

1 Introduction
Although their chemistry has long remained unexplored, trivalent lanthanide borohydrides Ln(BH4)3(THF)x (Ln = lanthanide, THF = tetrahydrofuran), as opposed to the commercially available lanthanides triflates and chlorides, are valuable starting reagents in 4f-elements chemistry. Indeed, several advantages are inherent to the use of a (BH4) ligand 〚1–9〛. First, it imparts quite some stability to the compounds and thus allows the preparation of products otherwise inaccessible. Although it is isosteric to the chloride anion, it behaves as a greater electron donor entity. Thus, sterically unsaturated complexes can be obtained and dimers, Lewis base adducts or “ate” species are not as readily observed as with chloride groups 〚8〛.
In contrast to the chloride and triflate ions, it can be clearly identified by 1H NMR spectroscopy. Its coordination mode through bridging hydrogen atoms in a bidentate or tridentate fashion can be determined by infrared analyses and eventually further confirmed by X-ray diffraction studies 〚1–2〛. While the borohydride ligand greatly promoted the actinide (especially uranium) chemistry over several decades (see for instance 〚3–9〛), its impact on the lanthanide congeners remains negligible. We thus investigated the chemistry of neodymium(III) borohydride derivatives, because this lanthanide element has an ionic radius (0.983 Å) close to that of uranium (1.025 Å) 〚10〛. We then used these new borohydride complexes as precursors to organometallic derivatives, especially f-elements cationic species. The reactivity of all these newly made compounds was studied in an effort to establish homologous series of neodymium and uranium products so as to set the bases for the comparison of lanthanide–actinide organometallic complexes.
2 Results and discussion
2.1 Neodymium borohydride complexes
Substitution of the borohydride groups in Nd(BH4)3(THF)3, 1, by several anionic reagents allowed the preparation of various organoneodymium derivatives. Upon reaction with cyclic ligands, such as the cyclopentadienyl and phospholyl potassium salts, KCp* (Cp* = η-C5Me5) and KP* (P* = η-PC4Me4), respectively, the first organometallic bisborohydride complexes of a trivalent lanthanide (Cp*)Nd(BH4)2(THF)2, 2, and 〚K(THF)〛〚(P*)2Nd(BH4)2〛, 3a, were quantitatively isolated, as illustrated in Fig. 1 〚11〛. While a neutral mono(pentamethylcyclopentadienyl) compound, 2, was obtained with the Cp* ligand, an anionic bis(tetramethylphospholyl) product, 3a, was isolated when the P* group was used. Although both ligands are isosteric, formation of this ‘ate’ derivative 3a is easily rationalised by the weaker electron donating ability of P* over Cp*. This feature of the phospholyl ligand has been previously highlighted in uranium coordination chemistry 〚12–14〛. Addition of a crown-ether to 3a allowed the preparation of 〚K(18-C-6)(THF)2〛〚(P*)2Nd(BH4)2〛, 3b, the monomeric structure of which has been determined by X-ray crystallography 〚15〛. Infrared spectra of 2 (strong: 2361, 2337, 2291, 2216 cm–1) and 3b (strong: 2415, 2363, 2332, 2198, 2130) are quite complex and suggest the presence of both bidentate and tridentate BH4 units, supporting a monomeric structure in the solid state 〚1〛. Complex 2 can be desolvated upon gentle heating under reduced pressure to form (Cp*)Nd(BH4)2, whose unique IR absorption at 2291 cm–1, typical of bridging borohydride groups, is indicative of a polymeric structure in the solid state 〚1,16〛. Complexes 3a and 3b are the latest addition to the series of bis(phospholyl) lanthanide(III) derivatives consisting of 〚P*2LnCl2〛– (Ln = Y, Nd, Sm, Lu), (P*)2Ln(CH{SiMe3}2) (Ln = Nd, Sm) and (P*)2NdH 〚17,18〛.
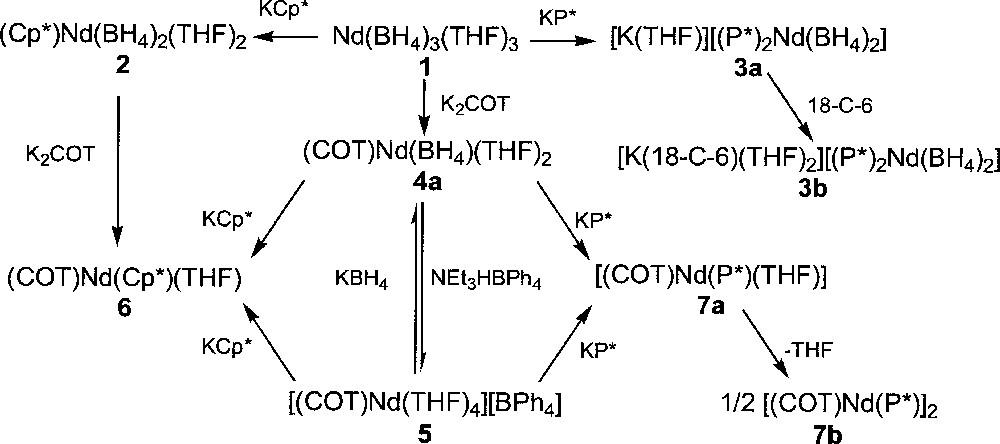
Syntheses of neodymium borohydrides.
