

1 Introduction
Heterocycles involving an arylsulfamido moiety have been reported to possess a variety of interesting biological activities 〚1–5〛. For example, 1,2-benzothiazines with basic side chains 〚6〛 are claimed to be diuretic agents. Antithrombotic and lipid-regulating properties are observed for 1,2-benzothiazine-3-carboxamides 〚7〛. Antibacterial activity was found for several penicillin derivatives containing the 1,2-benzothiazinyl fragment 〚8〛. Many reports have referred to the anti-inflammatory analgesic and antipyretic activities found for a variety of 1,2-benzothiazines 〚9〛. An initial report by Lombardino et al. 〚10〛 indicated that potent anti-inflammatory activity was present in a series of 4-hydroxy-2H-1,2-benzothiazin-3-carboxamide 1,1-dioxides 1 (Fig. 1).

Compounds 1–4.
Owing to these important biological properties of the benzo- and the heterocyclic ring-fused 1,2-thiazines, many publications have presented interesting methods of their synthesis 〚11–20〛. Meanwhile a few synthetic approaches to 1,2-naphtothiazine derivatives have been developed in the literature. Kaufmann and Zobel 〚21〛 first prepared 2,3-dihydro-3-oxonaphtho-〚1,8-d,e〛-1,2-thiazine 1,1-dioxides 2. More recently, this compound was prepared by Lombardino according to a short process 〚22〛. Both Trummlitz 〚23〛 and Steiner 〚24〛 have prepared independently naphtho〚2,1-e〛-1,2-thiazine 3 analogues of 1. Trummlitz and co-workers 〚23〛 also reported the synthesis of N-substituted 2H-naphtho〚2,1-c〛-1,2-thiazin-4(3H)-one 1,1-dioxides 4 by ring expansion of 3-bromomethylnaphth-〚2,1-d〛-isothiazole 1,1-dioxides.
As part of our continued efforts to develop synthetically useful anionic aromatic reactions for the synthesis of biologically active compounds 〚25〛, we report a general route based on the well known directed ortho-metalation reaction 〚20〛 in our synthetic approach to 3-substituted-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxides 7a–i (Fig. 2). These heterocycles are analogues of the 1,2-benzothiazines derivatives that were required for biological activities evaluations and as starting materials to prepare new drugs.
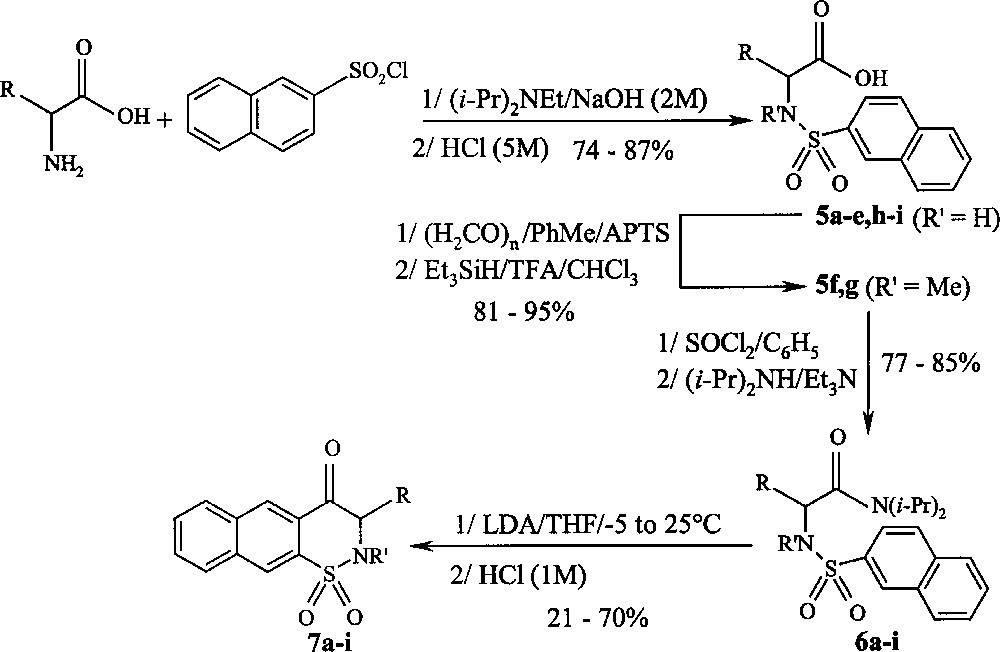
Synthesis of compounds 5a–i, 6a–i, and 7a–i.
2 Results and discussion
The aromatic directed ortho-metalation reaction has been developed into a broadly useful protocol for the regioselective construction of polysubstituted aromatics 〚26–29〛 and used in the efficient synthesis of several heterocyclic ring systems and bioactive compounds 〚30〛.
The process reported herein (Fig. 2), possibly driven by the Complex Induced Proximity Effect (CIPE) 〚31〛, constitutes a mild method for the LDA-mediated regiospecific conversion of N-(2-naphthylsulfonyl) amino amides 6a–i 〚32–36〛 readily available from commercial racemic or optically pure (S)-α-amino acids into a new 2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxides 7a–i. According to this process, it was possible to change substitution patterns at the C-3 position of compounds 7a–i.
In order to select the optimal reaction conditions for the synthesis of compounds 7a-i, we have carefully examined the ortho-metalation conditions carried out on aminoamides 6a or 6f by changing the base (nature and number of equivalents) and the temperature.
The results shown in Table 1 indicate the requirement of an excess of LDA for efficient ortho-lithiations and to improve the yields (entries 1 to 5). In addition of the possible α- and α’-amide carbanion formation, the excess of LDA also serves to displace the equilibrium between the complex formed by the association of the organolithium reagent and the Lewis basic functionality of the substrate and the coordinated ortho-lithiated species in favour of the latter complex 〚29,31〛.
