

1 Introduction
La nature et la position des groupements substitués à la périphérie du macrocycle porphyrine déterminent souvent une réactivité physico-chimique spécifique pour ces systèmes, en particulier leurs propriétés rédox. Les différentes fonctionnalisations du ligand porphyrine sont également à la base d’architectures moléculaires développées à partir de ces chromophores 〚1–4〛 et dans lesquelles les substituants vont servir de ponts entre les différentes unités. De ces liaisons vont dépendre en général les possibilités de transfert d’énergie et/ou de transfert de charges intramoléculaires 〚5,6〛. Il apparaît donc important de connaître les effets éventuels que ces substituants peuvent induire sur le comportement rédox des porphyrines qu’ils pontent. En général, les substituants périphériques, bien que pouvant profondément modifier la réactivité électrochimique spécifique à la porphyrine, ne sont pas eux-mêmes électroactifs et ne participent pas aux différents transferts de charges observés. Ces systèmes ont été abondamment étudiés, en particulier en ce qui concerne les relations entre les valeurs des potentiels rédox des porphyrines et les valeurs des paramètres de Hammett (effets électrodonneurs ou électroattracteurs) des substituants situés à la périphérie du macrocycle 〚7–10〛. En revanche, les systèmes porphyriniques portant des substituants périphériques électroactifs n’ont fait l’objet que de peu d’études, bien que pouvant présenter des possibilités intéressantes de réaction, comme dans le cas des électropolymérisations 〚11,12〛.
Nous nous proposons de présenter le comportement électrochimique en réduction de méso-tétraphénylporphyrines, base libre et complexes métalliques correspondants, substituées en position β par un groupement pyridinium (noté py+) (Fig. 1). Ce groupement, ainsi que le groupement bipyridinium (noté bipy+), de réactivité voisine, sont connus pour être électroréductibles aussi bien en milieu aqueux qu’en milieu organique 〚13–19〛. Les méso-tétraphénylporphyrines de référence de cette étude, notées MTPP (avec M = H2, Cu, Zn), ont un comportement électrochimique bien connu. Dans ces chromophores, où le métal est inerte électrochimiquement, le ligand est réductible en quatre étapes distinctes, dont les deux premières correspondent à des transferts de charge réversibles et monoélectroniques ; les deux étapes suivantes, irréversibles, se situent à des potentiels inférieurs à –2 V/ECS et traduisent des mécanismes rédox plus complexes 〚20–22〛.

Porphyrines étudiées MTPP-β-py+ (M = H2, Cu, Zn).
L’association de plusieurs sites électroactifs différents au sein d’une même molécule a déjà montré qu’elle pouvait engendrer des modifications notables des propriétés rédox spécifiques à chaque site 〚19,23,24〛. Nous avons abordé l’étude de la réduction électrochimique des porphyrines substituées en position β (notées MTPP-β-py+) en vue de déterminer de quelle manière les propriétés rédox des différents sites électroactifs pouvaient être affectées, et d’évaluer, en particulier, le degré d’interdépendance des deux systèmes rédox en présence, porphyrine et cation pyridinium.
2 Partie expérimentale
2.1 Réactifs
La méso-tétraphénylporphyrine base libre H2TPP a été synthétisée d’après la méthode de Adler 〚25〛 et les analogues ZnTPP et CuTPP ont été métallés d’après la méthode de Barnett 〚26〛. La méso-tétraphénylporphyrine base libre substituée en position β par un groupement pyridinium H2TPP-β-py+ a été synthétisée par voie électrochimique selon la méthode mise au point au laboratoire 〚27〛. Les analogues ZnTPP-β-py+ et CuTPP-β-py+ ont ensuite été préparés en métallant H2TPP-β-py+ selon les méthodes décrites ci-après.
