

1 Introduction
Les complexes triflates des métaux d et f sont des acides de Lewis très utilisés en synthèse organique et en catalyse 〚1–4〛. Ce sont également d’excellents précurseurs en synthèse inorganique et organométallique 〚5〛. Alors que les complexes triflates des lanthanides ont fait l’objet, ces dernières années, de très nombreuses études, notamment quant à leurs applications, quasiment rien n’a été publié concernant leurs homologues 5f, hormis la synthèse du triflate d’uranyle hydraté UO2(OTf)2(H2O)n et de quelques complexes triflates du thorium 〚6–8〛. Afin de pallier cette insuffisance, nous avons décidé de développer la chimie des complexes triflates des actinides, en nous focalisant en particulier sur l’uranium, qui est le métal le moins radioactif de la série, facile à manipuler, et qui possède en plus l’avantage de disposer d’une palette de degrés d’oxydation allant de +3 à +6.
2 Synthèse des complexes triflates organométalliques de l’uranium (IV)
Nous avons d’abord essayé d’utiliser les méthodes « classiques » pour synthétiser les complexes triflates de l’uranium 〚5〛. Nous avons tenté, en particulier, de remplacer les groupes halogénures de composés 〚U〛–(X)n par des triflates à l’aide d’agents de substitution courants, tels que AgOTf ou Me3Si–OTf. Nous avons également fait réagir des espèces 〚U〛–X (X = alkyle, amidure, halogénure) avec l’acide triflique. Si certaines de ces réactions se déroulent bien, ces méthodes se sont avérées toutefois peu sélectives et absolument pas générales. Nous avons alors trouvé que la protonation de complexes alkyles et amidures par le triflate de pyridinium, représentée par l’équation (1), constitue une excellente voie d’accès vers ces composés, obtenus facilement, avec de bons rendements 〚9〛.
Grâce à cette méthode, de nombreux composés triflates de l’uranium (IV), pour la plupart organométalliques, ont été isolés, ainsi que le montre la Fig. 1.
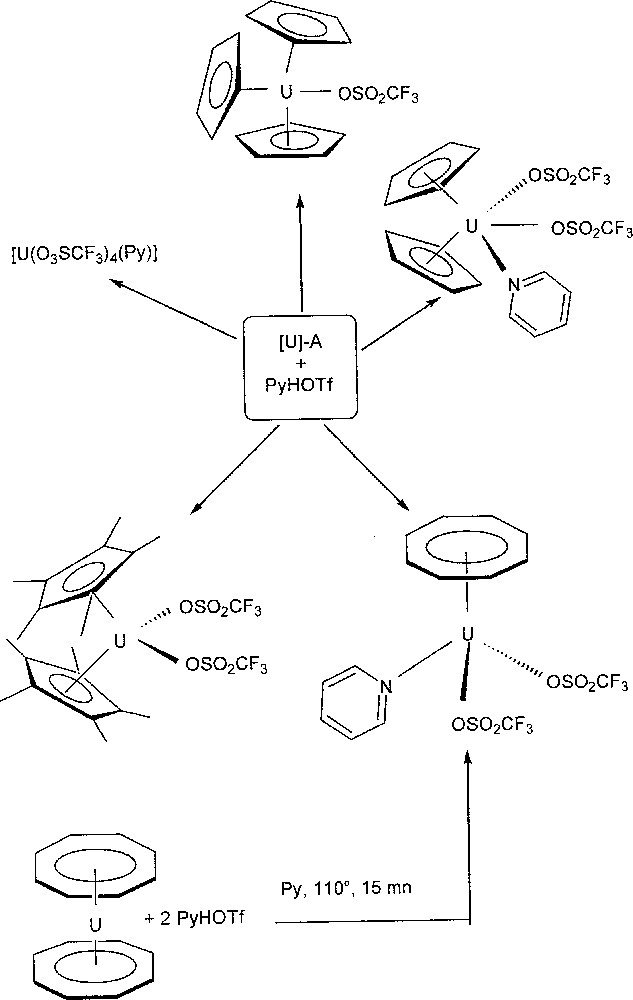
Synthèse de complexes triflates par protonation de précurseurs alkyles et amidures au moyen de PyH·OTf.
Il faut remarquer que le composé U(Cp)2(OTf)2(py)2 (py = pyridine) est stable dans les solvants coordinants ou aromatiques, contrairement à son homologue chloré U(Cp)2Cl2. Cette différence de stabilité entre espèces triflate et chlorure d’une même série met bien en évidence les influences électroniques distinctes de ces deux ligands et montre que la synthèse de précurseurs triflates devrait permettre le développement d’une chimie inaccessible avec les dérivés halogénures correspondants. C’est pourquoi nous avons synthétisé le composé U(OTf)4, isolé sous forme de monoadduit avec la pyridine, en protonant le métallacycle 〚U(N{SiMe3}2)2(N{SiMe3}{SiMeCH2})〛 avec quatre équivalents de PyHOTf.