Yields of products 7a and 7f (optimisition experiments).
| Entry | Substrate | BM (equivalent) | Yield (%)a |
| 1 | 6a | LDA (2) | 38 |
| 2 | 6a | LDA (3) | 47 |
| 3 | 6a | LDA (4) | 56 |
| 4 | 6f | LDA (2) | 61 |
| 5 | 6f | LDA (3) | 70 |
| 6 | 6f | LDA (3)b | 43 |
| 7 | 6f | LiHMDS (3) | 64 |
| 8 | 6f | sec-BuLi (3) | 33 |
The use of other bases, such as LiHMDS, sec-BuLi and n-BuLi, do not ameliorate the yield of products. With sec-BuLi, the cyclisation proceeds very poorly (entry 8), yielding a complex mixture of products. Moreover, either at 0 °C or at –78 °C, the exposure of (S)-N,N-diisopropyl-3-methyl-2-(N-2-naphthylsulfonylamino)butanamide 6a to n-BuLi gave the 2-methyl-3-(N-2-naphthylsulfonylamino)octan-4-one as a major product along with the starting material 6a. The ketone results from nucleophilic addition of n-BuLi to 6a before the directed ortho-lithiation reaction. In addition, we have found out that low temperature (–78 °C) decreases significantly the yield of the cyclisation reaction when LDA was used (compare entry 5 with entry 6).
Attempts of these different experimental conditions summarised in Table 1 led to the selection of the following procedure: treatment of compounds 6a–i with 〚LDA (four equivalents for N–H derivatives and three equivalents for N–Me derivatives)/THF /–5 to 25 °C over 1 h〛 gave the corresponding 2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxides 7a–i. However, the excess of LDA has racemised heterocycles prepared from enantiomerically pure intermediates 6a,c–h. Yields range between 21 and 70% (Table 2).
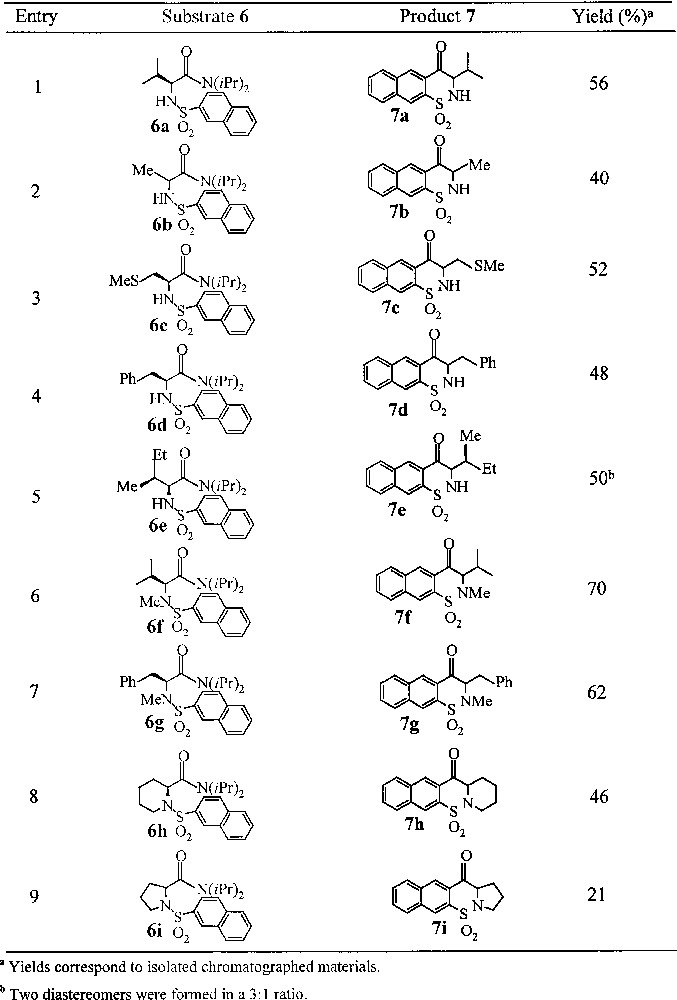
Synthesis of 3-substituted-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxides 7a–i.
Theoretically, ortho-lithiation of N-(2-naphthylsulfonyl)amino amides 6a–i can occur on carbon-1 and carbon-3 of the naphthalene ring. Characterisation of the reaction product proves the regiospecific cyclisation of substrates 5 to provide exclusively 2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxides 7a-i. To further prove the selectivity of H-3 deprotonation on expense of H-1 proton, lithiation–deuteration experiments with secondary and tertiary 2-naphthylsulfonamides 8 and 9 were undertaken (Fig. 3).

Lithiation–deuteration experiments with secondary and tertiary 2-naphthylsulfonamides 8 and 9, giving 10 and 11.
Thus, treatment of N-methyl-2-naphthylsulfonamide 8 with n-BuLi (2.2 equiv), followed by quench with CD3OD resulted in selective deuterium incorporation at carbon-3 (RMN 1H). Likewise, N,N-diethyl-2-naphthylsulfonamide 9 when subjected to similar n-BuLi (1.1 equiv) metalation–CD3OD quench conditions, afforded only the ortho-deuterated product at carbon-3 (Table 3). As metalation regioselectivity is influenced by variation of metalating agent 〚37〛, substrates 8 and 9 were metalated with 〚LDA (2.2 equiv for 8 and 1.1 equiv for 9)/THF/–5 °C to 25 °C〛. Aliquots were quenched at 0 °C and at room temperature during the course of the reaction by addition to CD3OD. Deuterium incorporation obtained in these experiments was the same as that obtained when n-BuLi was used.
Yields of products 10 and 11 (deuteration experiments).
| Product | RLi | Yield (%) |
| 10 | n-BuLi | 79 |
| 10 | LDA | 38 |
| 11 | n-BuLi | 87 |
| 11 | LDA | 42 |
Both results of ortho-lithiation–cyclisation with compounds 5 and ortho-lithiation–deuteration with compounds 8 and 9 demonstrate that the orientation of the sulfonamide group with respect to the proton removed during the ortho-lithiation significantly affects the reaction course 〚38〛. These results lead also to the suggestion of a naphthalenesulfonyl-base complex (Fig. 4), which places the LDA in the proximity of H-3 proton so as to exert a regio-control by a CIPE process.
