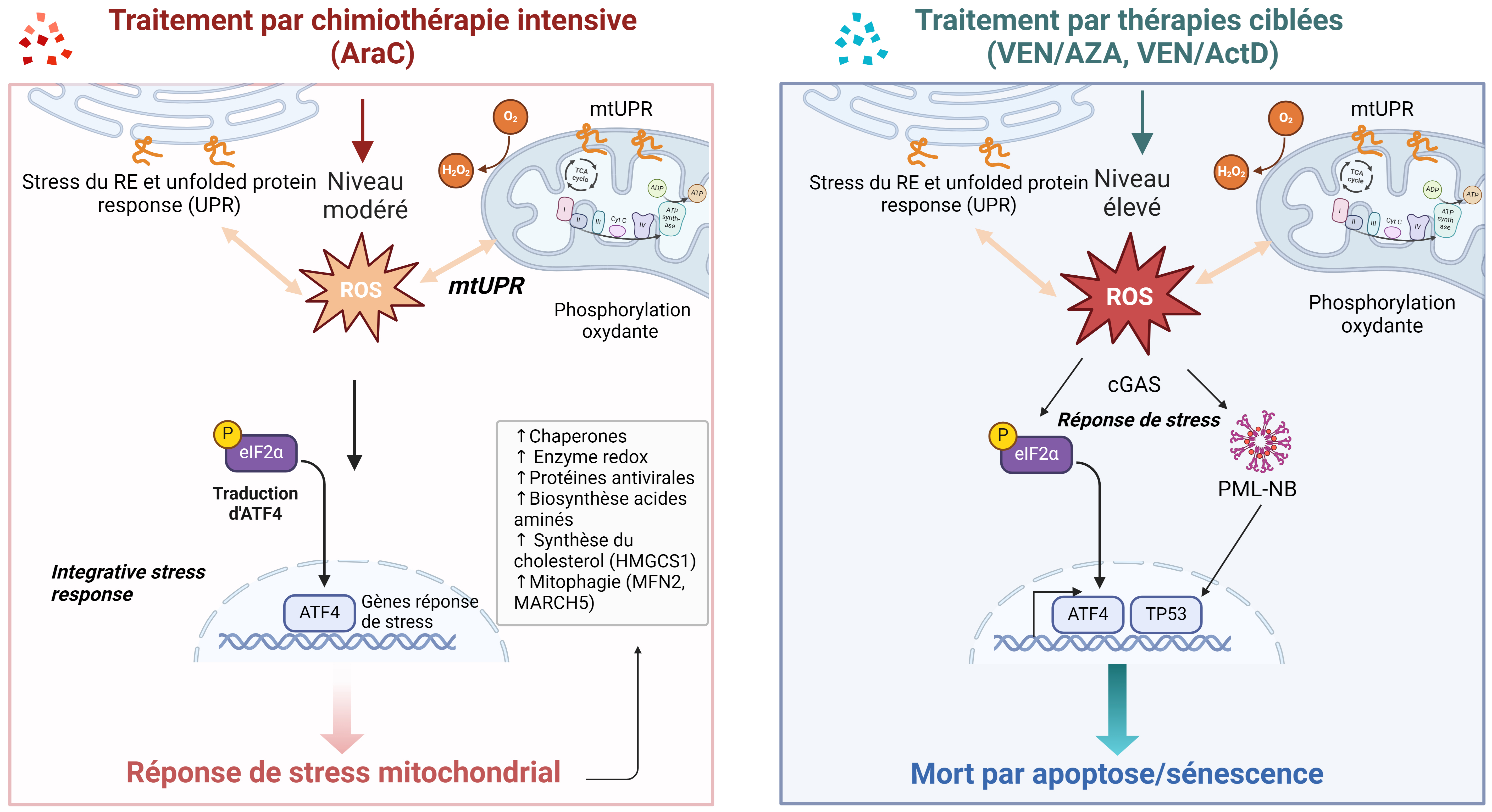
Régulation de la réponse aux stress mitochondriaux induits par les thérapies via le facteur de transcription ATF4 dans les LAM.