L’utilisation du triflate de pyridinium n’est pas seulement limitée à la protonation de liaisons simples uranium–carbone et uranium–azote. L’uranocène U(Cot)2, complexe stériquement encombré, réputé robuste et peu réactif, réagit très facilement en présence de 2 équiv de triflate de pyridinium, pour donner l’espèce monocyclooctatétraénylique U(Cot)(OTf)2(py) 〚10〛. La synthèse de tels composés U(Cot)(X)2(L)2 a constitué, pendant de nombreuses années, un véritable défi.〚11–13〛
L’ensemble de ces résultats montre que le triflate de pyridinium est un réactif de choix pour préparer des espèces triflates. Il présente plusieurs avantages par rapport à l’acide triflique. Cette poudre commerciale, stable, soluble dans le THF (THF = tétrahydrofuranne), peut être utilisée dans une grande gamme de solvants organiques, au contraire de l’acide triflique, qui est corrosif, très hygroscopique, dont l’utilisation nécessite une distillation préalable et qui ne peut être employé dans certains solvants, comme le THF, qu’il polymérise. Enfin, le triflate de pyridinium s’avère très réactif et beaucoup plus sélectif que TfOH dans les réactions de protonation.
Les structures cristallines des complexes U(Cp)3(OTf)(CNtBu) 〚9〛, U(Cp)2(OTf)2(py)2 〚10〛, U(Cp*)2(OTf)2(H2O) 〚9〛, et U(Cot)(OTf)2(OPPh3)2 sont représentées Figs. 2 à 5.
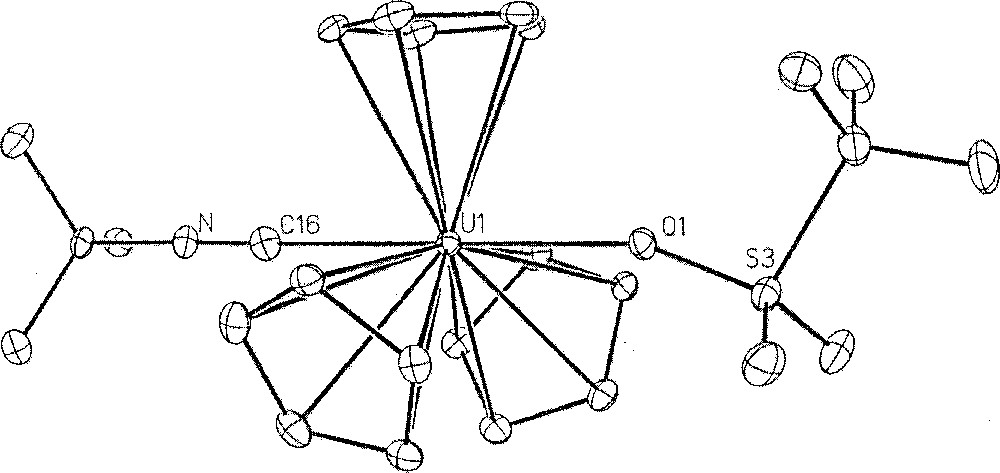
Structure cristalline du complexe U(Cp)3(OTf)(CNtBu). Reproduit avec l'autorisation de la Royal Society of Chemistry.