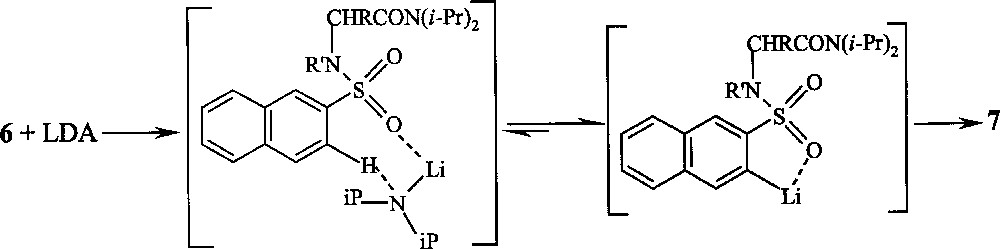
Chemical route from 6 to 7, through a naphthalenesulfonyl-base complex, which places the LDA in the proximity of H-3 proton and so exerts regio-control by a CIPE process.
3 Conclusion
In summary, a systematic study has provided optimised conditions for the synthesis of new 2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxides analogues of the 1,2-benzothiazine class. The synthetic approach applied to elaborate these heterocycles was based on the directed ortho-metalation procedure. This reaction has significant advantages over the more traditional alkoxide-mediated ring expansion of N-acyl-1,2-arenethiazole 1,1-dioxides derivatives 〚13, 24〛, since it constitutes a general, convenient and regiospecific method for the synthesis of substituted 1,2-naphthothiazines. The selectivity of ortho-lithiation reactions of 6, 8 and 9 is reasonably attributed to a favourable arrangement during the transfer of the proton to the lithiating reagent within a complex of sulfonamide with the organolithium reagent.
The continuous pharmaceutical interest in the arylsulfonamide compounds 〚25, 39–40〛 may justify further exploration of these results in the pharmacological field.
4 Experimental section
All reactions were carried out under argon in dried glassware, using syringe-septum cap techniques. Solvents were purified and dried by standard methods. Melting points were determined on a Büchi SMP-20 capillary apparatus and are uncorrected. TLC was carried out on Merck 60F-254 precoated silica gel plates (0.25 mm) and column chromatography was performed with Merck silica gel (70–230 mesh). NMR spectra were recorded on a Bruker 300 spectrometer (1H at 300 MHz and 13C at 75 MHz) with CDCl3 as solvent and TMS as internal standard reference. IR–TF spectra were recorded on a BIORAD FTS-6000 spectrometer. Mass spectra (EI) were recorded on a Hewlett Packard 5792 apparatus coupled to a GC HP-5890 chromatograph (column: 5% diphenyl, 95% dimethylpolysiloxane. 25 m × 0.25 mm ID, 0.35 μm film). Elemental analyses were carried out by the ‘Service de microanalyse’ of the University Paris-6. Optical rotations were measured at 21 °C on a Perkin-Elmer 241 polarimeter. n-BuLi and sec-BuLi were purchased from Acros as solutions in hexane stored in a resealable container, and titrated periodically against sec-butanol 〚41〛.
4.1 N-(2-Naphthylsulfonyl)-2-amino acids 5a-e and 5h,i. General procedure
To a solution of an α-amino acid (20 mmol) in aqueous NaOH (2M, 10 ml, 20 mmol) at 0 °C was added 2-naphthylsulfonyl chloride (21 mmol, 1.05 equiv), followed by EtN(i-Pr)2 (22 mmol, 1.1 equiv) and acetone (10 ml). The mixture was stirred at room temperature for 12 h. The reaction mixture was washed with Et2O (2 × 50 ml) and the combined washings extracted with aqueous NaOH (2M, 10 ml). The combined basic aqueous layers were cooled to 0 °C and acidified (pH 1) by the addition of concentrated HCl. The mixture was extracted with EtOAc (3 × 50 ml), dried over MgSO4, filtered and concentrated under reduced pressure to give N-(2-naphthylsulfonyl)-2-amino acids, which were purified by crystallisation.
4.2 (S)-3-Methyl-2-(N-2-naphthylsulfonylamino)butanoic acid: 5a
Yield 78%. mp 103–105 °C. 〚α〛D = –19.2 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 0.88 (d, 3H, J = 6.8 Hz), 0.97 (d, 3H, J = 6.8 Hz), 2.15 (m, 1H), 3.79 (dd, 1H, J = 4.6 Hz and 9.8 Hz), 5.27 (d, 1H, J = 9.8 Hz), 7.54–7.63 (m,2H), 7.77–7.94 (m, 4H), 8.35 (s, 1H), 9.45 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 16.64, 19.17, 28.23, 63.59, 123.94, 126.36, 127.32, 129.37, 130.56, 130.29, 130.96, 133.58, 133.80, 137.28, 173.55.
4.3 (±)-2-(N-2-Naphthylsulfonylamino)propanoic acid: 5b
Yield 83%. mp 151–105 °C. 1H NMR (300 MHz, CDCl3): δ = 1.22 (d, 3H, J = 7.4 Hz), 3.83 (qd, 1H, J = 7.4 Hz and J = 6.5 Hz), 5.56 (d, 1H, J = 6.5 Hz), 7.60–7.71 (m, 2H), 7.80–7.96 (m, 4H), 8.40 (s, 1H), 9.9 (br,1H). – 13C NMR (75 MHz, CDCl3): δ = 38.36, 57.03, 122.63, 126.59, 128.67, 129.11, 129.87, 131.03, 131.65, 134.78, 135.13, 138.43, 172.98.
4.4 (S)-3-Methylsulfanyl-2-(N-2-naphthylsulfonylamino)propanoic acid: 5c
Yield 77%. mp 93–94 °C. 〚α〛D = –15.6 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 3.28 (m, 2H), 3.75 (s, 3H), 4.23 (m, 1H), 6.32 (d, 1H, J = 9.8 Hz), 7.59–7.68 (m, 2H), 7.73–7.90 (m, 4H), 8.38 (s, 1H), 9.60 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 18.76, 41.46, 55.79, 124.15, 126.36, 128.65, 129.32, 130.56, 131.08, 131.98, 133.40, 134.74, 137.36, 172.36.