L’ensemble de ces résultats montre donc que, suite aux différents traitements anti-leucémiques, les cellules résistantes augmentent leur métabolisme mitochondrial grâce à l’activation d’une réponse de stress mitochondrial qui d’une part permet l’augmentation de la mitophagie pour éliminer les mitochondries défectueuses, et d’autre part augmente la traduction mitochondriale et la transcription de protéines chaperonnes pour restaurer la biogénèse mitochondriale et l’activité mitochondriale des cellules. L’inhibition de ces différentes voies permet de sensibiliser les cellules à la chimiothérapie ou aux thérapies ciblées, en activant alors une réponse de stress très soutenue qui n’est plus protectrice mais induit une mort par apoptose via le facteur de transcription ATF4 et la transcription du gène pro-apoptotique CHOP.
8. L’homéostasie des espèces réactives de l’oxygène dans le statut mitochondrial des cellules résistantes
Une des premières explorations liant les mtROS aux CSL et s’attachant à identifier des différences entre les CSH et les CSL a été la découverte de la surexpression de la glutathion peroxidase 3 (GPx3), un piégeur de ROS, dans les CSL des LAM de mauvais pronostic [72]. De plus, dans plusieurs travaux décrits ici précédemment, les CSL dépendantes de BCL-2, du catabolisme des acides aminés, de l’OxPHOS, contiennent un contenu en ROS faible et sont sensibles à la combinaison VEN/AZA [29, 39, 40]. Par ailleurs, les chimiothérapies intensives telles que 7 + 3 ou actinomycin D produisent en quelques heures des ROS et des mtROS dans les blastes et les CSL [71]. Le taux de ROS élevé et leur balance avec les protéines de défenses antioxydantes sont clés dans l’activation des réponses de stress mitochondrial et la mitohormesis détaillées ci-dessus. Ils jouent un rôle soit protecteur, soit inducteur de mort, en fonction de leur niveau d’activation. Lorsque le niveau de ROS est très élevé, la mort cellulaire par apoptose est induite. L’expression par les cellules leucémiques de protéines antioxydantes, telles que SOD2, catalase, myélopéroxydase [73], GPx3 or GPx4, permet de favoriser leur survie en réduisant le niveau des ROS. La déacétylase mitochondriale SIRT3 active SOD2, permettant de réduire la production des ROS, et entraîne une redirection du métabolisme mitochondrial vers l’OxPHOS alimenté par la FAO, contribuant ainsi à la chimiorésistance des cellules de LAM. L’inhibition de la FAO et de l’OxPHOS, par l’inhibition de SIRT3 entraîne ainsi une augmentation de la libération d’acides gras libres conduisant à une forte lipotoxicité, favorisant la mort des cellules leucémiques [74, 75]. En cohérence avec ces résultats, nos travaux montrent une signature FOXO3A des cellules persistantes qui est aussi activable par SIRT3 et qui augmente l’expression de SOD2 [17]. Ces observations confortent également l’intégration de l’axe sirtuine de l’UPRmt (comme décrit plus haut). De même, l’arsenic trioxide entraîne une forte augmentation des ROS, ce qui permet ainsi la synergie avec un traitement à VEN, qui induit quant à lui une diminution de l’anti-oxydant NRF2 [76]. Et l’inhibition de la myélopéroxydase, qui est surexprimée après un traitement par l’AraC, entraîne une forte accumulation des mtROS, à l’origine d’une altération de l’OxPHOS et de l’équilibre énergétique cellulaire. Cela entraîne la mort par apoptose et l’éradication sélective des cellules leucémiques exprimant fortement la myélopéroxydase [73]. L’utilisation d’anti-oxydants permet de protéger les cellules leucémiques et d’augmenter le statut OxPHOS des cellules. Ainsi, l’utilisation de pro-oxydants peut éradiquer les cellules tumorales en les poussant au-delà des limites protectives de la viabilité et être une nouvelle voie thérapeutique dans les LAM et dans les autres cancers [77]. Les ROS, selon leur intensité et selon les aptitudes antioxydantes intrinsèques des CSL, orchestrent différents mécanismes de la mitohormesis qui s’opposent à la mort cellulaire (Figure 3).
