

1 Introduction
Controlled radical polymerisation is becoming a heavily investigated new area of research, in view of the combined tolerance to polar functionalities and the ability to develop well-defined macromolecular architectures [1,2]. After the concept of the persistent radical effect (PRE) [3,4] was fruitfully applied to the development of so-called Stable Free Radical Polymerisation (SFRP) methodologies [5–11], the field of controlled radical polymerisation has experienced tremendous advances following the independent reports by the groups of Sawamoto and Matyjaszewski [12,13] that the PRE can be realized by an atom transfer process whereby a halogen-capped dormant chain and a metal complex are in fast equilibrium with a reactive free radical and a metal halide complex. This process has been termed Atom Transfer Radical Polymerisation (ATRP) (Fig. 1).
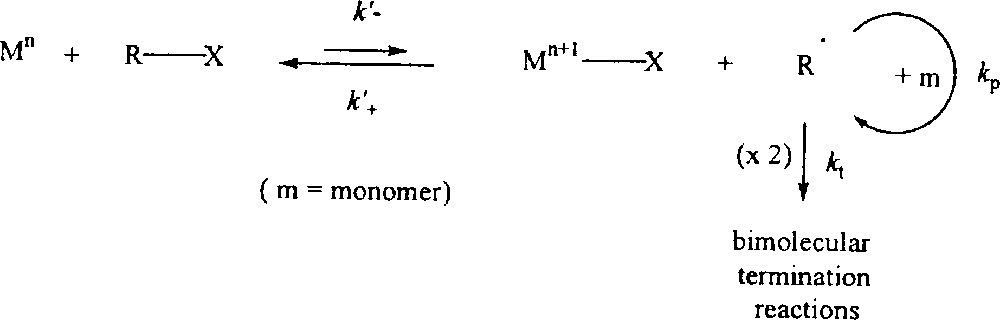
Scheme of Atom Transfer Radical Polymerisation (ATRP).
In a recent contribution [14], we have applied the analysis of homolytic Bond Dissociation Energies (BDE) to predict the ability of a metal complex to control radical polymerisation of styrene. We have shown that, in agreement with theoretical predictions, a family of MoIII/MoIV cyclopentadienyl complexes controls the radical polymerisation by both the SFRP and the ATRP protocols, while a system based on MoII/MoIII produces a largely insufficient radical concentration to sustain the polymerisation process. In addition, the polymerisation with MoIII/MoIV has been found to be polluted by a side-reaction of hydride transfer. The use of a simple kinetic model has allowed to rationalize the variable degree of this Catalytic Chain Transfer (CCT) reaction as a function of reaction conditions (notably the MoIII/MoIV ratio) when styrene polymerisation is conducted with butadiene-containing cyclopentadienyl–molybdenum complexes.
Since the variable of outmost importance for the regulation of the PRE, namely the BDE, appeared quite sensitive to the metal oxidation state, but not so much to the nature of the ancillary ligands,[14] we wondered whether the controlled radical polymerisation of styrene could also be achieved in the presence of cyclopentadienyl-free MoIII/MoIV systems. Numerous compounds of type MoCl3L3 and MoCl4Ln (L = ethers, thioethers, phosphines, etc.; n = 2 or 3) are known [15] and are more easily accessible than the cyclopentadienyl-containing analogues [16]. In this contribution, we report our initial studies indicating that, indeed, polystyrene is obtained by a controlled polymerisation procedure under a variety of conditions, with control being exerted by phosphine-containing MoIII/MoIV complexes.
2 Experimental section
2.1 General remarks
All manipulations were carried out under an atmosphere of dry and oxygen-free argon with standard Schlenk techniques. Styrene was washed with a aqueous NaOH solution (10%), neutralized with water, dried with MgSO4 and then distilled at 25 °C under reduced pressure. Chlorobenzene was bubbled with dry argon for more than 15 min immediately before use. Molecular weights and molecular weight distributions were measured with a Gynkoteck P580 apparatus using THF as eluent (1 ml min–1) and being equipped with a refractometer and 2 columns B Jordi DVB (range 1 000–1 000 000). MALDI–TOF mass spectrometric analyses were performed with a Perkin Elmer Voyager–DE STR. The matrix was the Dithranol and PS samples were cationised with silver salt (AgTFA). Compounds MoCl3(PMe3)3, 1 [17], and MoCl4(PMe3)3, 2 [16,18], were obtained according to previously described synthetic procedures. (1-Bromoethyl)benzene (BEB) and ethyl-2-bromoisobutyrate (BIB) were purchased from Aldrich Chemical Co. and degassed prior to use. Azobis(isobutyronitrile) (AIBN) was purchased from Janssen and recrystallized twice from MeOH before use.