4.5 (S)-3-Phenyl-2-(N-2-naphthylsulfonylamino)propanoic acid: 5d
Yield 84%. mp 133–134 °C. 〚α〛D = –43.2 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 3.15 (m, 2H), 3.98 (m, 1H), 5.59 (d, 1H, J = 10.0 Hz), 7.66–7.74 (m, 7H), 7.86–8.04 (m, 4H), 8.46 (s, 1H), 10.16 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 42.38, 57.17, 124.69, 125.61, 126.75, 127.14, 127.61, 128.07, 128.66, 129.14, 129.86, 131.55, 132.18, 135.73, 138.26, 139.10, 175.44.
4.6 (2S, 3S)-3-Methyl-2-(N-2-naphthylsulfonylamino)pentanoic acid: 5e
Yield 70%. mp 111–112 °C. 〚α〛D = –8.4 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 0.92 (d, 3H, J = 5.4 Hz), 1.18 (t, 3H, J = 3.5 Hz), 1.25–1.39 (m, 2H), 2.48–2.55 (m, 1H), 4.12–4.20 (dd, 1H, J = 3.3 and J = 10.0 Hz), 5.25–5.30 (d, 1H, J = 10.0 Hz), 7.58–7.69 (m, 2H), 7.81–7.96 (m, 4H) 8.28 (s, 1H), 9.6 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 12.03, 14.52, 25.71, 34.36, 62.41, 122.69, 125.33, 127.74, 128.72, 129.13, 130.48, 131.11, 133.67, 134.10, 137.15, 173.19.
4.7 (S)-N-(2-Naphthylsulfonyl)pipecolinic acid: 5h
Yield 75%. mp 131–132 °C. 〚α〛D = –35.0 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 0.9–1.1 (m, 1H), 1.4–1.6 (m, 4H), 2.5–2.7 (m, 2H), 3.39 (dt, 1H, J = 3.5 Hz and J = 10.8 Hz), 5.34 (t, 1H, J = 3.5 Hz), 7.53–7.64 (m, 2H), 7.79–7.98 (m, 4H), 8.42 (s, 1H), 9.95 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 20.17, 24.15, 24.36, 45.59, 61.82, 123.63, 126.28, 128.93, 129.71, 130.79, 131.09, 131.69, 133.73, 134.44, 137.48, 174.42.
4.8 (±)-N-(2-Naphthylsulfonyl)proline: 5i
Yield 74%. mp 128–130 °C. 1H NMR (300 MHz, CDCl3): δ = 1.76–1.82 (m, 2H), 2.18–2.32 (m, 2H), 3.34–3.52 (m, 2H), 4.30 (dt, 1H, J = 4.1 Hz and J = 10.7 Hz), 7.61–7.72 (m, 2H), 7.83–8.02 (m, 4H), 8.28 (s, 1H), 9.7 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 24.96, 31.18, 47.96, 59.42, 123.80, 126.65, 128.73, 129.51, 130.39, 130.36, 131.31, 133.49, 134.33, 137.47, 172.52.
4.9 N-Methyl-N-(2-naphthylsulfonyl)-2-amino acids 5f,g. General procedure
N-(2-naphthylsulfonyl)-2-amino acid was suspended in 150 ml of toluene, and paraformaldehyde (2 g) and p-toluenesulfonic acid (200 mg) were added. The mixture was refluxed for 30 min with azeotropic water removal. The solution was then cooled, washed with 1 N aqueous NaHCO3 (2 × 50 ml) and dried over MgSO4. Concentration in vacuo gave the corresponding N-(2-naphthylsulfonyl)oxazolidinone, without further purification was dissolved in 20 ml of CHCl3 and 20 ml of trifluoroacetic acid and triethylsilane (30 mmol) were added. The solution was stirred at room temperature for 24 h; subsequent concentration in vacuo and purification by crystallisation gave pure N-methyl-N-(2-naphthylsulfonyl)-2-amino acid.
4.10 (S)-3-Methyl-2-(N-methyl-N-2-naphthylsulfonylamino)butanoic acid: 5f
Yield 81%. mp 116–117 °C. 〚α〛D = –25.1 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 0.89 (d, 3H, J = 7.2 Hz), 1.13 (d, 3H, J = 7.2 Hz), 2.48 (m, 1H), 2.52 (s, 3H), 4.36 (d, 1H, J = 3.3 Hz), 7.54–7.64 (m,2H), 7.81–7.97 (m, 4H), 8.37 (s, 1H), 10.5 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 17.19, 19.38, 27.86, 38.60, 63.19, 123.78, 126.46, 128.73, 128.98, 130.20, 130.52, 131.25, 133.49, 134.35, 136.88, 172.23.
4.11 (S)-3-Phenyl-2-(N-methyl-N-2-naphthylsulfonylamino)propanoic acid: 5g
Yield 95%. mp 139–141 °C. 〚α〛D = –48.5 (c1, MeOH). 1H NMR (300 MHz, CDCl3): δ = 2.20 (s, 3H), 3.19 (d, 2H, J = 3.9 Hz), 4.24 (t, 1H, J = 3.9 Hz), 7.37–7.58 (m, 7H), 7.78–7.94 (m, 4H), 8.27 (s, 1H), 9.40 (br, 1H). 13C NMR (75 MHz, CDCl3): δ = 37.56, 41.61, 56.31, 128.53, 128.94, 129.09, 129.58, 130.01, 130.75, 131.29, 131.96, 133.01, 133.87; 134.25, 134.86, 135.75, 136.93, 173.15.