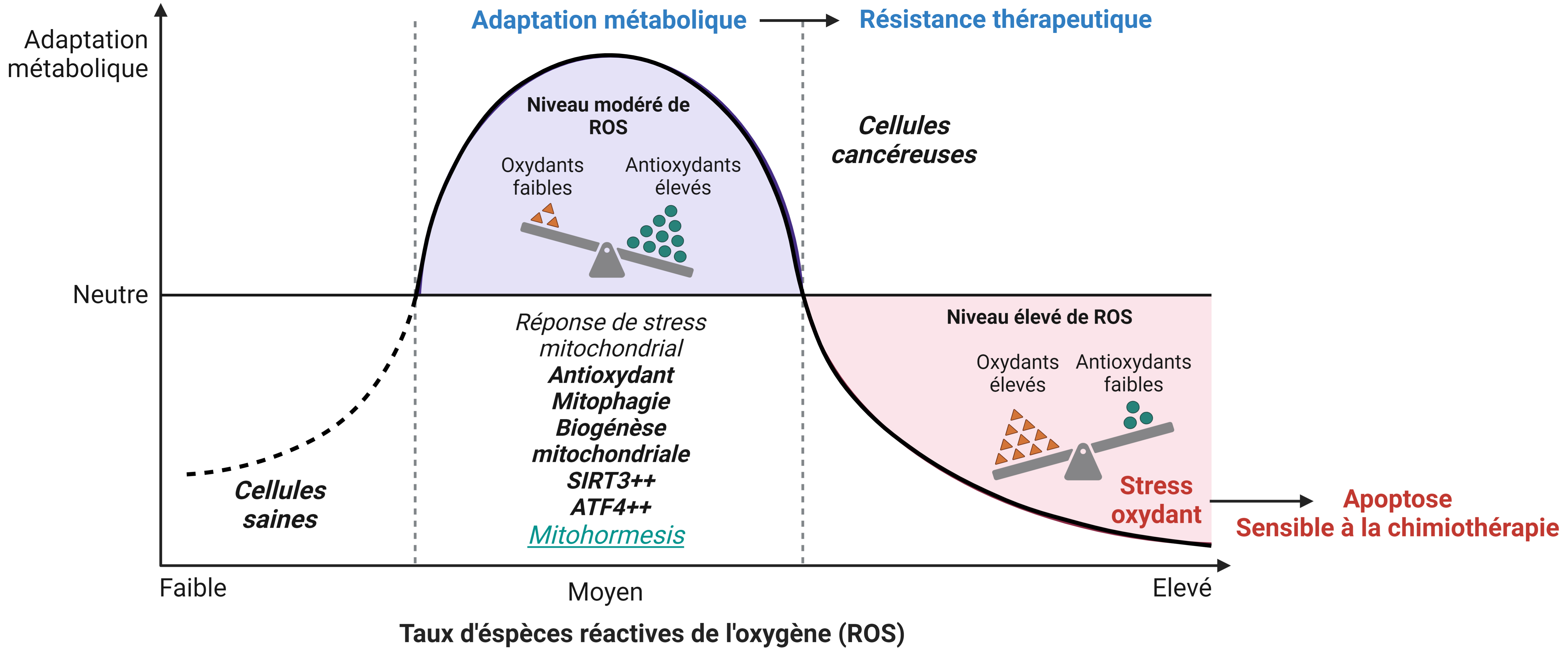
Rôle de l’homéostasis redox et de l’impact du niveau d’espèces réactives de l’oxygène sur la réponse thérapeutique dans les LAM.