2.1.1 ZnTPP-β-py+
H2TPP-β-py+ est dissous dans un mélange CH2Cl2/CH3OH (1:1) avec un excès d’acétate de zinc (Zn(CH3COO)2·2 H2O) (10 équiv). Le mélange, maintenu sous reflux pendant 3 h, est traité avec un excès de tétraéthylammonium hexafluorophosphate (TEAPF6) (100 équiv) après retour à température ambiante. Après évaporation du solvant, le résidu est dissout dans un minimum de CH2Cl2 et lavé à l’eau trois fois. La phase organique est ensuite concentrée, puis chromatographiée sur colonne de silice (éluant : CH2Cl2/CH3OH, 98 :2) pour donner la métalloporphyrine désirée. Après séchage, ZnTPP-β-py+ est obtenu sous la forme d’une poudre verte, avec un rendement de 85 %.
UV–vis (CH2Cl2) λmax(nm) (ϵ, M–1cm–1) : 596 (9834), 556 (13863), 520 ep. (4621), 423 (229265).
RMN 1H (300 MHz, (CD3)2CO) δ (ppm) : 9,51 (d, 2H, J = 5,5 Hz, 3–5 py+) ; 9,43 (s, 1H, β-H adjacent à py+) ; 8,91–8,75 (m, 6H, β-H et 4 py+) ; 8,64 (d, 1H, J = 4,7 Hz, β-H) ; 8,29-8,13 (m, 8H, o-H) ; 8,05 (d, 2H, J = 5 Hz, 2–6 py+) ; 7,88–7,79 (m, 9H, m- et p-H) ; 7,53–7,40 (m, 3H, m- et p-H du phényle voisin de py+). FAB-SM, matrice NBA m+/z = 754,1 : (C49H31N5Zn)+. Anal. calc. pour ZnC49H32N5PF6 : C 65,31 ; H 3,58 ; N 7,77 ; tr. : C 65,24 ; H 3,58 ; N 7,76 %.
2.1.2 CuTPP-β-py+
La même procédure appliquée à H2TPP-β-py+ en présence d’un excès d’acétate de cuivre (Cu(CH3COO)2·2 H2O) (10 équiv) conduit à l’obtention de CuTPP-β-py+ sous la forme d’une poudre rouge, avec un rendement de 90 %.
UV–vis (CH2Cl2) λmax(nm) (ϵ, M–1cm–1) : 585 (12053), 546 (16751), 515 ep. (7150), 418 (221450). FAB-SM, matrice NBA m+/z = 753,1 : (C49H31N5Cu)+. Anal. calc. pour CuC49H32N5PF6 : C 65,44 ; H 3,59 ; N 7,79 ; tr. : C 65,37 ; H 3,59 ; N 7,78 %.
Le solvant d’étude (DMF, Fluka, ultra-pur) ainsi que l’électrolyte support (tétrabutylammonium hexafluorophosphate TBAPF6, Fluka) ont été utilisés sans purification préalable. La solution de travail a été dégazée avec de l’argon qualité U et maintenue sous atmosphère inerte pendant toute la durée des expériences.
2.2 Appareillage
Les voltampérométries cycliques et stationnaires ont été réalisées avec un potentiostat DEA 101 muni d’une interface IMT 102 (Radiometer) dans une cellule électrochimique comportant un montage standard à trois électrodes. L’électrode de travail était une électrode tournante avec un disque de platine de surface 3,14 mm2. L’électrode de référence était une électrode au calomel saturée (ECS), munie d’un pont de jonction rempli de l’électrolyte et du solvant correspondants. L’électrode auxiliaire était un fil de platine. Les voltamogrammes ont été traités grâce au logiciel Volta Master 3® (Radiometer).
Les coulométries exhaustives ont été réalisées avec un potentiostat PJT 120-1 (Tacussel) et le courant mesuré par un enregistreur L250E (Linseis). Le compartiment anodique était séparé du compartiment cathodique par un disque de verre fritté (porosité 4) et seul le compartiment cathodique était maintenu sous atmosphère inerte. L’électrode de travail était une spirale de platine. L’électrode de référence était une électrode au calomel saturée (ECS) munie d’un pont de jonction rempli de l’électrolyte et du solvant correspondants. L’électrode auxiliaire était constituée d’une spirale de platine de grande surface.
Les spectres d’absorption UV–visible ont été enregistrés avec un spectrophotomètre Shimazu UV-260.