Two borohydride groups in 1 can be simultaneously exchanged upon reaction with K2COT (COT = η-C8H8) to afford the unique cyclooctatetraenyl borohydride compound of a lanthanide, (COT)Nd(BH4)(THF)2, 4a, in 98% yield (Fig. 1) 〚11,19〛. Again, the binding mode of the BH4 unit was determined from the IR spectrum, which showed it to be tridentate non-bridging (sharp: 2424; broad: 2000 cm–1) in a monomeric structure. During the crystallization of 4a in benzene, one THF ligand dissociated upon dimerisation into 〚(COT)Nd(BH4)(THF)〛2, 4b, the crystal structure of which (Fig. 2) has been previously described in details 〚19〛. The most distinctive feature of this structure is the unusual coordination mode of the bridging BH4 ligands in a (μ3-H)2B(μ2-H)2 fashion, only once encountered in 〚(C5H3tBu2)2Ce(BH4)〛2 〚16〛 and further confirmed by the IR data (a unique broad strong band at 2255 cm–1). Indeed, in f-element chemistry, bridging borohydride ligands are rather rare and usually coordinate the metallic centres in a (μ2-H)2B(μ2-H)2 mode 〚4, 20, 21〛.
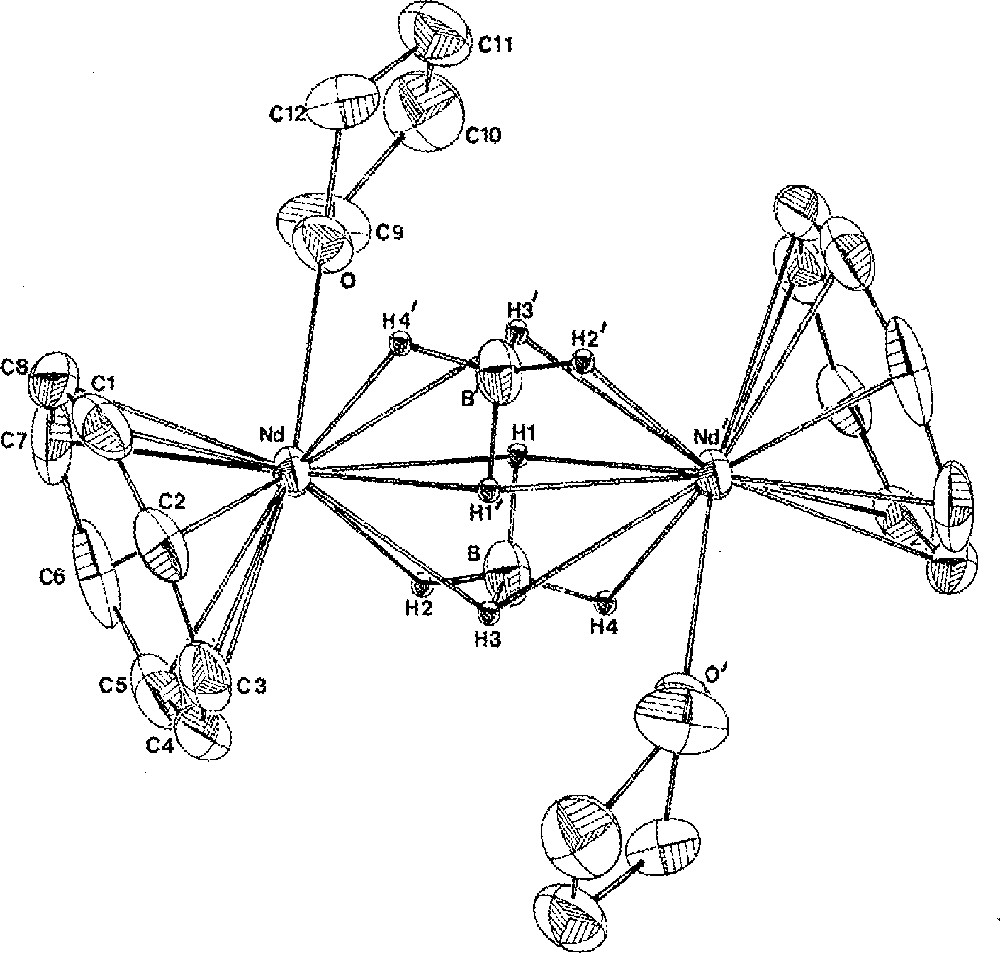
Molecular structure of 〚(COT)Nd(BH4)(THF)〛2, 4b, with thermal ellipsoids drawn at the 30% probability level. Atoms labelled with primes are related to the non-primed atoms by the centre of symmetry. Reprinted with permission from Ref. 〚19〛. Copyright The American Chemical Society.
2.2 Neodymium cyclooctatetraenyl complexes
The most important reactivity of these new neodymium borohydride complexes 2–4 has been unveiled by the protonolysis reaction of (COT)Nd(BH4)(THF)2, 4a, with the acidic ammonium salt NEt3HBPh4 which afforded 〚(COT)Nd(THF)4〛〚BPh4〛, 5, in 75% yield (Fig. 1). The X-ray structure of this neodymium cation has been previously reported 〚19〛. Direct syntheses of complexes 4a or 5 from the triflate or chloride congeners of 1 were less selective than those involving borohydride ligands as a consequence of the greater solubility of triflate or chloride salts. Compound 5 could neither be prepared via cleavage of the Nd–BH4 bond in 4a by TlBPh4 or AgBPh4, a previously established route to cationic species from halide precursors 〚22–24〛. The cationic derivative 5 was transformed back into 4a by addition of a borohydride ligand (Fig. 1). The formation of 〚(COT)Nd(THF)4〛〚BPh4〛, 5, the sole example of a cationic (cyclooctatetraenyl)lanthanide complex, from the borohydride reagent 4a clearly revealed the superiority of the borohydride ligand in 4f-element chemistry.