4.12 N,N-Diisopropyl-2-amino amides 6a–i. General procedure
To a stirred solution of N-(2-naphthylsulfonyl)-2-amino acid or N-methyl-N-(2-naphthylsulfonyl)-2-amino acid (10 mmol) in dry benzene (70 ml), was added at room temperature SOCl2 (12 mmol, 1.2 equiv). The mixture was then refluxed for 3 h, cooled and concentrated to oil. The oil was dissolved in CH2Cl2 and reconcentrated two times. The corresponding acid chloride obtained, without further purification, was dissolved in 50 ml of CH2Cl2 and added to a stirred solution of diisopropylamine (11 mmol) and triethylamine (11 mmol) in CH2Cl2 (30 ml). The mixture was stirred at 0 °C for 2 h and then poured into 100 ml of a diluted HCl solution. The N,N-diisopropyl-2-amino amide was extracted with CH2Cl2 and the organic phase was washed with saturated NaCl solution, dried over MgSO4, filtered and concentrated. The residue was purified by silica gel chromatography 〚SiO2; (EtOAc:hexane)〛 to afford the N,N-diisopropyl-2-amino amide.
4.13 (S)-N,N-Diisopropyl-3-methyl-2-(N-2-naphthylsulfonylamino)butanamide: 6a
Yield 84%. mp 118–121 °C. 〚α〛D = –31.9 (c1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 0.85 (d, 3H, J = 7.0 Hz), 1.11 (d, 3H, J = 7.0 Hz), 1.14–1.29 (m, 12H), 2.88 (m, 1H), 3.05–3.13 (m, 2H), 4.36 (dd, 1H, J = 4.1 Hz and 10.18 Hz), 5.50 (d, 1H, J = 10.1 Hz), 7.61–7.71 (m, 2H), 7.82–8.9 (m, 4H), 8.44 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.05, 12.27, 12.86, 13.51, 16.71, 19.20, 29.02, 40.63, 41.37, 67.84, 123.86, 127.09, 128.73, 129.41, 130.17, 130.52, 131.42, 133.60, 134.19, 137.33, 168.61. C15H15NO3S (390.54): calcd C 64.58, H 7.74; found C 64.47, H 7.81.
4.14 (±)-N,N-Diisopropyl-2-(N-2-naphthylsulfonylamino)propanamide: 6b
Yield 78%. mp 113–114 °C. 1H NMR (300 MHz, CDCl3): δ = 1.0–1.21 (m, 12H), 1.28 (d, 3H, J = 7.2 Hz), 3.11–3.25 (m, 2H), 3.84 (qd, 1H, J = 7.2 Hz and J = 6.5 Hz), 5.53 (d, 1H, J = 6.5 Hz), 7.73–7.85 (m, 2H), 7.80–7.98 (m, 4H), 8.32 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.13, 12.35, 13.06, 13.66, 38.31, 40.49, 41.32, 56.84, 123.62, 126.67, 128.70, 129.43, 130.37, 131.12, 131.53, 134.18, 134.62, 138.19, 167.79. C15H15NO3S (362.49): calcd C 62.95, H 7.23; found C 62.89, H 7.26.
4.15 (S)-N,N-Diisopropyl-3-methylsulfanyl-2-(N-2-naphthylsulfonylamino)propanamide: 6c
Yield 79%. mp 102–103 °C. 〚α〛D = –35 (c1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 1.09–1.26 (m, 12H), 3.02–3.24 (m, 2H), 3.37 (m, 2H), 3.68 (s, 3H), 4.40 (m, 1H), 6.53 (d, 1H, J = 10.5 Hz), 7.59–7.72 (m, 2H), 7.82–8.01 (m, 4H), 8.28 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.10, 12.33, 13.04, 13.47, 18.69, 40.58, 41.36, 41.55, 54.68, 123.61, 126.78, 128.73, 129.24, 130.42, 131.50, 132.25, 133.36, 134.25, 137.38, 168.39. C15H15NO3S (376.51): calcd C 63.80, H 7.49; found C 63.96, H 7.43.
4.16 (S)-N,N-Diisopropyl-3-phenyl-2-(N-2-naphthylsulfonylamino)propanamide: 6d
Yield 80%. mp 153–155 °C. 〚α〛D = –46.3 (c1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 0.89–1.02 (m, 12H), 3.02–3.18 (m, 2H), 3.47 (m, 2H), 4.58 (m, 1H), 6.52 (d, 1H, J = 10.1 Hz), 7.23–7.62 (m, 7H), 7.75–7.91 (m, 4H), 8.25 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.24, 12.43, 13.07, 13.60, 40.81, 41.56, 41.69, 53.79, 125.56, 125.78, 126.77, 127.15, 127.61, 128.31, 129.24, 129.69, 130.17, 131.82, 132.54, 135.70, 138.36, 139.58, 168.74. C15H15NO3S (438.58): calcd C 68.46, H 6.89; found C 68.38, H 6.94.
4.17 (2S, 3S)-N,N-Diisopropyl-3-methyl-2-(N-2-naphthylsulfonylamino)pentanamide: 6e
Yield 78%. mp 106–107 °C. 〚α〛D = –29.7 (c1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 0.95–1.29 (m, 15H), 1.32–1.49 (m, 5H,), 2.72–2.80 (m, 1H), 3.05–3.18 (m, 2H), 4.61–4.73 (dd, 1H, J = 3.2 and J = 8.5 Hz), 5.33–5.46 (d, 1H, J = 10.1 Hz), 7.60–7.72 (m, 2H), 7.84–8.02 (m, 4H) 8.39 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.16, 11.49, 12.32, 12.91, 13.62, 15.28, 25.83, 34.66, 40.82, 42.05, 67.68, 123.16, 125.59, 127.87, 128.59, 129.69, 130.99, 131.63, 133.43, 134.46, 137.21, 168.32. C15H15NO3S (404.57): calcd C 65.31, H 7.97; found C 65.42, H 7.91.