2.2 ATRP polymerisations
All ATRP polymerisation reactions were performed following the same experimental procedure. The complex was added to a 50 ml Schlenk tube equipped with a stirring bar. Styrene and the initiator (BEB or BIB) were added to the reaction flask by a syringe. The Schlenk tube was then immersed in an oil bath heated at 90 °C. Aliquots were withdrawn periodically for a reaction monitoring. These were dried by room temperature evaporation followed by oven drying at 100 °C for 8 h for the calculation of the conversion. The GPC analyses were carried out on the crude dried polymers after redissolution in THF.
2.3 Reverse ATRP polymerisation
Complex 2 and AIBN were added to a 50 ml Schlenk tube equipped with a stirring bar. Chlorobenzene and styrene were added by a syringe. The Schlenk tube was immersed in an oil bath heated at 120 °C for 15 min, then it was transferred to another bath at 90 °C. Aliquots were withdrawn periodically for a reaction monitoring by GPC, as described above.
3 Results and discussion
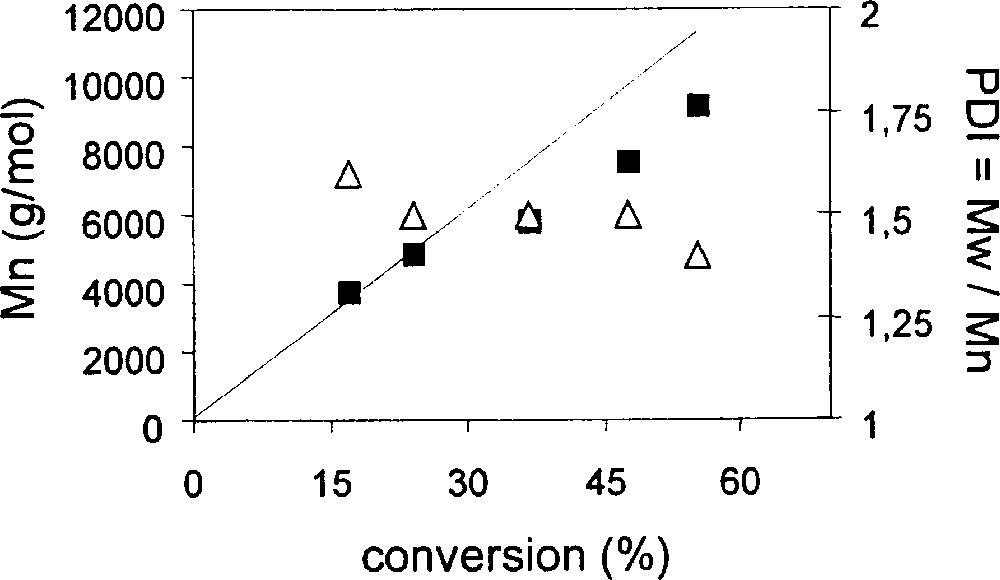
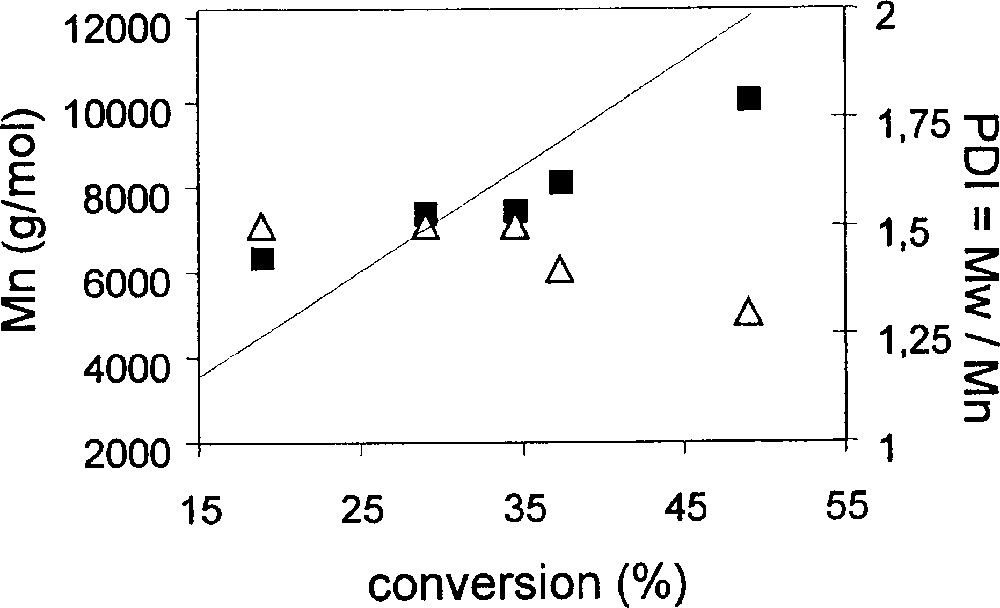
Styrene is smoothly polymerised in the presence of compound 1 and PhCH(CH3)Br (BEB) in a 1:1 ratio at 90° in bulk. The progression of polydispersity and number average molecular weight as a function of conversion is shown in Table 1 and graphically in Fig. 2. The continuous decrease of PDI as the conversion increases, reaching values under 1.5 at 50% conversion, is as expected in the presence of a PRE. Mn is quite close to the theoretical values. A