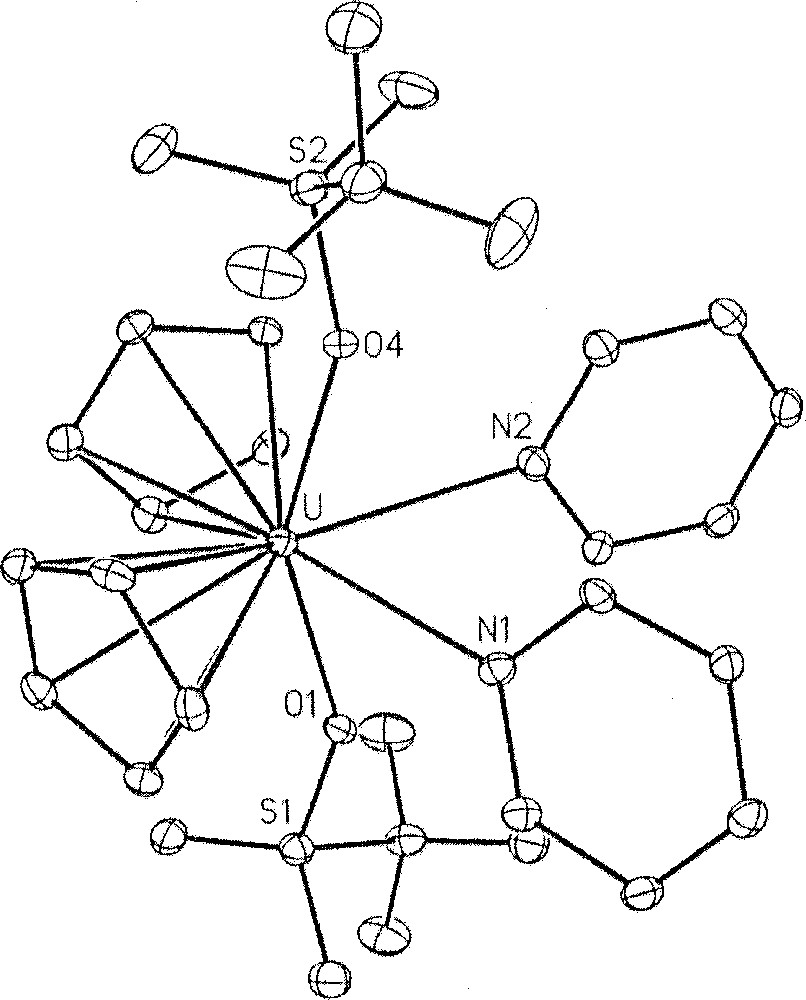
Structure cristalline du complexe U(Cp)2(OTf)2(py)2.
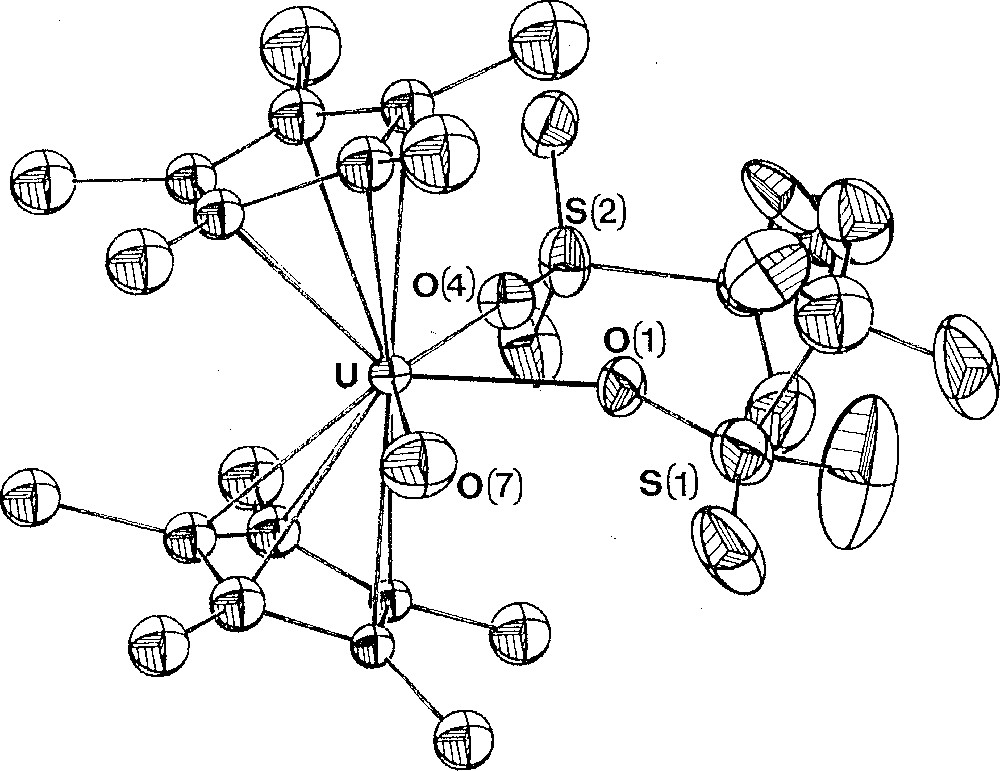
Structure cristalline du complexe U(Cp*)2(OTf)2(H2O). Reproduit avec l'autorisation de la Royal Society of Chemistry.

Structure cristalline du complexe U(Cot)(OTf)2(OPPh3)2. Pour la clarté du dessin, seuls les carbones ipso des groupes phényl sont représentés.