The mixed cyclooctatetraenyl–cyclopentadienyl or –phospholyl complexes (COT)Nd(Cp*)(THF), 6, and 〚(COT)Nd(P*)(THF)〛, 7a, were formed either by metathesis reaction of 2 with K2COT or 4a with KCp*, and 4a with KP*, respectively, or by addition of these latter two potassium salts to 5 (Fig. 1). The phospholyl product 7a lost the one THF molecule upon drying under vacuum and was thus isolated as a dimer 〚(COT)Nd(P*)〛2, 7b (Fig. 1) 〚25〛. This result is in agreement with the weaker electron-donating power of the P* group relative to the isosteric Cp*; desolvation of 7a renders the neodymium centre electron-deficient, which consequently couples to another 〚(COT)Nd(P*)〛 fragment to form 7b. These two compounds 6–7 represent the only mixed sandwich (cyclooctatetraenyl)neodymium complexes reported to date.
The first cyclooctatetraenyl neodymium complexes incorporating alkoxide and thiolate ligands 〚(COT)Nd(OEt)(THF)〛2, 8, 〚Na〛〚(COT)Nd(BH4)(StBu)〛, 9, (COT)Nd(StBu)(THF)x, 10, 〚Na〛〚(COT)Nd(StBu)2〛, 11, and 〚Na(THF)2〛〚{(COT)Nd}2(StBu)3〛, 12, were similarly obtained upon treatment of 4a or 5 with NaOEt or NaStBu (Fig. 3) 〚11〛. While the dimeric alkoxide 8 could be isolated via both ways, the anionic 9 and neutral 10 monothiolate complexes were observed as intermediate during the formation of 11 from 4a or 5, respectively. The greater electron-donating ability of the alkoxide group compared to the thiolate ligand accounts for the anionic nature of 11 in contrast to 8. The above two different routes to the neodymium alkoxide 8 represent alternative pathways to the previously established metathetical exchange between (COT)LnCl(THF)x and alkoxide anions 〚26–27〛. The only three crystallographically characterised monocyclooctatetraenyl lanthanide alkoxides, 〚(COT)Ln(OR)(THF)〛2 (Ln = Nd, R = Et, 8, previously presented 〚11〛; Ln = Dy, R = (CH2)3CH=CH2) and Ln = Y, R = Ph 〚13〛); all exhibit a dimeric structure with two bridging alkoxide ligands. The sole organometallic neodymium thiolate complex 11 looses one StBu group and dimerises upon THF/toluene crystallisation to afford 〚Na(THF)2〛〚{(COT)Nd}2(StBu)3〛, 12, the X-ray structure of which is illustrated in Fig. 4 〚11〛. The particularity lies in the presence of three StBu groups bridging the two neodymium centres, of which two StBu are coordinated to the 〚Na(THF)2〛 moiety. The sodium salt in 12 remained firmly bound, compound 12 not evolving toward the formation of 10.
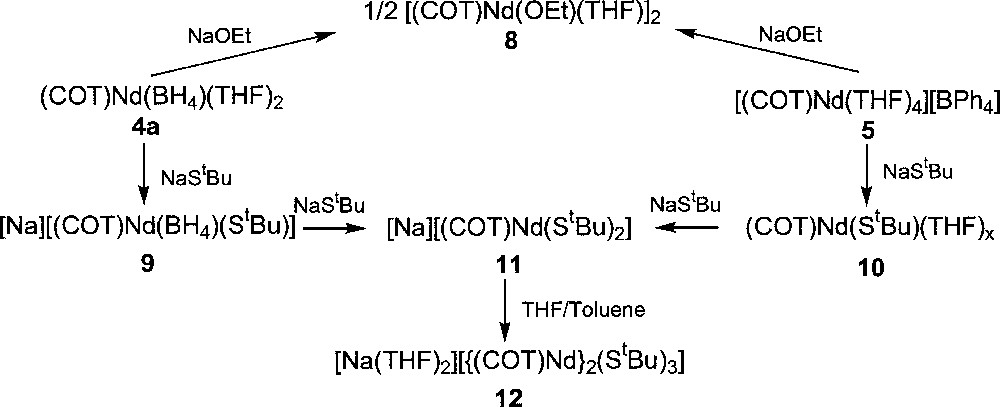
Syntheses of cyclooctatetraenyl neodymium alkoxides and thiolates complexes.