4.18 (S)-N,N-Diisopropyl-3-methyl-2-(N-methyl-N-2-naphthylsulfonylamino)butanamide: 6f
Yield 85%. mp 121–123 °C. 〚α〛D = –27.3 (c 1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 0.93 (d, 3H, J = 7.2 Hz), 1.10 (d, 3H, J = 7.2 Hz), 1.13–1.29 (m, 12H), 2.57 (m, 1H), 3.01 (s, 3H), 3.14–3.28 (m, 2H), 4.49 (d, 1H, J = 3.5 Hz), 7.66–7.71 (m, 2H), 7.72–7.89 (m, 4H), 8.37 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.32, 12.18, 12.88, 13.57, 17.19, 19.24, 27.86, 38.76, 41.09, 42.11, 68.01, 123.82, 126.58, 128.25, 129.39, 130.52, 131.28, 131.85, 133.48, 134.68, 137.72, 169.14. C15H15NO3S (404.57): calcd C 65.31, H 7.97; found C 65.40, H 7.94.
4.19 (S)-N,N-Diisopropyl-3-phenyl-2-(N-methyl-N-2-naphthylsulfonylamino)propanamide: 6g
Yield 77%. mp 129–130 °C. 〚α〛D = –31.5 (c 1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 0.92–1.11 (m, 12H), 2.34 (s, 3H), 3.12–3.22 (m, 2H), 3.41 (d, 2H, J = 3.7 Hz), 4.37 (t, 1H, J = 3.7 Hz), 7.47–7.62 (m, 7H), 7.86–7.90 (m, 4H), 8.29 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.29, 12.37, 12.74, 13.71, 37.15, 40.93, 41.73, 42.57, 54.39, 128.17, 128.24, 128.69, 129.10, 129.49, 130.08, 130.96, 131.87, 132.11, 133.27; 133.65, 134.54, 135.69, 136.81, 167.58. C15H15NO3S (452.61): calcd C 68.99, H 7.12; found C 68.90, H 7.21.
4.20 (S)-N,N-Diisopropyl-2-(N-2-naphthylsulfonyl)pipecolinamide: 6h
Yield 82%. mp 138–140 °C. 〚α〛D = –16.4 (c 1, CHCl3). 1H NMR (300 MHz, CDCl3): δ = 1.03–1.28 (m, 12H), 1.32–1.37 (m,1H), 1.57–1.79 (m, 4H), 2.37–2.65 (m, 2H), 3.10–3.19 (m, 2H), 3.64 (dt, 1H, J = 4.2 Hz and J = 11.9 Hz), 5.46 (t, 1H, J = 4.2 Hz), 7.55–7.66 (m, 2H), 7.91–8.10 (m, 4H), 8.47 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.23, 12.29, 12.82, 13.67, 21.10, 24.37, 25.21, 40.80, 41.93, 46.31, 62.72, 123.69, 127.41, 128.74, 129.65, 130.46, 130.59, 131.37, 133.76, 134.41, 137.49, 169.04. C15H15NO3S (402.55): calcd C 65.64, H 7.51; found C 65.68, H 7.48.
4.21 (±)-N,N-diisopropyl-2-(N-2-naphthylsulfonyl)prolinamide: 6i
Yield 78%. mp 134–135 °C. 1H NMR (300 MHz, CDCl3): δ = 0.98–1.21 (m, 12H), 1.53–1.67 (m, 2H), 2.16–2.35 (m, 2H), 3.02–3.18 (m, 2H), 3.56–3.78 (m, 2H), 4.87 (dt, 1H, J = 3.7 Hz and J = 11.0 Hz), 7.64–7.70 (m, 2H), 7.84–7.99 (m, 4H), 8.39 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.09, 12.37, 13.05, 13.74, 23.66, 32.17, 48.32, 56.41, 123.98, 126.53, 128.87, 130.28, 130.86, 131.11, 131.73, 133.68, 134.69, 137.79, 168.31. C15H15NO3S (388.52): calcd C 64.92, H 7.26; found C 65.09, H 7.30.
4.22 2H-1,2-Naphthothiazin-4(3H)-one 1,1-dioxides 7a–i. General procedure
To a solution of LDA (4 equiv for N–H derivatives and 3 equiv for N–Me derivatives of 6) freshly prepared in THF at –5 °C was added dropwise a solution of N-(2-naphthylsulfonyl)amino amide (5 mmol). The reaction mixture was allowed to warm to room temperature over 1 h, and then quenched with a hydrochloric acid solution (1 M, 25 ml). The organic layer was separated and the aqueous layer extracted with methylene chloride (3 × 20 ml). The organic extracts were dried (Na2SO4), and evaporated. The residue was purified by silica gel chromatography 〚SiO2; (EtOAc:hexane)〛 to afford the 2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxide.
4.23 3-Isopropyl-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7a
Yield 56%. mp 145–147 °C. 1H NMR (300 MHz, CDCl3): δ = 0.98 (d, 3H, J = 6.9 Hz), 1.17 (d, 3H, J = 6.9 Hz), 2.85 (m, 1H), 4.48 (dd, 1H, J = 3.6 Hz and 10.2 Hz), 5.47 (d, 1H, J = 10.2 Hz), 7.65 (m,2H), 7.85 (m, 2H), 8.15 (s, 1H), 8.45 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 16.66, 19.19, 28.26, 68.45, 123.90, 126.56, 128.79, 129.27, 130.18, 130.36, 131.11, 133.63, 134.10, 137.21, 191.91. IR (KBr): ν = 1679 (C=O), 1128, 1323 (SO2N), 1552 (NH), 1044 (SO2). C15H15NO3S (289.33): calcd C 62.26, H 5.22; found C 62.34, H 5.30.
4.24 3-Methyl-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7b
Yield 40%. mp 126–128 °C. 1H NMR (300 MHz, CDCl3): δ = 1.33 (d, 3H, J = 7.2 Hz), 3.92(qd, 1H, J = 7.2 Hz and J = 6.6 Hz), 5.63 (d, 1H, J = 6.6 Hz), 7.70 (m, 2H), 7.82 (m, 2H), 8.12 (s, 1H), 8.51 (s,1H). 13C NMR (75 MHz, CDCl3): δ = 38.49, 56.94, 123.91, 126.51, 128.72, 129.32, 130.29, 130.33, 131.22, 134.01, 134.72, 138.13, 192.90. IR (KBr): ν = 1669 (C=O), 1126, 1339 (SO2N), 1569 (NH), 1048 (SO2). C13H11NO3S (261.28): calcd C 59.75, H 4.23; found C 59.62, H 4.31.