similar behaviour is observed when the polymerisation is conducted in chlorobenzene (Table 2 and Fig. 3). The polymers obtained in solution have slightly lower PDI than those obtained in bulk. The rate of polymerisation, on the other hand, is greater for the bulk experiment (Fig. 4), as expected from the increased monomer and radical concentrations.
Results of the ATRP of styrene in bulk with MoCl3(PMe3)3 and BEB. T = 90 °C, [styrene]/[BEB]/[Mo] = 196/1/1.
| Time (min) | Conversion (%) | Mn (experimental) | Mn (theoretical) | PDI |
| 262 | 17.0 | 3752 | 3460 | 1.6 |
| 580 | 24.3 | 4867 | 4968 | 1.5 |
| 1 025 | 36.8 | 5873 | 7505 | 1.5 |
| 1 317 | 47.5 | 7538 | 9698 | 1.5 |
| 1 625 | 55.4 | 9197 | 11301 | 1.4 |
Plot of Mn (black squares) and PDI (open triangles) vs conversion for the ATRP of styrene in bulk with complex 1. The straight line corresponds to the theoretical Mn for a living polymerisation. Conditions are as in figure 4.
Results of the ATRP of styrene in PhCl solution (30% v/v) with MoCl3(PMe3)3 and BEB. T = 90 °C, [styrene]/[BEB]/[Mo] = 235/1/1.
| Time (min) | Conversion (%) | Mn (experimental) | Mn (theoretical) | PDI |
| 385 | 18.9 | 6388 | 4617 | 1.5 |
| 1 072 | 24.3 | 7396 | 7108 | 1.5 |
| 1 332 | 34.5 | 7473 | 8442 | 1.5 |
| 1 702 | 37.3 | 8103 | 9129 | 1.4 |
| 3 181 | 49 | 10095 | 12000 | 1.3 |
Plot of Mn (black squares) and PDI (open triangles) vs conversion for the ATRP of styrene in PhCl solution with complex 1. The straight line corresponds to the theoretical Mn for a living polymerisation. Conditions are as in Fig. 4.