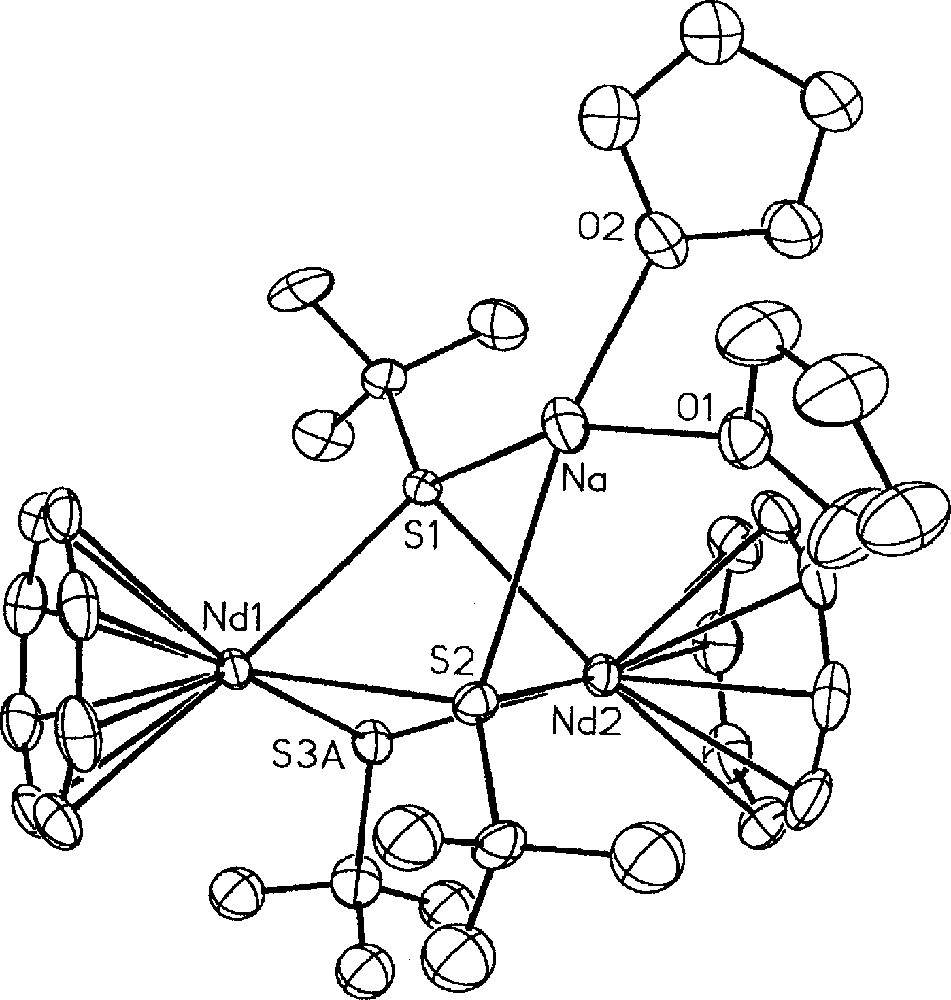
Molecular structure of 〚Na(THF)2〛〚{(COT)Nd}2(StBu)3〛, 12, with thermal ellipsoids drawn at the 20% probability level. Only one of the two disordered tBu and StBu groups is represented. Reprinted with permission from Ref. 〚11〛. Copyright The American Chemical Society.
Reactions of the cationic complex 〚(COT)Nd(THF)4〛〚BPh4〛, 5, with these various substrates (NaCp*, KP*, NaOEt, NaStBu) gave high yields and were generally more selective than those involving the borohydride 4a. Such an observation on the superiority of cationic reagents has already been made in uranium chemistry 〚28–29〛. Complex 5 thus appears as a versatile precursor to organometallic derivatives. Hence, it seemed important to further develop cationic derivatives of f-element in general. Besides, cationic species are now well recognised as key intermediates in many catalytic and polymerisation processes (see for instance 〚30–33〛).
2.3 Neodymium and uranium cationic complexes
While organo-f-element cations have so far been most commonly formed by oxidation of divalent products, heterolytic rupture of a metal–halide or metal–carbon bond or protonolysis of a metal–carbon or metal–nitrogen bond 〚22–24,34–43〛, most uranium cations have been prepared following this latter approach 〚44–47〛. The inorganic 〚U(NEt2)3〛〚BPh4〛 and 〚U(NEt2)2(THF)3〛〚BPh4〛2 and the organometallic 〚(Cp*2)U(NMe2)(THF)〛〚BPh4〛 and 〚(COT)U(NEt2)(THF)2〛〚BPh4〛 cationic amide compounds were thus obtained by protonolysis of the U–NEt2 bond 〚44–47〛. As mentioned above, protonolysis of the metal–borohydride bound in (COT)Nd(BH4)(THF)2, 4a, by the ammonium salt NEt3HBPh4, afforded the unique example of a 〚(COT)Ln〛+ complex, 〚(COT)Nd(THF)4〛〚BPh4〛, 5. In order to generalise this method to the synthesis of 4f- and 5f-element cations, we similarly transformed uranium borohydride precursors by protonolytic cleavage of the U–BH4 bond.
Using (COT)U(BH4)2(THF) 〚48〛 as starting reagent, we successfully isolated, upon treatment with NEt3HBPh4 in THF, the cyclooctatetraenyl uranium cations 〚(COT)U(BH4)(THF)2〛〚BPh4〛, 13, and 〚(COT)U(HMPA)3〛〚BPh4〛2, 14 (HMPA = hexamethylphosphoramide) in 87% and 75% yield, respectively (Fig. 5) 〚49〛. The monoprotonolysis experiment of (COT)U(BH4)2(THF) to make the monocation 13 had to be performed at reflux temperature to speed up the reaction, which only reached completion after three weeks at ambient temperature. Yet, a default of the ammonium reactant had to be used to ensure that the protonolysis would not proceed any further towards the formation of the dication 14. Reaction times were lowered upon addition of a stronger Lewis base (L = OPPh3, OP(NMe2)3) to the reaction mixture and the compounds analogues to 13, 〚(COT)U(BH4)(L)3〛〚BPh4〛 (L = OPPh3, HMPA), were observed as monitored by NMR spectroscopy. However, the selectivity was greatly affected by this modification: an inseparable mixture of the monocation and dication species was commonly observed in NMR spectra. Thus, only the THF adduct 〚(COT)U(BH4)(THF)2〛〚BPh4〛, 13, could be isolated 〚49〛. The IR spectrum of 13 displayed a strong absorption band centred at 2462 cm–1, which is diagnostic of a bidentate BH4 ligand 〚1〛. Further reaction of the monocations 〚(COT)U(BH4)(L)x〛〚BPh4〛 ((L)x = (THF)2, (OPPh3)3, (HMPA)3), with an excess of NEt3HBPh4 in refluxing THF afforded the corresponding dications 〚(COT)U(L)x〛〚BPh4〛2. As mentioned for the preparation of the monocations 〚(COT)U(BH4)(L)x〛〚BPh4〛 ((L)x = (THF)2, (OPPh3)3, (HMPA)3), the formation of the dication was faster with HMPA than with OPPh3 or THF. For similar solubility properties, the dications could not be obtained pure from the corresponding monocations, themselves never fully consumed, except for the HMPA-solvated product 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, for this latter precipitates in THF. Complex 14 could also be directly and cleanly prepared by protonolysis of both U-BH4 bonds in the bisborohydride precursor (COT)U(BH4)2(THF) (Fig. 5) 〚49〛. The crystal structure of this dication 14 was successfully resolved, as illustrated in Fig. 6, and has already been discussed 〚49〛.
