

1 Introduction
This paper summarises the results of a collaboration between the groups led by Osborn in Strasbourg, and Ivin and Rooney in Belfast, contained in nine original papers 〚1–9〛 and four conference papers 〚10–13〛, published between 1985 and 1992.
In 1970 Hérisson and Chauvin 〚14〛 had put forward the hypothesis that the ROMP of cycloalkenes was initiated by the addition of monomer to a metal–carbene complex that had been produced in some way from the catalyst/monomer system, the reaction then being propagated by addition of further monomer to the resulting metal–carbene complex. This hypothesis accounted for various observations and was later supported by the fact that a number of stable metal–carbene complexes, such as those prepared by Fischer 〚15〛 or Casey 〚16〛, could act as initiators of ROMP when activated by heat 〚17–21〛, light 〚22,23〛, or the presence of a cocatalyst 〚24–26〛. From 1974 〚27〛, Schrock developed a new family of high-oxidation state metal–carbene complexes. In 1980, he described well-defined tungsten–carbene complexes that were active on their own, and found that the propagating tungsten–carbene complexes in the metathesis of acyclic alkenes could be observed directly by 1H NMR spectroscopy 〚28〛. In 1982–1983, Osborn reported similar results involving highly active catalysts 〚29,30〛. This caused Ivin and Rooney, who had been working on the ROMP of cycloalkenes and bicycloalkenes catalysed by WCl6, MoCl5, RuCl3, OsCl3, IrCl3 etc. 〚31〛, to wonder whether, using these complexes as initiators, one would be able to observe the propagating species in the ROMP of these substrates. The collaboration was initiated at the 5th International Symposium on Metathesis (ISOM 5) held in Graz in 1983, and continued during several years.
2 Detection of propagating metal–carbene complexes in ROMP
Qualitative experiments were first carried out using a solution of the Lewis acid adduct W(=CHBut)(OCH2But)2Br2·GaBr3 as initiator in C6D5Br, contained in an NMR tube 〚1〛. A ten-fold excess of monomer was added to the initiator solution at 240 K, the tube briefly warmed to room temperature to ensure complete polymerisation, and then re-cooled to 240 K to record the 1H NMR spectrum. The monomers used were norbornene (1) and a number of its derivatives: endo-5-methyl- (2), 5,5-dimethyl- (3), 1-methyl- (4), a mixture of syn-7-methyl- (5) and anti-7-methyl- (6), exo,exo-5,6-dimethyl- (7), and endo,endo-5,6-dimethyl- (8). In all cases the NMR spectrum of the resulting solution showed in particular that the carbene proton signal of the initiator (singlet, δ 11.57) had been partially replaced by that of stable tungsten–carbene products: one doublet (J = 10 Hz) was observed for each one of the products from 1, 7, and 8, two doublets each from the head and tail products of 2 and 3, two doublets for the products derived from a mixture of 5 and 6, and a singlet and doublet from the head and tail structures produced respectively by the two modes of addition of 4 (Fig. 1). For the unsymmetrical monomers (2, 3 and 4), the head product, with the methyl substituent(s) closer to the metal centre than in the tail product, was always present in greater proportion, indicating that its reactivity towards addition of further monomer was lower than for the tail product.
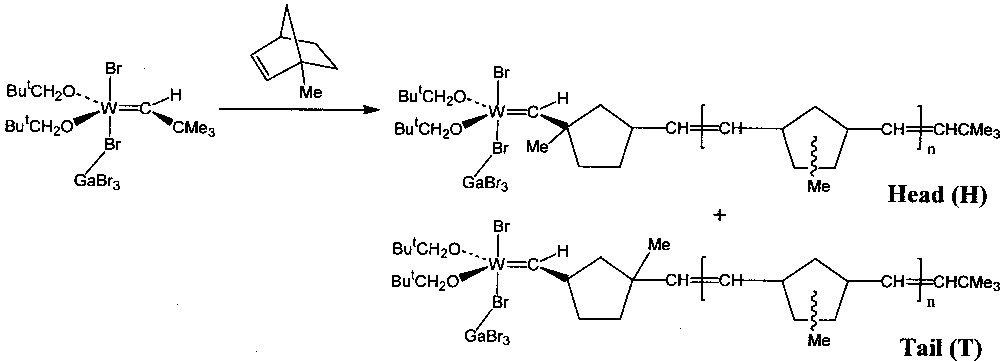
Propagating tungsten–carbene complexes obtained from 4.