Dans tous ces composés, les ligands triflate sont coordonnés de façon monodentate. Les distances U–O(OTf) sont de l’ordre de 2,38 Å, sauf pour le complexe triscyclopentadiénylique où celle-ci est plus longue (2,485(9) Å), reflétant certainement l’effet stérique et électronique des ligands auxiliaires. Ces longueurs de liaison sont plus grandes que les distances uranium–oxygène des complexes alcoxydes (ca 2,0–2,3 Å), ce qui suggère que le ligand triflate est moins solidement lié au métal. Cependant, le fait que, dans l’ensemble de ces structures, aucun des groupes triflate ne se trouve dissocié, quel que soit le solvant coordinant employé (pyridine, THF) ou la base de Lewis introduite en excès (OPPh3, tBuNC), molécules qui présentent pourtant une forte affinité pour l’uranium, montre que le ligand triflate est assez fortement lié aux actinides.
Nous voyons ainsi que l’utilisation du triflate de pyridinium s’avère intéressante pour la préparation de complexes triflates. Néanmoins, cette méthode repose sur la protonation de complexes alkyles et amidures, dont les synthèses nécessitent souvent plusieurs étapes.
3 Synthèse des complexes homoleptiques U(OTf)3 et U(OTf)4
La préparation en grande quantité des espèces homoleptiques U(OTf)n (n = 3–6) selon des méthodes simples et rapides ferait de ces composés des précurseurs de choix pour le développement de la chimie de l’uranium, au même titre que UI3(THF)4 et UCl4, qui sont pratiquement les seuls précurseurs solubles disponibles à ce jour 〚14,15〛.
Nous avons trouvé plusieurs voies de synthèse des composés U(OTf)3 et U(OTf)4 ; celles-ci sont résumées sur la Fig. 6 〚16〛.

Synthèse des complexes triflates homoleptiques U(OTf)n (n = 3, 4).
La première méthode, « classique », car souvent utilisée dans le cas des complexes des métaux p et d, consiste à traiter les chlorures correspondants par un large excès d’acide triflique pur 〚5〛. Les réactions de UCl3 ou UCl4 procèdent à 120 °C, provoquant un dégagement violent de chlorure d’hydrogène ; les triflates U(OTf)n (n = 3, 4) sont récupérés simplement après évaporation de l’acide en excès. Cette voie nécessite toutefois l’obtention des précurseurs chlorés, dont les préparations restent délicates 〚15,17〛.
Nous avons trouvé une nouvelle voie d’accès, originale et beaucoup plus pratique, en traitant par TfOH des copeaux d’uranium métallique préalablement activés. Les composés U(OTf)3 ou U(OTf)4 sont formés de façon quantitative et sélective en contrôlant la température du milieu. À 120 °C, seul U(OTf)3 se forme ; celui-ci est facilement oxydé en U(OTf)4 par chauffage à 180 °C dans l’excès d’acide. Ces synthèses nécessitent plusieurs jours, mais ce délai peut être considérablement réduit en partant d’une poudre finement divisée du trihydrure métallique UH3, aisément préparé par hydrogénation à chaud de tournures d’uranium. Dans ce cas, le traitement de UH3 par l’acide triflique conduit, en quelques minutes à la température ambiante, au triflate de l’uranium (III). Les rendements de ces synthèses sont quasiment quantitatifs. Les composés homoleptiques sont solubles dans les solvants organiques classiques (THF, pyridine, DME – diméthoxyéthane –...) ; néanmoins, la rétention de traces d’acide triflique dans les produits conduit à la polymérisation du THF, si on utilise directement ces complexes dans ce solvant. Enfin, ces triflates d’uranium sont des acides de Lewis qui forment des adduits stables en présence de bases de Lewis. Dans la pyridine en présence d’un large excès d’oxyde de triphényle phosphine, U(OTf)3 et U(OTf)4 conduisent aux dérivés 〚U(OTf)2(OPPh3)4〛〚OTf〛 et U(OTf)4(OPPh3)3, dont les structures cristallines sont représentées sur les Figs. 7 et 8 〚10,16〛.

Structure cristalline du complexe 〚U(OTf)2(OPPh3)4〛〚OTf〛. Pour la clarté du dessin, seuls les atomes de carbone ipso des groupes phényl sont représentés. Reproduit avec l'autorisation de Wiley-VCH.