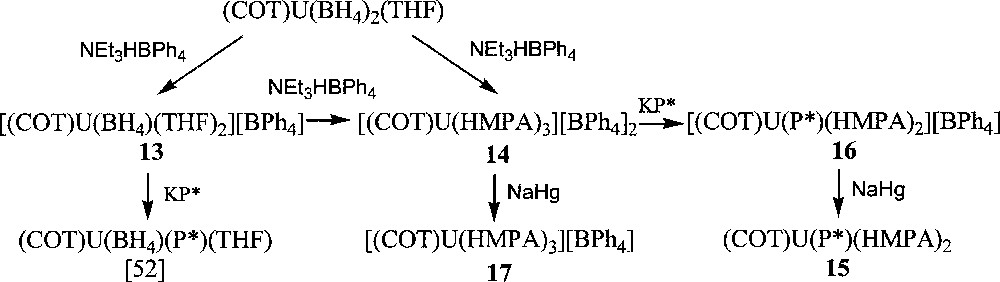
Syntheses of cyclooctatetraenyl uranium compounds.
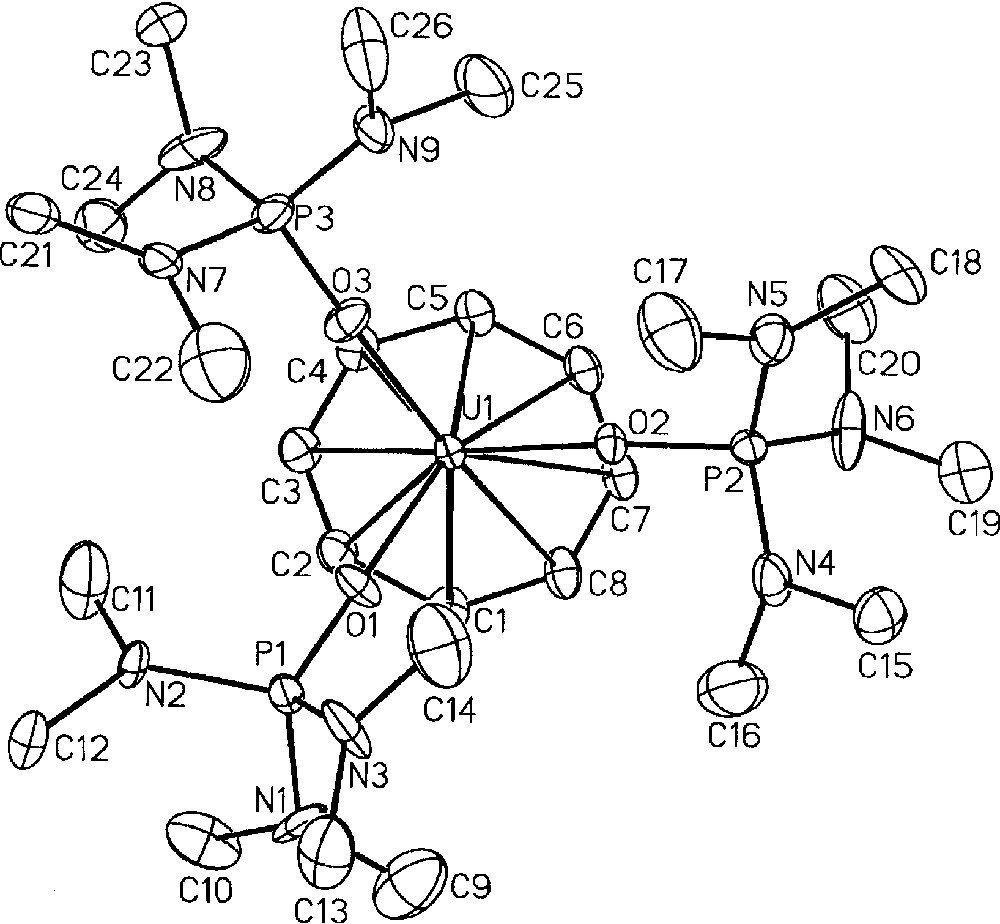
Molecular structure of one of the two independent dications 〚(COT)U(HMPA)3〛+2, 14, with thermal ellipsoids drawn at the 30% probability level. Reprinted with permission from Ref. 〚49〛. Copyright The American Chemical Society.