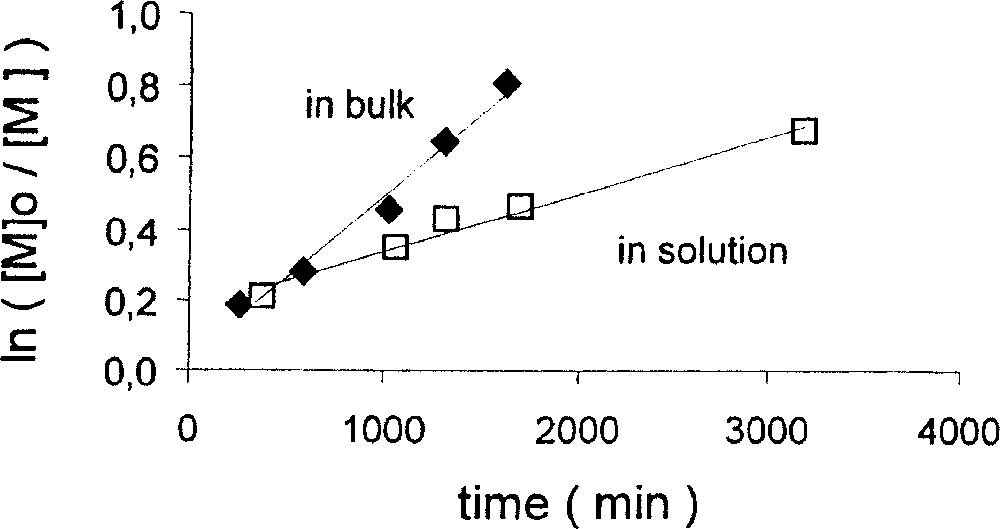
Plots of ln([M]0/[M]) vs time for the bulk and solution (30% v/v in PhCl) ATRP of styrene with complex 1. Conditions: T = 90 °C; [styrene]/[BEB]/[Mo] = 196/1/1 (bulk), 235/1/1 (PhCl solution).
The absence of chain transfer in this polymerisation process has been confirmed by the NMR and MS analyses of the polymer. The 1H NMR spectrum on the polymers obtained at small conversions shows clearly the presence of brominated chain-ends (peak at 4.3–4.6 ppm) [19] and the absence of chains terminated by vinyl groups (for which a resonance would be expected in the 6.0–6.4 ppm range) [20].
A MALDI–TOF-MS analysis of the polymer provides supporting evidence for the presence of Br-terminated chains, but shows also three additional families of polymer chains (figure 5a). The position and relative intensities of the four series of peaks are identical with those of the polymer obtained by using the CpMoCl2(PMe3)2/BEB system and analysed under the same instrumental conditions [14]. The fours peaks at m/z = 3 321.7, 3 337.9, 3 353.5, and 3 379.6 in figure 5a, for instance, are attributed to the formulas MeCH(Ph)–(Sty)n–CH2CH(Ph)Br + Ag+, MeCH(Ph)–(Sty)n+1–CH=CH(Ph) + Ag+, MeCH(Ph)–(Sty)n+2–CH=CH(Ph)+Na+, and MeCH(Ph)–(Sty)n+1-CH2CH(Ph)Cl + Ag+, respectively, with Sty = CH2CHPh and n = 28. The presence of Cl-terminated chains is not surprising, since the radical forming equilibrium (equation (1)) can be reversed in principle by abstraction of either a bromine or a chloride atom. However, the Mo–Cl BDE is stronger than the Mo–Br BDE according to our calculations [14], consequently favouring the bromine atom transfer. Indeed, the Cl-terminated chains are less abundant than the Br-terminated chains. The most intense peaks, on the other hand, are those related to vinyl-terminated chains with either Ag+ or Na+ being provided by the matrix. Sodium contamination is a common phenomenon for MALDI–TOF analyses of polymers [21,22]. The related Na peaks of the Br- or Cl-capped polymer chains are either masked by other peaks or too small to be observed.