The stability of the product metal–carbene complexes indicated that they could be regarded as ‘living’ propagating species and that these systems could be used to make block copolymers by successive additions of different monomers, provided that the initiation process and cross-propagation processes were reasonably efficient. Successive additions of 1, 2, and 1 to the initiator did indeed confirm that the propagating species could be interconverted, though not with 100% efficiency 〚1〛. Experiments using the more efficient, but less active, initiator W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2 showed that within certain limitations AB and ABA type block copolymers could be prepared in this way 〚5〛. Schrock 〚32〛 described the preparation of other block copolymers by this method using molybdenum–carbene complex catalysts, and many other examples have since been reported 〚33〛.
3 Detection of intermediate tungstacyclobutane complexes
In the early experiments described above, W(=CHBut)(OCH2But)2Br2·GaBr3 was never completely consumed and it was thought that this may have been due, at least in part, to the combined effect of rapid polymerisation and relatively inefficient mixing of monomer and initiator. It was therefore decided to switch to CD2Cl2 as solvent to enable mixing to be carried out at lower temperature (195 K) where the polymerisation would be much slower. To our great surprise, a much more complicated 1H NMR spectrum was observed in the initial stages of reaction at the lower temperature, but on warming to room temperature this simplified to that of the propagating metal–carbene complex 〚2〛. However, there was still residual initiator, even when the molar ratio of monomer:initiator was 50, indicating that the rate constant for propagation (kp) was greater than for initiation (ki). Quantitative work using various monomer/initiator systems has since shown that kp/ki values can range from 0.26 to more than 200 〚34〛.
Detailed analysis of the NMR spectrum of the reaction mixtures at low temperature showed that the additional fine structures were in fact due to the presence of the intermediate tungstacyclobutane complexes, formed by 〚2 + 2〛 addition of the metal–carbene complexes to norbornene, one of the two central steps of the catalytic mechanism (Figs. 2 and 3). These intermediates are best described as ion pairs resulting from transfer of a Br– ligand from tungsten to gallium to form a cationic complex and the GaBr4– counter-ion. Previously, it had been uncertain whether these were true intermediates or whether they should be regarded as the transition state complexes. Around the same time Grubbs 〚35,36〛 and Schrock 〚37〛 found that stable titanacyclobutane and tantalacyclobutane complexes respectively would initiate the ROMP of norbornene at 60 °C, but that in these systems the intermediate metal–carbene complexes could not be detected.
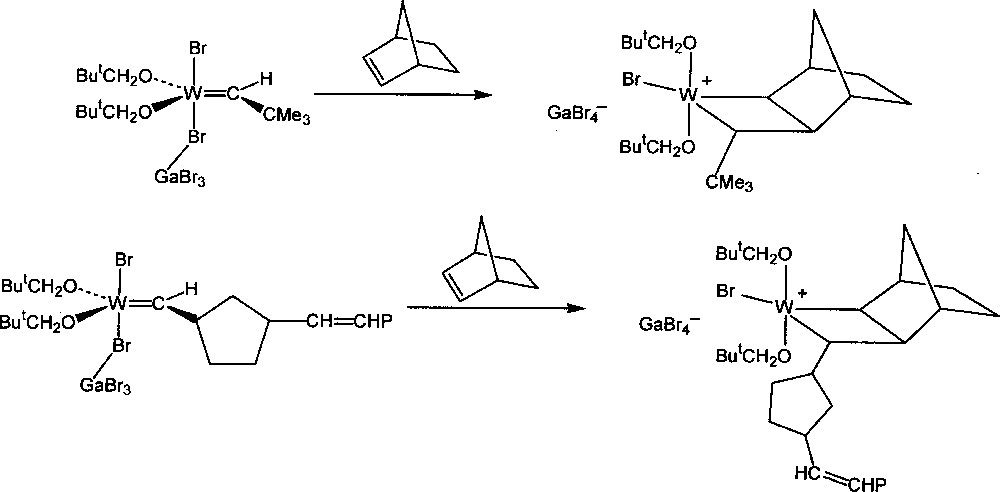
Formation of the initial and propagating tungstacyclobutane intermediates.