The mixed ring compound (COT)(Cp)U(BH4)(THF) 〚48〛 was also found to be easily and quantitatively transformed into the cation 〚(COT)(Cp)U(THF)2〛〚BPh4〛 (Cp = η-C5H5, 〚47〛) (equation (1)) 〚50〛. All these protonolysis reactions occurred with gas evolution,
presumably hydrogen, along with the formation of the borane BH3·NEt3. In contrast, protonolysis of the uranium–amide bond in (COT)U(NEt2)2(THF) by NEt3HBPh4 only occurred once, precluding the formation of the dication 〚(COT)U〛+2 in this manner, and further highlighting the great potential of borohydride precursors in the synthesis of organometallic complexes. Besides these thorough investigations on the preparation of the organometallic cations 〚(COT)Nd(THF)4〛〚BPh4〛, 5, 〚(COT)U(BH4)(THF)2〛〚BPh4〛, 13, and 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, by protonolysis of the metal–borohydride bond 〚11,19,49〛, the inorganic 〚U(BH4)2(THF)5〛〚(BPh4)〛 product was similarly prepared 〚51〛. Compounds 〚(COT)U(BH4)(THF)2〛〚BPh4〛, 13, and 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, represent the sole example of a cationic organometallic borohydride and the unique organometallic dication of an f-element, respectively. Both cations 13 and 14 are valuable reagents for obtaining new neodymium and uranium derivatives (see below) 〚25,49,52,53〛.
Still using the same ammonium salt NEt3HBPh4, the metal-P* and metal-PPh2 bonds were successfully cleaved in THF to make cationic derivatives 〚50〛. Thereby, 〚(COT)Nd(P*)(THF)〛, 7a, and its uranium analogue (COT)U(P*)(HMPA)2, 15, as well as the cationic congener 〚(COT)U(P*)(HMPA)2〛〚BPh4〛, 16, (see thereafter) easily gave, at room temperature, the corresponding cation, 〚(COT)Nd(THF)4〛〚BPh4〛, 5, and 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, respectively, as observed from NMR analyses (equations (2)–(4)). In the case of the neutral phospholyl precursor 15, the monocation 16, initially formed,
further reacted with the acidic reactant to finally give the dication 14. Protonolysis of the mixed borohydride-phospholyl complex (COT)U(BH4)(P*)(THF) (equation (5)) 〚52〛, showed the uranium–phospholyl bond to be protonated more easily than the uranium-borohydride one,
as the cationic borohydride complex 13 was formed selectively 〚50〛. This trend should certainly be further confirmed on more complexes before any generalisation is driven. Finally, the uranium–phosphide bonds in complexes (Cp*)2U(PPh2)2 and U(NEt2)3(PPh2) 〚54〛 were also successfully protonated by NEt3HBPh4 to give the known compounds 〚(Cp*)2U(THF)2〛〚BPh4〛 〚55〛 and 〚U(NEt2)3〛〚BPh4〛 〚44〛, as monitored by NMR spectroscopy (equations (6) and (7)) 〚50〛. Formation of 〚(Cp*)2U(THF)2〛+ proceeds through the initial
uranium(IV) phosphido cation 〚(Cp*)2U(PPh2)(THF)x〛〚BPh4〛, which undergoes a reductive elimination of Ph2P–PPh2. Protonolysis of f element–borohydride–phospholyl or –phosphide bond by NEt3HBPh4 has been shown to allow the access to a wide variety of cationic species, as illustrated with neodymium and uranium complexes in Figs 1 and 5 and equations (1)–(7).
2.4 Reactivity of the uranium dicationic complex
Besides checking the effectiveness of the protonolysis reaction of the metal–borohydride in making cations, the U(IV) dication, 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, was also prepared with the hope to access the previously never observed (cyclooctatetraenyl)U(III) derivatives. Reduction of the orange dication 14, by sodium amalgam in THF smoothly afforded at 20 °C the monocation 〚(COT)U(HMPA)3〛〚BPh4〛, 17, as a green product in 88% yield 〚53〛. This is the first cationic cyclooctatetraenyl trivalent uranium complex, the only other two 〚(COT)U(III)〛 derivatives being either anionic 〚K(diglyme)〛〚U(η-C8H7Me)2〛 〚56〛, or neutral (COT)U(Cp*)(L) (L = HMPA, THF, 4,4’-dimethyl-2,2’-bipyridine) 〚25,57〛. To date, the other cationic organometallic uranium(III) examples are limited to 〚(Cp*)2U(THF)2〛〚BPh4〛 〚55〛, 〚U(η-2,4-Me2-C5H3)2〛〚BPh4〛 〚58〛 and the trinuclear 〚U3(η-C6Me6)3(AlCl4)3Cl5〛〚AlCl4〛 〚59〛. The crystal structure of the monocation 17 is presented in Fig. 7 (vide infra) 〚53〛. Addition in THF of KP* to the dicationic uranium compound 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, readily allowed the preparation of the sole cationic phospholyl uranium species 〚(COT)U(P*)(HMPA)2〛〚BPh4〛, 16, in 81% yield (Fig. 5) 〚52〛. The similar reduction of 16 by Na/Hg amalgam in THF gave the first example of cyclooctatetraenyl uranium(III) phospholyl complex (COT)U(P*)(HMPA)2, 15, in 57 % yield 〚25,52〛.