9. Le rôle du micro-environnement dans le statut OxPHOS et la résistance des blastes
L’interaction entre les cellules stromales mésenchymateuses et les cellules leucémiques dans la moelle osseuse contribuent de manière importante à la chimiorésistance et aux rechutes de la maladie [78]. Les blastes s’adaptent continuellement aux fluctuations dans la disponibilité des nutriments et de l’oxygène depuis le micro-environnement de la moelle osseuse. Plusieurs équipes ont démontré grâce à un système de co-culture organotypique de la niche médullaire, que les cellules stromales de la moelle osseuse transfèrent les mitochondries aux blastes [78, 79]. Ce transfert unidirectionnel est augmenté par l’AraC et nécessite des contacts directs entre les cellules, passant par une voie endocytique. Cela participe à l’augmentation de l’OxPHOS observée dans les cellules résistantes. L’utilisation de la metformine, le médicament le plus couramment prescrit pour le diabète de type 2, également connu pour sa fonction inhibitrice du complexe I de la chaîne de transport des électrons, augmente de manière significative la chimiosensibilité des cellules de LAM in vitro et in vivo [80]. L’effet chimiosensibilisant de la metformine se traduit, entre autres, par une réduction du transfert mitochondrial et de l’OxPHOS dans les cellules de LAM. Cependant, cela a pour conséquence d’induire l’effet Pasteur (c’est-à-dire une augmentation de la consommation du glucose et du flux glycolytique) et l’acidification du micro-environnement, ce qui limite ainsi l’efficacité de la metformine. Dans l’ensemble, ces études montrent que le ciblage du transfert mitochondrial pourrait représenter une cible thérapeutique future pour le traitement des LAM [80, 81].
D’autres interactions entre les cellules stromales médullaires et les blastes peuvent être bidirectionnelles, c’est-à-dire que ce ne sont pas seulement les cellules stromales qui exercent une action sur les blastes pour soutenir leur survie et leur prolifération, mais que les cellules leucémiques dialoguent en retour avec leur micro-environnement, pour le reprogrammer et le rendre favorable au développement leucémique. La production d’aspartate par les cellules stromales médullaires permet aux blastes leucémiques de soutenir leur production de pyrimidine [82] et, les cellules de LAM entraînent également la production d’interleukine IL-6 par les cellules stromales leur permettant ainsi la phosphorylation de STAT3, pouvant se lier à la membrane mitochondriale ce qui conduit à l’augmentation de l’OxPHOS et leur chimiorésistance [83]. L’IL-6 permet également l’entrée des acides gras dans les cellules de LAM par l’intermédiaire du récepteur membranaire CD36, grâce à l’activation de STAT3 soutenant l’oxydation des acides gras [84] et la fusion mitochondriale par l’augmentation de la mitofusin 1, participant à l’augmentation de l’OxPHOS [85].
Comme précédemment discuté, le métabolisme des acides gras joue un rôle clé dans la résistance à l’AraC. L’utilisation seul d’un inhibiteur de la FAO, l’avocatin B, un lipide dérivé du fruit de l’avocat, induit la mort des cellules leucémiques en monoculture [86]. Cependant, lorsque les cellules leucémiques sont en contact avec les adipocytes de la moelle osseuse, l’inhibiteur conduit à la libération d’acides gras libres par les adipocytes. Ceux-ci sont captés par les blastes grâce à la surexpression de la fatty acid binding protein 4 (FABP4), entraînant une réorientation du métabolisme vers la glycolyse en augmentant l’entrée du glucose dans les cellules leucémiques, qui sont ainsi protégées de la mort induite par l’inhibition de la FAO par l’avocatin B. L’ajout de l’AraC permet la synergie avec l’avocatin B, de par la dépendance des blastes à la FAO et la forte production de ROS qui est cytotoxique [87]. Par ailleurs, le tissu adipeux sert également de réservoir à une sous-population de CSL surexprimant CD36, permettant l’entrée des acides gras et leur résistance thérapeutique [88]. CD36 est positivement associé à la dissémination extramédullaire des blastes leucémiques, et à un risque accru de rechute après une chimiothérapie intensive [89]. Il favorise la migration des blastes en se liant à la thrombospondine-1, et l’inhibition de CD36 réduit les métastases des blastes et prolonge la survie des souris traitées par chimiothérapie in vivo [89].