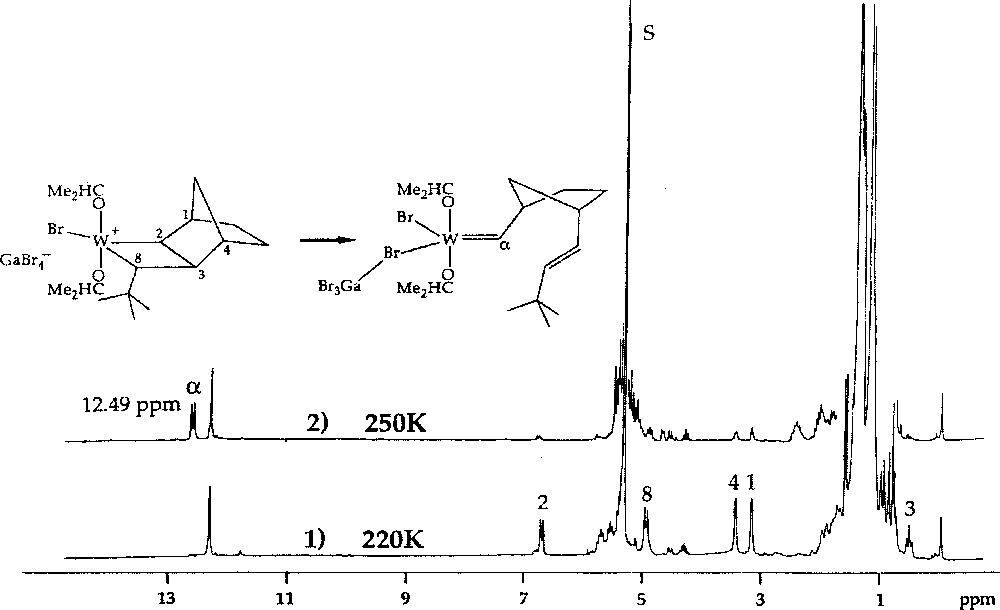
1H NMR spectra of the reaction of W(=CHBut)(OCHMe2)2Br2·GaBr3 with norbornene (2 equiv) in CD2Cl2, showing (1) conversion at 220 K of ca 55% of the initial neopentylidene complex into the initial tungstacyclobutane complex, and (2) conversion at 250 K of the initial tungstacyclobutane complex into the first-insertion tungsten–carbene complex.
Further studies were made of the intermediate tungstacyclobutanes obtained by reaction of various of the monomers 1-8 with W(=CHR)(OCH2But)2X2·GaX3 (R = But, Bui, etc.; X = Br, Cl), W(=CR’R”)(OCH2But)2Br2·GaBr3 (R’,R” ≠ H) or W(=CHR)(OR’)2Br2·GaBr3 (R’ = Pri, etc.) 〚4,7〛. Only transoid tungstacyclobutanes (Fig. 2) were detected as intermediates although the polymer formed generally contains both cis and trans double bonds. It may be concluded that the intermediate cisoid tungstacyclobutanes have a shorter life than the transoid tungstacyclobutanes at a given temperature, their stationary concentration being too small to be observed. This instability may be attributed to the repulsion between the cis substituents in the cisoid intermediate 〚4〛, in agreement with the non-observation of the tungstacyclobutanes derived from the carbene complexes containing the (=CR’R”) function.
The stability of the resulting transoid tungstacyclobutanes was found to depend on the norbornene and carbene substituents, as well as on the nature of ligands X and OR’. The intermediate formed by the reaction of W(=CHBut)(OCH2But)2Br2·GaBr3 with endo,endo-5,6-dimethylnorbornene 8 (3 equiv) in CD2Cl2 at 235 K was unusually stable. As much as 70% of the initiator could be converted into the first propagating transoid tungstacyclobutane. Its rearrangement to the corresponding metal–carbene product (Fig. 4), which corresponds to the second central step of the catalytic mechanism, could be measured at 245, 250, and 255 K, giving half-lives of 43, 18, and 10.5 min respectively (activation energy 72 kJ mol–1). With unsymmetrical monomers such as endo-5-methylnorbornene 2, two initial transoid tungstacyclobutanes are formed. The methyl group can have two possible positions: either adjacent to (tail form) or away from (head form) the bond that becomes a trans double bond on rearrangement. The two forms show quite different stabilities, that of the tail form being similar to that formed from 8, while the less stable head form has a half-life similar to that formed from norbornene. This effect is specific and is not induced by methyl groups in other positions 〚4〛.
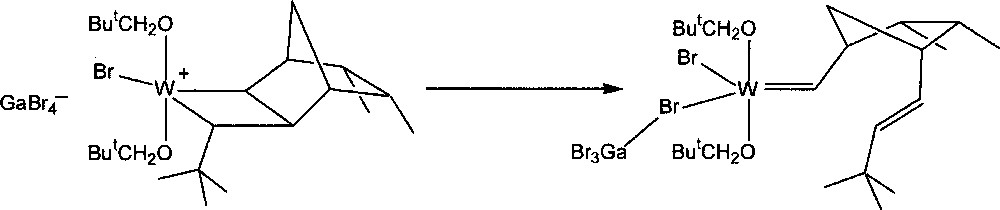
Conversion of a tungstacyclobutane intermediate into a tungsten–carbene complex.