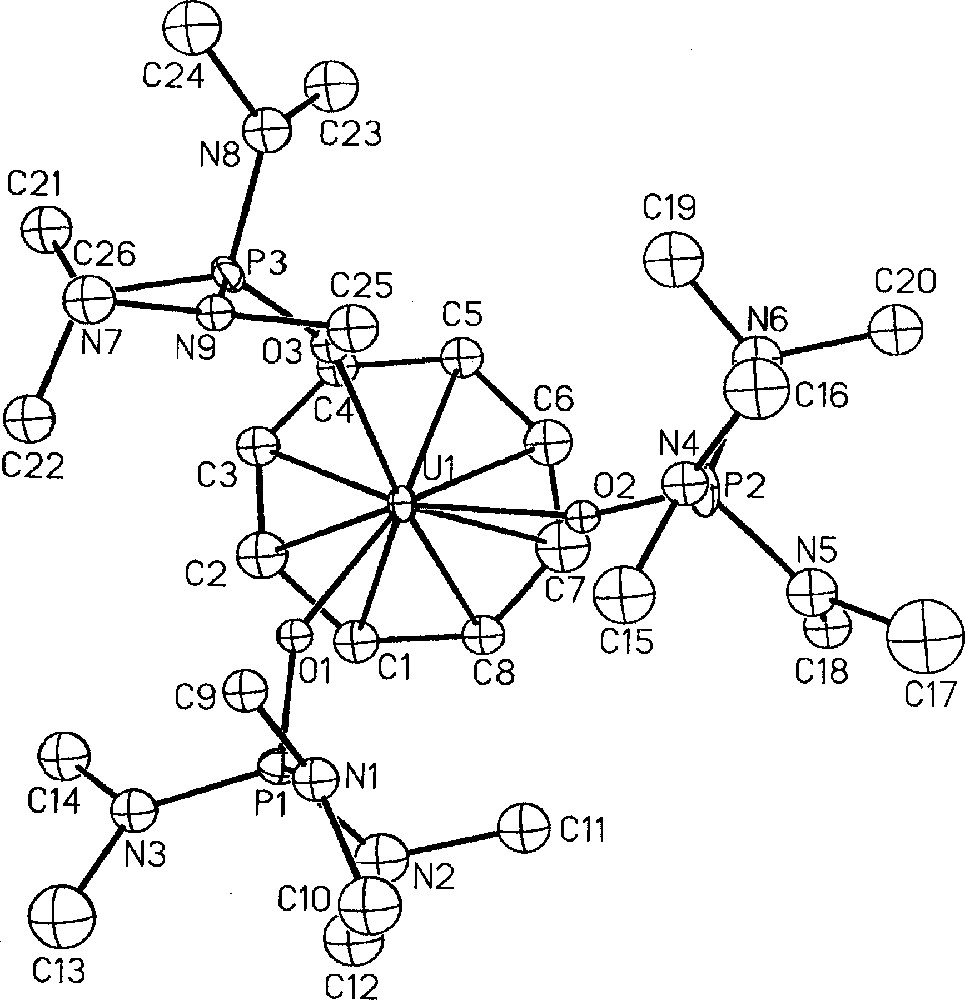
Molecular structure of one of the two independent cations 〚(COT)U(HMPA)3〛+, 17, with thermal ellipsoids drawn at the 30% probability level. Reprinted with permission from Ref. 〚53〛. Copyright Wiley-VCH.
2.5 Comparison of neodymium and uranium complexes
In an effort to compare structural and chemical properties of homologous 4f and 5f compounds, we prepared additional products to complete the series of 〚(COT)M(HMPA)3〛+n cations (n = 1, M = Nd, 18, U, 17; n = 2, M = U, 14) 〚49, 53〛. The neodymium compound 18 was synthesised by substitution of the THF ligands in 〚(COT)Nd(THF)4〛〚BPh4〛 〚11,19〛 and crystallised as a THF solvate 18·THF 〚53〛. All three complexes 14, 17, 18·THF have been characterized by their crystal structures, which are very similar, as illustrated in Figs 6, 7 and 8, respectively. This allowed the direct comparison of the Nd(III) and U(III) compounds as well as that of U(III) and U(IV) derivatives 〚49, 53〛. The most significant averaged parameters are reported in Table 1. All three cations adopt a three-legged piano-stool configuration, in which the angles do not greatly differ from one structure to another, with an average O–M–O angle of 85° and COT–M–O angle of 128° (COT = centroid of the C8H8 ring). The average metal–COT, metal–carbon and metal–oxygen bond distances are all shorter for Nd(III) compared to U(III) as well as for the U(IV) and U(III) couple, respectively. Comparison of the structures of the U(IV), 14, and U(III), 17, cations, whose metals display a 0.14 Å difference in their ionic radius, shows a significant difference in the bond lengths, from 0.1 to 0.2 Å. Note that this same difference between the Nd(III), 18·THF, and the U(III), 17, complexes is of only 0.05 Å with a similar value for the difference in the ionic radii (0.04 Å). These trends follow the variation in the ionic radii of the metals Nd(III) (0.983 Å), U(III) (1.025 Å) and U(IV) (0.89 Å) 〚10〛. Such comparative studies have previously been performed on a limited number of isostructural derivatives 〚60–63〛. A better assessment of the differences in the nature of the chemical bonds in lanthanide and actinide compounds will require much more results. Also, a comparison of 〚(COT)M(Cp*)(HMPA)〛 and 〚(COT)M(P*)(HMPA)〛 complexes (M = Nd, U) will be presented in a forthcoming paper 〚25〛.