10. La mitothérapie dans les leucémies aiguës myéloïdes
Au vu du rôle central des adaptations métaboliques et mitochondriales dans la résistance thérapeutique, le ciblage de l’OxPHOS (seul ou en combinaison avec d’autres voies métaboliques), appelé mitothérapie, pourrait constituer une stratégie thérapeutique intéressante pour sensibiliser aux agents chimiothérapeutiques conventionnels et aux thérapies ciblées afin d’améliorer la survie des patients atteints de LAM [90]. De nombreux essais cliniques ont ainsi été menés avec des résultats prometteurs, malgré l’apparition de nouveaux mécanismes de résistance. Comme nous l’avons vu, le cycle de Krebs, la β-oxydation des acides gras et les complexes de la chaîne de transport des électrons permettent de maintenir l’homéostasie redox et de générer l’ATP nécessaire à la survie et à la prolifération des cellules leucémiques, en créant ainsi une dépendance plus importante de ces cellules à une de ces voies métaboliques. Deux stratégies principales d’inhibition directe ou indirecte de l’OxPHOS sont donc étudiées actuellement.
Tout d’abord, des inhibiteurs ciblant spécifiquement les complexes de la chaîne de transport des électrons ont été développés. Ceux-ci ciblent différentes protéines ou enzymes nécessaires au fonctionnement de la chaîne de transport des électrons. C’est principalement le complexe I de la chaîne respiratoire qui est ciblé en utilisant l’IACS-010759 en essai clinique de phase I ou le mubritinib qui cible spécifiquement les CSL en diminuant l’OxPHOS [27, 40]. Plusieurs inhibiteurs sont actuellement testés en études pré-cliniques et montrent des effets anti-leucémiques prometteurs comme l’EVT-701 ou le pyrvinium [91, 92]. L’inhibition de la DHODH qui permet le transfert d’électrons à l’ubiquinone dans la chaîne respiratoire grâce à l’oxydation du dihydro-orotate en orotate est également une stratégie prometteuse de ciblage du complexe III, permettant la différenciation des cellules, actuellement en essais cliniques de phase I et II dans les LAM (Tableau 1) [93, 94].
Description des essais cliniques évaluant différents inhibiteurs mitochondriaux pour le traitement des patients atteints de LAM
| Molécule | Cible thérapeutique | Essai clinique | Phase |
|---|---|---|---|
| IACS-010759 | ETCI | NCT02882321 | Phase I LAM réfractaires ou rechute |
| Atovaquone | ETCIII | NCT03568994 | Phase I LAM de novo (en association avec chimiothérapie induction standard) |
| ONC-201 | Protéase mitochondriale ClpP | NCT02392572 | Phase I/II LAM réfractaires ou rechute |
| CPI-613 | Pyruvate déshydrogénase | NCT03504410 | Phase III (en association avec forte dose cytarabine et mitoxantrone) |
| α-Cétoglutarate déshydrogénase | NCT05854966 | Phase II LAM réfractaires ou rechute (en association avec la metformine) | |
| NCT01520805 | Phase IIa LAM réfractaires aux agents hypométhylants | ||
| CB-839 | Glutaminase | NCT02071927 | Phase II LAM réfractaires ou rechute |
| Erwinase | Asparaginase | NCT02283190 | Phase I |
| GRASPA | L-asparaginase | NCT01810705 | Phase II patients agés/non éligible chimiothérapie intensive (en association avec faible dose cytarabine) |
| PEG-BCT-100 | Arginase1 | NCT02899286 | Phase II LAM réfractaires ou rechute |
| ADI-PEG 20 | Arginine deiminase | NCT05001828 | Phase I (en association avec VEN/AZA) |
| BAY2402234 | DHODH | NCT03404726 | Phase I |
| Brequinar | DHODH | NCT03760666 | Phase II LAM réfractaires ou rechute |