Les spectres RMN 1H ont été enregistrés sur un appareil Brucker AC 300 (300 MHz).
Les spectres de masse (FAB–SM) ont été obtenus sur un spectromètre de masse ZAB-HF utilisant une matrice NBA (3-nitrobenzyl alcool, 153 g mol–1).
Les analyses élémentaires ont été effectuées par le service de microanalyse du département de chimie de l’IUT Strasbourg-Sud.
3 Résultats et discussion
Sur l’intervalle de potentiels étudié (0 à –2 V/ECS), la réduction du ligand des porphyrines MTPP (M = H2, Cu, Zn) a lieu en deux étapes réversibles monoélectroniques et génère respectivement les radicaux anions et les dianions. Les résultats obtenus (Tableau 1) sont en accord avec les données de la littérature 〚8–10,20–22〛. Dans les mêmes conditions expérimentales, les porphyrines bêta-substituées correspondantes, MTPP-β-py+, présentent une étape de réduction supplémentaire (Fig. 2) à des potentiels compris entre –0,79 et –1,01 V/ECS, les étapes de réduction suivantes se situant à des potentiels voisins de ceux mesurés pour les analogues non-substitués (Tableau 1).
Potentiels de réduction en V/ECS obtenus en milieu DMF/TBAPF6 0,1 M en voltampérométrie stationnaire. Électrode de travail : Pt. v = 5 mV s–1 ; ω = 4000 π rad s–1.
| Porphyrines | ||||
| H2TPP | – | –1,06 | –1,51 | 0,45 |
| H2TPP-β-py+ | –0,79 | –1,11* | –1,69* | 0,58 |
| CuTPP | – | –1,18 | –1,70 | 0,52 |
| CuTPP-β-py+ | –0,91* | –1,18 | –1,70 | 0,52 |
| ZnTPP | – | –1,34 | –1,77 | 0,43 |
| ZnTPP-β-py+ | –1,01* | –1,32 | –1,78 | 0,46 |
* Transferts de charge irréversibles.

Voltampérométrie stationnaire de ZnTPP-β-py+·PF6– dans DMF/TBAPF6. Électrode de travail : Pt ; v = 5 mV s–1 ; ω = 4000 π rad min–1.
En comparant les hauteurs des différentes vagues enregistrées en voltampérométrie stationnaire (VS) à celles de systèmes connus (porphyrines non substituées correspondantes), les voltamogrammes obtenus indiquent que tous les transferts de charge observés sont monoélectroniques. De nombreuses études antérieures, consacrées au comportement électrochimique des porphyrines, substituées ou non, ont mis en évidence plusieurs paramètres spécifiques, aussi bien en série méso-tétraphénylporphyrine qu’en série octaéthylporphyrine. En particulier, une caractéristique typique du comportement électrochimique en réduction de ces systèmes est donnée par la quasi-invariance de l’écart entre les valeurs des deux premiers potentiels de réduction du ligand porphyrine 〚22,28,29〛. Cet écart ΔEred1/2, relativement peu affecté par la nature ou le degré de substitution de la porphyrine ainsi que par la nature du métal central dans le cas des métalloporphyrines, est justifié tant que seuls les effets polaires des substituants périphériques restent prépondérants vis-à-vis d’influences de types différents.
Dans l’évolution décrite précédemment (Tableau 1) pour les porphyrines substituées MTPP-β-py+, nous constatons que seul l’écart ΔEred1/2 entre les deuxième et troisième vagues de réduction présente une quasi-invariance, qui peut être corrélée à celle mesurée pour les porphyrines non substituées correspondantes. Ce résultat suggère que la première étape de réduction observée ne correspond pas à un transfert de charge affectant le ligand porphyrine, mais est attribuable à la réduction du groupe pyridinium substitué en position β de la porphyrine. Cette attribution est en accord avec les données de la littérature concernant la valeur du potentiel de réduction des groupements pyridinium en milieu non aqueux 〚16–19〛, qui indiquent une première étape de réduction monoélectronique voisine de –1 V/ECS. De plus, les valeurs mesurées des potentiels correspondant aux deuxième et troisième étapes de réduction des porphyrines substituées MTPP-β-py+ sont proches des valeurs des potentiels de demi-vague mesurés pour les porphyrines correspondantes non substituées MTPP. Ce résultat est, a priori, cohérent avec la présence d’un groupement substitué neutre (pyridinium réduit), où l’effet d’une charge positive sur le système π de la porphyrine serait absent. Un résultat similaire a été décrit dans le cas de la réduction de l’octaéthylporphyrine de zinc tétrasubstituée par des groupements pyridinium 〚30〛.