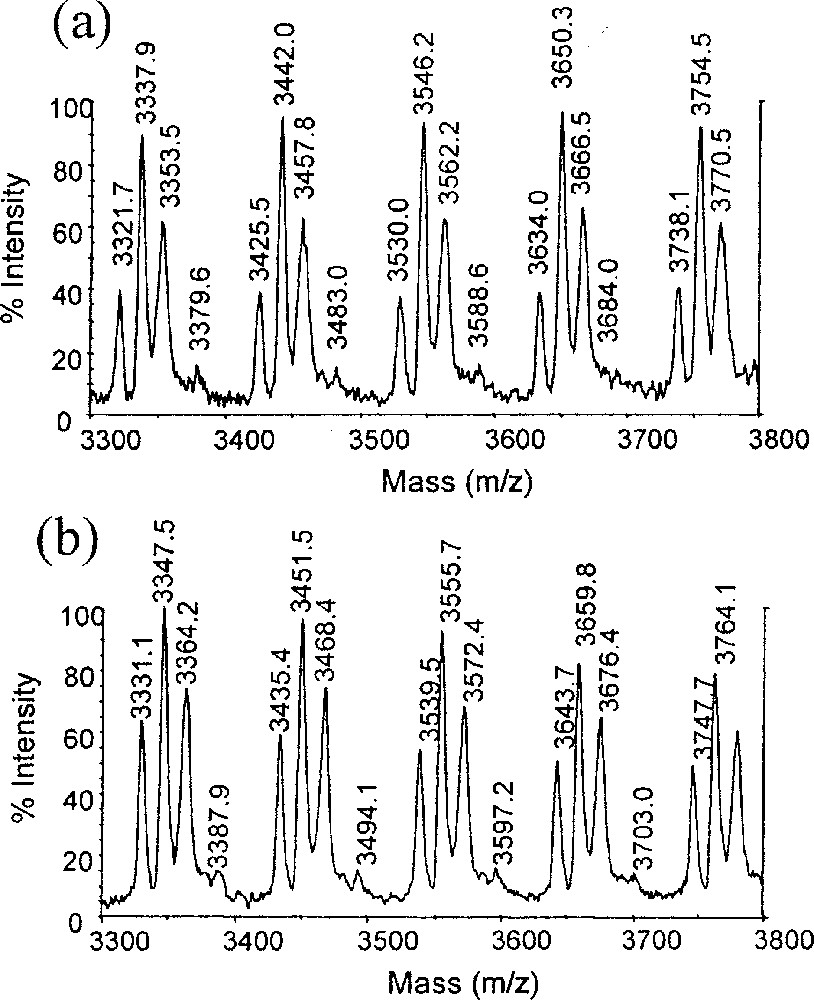
Details of the MALDI–TOF spectra for polystyrene obtained using BEB (a) or BIB (b) as initiator.
| 1 |
The question arises as to the origin of these vinyl-terminated chains. Dehydrobromination processes are known to occur readily for Br-terminated polymers upon contact with the silver salt and the Cl-terminated chains could analogously be dehydrochlorinated. This possibility seems also confirmed by the inability to observe vinyl-terminated end group resonances in the NMR. However, the alternative of a CCT process remains a possibility, since after the transfer process the resulting metal hydride would, upon insertion of the first styrene monomer, yield the same CH3CH(Ph)- end group provided by the BEB initiator. In order to settle this point, the polymerisation was repeated with a different initiator, BIB, which differs from BEB in having 10 additional mass units. The result of the MALDI–TOF–MS analysis of the resulting polymer (figure 5b) shows quite clearly that all four families of polymer chains are shifted by exactly ten mass units and consequently demonstrates that they are all initiated by BIB. In other words, there is no CCT in this polymerisation process.
A common test for a living polymerisation process is also the so-called ‘stop-and-go’ experiment (chain extension). In a genuine ‘living’ polymerisation, after the chain growth process is stopped by lowering the temperature or by the complete monomer consumption, resuming the polymerisation by warming or by adding new monomer restores the growth of the previously formed chains without initiating any new ones. The results shown in Table 3 and Fig. 6 for a polymerisation of styrene with 1/BEB clearly show that (i) the polymerisation process takes place with the same rate before and after being stopped for ca 14 h, indicating that the total number of chains is conserved; (ii) the Mn of the polymers before and after the stop fall on the same line and are close to the theoretical masses; (iii) the PDI continues to decrease after resumption of the polymerisation.
Results of the stop-and-go ATRP of styrene in bulk with MoCl3(PMe3)3 and BEB. T = 90 °C, [styrene]/[BEB]/[Mo] = 239/1/1a.
| Time (min) | Conversion (%) | Mn (experimental) | Mn (theoretical) | PDI |
| 150 | 11.7 | 4097 | 2903 | 2 |
| 303 | 24.3 | 4693 | 3666 | 1.7 |
| 425 | 17.6 | 4747 | 4365 | 1.8 |
| 565 | 20.1 | 5682 | 4994 | 1.7 |
| 740 | 23 | 6574 | 5717 | 1.6 |
| 1580 | 22.9 | 5719 | 5696 | 1.7 |
| 1775 | 26.9 | 7119 | 6680 | 1.6 |
| 1985 | 30.3 | 7981 | 7526 | 1.5 |
| 2105 | 31.9 | 7706 | 7938 | 1.5 |
a The polymerisation was stopped after 740 min and resumed after 1580 min (see Fig. 6a).