4.25 3-Methylsulfanylmethyl-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7c
Yield 52%. mp 175–177 °C. 1H NMR (300 MHz, CDCl3): δ = 3.32 (m, 2H), 3.91 (s, 3H), 4.45 (m, 1H), 6.60 (d, 1H, J = 10.6 Hz), 7.62 (m, 2H), 7.83 (m, 2H), 8.21 (s, 1H), 8.50 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 18.78, 41.57, 53.98, 123.89, 126.46, 128.61, 129.34, 130.28, 130.38, 131.25, 133.56, 134.18, 137.30, 198.18. IR (KBr): ν = 1695 (C=O), 1125, 1323 (SO2N), 1570 (NH), 1032 (SO2). C14H13NO3S2 (307.38): calcd C 54.70, H 4.25; found C 54.81, H 4.29.
4.26 3-Benzyl-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7d
Yield 48%. mp 132–134 °C. 1H NMR (300 MHz, CDCl3): δ = 3.41 (m, 2H), 4.46 (m, 1H), 6.66 (d, 1H, J = 10.2 Hz), 7.26–7.61 (m, 7H), 7.87–7.94 (m, 2H), 8.06 (s, 1H), 8.10 (s, 1H). – 13C NMR (75 MHz, CDCl3): δ = 41.56, 54.08, 125.62, 125.71, 126.81, 127.12, 127.71, 128.16, 128.59, 128.74, 129.00, 131.62, 132.04, 135.90, 138.18, 139.03, 198.08. IR (KBr): ν = 1671 (C=O), 113, 1326 (SO2N), 1549 (NH), 1050 (SO2). C19H15NO3S (337.38): calcd C 67.64, H 4.47; found C 67.48, H 4.34.
4.27 3-(1-(S)-Methylpropyl)-2H-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7e
Yield 50%. mp 112–114 °C. 1H NMR (300 MHz, CDCl3): δ = 0.95 (d, 3H, J = 5.2 Hz), 1.01 (t, 3H, J = 3.5 Hz), 1.09–1.31 (m, 2H), 2.58–2.71 (m, 1H), 4.54–4.72 (dd, 1H, J = 3.2 and J = 10.1 Hz), 5.21–5.33 (d, 1H, J = 10.1 Hz), 7.64–7.76 (m, 2H), 7.95–8.01 (m, 2H) 8.25 (s, 1H), 8.56 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 11.5, 14.62, 25.92, 34.72, 67.45, 122.90, 125.55, 127.76, 128.69, 129.24, 130.35, 131.14, 133.53, 134.18, 137.01, 194.85. IR (KBr): ν = 1687 (C=O), 1139, 1313 (SO2N), 1558 (NH), 1034 (SO2).
4.28 3-Isopropyl-N-methyl-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7f
Yield 70%. mp 152–154 °C. 1H NMR (300 MHz, CDCl3): δ = 0.96 (d, 3H, J = 7.0 Hz), 1.18 (d, 3H, J = 7.0 Hz), 2.40 (m, 1H), 2.82 (s, 3H), 4.47 (d, 1H, J = 3.5 Hz), 7.67 (m,2H), 7.86 (m, 2H), 8.17 (s, 1H), 8.46 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 16.49, 19.03, 27.96, 38.65, 68.07, 123.88, 126.49, 128.66, 129.41, 130.22, 130.40, 131.09, 133.58, 134.18, 137.11, 191.72. IR (KBr): ν = 1672 (C=O), 1138, 1342 (SO2N), 2810 (NCH3), 1040 (SO2). C16H17NO3S (303.36): calcd. C 63.34, H 5.64; found C 63.22, H 5.61.
4.29 3-Benzyl-N-methyl-1,2-naphthothiazin-4(3H)-one 1,1-dioxide: 7g
Yield 62%. mp 138–140 °C. 1H NMR (300 MHz, CDCl3): δ = 2.29 (s, 3H), 3.38 (d, 2H, J = 3.8 Hz), 4.33 (t, 1H, J = 3.8 Hz), 7.25–7.77 (m, 7H), 7.87–7.93 (m, 2H), 8.08 (s, 1H), 8.10 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 37.11, 42.06, 54.21, 128.03, 128.34, 128.49, 129.12, 129.21, 129.65, 130.86, 131.76, 132.21, 133.03; 133.75, 134.00, 135.35, 136.51, 197.71. IR (KBr): ν = 1675 (C=O), 1132, 1340 (SO2N), 2817 (NCH3), 1047 (SO2). C20H17NO3S (351.40): calcd C 68.35, H 4.87; found C 68.48, H 5.01.
4.30 2,3,4,5-tetrahydropyrido〚1,2-b〛〚1,2〛naphthothiazin-7-one 12,12-dioxide: 7h
Yield 46%. mp 98–101 °C. 1H NMR (300 MHz, CDCl3): δ = 1.1–1.4 (m, 1H), 1.5–1.8 (m, 4H), 2.4–2.7 (m, 2H), 3.66 (dt, 1H, J = 3.6 Hz and J = 11.4 Hz), 5.23 (t, 1H, J = 3.6 Hz), 7.61–7.66 (m, 2H), 7.82–7.86 (m, 2H), 8.12 (s, 1H), 8.47 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 20.10, 24.22, 24.31, 46.16, 63.92, 123.81, 127.01, 128.82, 129.55, 130.29, 130.46, 131.19, 133.78, 134.30, 137.42, 192.94. IR (KBr): ν = 1692 (C=O), 1127, 1343 (SO2N), 1042 (SO2). C16H15NO3S (301.35): calcd C 63.77, H 5.01; found C 63.54, H 5.11.