En voltampérométrie cyclique (VC), nous observons, pour les complexes métalliques, que l’étape de réduction attribuée au cation pyridinium est irréversible (Fig. 3). Ce résultat est en accord avec les résultats antérieurs de la littérature, qui précisent que cette réduction est irréversible lorsque les positions 2, 4, 6 du groupement pyridinium ne portent pas de substituants 〚13,14,17〛. Les étapes de réduction suivantes attribuées au macrocycle porphyrine sont, quant à elles, réversibles. Seule la base libre H2TPP-β-py+ présente un comportement rédox totalement différent. L’étape de réduction du cation pyridinium est réversible même aux faibles vitesses de balayages (20 mV s–1), tandis que les deux transferts de charge suivants concernant le macrocycle porphyrine perdent de leur réversibilité (Fig. 3). Ce comportement rédox inattendu indique, d’une part, une bonne stabilité du radical py(0) formé, bien que celui-ci ne soit pas substitué sur les positions 2, 4, 6, et, d’autre part, pour le ligand porphyrine, une forte réactivité du radical anion et du dianion générés.
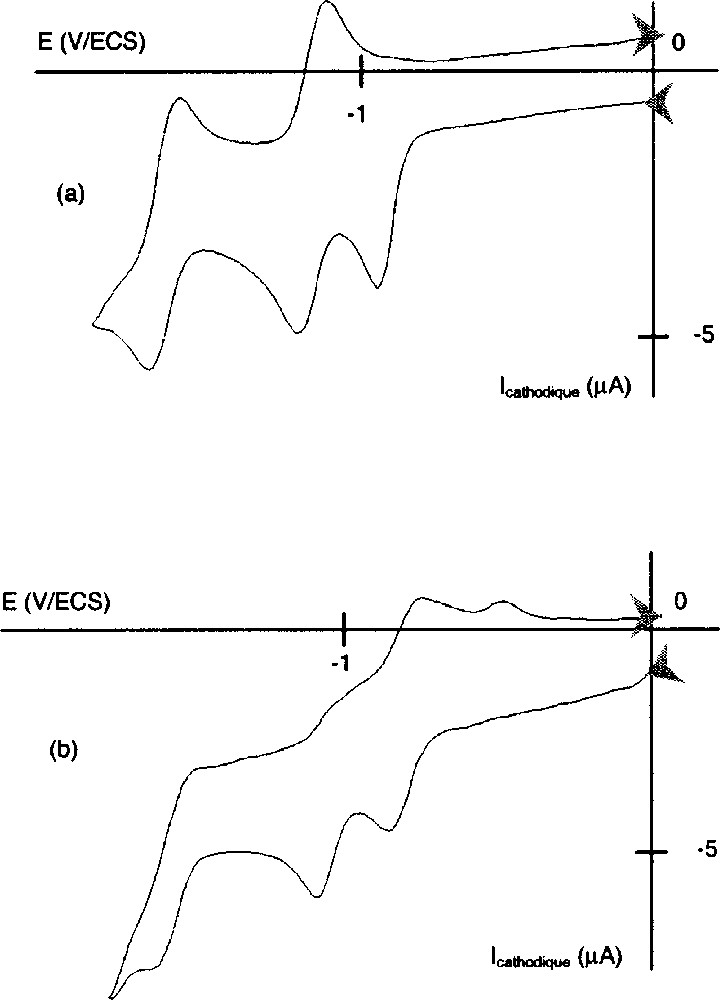
Voltampérométries cycliques en milieu DMF/TBAPF6. Électrode de travail : Pt. v = 100 mV s–1. (a): CuTPP-β-py+·PF6–, (b): H2TPP-β-py+·PF6–.