If the initiator contains a chiral centre at the β position of the carbene ligand, as for example (=CHC*HMeEt), the initial transoid tungstacyclobutane formed from norbornene consists of two diastereoisomers, giving rise to separate H-2 doublets (J = 9 Hz) at δ 7.06 and 6.80 and showing different stabilities 〚7〛. For the propagating tungstacyclobutanes containing two or more monomer units, that are similarly derived from a carbene with a chiral β carbon (Fig. 2), more than two diastereoisomers can be distinguished, the chemical shift being additionally sensitive to more remote structural differences (cis/trans, head/tail or m/r). This is particularly the case for endo-5-methylnorbornene or dicyclopentadiene, for which eight distinct structures can be observed (Figs. 5 and 6) 〚7〛.
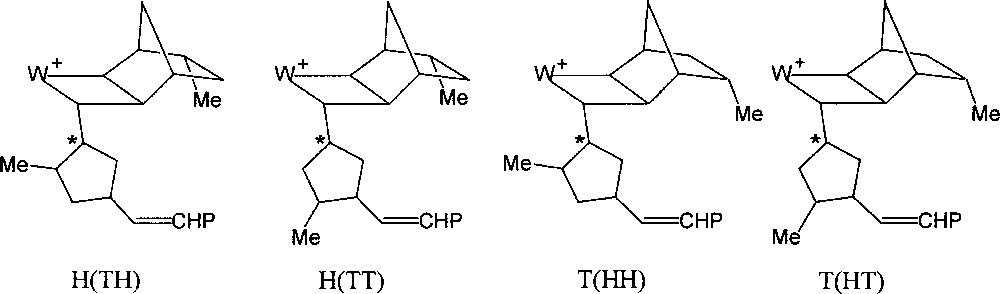
The eight propagating tungstacyclobutanes expected from 2 (each regioisomer exists as a diastereomeric pair).
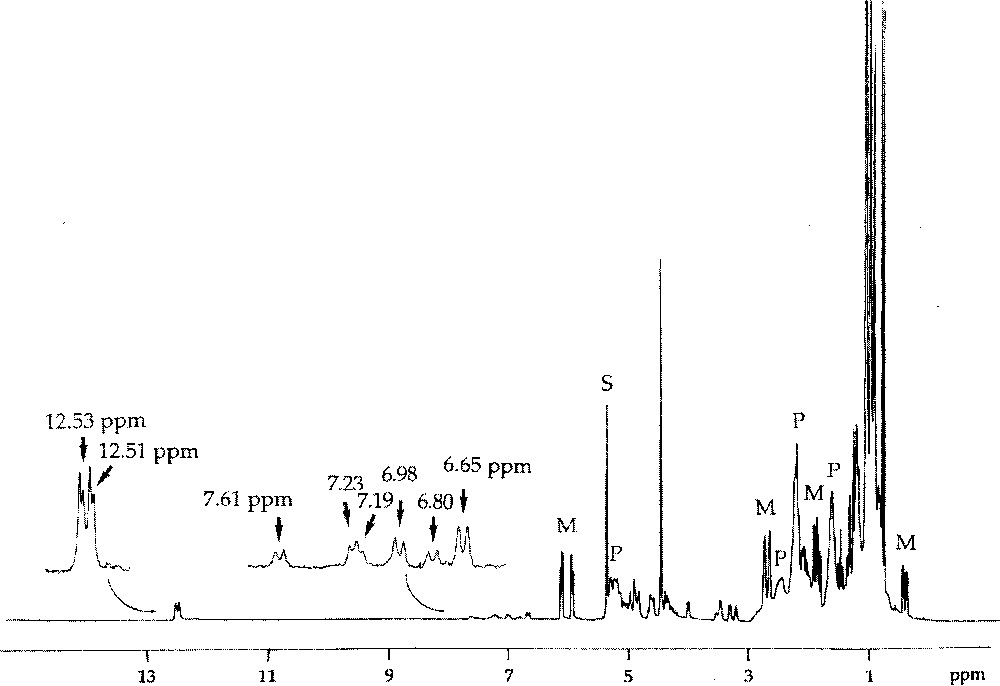
1H NMR spectrum of the reaction mixture obtained after addition of 5-endo-Me-NBE (4 equiv, M) to W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2·GaBr3 in CD2Cl2 at 235 K, showing formation of the propagating tungsten–carbene complexes (characterised by the doublets at δ 12–13 ppm), of six propagating tungstacyclobutane intermediates (characterised by the doublets at δ 6–8 ppm), and of the polymer chains (P).