4.31 2,3,4- trihydropyrro〚1,2-b〛〚1,2〛naphthothiazin-6-one 11,11-dioxide: 7i
Yield 21%. mp 86–88 °C. 1H NMR (300 MHz, CDCl3): δ = 1.80–1.92 (m, 2H), 2.05–2.22 (m, 2H), 3.45–3.62 (m, 2H), 4.70 (dt, 1H, J = 3.9 Hz and J = 10.9 Hz), 7.63–7.68 (m, 2H), 7.83–7.87 (m, 2H), 8.16 (s, 1H), 8.49 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 24.76, 31.14, 48.17, 57.43, 123.90, 126.93, 128.75, 129.48, 130.16, 130.37, 131.15, 133.61, 134.23, 137.51, 193.16. IR (KBr): ν = 1689 (C=O), 1130, 1335 (SO2N), 1040 (SO2). C15H13NO3S (287.32): calcd C 62.70, H 4.55; found C 62.75, H 4.42.
4.32 N-methyl-2-naphthylsulfonamide: 8
To a solution of 2-naphthylsulfonyl chloride (10 mmol) in CH2Cl2 (50 ml) stirred at 0 °C was added dropwise a solution of methylamine 30% in water (22 mmol). The reaction mixture was allowed to warm to room temperature over 1 h. Water (30 ml) was then added, and the aqueous phase was extracted with CH2Cl2 (3 × 30 ml). The extract was dried (Na2SO4), subjected to filtration and concentrated. The residue was purified by flash column chromatography 〚SiO2; (EtOAc:cyclohexane)〛.
Yield 88%. mp 101–102 °C. 1H NMR (300 MHz, CDCl3): δ = 2.76 (d, 3H, J = 5.3 Hz), 5.45 (q, 1H, J = 5.3 Hz), 7.56–7.67 (m, 2H), 7.78–7.95 (m, 4H), 8.32 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 29.91, 122.69, 127.33, 127.73, 128.25, 128.60, 128.88, 129.12, 131.86, 134.23, 136.61.
4.33 N,N-Diethyl-2-naphthylsulfonamide: 9
To a solution of diethylamine (12 mmol) and triethylamine (12 mmol) in CH2Cl2 (100 ml) stirred at 0 °C was added dropwise a solution of 2-naphthylsulfonyl chloride (10 mmol) in CH2Cl2 (30 ml). The reaction mixture was allowed to warm to room temperature over 1 h. Water (30 ml) was then added, and the aqueous phase was extracted with CH2Cl2 (3 × 30 ml). The extract was dried (Na2SO4), subjected to filtration and concentrated. The residue obtained was purified by flash column chromatography 〚SiO2; (EtOAc:cyclohexane)〛.
Yield 92%. mp 89–91 °C. 1H NMR (300 MHz, CDCl3): δ = 1.15 (t, 6H, J = 6.9 Hz), 3.24 (q, 4H, J = 6.9 Hz), 7.65–7.78 (m, 2H), 7.84–7.98 (m, 4H), 8.25 (s, 1H). 13C NMR (75 MHz, CDCl3): δ = 16.38, 37.42, 122.58, 127.41, 127.86, 128.11, 128.74, 129.05, 129.32, 131.56, 134.85, 135.96.
4.34 Deuteration experiments
4.34.1 Using n-BuLi. General procedure
A solution of 8 or 9 in THF (0.5 M) was cooled to –5 °C under argon atmosphere and treated with n-BuLi in hexane (2.2 and 1.1 equiv, respectively). After stirring at –5 °C for 20 min, the reaction mixture was warmed quickly to room temperature and stirred for an additional 1 h before quenching with CD3OD (10 equiv). The resulting solution was stirred for 1 h and quenched with saturated NH4Cl solution. The aqueous portion was extracted with CH2Cl2, and the extract was dried (Na2SO4), subjected to filtration, and concentrated. The residue was purified by flash column chromatography 〚SiO2; (EtOAc:cyclohexane)〛.
10. Use of the general procedure with the following materials 〚8 (0.221 g, 1 mmol), n-BuLi (0.88 ml, 2.5 M, 2.2 mmol), CD3OD (0.40 ml, 10 mmol) in THF (10 ml)〛, followed by flash column chromatography (EtOAc/cyclohexane 2:8) afforded a light yellow solid. Yield 79%. 1H NMR (300 MHz, CDCl3): δ = 2.78 (d, 3H, J = 5.3 Hz), 5.21 (bs, 1H), 7.54–7.61 (m, 2H), 7.87–7.94 (m, 2H), 8.06 (s, 1H), 8.15 (s, 1H).
11. Use of the general procedure with the following materials 〚9 (0.263 g, 1 mmol), n-BuLi (0.44 ml, 2.5 M, 1.1 mmol), CD3OD (0.40 ml, 10 mmol) in THF (10 ml)〛, followed by flash column chromatography (EtOAc/cyclohexane 2:8) afforded a light yellow solid. Yield 87%. 1H NMR (300 MHz, CDCl3): δ = 1.14 (t, 6H, J = 6.9 Hz), 3.22 (q, 4H, J = 6.94 Hz), 7.62–7.77 (m, 2H), 7.80–7.96 (m, 2H), 8.10 (s, 1H), 8.18 (s, 1H).
4.34.2 Using LDA. General procedure
A solution of 8 or 9 in THF (0.2 M) was cooled to –5 °C under argon atmosphere and treated with LDA (2.2 and 1.1 equiv, respectively, prepared by dropwise addition of n-BuLi to a solution of HN(i-Pr)2 in THF (1 M) precooled to 0 °C). After stirring for 20 min, a 10 ml aliquot was quenched by addition to CD3OD precooled to –5 °C. The reaction mixture was warmed to room temperature and stirred for an additional 1 h before quenching with CD3OD. Each aliquot was extracted with CH2Cl2, and the extract was dried (Na2SO4), subjected to filtration, and concentrated. The residue was purified by flash column chromatography 〚SiO2; (EtOAc:cyclohexane)〛.
Acknowledgements
We gratefully thank the DGRST, CMCU organisms and the ‘Ministère de l’Enseignement supérieur’ (Tunisia) for their generous support of our program.