Le pic d’oxydation observé à environ –0,5 V/ECS n’apparaît que pour des balayages à des potentiels inférieurs à –1,69 V/ECS, c’est-à-dire lorsqu’on génère le dianion de la porphyrine. On peut raisonnablement s’attendre à ce que le dianion formé réagisse rapidement avec le milieu d’étude, pour former une espèce électroactive à ce potentiel.
Dans le cas des transferts de charge réversibles, pour des vitesses de balayage allant jusqu’à 100 mV s–1, nous mesurons des écarts de pics ΔEp = Epc – Epa voisins de 60 mV et des rapports de courant de pics Ipc/Ipa proches de l’unité. Les valeurs des potentiels de réduction déduites des résultats de voltampérométrie cyclique, à savoir sont en accord avec celles mesurées par voltampérométrie stationnaire.
L’étape de réduction supplémentaire, correspondant au premier transfert de charge, peut être décrite par le mécanisme suivant :
Les valeurs des potentiels de réduction du cation pyridinium (Tableau 1) mesurées pour les transferts de charge irréversibles (M = Cu, Zn) peuvent être raisonnablement comparées à celle obtenue lorsque le transfert de charge est réversible (M = H2), dans la mesure où on l’estime que la constante de vitesse de la réaction chimique postérieure à la réaction électrochimique reste faible devant la constante de vitesse du transfert de charge. Ainsi, l’analyse des variations du potentiel de première réduction des trois systèmes étudiés permet de mettre en évidence différents effets, attribuables à la porphyrine en tant que substituant du groupement pyridinium. Nous constatons que cette valeur potentiel de réduction du cation pyridinium, est la plus négative lorsqu’il est substitué par ZnTPP, qui peut être considéré comme le système le plus électronégatif ; le groupe pyridinium, portant la base libre H2TPP, système le plus électropositif, est au contraire le plus facile à réduire. Cette évolution (Fig. 4) suit l’ordre d’électronégativité croissante attribuable à ces trois porphyrines (H2TPP, CuTPP, ZnTPP) 〚28〛, qui peut être aussi illustré par les valeurs de leurs potentiels de réduction respectifs. Cette évolution du potentiel de réduction du groupe pyridinium, qui traduit l’effet électrodonneur de la porphyrine vis-à-vis du groupe pyridinium ainsi que la conjugaison qui existe entre les deux systèmes 〚15,18〛, démontre clairement l’interdépendance des deux sites, porphyrine–pyridinium. L’examen des valeurs du premier potentiel de réduction du ligand porphyrine des différents systèmes MTPP-β-py+ (deuxième étape de réduction) ne met en évidence aucune évolution significative par rapport à la porphyrine non substituée.

Évolution du potentiel de première réduction des porphyrines substituées MTPP-β-py+ en fonction du potentiel de première réduction des porphyrines non substituées MTPP correspondantes.
Les études antérieures concernant la réduction électrochimique des cations pyridinium ont clairement montré l’influence cruciale de la nature des groupements portés par les positions 1, 2, 4, 6 sur le mécanisme de transfert de charge 〚13–19〛. Lorsque les positions 2, 4 et 6 du cation ne portent pas de substituant, l’instabilité du radical neutre formé conduit à des réactions chimiques comme la dimérisation, et l’étape de réduction est alors irréversible 〚13,14,23〛. La substitution par au moins un groupe phényle (sur la position 4 du cation pyridinium) permet de rendre la réduction réversible, la stabilité du radical neutre obtenu augmentant 〚18〛 avec le nombre des substituants portés sur les positions 2, 4, 6. Cependant, deux cas particuliers dérogent à ce dernier comportement. Lorsque la position 1 porte un groupement benzyle ou allyle, la réduction devient irréversible, même avec un groupe phényle substitué en position 4, et conduit à la rupture exocyclique de la liaison C–N 〚18,23〛. Pour les systèmes de notre étude, MTPP-β-py+, la conjugaison, même partielle, entre les deux sites aromatiques porphyrine–cation pyridinium va permettre une délocalisation de l’électron célibataire du radical py(0) généré plus ou moins importante, suivant la nature de la porphyrine. Nous pouvons alors envisager une interprétation des différences de réversibilité observées entre les complexes métalliques et la base libre, en prenant en compte, en particulier, les interactions d–π existant dans les métalloporphyrines 〚31,32〛. Il est raisonnable de postuler que, pour les métalloporphyrines MTPP-β-py+, les interactions d–π vont renforcer une localisation de l’électron célibataire sur le cation pyridinium réduit py(0) et accroître sa réactivité, pour conduire au comportement décrit 〚13,14〛 lorsqu’il n’est pas substitué sur les positions 2, 4 et 6. L’hypothèse d’une dimérisation du pyridinium réduit n’est pas incompatible avec une même amplitude des trois vagues enregistrées, car il est peu probable d’avoir une différence significative des valeurs des coefficients de diffusion respectifs des espèces monomères et dimères.