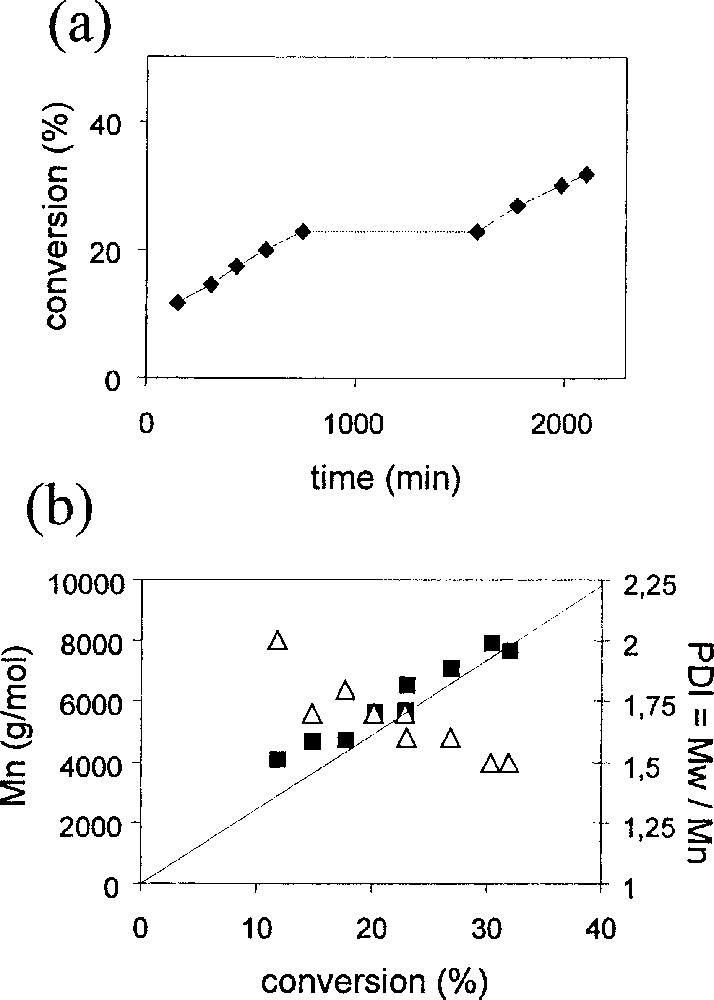
Plots of (a) conversion vs. time and (b) Mn (black squares) and PDI (open triangles) vs conversion for the stop-and-go ATRP of styrene in bulk. The straight line corresponds to the theoretical Mn for a living polymerisation. Conditions: T = 90 °C, [styrene]/[BEB]/[Mo] = 239/1/1.
A final test for the involvement of equation (1) in determining the PRE for the living polymerisation process has been the establishment of a controlled polymerisation under ‘reverse’ conditions. That is, equilibrium (1) can in principle be equally established starting from the left hand side, as shown above (regular ATRP process), or from the right hand side by exposing the preformed Mo(IV) complex to a radical (reverse ATRP). Complex MoCl3Br(PMe3)3 does not exist to the best of our knowledge and out attempts to synthesize it by Br2 oxidation of 1 have only yielded analytically impure products. However, the related complex MoCl4(PMe3)4, 2, is known [18] and we have recently improved its preparation procedure [16]. A reverse ATRP experiment, the initial radical being provided by the thermal decomposition of AIBN, confirms that the polymerisation process is controlled under these conditions, see Table 4 and Fig. 7. The control of the radical concentration would be established by equilibrium (2). The experimental molecular weight is higher than theoretical molecular weight, indicative that the initiator efficiency (AIBN) is less than 1. In addition, PDI decreases with conversion, as expected. The successful controlled polymerisation under ‘reverse’ ATRP conditions demonstrates that the PRE is established by use of the MoIII/MoIV couple.