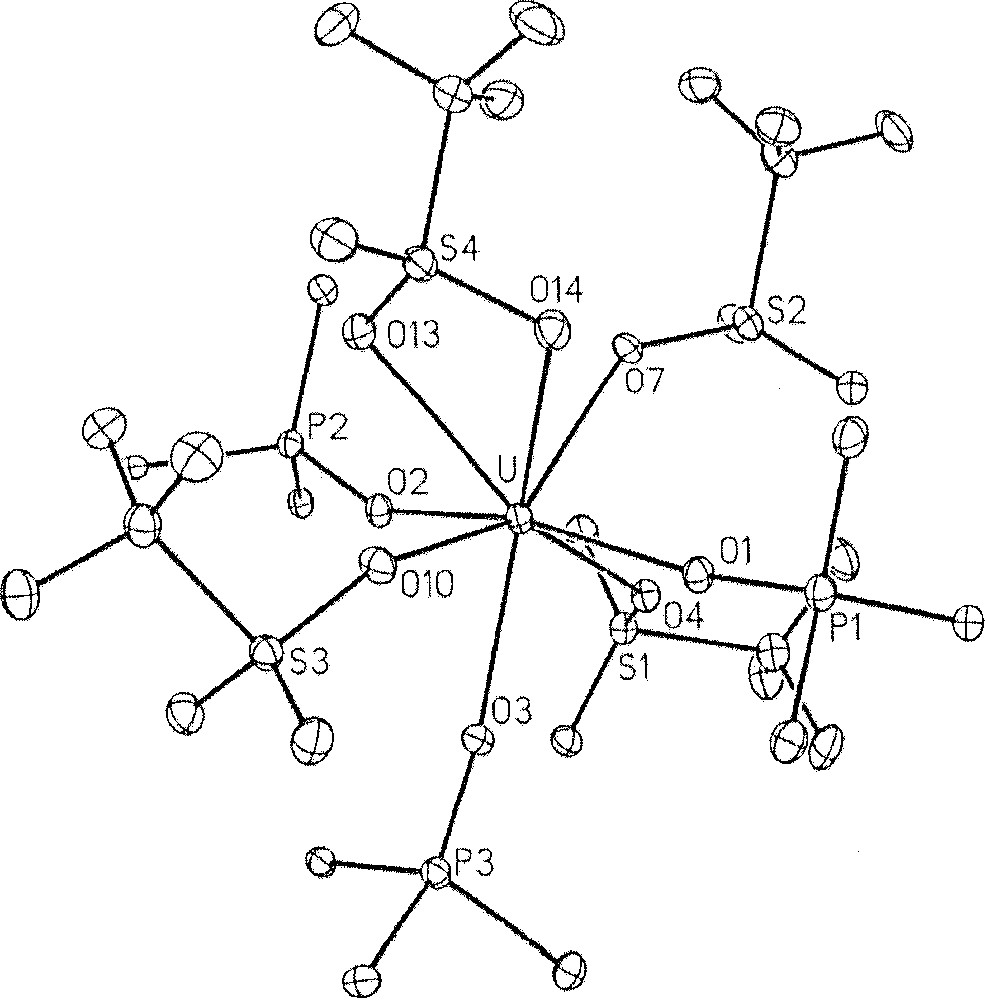
Structure cristalline du complexe U(OTf)4(OPPh3)3. Pour la clarté du dessin, seuls les atomes de carbone ipso des groupes phényl sont représentés.
Le complexe de l’U(III) cristallise sous forme de monocation, du fait de la dissociation d’un des ligands triflate. Le métal se trouve coordonné par quatre molécules de OPPh3 et deux ligands triflate, présentant chacun un mode de coordination différent (monodenté et bidenté). La distance uranium–oxygène du triflate monodenté, égale à 2,446(4) Å, est plus courte de près de 0,2 Å que la distance moyenne des liaisons U–O du triflate bidenté. On peut noter que l’excès d’oxyde de triphényle phosphine présent dans la solution lors de la cristallisation n’entraîne pas la dissociation de tous les ligands triflate et la formation d’une entité tricationique 〚U(OPPh3)n〛3+. De même, aucune dissociation de ligand triflate n’est observée dans le complexe U(η1-OTf)3(η2-OTf)(OPPh3)3 (Fig. 6), dont la structure montre, ici encore, que les distance U–O des triflates monodentés sont plus courtes que celles du triflate bidenté. De tels complexes, présentant dans leur structure cristalline des ligands triflate bidentés non pontants, sont rares 〚18–20〛. On en compte quelques exemples avec les métaux d et les alcalino-terreux, et un seul cas avec les lanthanides. Il s’agit du complexe cationique du lanthane, 〚La(η1-OTf)(η2-OTf)(HMPA)4〛〚OTf〛 (HMPA = OP{NMe2}3), dont la structure est très similaire à celle du complexe de l’U(III) 〚21〛.
4 Synthèse des complexes triflates non solvatés UO2(OTf)2 et Ce(OTf)4
Par analogie avec la synthèse des complexes triflates des lanthanides Ln(OTf)3, obtenus en traitant en milieu aqueux les oxydes Ln2O3 par l’acide triflique 〚22〛, nous espérions préparer de la même façon le complexe U(OTf)6 à partir du trioxyde d’uranium (VI), UO3. L’obtention de U(OTf)6 nous semble intéressante, car celui-ci pourrait être un précurseur pour le développement de la chimie de l’uranium (VI) autre que celle de l’uranyle et présenter une acidité de Lewis très marquée, permettant son utilisation en synthèse organique et en catalyse.