Dans le cas de la base libre H2TPP-β-py+, ces interactions d–π n’existent pas et une plus grande délocalisation de l’électron célibataire du radical formé sur l’ensemble du système porphyrine–pyridine est possible, avec pour conséquence, d’une part, une moindre réactivité chimique du groupe pyridinium réduit, d’autre part, pour le ligand porphyrine rendu plus basique par la présence de l’électron délocalisé (accroissement de la basicité pouvant expliquer l’augmentation de la valeur de l’écart ΔEred1/2), une augmentation de l’instabilité des espèces réduites générées (anion et dianion), d’où la perte de réversibilité observée des transferts électroniques sur le ligand. Cette hypothèse ne permet cependant pas d’exclure la possibilité d’une rupture exocyclique de la liaison C–N, qui peut découler aussi bien d’une augmentation de la densité électronique sur le macrocycle porphyrine que de réactions chimiques dues à l’évolution du radical py(0).
Des différences de comportement électrochimique à propos de systèmes porphyriniques base libre et métallés ont déjà été interprétées 〚33〛 à partir de telles interactions d–π. Pour mieux comprendre ces différents comportements et l’évolution de l’espèce monoréduite MTPP-β-py(0), nous avons entrepris une série de coulométries exhaustives des porphyrines MTPP-β-py+ au potentiel de première réduction (Tableau 1). Les premiers résultats obtenus ont fourni des valeurs du nombre d’électrons échangés n comprises entre 2 et 3. Cette non-concordance avec la valeur n = 1, établie à partir des mesures effectuées par voltampérométrie cyclique et voltampérométrie stationnaire, montre que le mécanisme global de la réaction électrochimique de réduction du cation pyridinium dépasse le simple transfert de charge monoélectronique. Les valeurs mesurées de n suggèrent, soit la présence de réactions secondaires générant des composés électroactifs à ces potentiels, soit une évolution du dimère formé vers des produits électroréductibles.
4 Conclusion
La réduction électrochimique de méso-tétraphénylporphyrines substituées en position bêta par un cation pyridinium a montré que les transferts de charges se font successivement sur deux sites différents. Une première étape de réduction concerne le cation pyridinium, pour conduire à la formation d’un radical neutre après un transfert de charge monoélectronique. L’évolution des valeurs du potentiel de réduction correspondant est en corrélation avec le pouvoir donneur attribué aux différentes porphyrines. Les transferts de charges ultérieurs concernent le ligand porphyrine. L’importance de la présence d’un système hautement conjugué – la porphyrine – en position 1 du cation pyridinium apparaît clairement dans les résultats obtenus en voltampérométrie cyclique, qui mettent en évidence une réactivité électrochimique de la base libre radicalement différente de celle des complexes métalliques correspondants, conduisant en particulier à la formation quasi irréversible du radical anion et du dianion. Ces différences de comportement rédox confirment l’interdépendance des deux sites réactionnels pyridinium–porphyrine et peuvent être interprétées en partie à partir de l’effet des interactions d–π existant dans les métalloporphyrines.
Remerciements
Ce travail a été supporté financièrement par le Centre national de la recherche scientifique.