| ASLAN003 | DHODH | NCT03451084 | Phase II LAM patients inéligibles à un traitement standard (en combinaison avec l’AZA) |
| JNJ-74856665 | DHODH | NCT04609826 | Phase I (seul ou en combinaison avec VEN/AZA) |
| PTC299 | DHODH | NCT03761069 | Phase Ib LAM réfractaires ou rechute |
D’autre part, l’inhibition de la traduction mitochondriale, en utilisant la tigécycline, actuellement en phase I, permet d’inhiber la synthèse des complexes de la chaîne respiratoire et de cibler spécifiquement les CSL par rapport aux CSH [29]. De plus, comme décrit précédemment, la protéase mitochondriale ClpP régule l’activité des enzymes de la chaîne respiratoire responsable de la forte activité OxPHOS [95]. De ce fait, des inhibiteurs ont été développés réduisant ainsi l’activité du complexe II de la chaîne respiratoire ciblant spécifiquement les CSL. L’activation de la ClpP, par des imipridones comme ONC-201 et ONC-212 qui se lient à la ClpP permet d’éradiquer les CSL, montrant ici que la régulation positive ou négative des ClpP permet de réguler à la baisse l’OxPHOS des cellules [96]. Récemment, une étude a également montré le rôle d’une imipridone, ONC-213, dans la diminution de l’activité d’une enzyme essentielle à la production de NADH dans le cycle de Krebs, l’αKGDH. Cette inhibition entraîne une réponse aux stress mitochondriaux via ATF4, conduisant à une diminution de la protéine anti-apoptotique MCL1 conduisant à la mort des cellules [97]. Autre exemple, le CPI-613, utilisé en essai clinique de phase II et III en association avec l’AraC, cible deux enzymes du cycle de Krebs : la pyruvate déshydrogénase (PDH) et de l’α-KG déshydrogénase. Ces essais montrent des résultats mitigés sur les patients atteints de LAM réfractaires ou post-rechutes, avec un taux de rémission complète ou incomplète (CR/CRi) de 50 % et la présence de toxicité [96]. Enfin, l’utilisation du CB-839 (télaglénastat), un inhibiteur de la glutaminase (l’enzyme responsable de la conversion de la glutamine en glutamate, permettant d’alimenter le cycle de Krebs et de diminuer la production de glutathion), cause l’accumulation de ROS et l’apoptose des cellules de LAM ; ce médicament est actuellement en essai clinique de phase I dans les LAM réfractaires et en rechute [98, 99].
Dans l’ensemble, ces données démontrent que le ciblage des voies métaboliques nécessaires à la synthèse des intermédiaires du cycle de Krebs ou à la régulation des enzymes de l’ETC sont des stratégies thérapeutiques très prometteuses pour cibler les CSL. Il est important de noter que l’inhibition directe et indirecte de l’OxPHOS cible des CSL ayant des antécédents génétiques divers [30, 95, 39, 40]. Ces données suggèrent donc que la dépendance à l’OxPHOS est au moins partiellement indépendante du génotype du cancer et peut ainsi représenter une vulnérabilité largement conservée des CSL.
Parmi les thérapies ciblées actuellement utilisées en clinique, les inhibiteurs de BCL-2 ou l’inhibition d’IDH1 et IDH2 par l’ivosidénib et l’énasidénib permettent de réduire l’OxPHOS et le cycle de Krebs respectivement, ce qui est responsable en partie de leur activité anti-leucémique. En plus de leur effet sur l’inhibition de l’adaptation mitochondriale, les mitothérapies permettent de diminuer la cardiotoxicité de la doxorubicine [100] et entraînent également une diminution de l’autophagie ainsi que de la réponse inflammatoire [101], responsable en partie de la chimiorésistance des cellules leucémiques.