Cependant, comme le montre la Fig. 9, la réaction d’un excès d’acide triflique avec UO3 solide ou en suspension dans l’eau (réactions (a) et (b)) ne conduit pas à la formation de U(OTf)6 mais à celle du triflate d’uranyle non solvaté. En solution aqueuse, l’espèce UO2(OTf)2+n(H3O)n est formée et mène au composé UO2(OTf)2, après un chauffage prolongé sous vide permettant la sublimation du sel H3O·OTf 〚23〛.
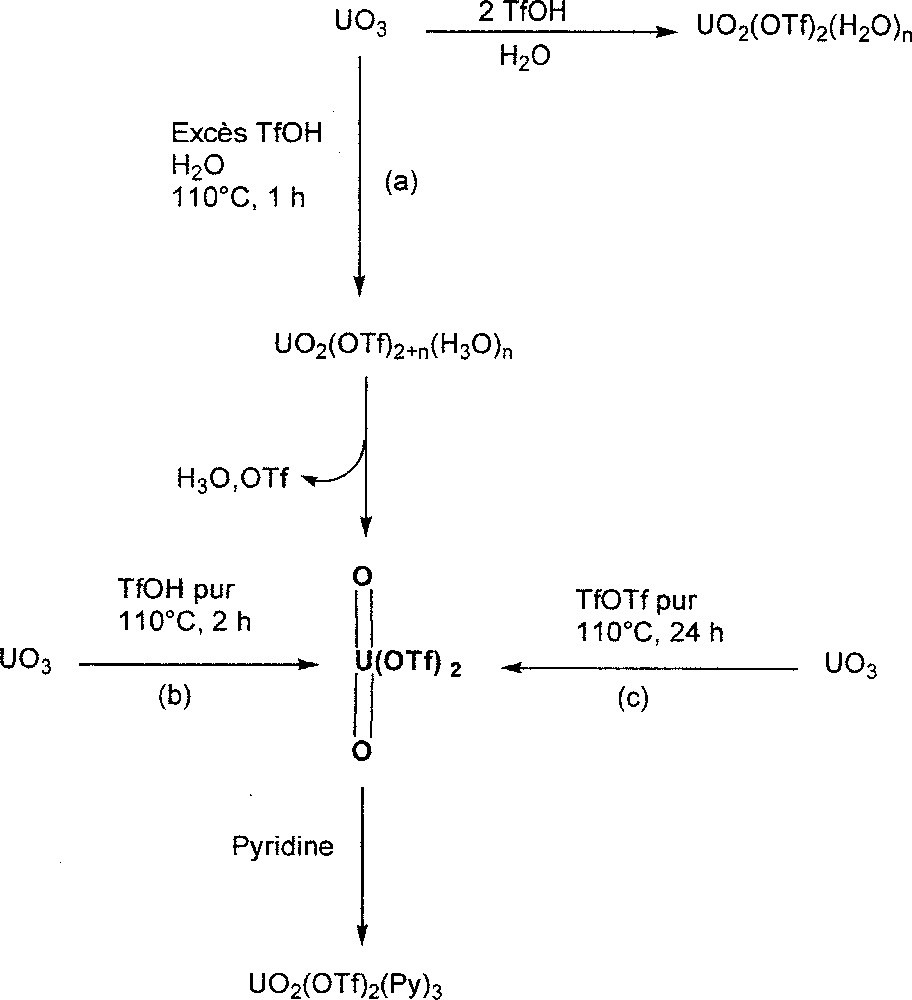
Synthèse du triflate d'uranyle non solvaté UO2(OTf)2.
Si les réactions (a) et (b) de la Fig. 9 sont efficaces, les étapes finales d’élimination de l’acide triflique et de sublimation de H3O·OTf sont longues, difficiles, ce qui limite leur portée pratique. Le traitement thermique à 110 °C pendant quelques heures d’une suspension de UO3 dans l’anhydride triflique (réaction (c) de la Fig. 9) constitue une voie d’accès bien supérieure aux deux précédentes. Une simple évaporation sous vide de l’excès de réactif conduit rapidement au triflate d’uranyle anhydre. Ces différences de facilité des synthèses (a), (b) et (c) sont directement liées aux propriétés physiques distinctes des réactifs TfOH et TfOTf. L’acide triflique est en effet un réactif coordinant à haut point d’ébullition (162 °C), tandis que l’anhydride triflique ne présente pas de propriétés coordinantes et possède une température d’ébullition relativement basse (80 °C).