Results of the reverse ATRP of styrene in bulk with MoCl4(PMe3)3 and AIBN. T = 120 °C for 15 min, then T = 90 °C; [styrene]/[AIBN]/[Mo] = 240/0.5/1.
| Time (min) | Conversion (%) | Mn (experimental) | Mn (theoretical) | PDI |
| 488 | 18.2 | 7 895 | 4 147 | 2.3 |
| 1 373 | 29.6 | 9 350 | 7 382 | 2.0 |
| 2 123 | 38.1 | 11 206 | 9 507 | 1.9 |
| 5 643 | 55.2 | 15 080 | 13 764 | 1.6 |
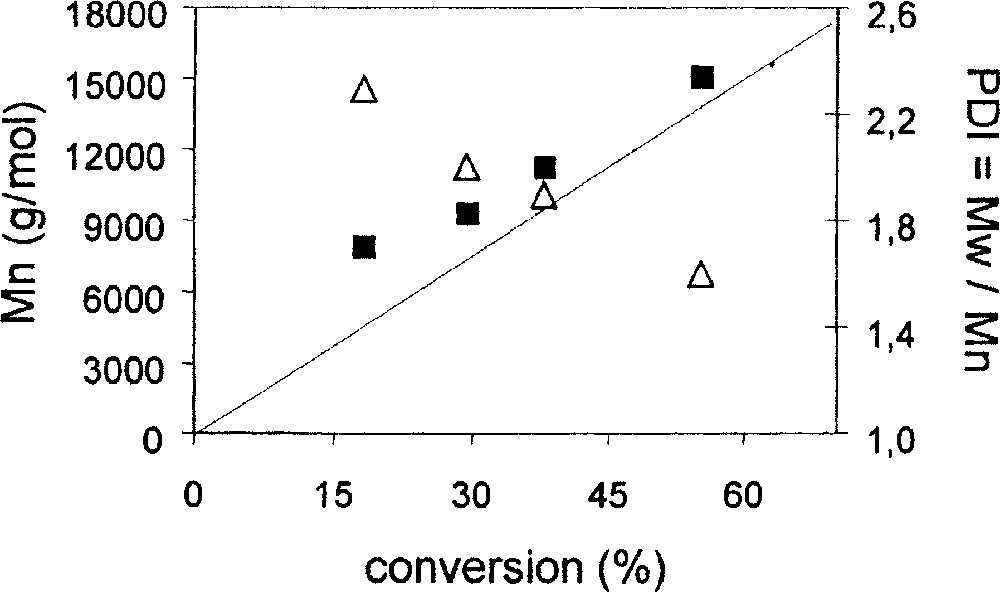
Plot of Mn (black squares) and PDI (open triangles) vs conversion for the reverse ATRP of styrene in bulk. The straight line corresponds to the theoretical Mn for a living polymerisation. Conditions: T = 120 °C for 15 min, then T = 90 °C; [styrene]/[AIBN]/[Mo] = 240/0.5/1.
| 2 |
Complex MoCl3(THF)3, i.e. the synthetic precursor of complex 1, does not exert any control in the polymerisation of styrene. In fact, a faster polymerisation process having all the characteristics of cationic polymerisation is observed when using this complex. We presume that the Lewis acidity of the Mo(III) centre in this coordination compound may favour the opening process of the THF ligand to afford a MoIV–OCH2CH2CH2CH2+ moiety faster than the atom transfer process. The zwitterionic species formed would then trigger the cationic polymerisation mechanism.
No degree of control is witnessed in the radical polymerisation of MMA with 1. The PMMA obtained at low conversions is characterized by a high Mn and a broad PDI. Our interpretation of this result is that equilibrium (1) is shifted to a greater extent toward the right end side when R• = (polymer–CH2)(CH3)C•–COOCH3, in agreement with a lower BDE of the corresponding C–Br bond and an uncontrolled radical polymerisation caused by a large radical concentration.
4 Conclusions and perspectives
In conclusion, we have reported here for the first time that the ATRP polymerisation of styrene can be accomplished by simple coordination compounds of molybdenum, the PRE being established by a redox couple containing MoIII and MoIV species. A controlled radical polymerisation of styrene has been accomplished by using the lithium molybdate(V) complexes [LiMo(NAr)2(C–N)R] (C–N = C6H4(CH2NMe2)-2; Ar = C6H3Pri2-2,6; R = C–N, Me, CH2SiMe3, p-Tol) and benzyl chloride as initiator [23]. The direct implication of the Mo complex in the polymerisation control mechanism was not unambiguously proven in that case, however. Current studies are aimed at the modification of the metal coordination sphere for achieving control in the radical polymerisation of other monomers.
Acknowledgements
We are grateful to the CNRS for supporting this work (programme ‘Catalyse pour l’industrie et l’environnement’) and for a BDI PhD fellowship to F.S. Support from the Ministry of Research and COST D17 (Working group on Transition Metal Mediated Living Radical Polymerisation) is also gratefully acknowledged.