Cependant, un traitement prolongé inhibant les ETC et l’OxPHOS peut induire plusieurs types de toxicités. L’inhibition de l’OxPHOS entraîne une adaptation métabolique des cellules saines de l’organisme, avec une augmentation compensatoire de la glycolyse qui conduit à une acidose lactique, dont les symptômes peuvent être sévères. De plus, des mécanismes de résistance ont également été observés avec des redirections des flux métaboliques en réponse à l’inhibition de l’OxPHOS. En effet, l’utilisation de ces différents inhibiteurs entraîne dans la majorité des cas une réponse de stress mitochondrial comme précédemment décrite, plus ou moins importante selon les inhibiteurs [102]. Il a été montré que, suite à l’utilisation de l’IACS-010759, de la même manière qu’avec l’AraC, les cellules stromales interagissent de façon directe avec les blastes et transfèrent des mitochondries fonctionnelles, entraînent une augmentation de la fission mitochondriale et de la mitophagie ce qui permet de garder les mitochondries fonctionnelles [103]. Les cellules leucémiques ont également la capacité de rediriger leur métabolisme suite à différents inhibiteurs de l’OxPHOS vers d’autres voies énergétiques. Ce qui est notamment le cas suite à l’inhibition de MPC1 (Mitochondrial Pyruvate Carrier), qui bloque l’entrée du pyruvate dans les mitochondries, baissant ainsi l’OxPHOS des cellules. Un mécanisme d’adaptation se met en place en alimentant cette fois-ci l’OxPHOS grâce à la glutaminolyse, suite à la transcription du miR-1, permettant d’alimenter le TCA cycle via la glutamine et le fumarate [104]. Ces études montrent ainsi l’intérêt de cibler plusieurs voies métaboliques pour empêcher ces adaptations ou utiliser des inhibiteurs mitochondriaux qui n’activent pas ces réponses.
Enfin, l’une des stratégies à étudier pour éviter la survenue d’effets indésirables graves suite aux mitothérapies serait d’alterner différents inhibiteurs selon une stratégie « in and out » qui éviterait à l’organisme d’être soumis à de trop fortes doses de traitement sur une longue période. Par ailleurs, l’un des problèmes majeurs de l’utilisation des inhibiteurs de l’OxPHOS en clinique est la toxicité (acidose lactique et neurotoxicité) et la capacité d’adaptation métabolique des cellules leucémiques, qui parviennent à échapper aux traitements en redirigeant leur trajectoire métabolique vers d’autres voies. Les cellules parvenant à s’adapter deviennent ainsi résistantes, épuisant les stratégies thérapeutiques envisageables. Pour y remédier, la stratégie principale serait la mise en place de thérapies combinatoires, associant des agents cytotoxiques (chimiothérapie, thérapies ciblées) à des thérapies métaboliques diverses ciblant à la fois l’OxPHOS et d’autres voies de support énergétique comme le métabolisme des acides aminés, des acides gras ou la glycolyse. Cela permettrait d’épuiser la capacité de reprogrammation métabolique des cellules qui, n’ayant plus la capacité de s’adapter, mourront. Suivre le statut métabolique des cellules leucémiques, pour comprendre leur vulnérabilité et proposer ainsi la meilleure association de traitement, et faire varier les cibles métaboliques à chaque cycle de traitement en fonction des dépendances observées, permettrait ainsi d’épuiser toutes les voies de support métabolique, pour empêcher que les cellules s’adaptent pour résister aux traitements et initient in fine des rechutes. Le design des futurs essais cliniques devra être affiné suivant ces critères-là, en utilisant des biomarqueurs pertinents du statut mitochondrial tumoral et du statut métabolique de l’hôte.
Déclaration d’intérêts
Les auteurs ne travaillent pas, ne conseillent pas, ne possèdent pas de parts, ne reçoivent pas de fonds d’une organisation qui pourrait tirer profit de cet article, et n’ont déclaré aucune autre affiliation que leurs organismes de recherche.