Le triflate d’uranyle est un acide de Lewis qui coordonne très facilement des bases de Lewis, comme la pyridine avec laquelle il forme l’adduit neutre UO2(OTf)2(py)3, dont la structure cristalline est représentée sur la Fig. 10 〚23〛.
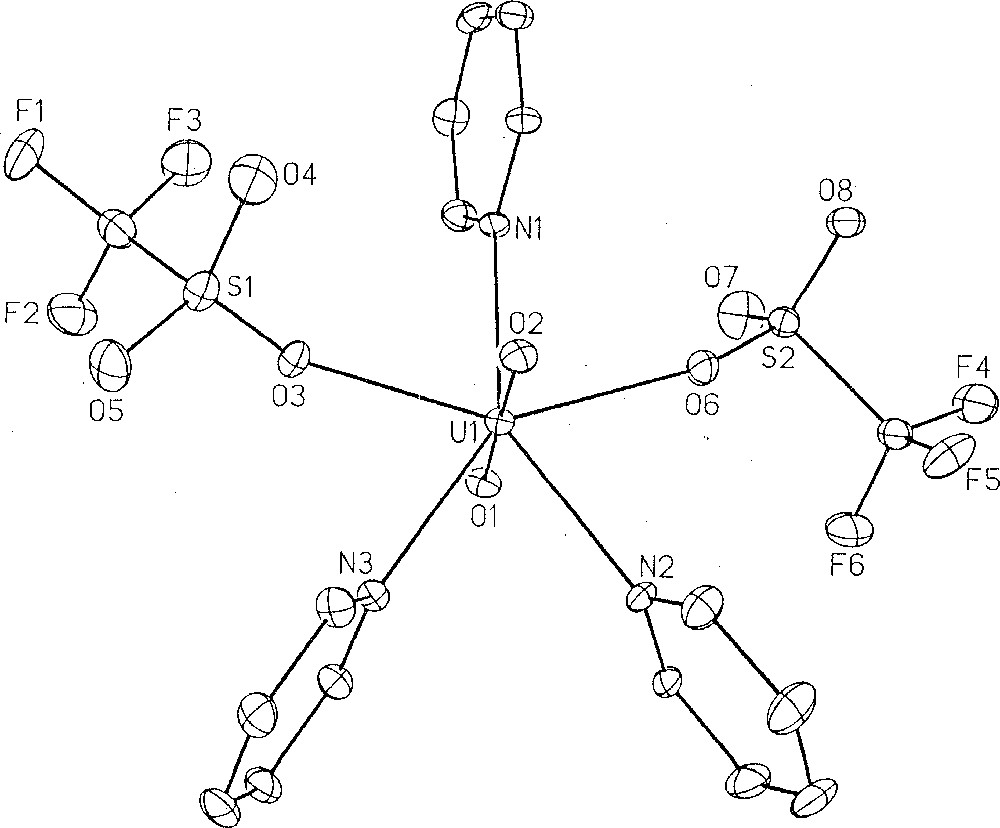
Structure cristalline du complexe UO2(OTf)2(py)3. Reproduit avec l'autorisation de Wiley-VCH.
Ce complexe très hygroscopique s’hydrate immédiatement à l’air ou en présence de traces d’eau pour donner l’ion aquo UO2(H2O)n2+. Ces espèces hydratées ne peuvent être utilisées directement en milieu organique basique, comme la pyridine, où elles évoluent en dérivés oxo tel que celui représenté sur la Fig. 11 〚23〛. Afin d’éliminer ces molécules d’eau, dont la décomplexation est difficile 〚24〛, et pouvoir développer la chimie de l’uranyle en milieu organique strictement anhydre, une méthode de déshydratation simple et rapide de ces espèces était hautement souhaitable. En fait, l’anhydride triflique (TfOTf) est un excellent agent de déshydratation du triflate d’uranyle. Ainsi, le chauffage de UO2(OTf)2(H2O)n en suspension dans TfOTf, permet d’obtenir UO2(OTf)2 avec libération, comme seul sous-produit, de l’acide triflique, qui est entraîné avec l’anhydride par évaporation sous vide (équation (2)).