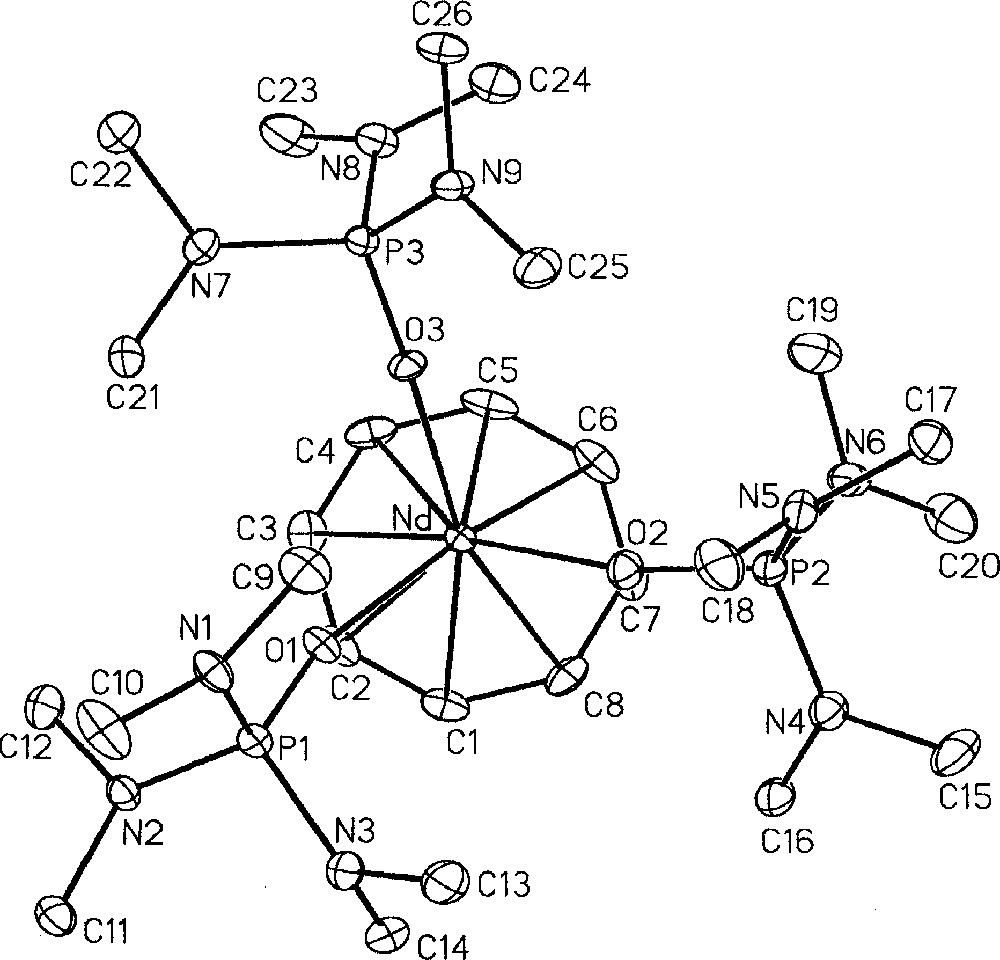
Molecular structure of 〚(COT)Nd(HMPA)3〛+, 18·THF, with thermal ellipsoids drawn at the 30% probability level 〚53〛.
Geometrical parameters of the cations 〚(COT)Nd(HMPA)3〛+, 18, 〚(COT)U(HMPA)3〛+, 17, and 〚(COT)U(HMPA)3〛+2, 14 (COT = centroid of the C8H8 ring; M–C = metal–C8H8 ring carbon distance).
| 〚(COT)Nd(HMPA)3〛+ | 〚(COT)U(HMPA)3〛+ | 〚(COT)U(HMPA)3〛+2 | |
| M–COT (Å) | 1.98(1) | 2.00(1) | 1.92(2) |
| <M–C> (Å) | 2.70(2) | 2.71(4) | 2.65(2) |
| <M–O> (Å) | 2.36(3) | 2.41(4) | 2.22(1) |
| <O–M–O> (°) | 85(1) | 85(2) | 87(1) |
| <COT–M–O> (°) | 129(3) | 129(3) | 127(1) |
3 Conclusion
The borohydride ligand has been very valuable in developing organometallic complexes of neodymium(III). Substitution of the BH4 ligand with alkali metal salts of various anionic reagents allowed the isolation of several pentamethylcyclopentadienyl, tetramethylphospholyl and cyclooctatetraenyl products, 2–12. Both (COT)Nd(BH4)(THF)2, 4a and 〚(COT)Nd(THF)4〛〚BPh4〛, 5, were largely employed to extend the chemistry of mono(COT) neodymium compounds. The main advantage in using borohydride reagents was revealed by their protonolysis, which allowed access to highly valuable cationic species, such as the borohydride 〚(COT)U(BH4)(THF)2〛〚BPh4〛, 13, the dication 〚(COT)U(HMPA)3〛〚BPh4〛2, 14, and trivalent uranium complex 〚(COT)U(HMPA)3〛〚BPh4〛, 17. This new method for preparing cations by protonolysis of a metal–BH4 bond by the ammonium salt NEt3HBPh4, was further extended to the protonolysis of metal–P* and metal–PPh2 bonds. The first cyclooctatetraenyl–uranium (III) derivatives, 〚(COT)U(HMPA)3〛〚BPh4〛, 17, and (COT)U(P*)(HMPA)2, 15, were formed by reduction of parent tetravalent uranium precursors 14 and 〚(COT)U(P*)(HMPA)2〛〚BPh4〛, 16. All the new products 2–18 were fully characterised, especially by IR, 1H and 31P NMR, elemental analyses and also by determination of the X-ray structure in many cases (3b, 4a, 5, 6, 8, 12, 14, 17, 18·THF 〚11,15,19,25,49,53〛). Finally, homologous organometallic complexes (14, 17, 18) allowed the direct evaluation of chemical and structural differences between a 4f and a 5f element, an ongoing research 〚25〛.