4 ROMP of pairs of isomeric monomers
4.1 Exo and endo adducts of cyclopentadiene and maleic anhydride
The ROMP of the exo adduct proceeds at measurable speed at 25 °C in CD2Cl2 when initiated by the tungsten cyclopentylidene complex W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2 in the absence of GaBr3 〚3〛. The propagating carbene complex shows a carbene proton doublet (J = 10 Hz) at δ 11.245. The initiator (I) is totally consumed in the early stages of reaction and the remaining monomer (M) then disappears according to a first-order law with a half-life of 74 min when the concentration of propagating species is 5.0 × 10–2 mol dm–3, whence kp = 3.15 × 10–3 dm3 mol–1 s–1. Furthermore, when the initial monomer/initiator ratio is not too high, the OCH2CMe3 proton signal of the initial propagating species (P1) can be readily distinguished from that of the subsequent propagating species (Pn). The concentration of P1 rises to a maximum and then falls away as it is converted into P2. At the maximum, ki〚I〛〚M〛 = kp〚P1〛〚M〛, and so from the value of 〚I〛/〚P1〛 at the maximum one can obtain kp/ki = 0.26. This low value indicates that the cyclopentylidene ligand in the initiator offers less resistance to approach of the monomer than does the more bulky carbene ligand in the propagating species.
It was also found that after the exo monomer had disappeared a second carbene proton doublet (J = 10 Hz) began to appear slowly at δ 11.59. This was due to the very slow addition of the much less reactive endo monomer, present as an impurity in the exo monomer. On adding more endo monomer to the reacting system, the doublet at δ 11.59 became stronger, eventually overtaking that at δ 11.245. The resulting product must therefore be an AB-type block copolymer containing short lengths of units derived from the endo monomer at the end of the chains of units derived from the exo monomer. The ROMP of the endo monomer had not been previously observed.
4.2 Syn-7-methylnorbornene/anti-7-methylnorbornene
Exposure of a 1:1 mixture of syn- and anti-7-methylnorbornene to W(=CHBut)(OCH2But)2Br2 (in the absence of GaBr3) in CD2Cl2 resulted in the selective ROMP of the anti isomer, allowing the recovery of the residual syn isomer, which could then be polymerised independently for the first time using the more active W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2·GaBr3 as initiator, giving a 90% trans polymer 〚8〛. A near-perfect AB-type block copolymer of the anti and syn isomers can be made by first adding W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2 to a 1:1 mixture of monomers, resulting in selective polymerisation of the anti monomer, and then adding an equivalent of GaBr3 to enhance the reactivity towards the syn isomer. If instead one initiates the ROMP of the 1:1 mixture with W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2·GaBr3, the anti monomer is consumed more rapidly than the syn isomer at the beginning of the reaction, but polymerisation of the syn isomer becomes significant before all the anti isomer has been used up, and the result is a tapered block copolymer. At low temperature the intermediate transoid tungstacyclobutane can be detected for the adduct of the anti monomer, but not for the adduct of the syn monomer.
5 The ROMP of 5-methoxycarbonyl and 5,6-bis(methoxycarbonyl) derivatives of norbornene
The ROMP of the two 5-methoxycarbonyl derivatives (exo- and endo-) and the three 5,6-bis(methoxycarbonyl) derivatives (exo,exo-, endo,exo-, and endo,endo-) were studied using W〚=C(CH2)3CH2〛(OCH2CMe3)2Br2 as initiator in CD2Cl2 at 290–320 K 〚9〛. In the 1H NMR spectrum of the reaction mixture the initial tungsten–carbene adduct is usually distinguishable from the subsequent adducts. The initiation rate constant is generally at least as great as the propagation rate constant. The latter is two to four times higher for the monosubstituted monomers than for the disubstituted monomers.
6 Conclusions
We have reviewed how, starting from the early detection of the propagating tungsten–carbene complexes in catalytic ROMP reactions of norbornenes, our NMR studies could lead to the detailed characterisation of the tungstacyclobutane intermediates, and the direct observation and study, for the first time, of the interconversion of the two types of species in a catalytic system. Advantage could be taken of such intimate mechanistic information to make a variety of block copolymers. The high activity of the catalysts allowed us furthermore to carry out polymerisation reactions that had not been reported previously.
Acknowledgements
KJI thanks John Osborn for invitation to continue the collaboration upon his retirement in 1986. Funds were provided by the SERC and the Leverhulme Trust (UK), and CNRS and the Ministry of Education (France).