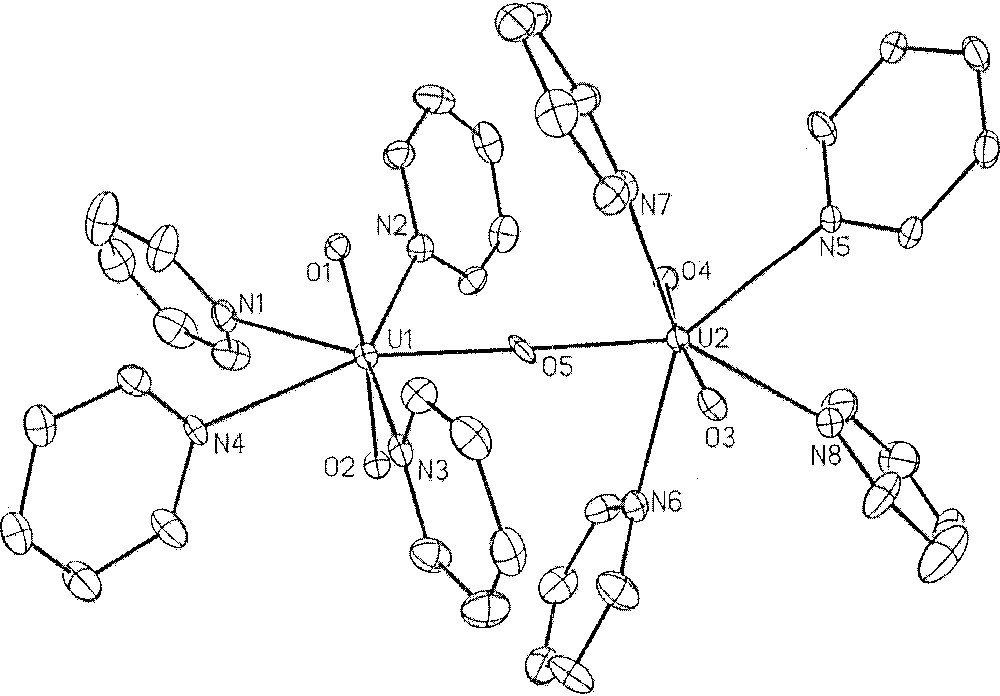
Structure cristalline du dication 〚{UO2(py)4}2(μO)〛〚OTf〛2. Reproduit avec l'autorisation de Wiley-VCH.
Cette méthode peut être généralisée à la déshydratation des triflates métalliques et a été appliquée, en particulier, à la synthèse du triflate de cérium (IV), Ce(OTf)4, à partir du triflate commercial hydraté. L’obtention du triflate de cérium (IV) anhydre était jusqu’à présent entravée par la sensibilité thermique du composé, qui se réduit en Ce(III) vers 120 °C 〚25〛.
5 Conclusion et perspectives
Ce travail a permis de trouver plusieurs méthodes de préparation de complexes triflates de l’uranium(III), (IV) et (VI). En ce qui concerne les composés organométalliques, une bonne voie de synthèse consiste à protoner des précurseurs alkyles ou amidures par le triflate de pyridinium. Cette réaction de protonation, facile à réaliser et très sélective, s’avère bien supérieure à celle utilisant l’acide triflique. Les complexes triflates homoleptiques de l’uranium (III) et (IV), U(OTf)n (n = 3, 4) ont été préparés pour la première fois, selon des voies originales, par réaction de l’acide triflique avec des tournures d’U(0) ou UH3. Le complexe de l’uranium (III) ou celui de l’uranium (IV) peut être obtenu sélectivement, en ajustant simplement la température du milieu réactionnel.
Le triflate d’uranyle non solvaté est facilement préparé par traitement de l’oxyde d’uranium (VI), UO3, ou par déshydratation de UO2(OTf)2(H2O)n, au moyen de l’anhydride triflique. Ce réactif s’avère un excellent agent de déshydratation des complexes triflates. La chimie de l’ion uranyle étant pour l’essentiel limitée aux solutions aqueuses, la formation aisée de UO2(OTf)2 avec celle récemment décrite de UO2Cl2(THF)3 〚26〛 inaugurent le développement plus facile de la chimie de cet ion en milieu organique.
Les composés triflates de l’uranium, acides de Lewis forts, pourraient être utilisés en synthèse organique et en catalyse. Il sera intéressant de comparer la réactivité de U(OTf)3 à celle des homologues halogénés UX3 〚17,27〛, d’une part, et à celle des triflates de lanthanides Ln(OTf)3 〚22〛, d’autre part. La comparaison entre U(OTf)3 et Ln(OTf)3 sera également riche d’enseignements dans le cadre des études de complexation sélective des lanthanides et actinides trivalents par des molécules extractantes telles que les bases polyazotées. Ces recherches à caractère fondamental visant à définir le rôle respectif des électrons 4f et 5f dans la liaison métal–ligand sont d’une importance capitale pour une meilleure compréhension des processus de séparation des déchets nucléaires.
Remerciements
Les auteurs remercient Monique Lance du laboratoire de cristallochimie pour son aide continuelle lors de ce travail.